

Abstract
Increasing evidence supports a role of neuroinflammation in the pathogenesis of Alzheimer's disease (AD). Previously, we identified a neuron–glia signaling pathway whereby Aβ acts as an upstream activator of astroglial nuclear factor kappa B (NF-κB), leading to the release of complement C3, which acts on the neuronal C3a receptor (C3aR) to influence dendritic morphology and cognitive function. Here we report that astrocytic complement activation also regulates Aβ dynamics in vitro and amyloid pathology in AD mouse models through microglial C3aR. We show that in primary microglial cultures, acute C3 or C3a activation promotes, whereas chronic C3/C3a treatment attenuates, microglial phagocytosis and that the effect of chronic C3 exposure can be blocked by cotreatment with a C3aR antagonist and by genetic deletion of C3aR. We further demonstrate that Aβ pathology and neuroinflammation in amyloid precursor protein (APP) transgenic mice are worsened by astroglial NF-κB hyperactivation and resulting C3 elevation, whereas treatment with the C3aR antagonist (C3aRA) ameliorates plaque load and microgliosis. Our studies define a complement-dependent intercellular cross talk in which neuronal overproduction of Aβ activates astroglial NF-κB to elicit extracellular release of C3. This promotes a pathogenic cycle by which C3 in turn interacts with neuronal and microglial C3aR to alter cognitive function and impair Aβ phagocytosis. This feedforward loop can be effectively blocked by C3aR inhibition, supporting the therapeutic potential of C3aR antagonists under chronic neuroinflammation conditions.
SIGNIFICANCE STATEMENT The complement pathway is activated in Alzheimer's disease. Here we show that the central complement factor C3 secreted from astrocytes interacts with microglial C3a receptor (C3aR) to mediate β-amyloid pathology and neuroinflammation in AD mouse models. Our study provides support for targeting C3aR as a potential therapy for Alzheimer's disease.
Keywords: Alzheimer's disease, amyloid, C3, C3a receptor, mice, microglia
Introduction
Although less well studied than the archetypical amyloid plaques and neurofibrillary tangles, Alzheimer's disease (AD) is also characterized by prominent neuroinflammation marked by reactive astrogliosis, microgliosis, and elevated levels of proinflammatory cytokines (Hensley, 2010; Heneka et al., 2015). The role of neuroinflammation in disease progression has been controversial. Both positive and negative effects have been reported, which likely depend on the cellular or molecular alterations, either unique to the specific immune cells in the CNS or through cross talk among these cell types (Hensley, 2010; Philips and Robberecht, 2011; Orsini et al., 2014). Given the complexity of this response, it is perhaps not surprising that attempts to use anti-inflammatory drugs in the treatment of AD have largely been unsuccessful. Harnessing the inflammatory response for good, or alternatively stopping it from harm, requires a much clearer understanding of the cellular players and molecular signals involved, as well as their upstream activators and downstream consequences.
The complement system is an evolutionarily conserved branch of the innate immune system. The mammalian complement family consists of >30 soluble and cell-associated factors that contribute to one of three signaling cascades: the classical pathway, the alternative pathway, and the lectin pathway, each of which culminate in the cleavage of C3 to release the anaphylactic peptide C3a and the opsonin C3b, followed by a cascade of downstream events (Zipfel and Skerka, 2009). In the CNS, complement factors C3 and C1q serve not only as mediators of innate immunity, but also coordinate microglial engulfment of supernumerary spines to ensure synaptic refinement and neuronal survival during circuit development (Stevens et al., 2007; Shinjyo et al., 2009; Benoit and Tenner, 2011). In disease, the complement system is strongly activated by neuronal damage, consistent with the observation of prominent complement protein expression in response to Aβ exposure and in AD brain tissue (Bradt et al., 1998; Tacnet-Delorme et al., 2001; Fan and Tenner, 2004; Loeffler et al., 2008; Wang et al., 2011). The identification of complement receptor 1 as a genetic risk factor in AD (Lambert et al., 2009; Crehan et al., 2012) raises the possibility that complement activation may be an active participant in AD pathogenesis rather than a passive consequence of amyloid deposition.
Previously, we discovered a key role for C3 protein as the secreted mediator of astroglial NF-κB activation on neuronal morphology and synaptic function (Lian et al., 2015). However, microglia are a known cell type for C3a receptor (C3aR) expression (Zhang et al., 2014), and their function could also be impacted by astrocytic release of C3 following exposure to Aβ. Here we reveal an astrocytic C3/C3a–microglial C3aR axis that governs Aβ dynamics and AD neuropathology.
Materials and Methods
Mouse breeding.
Mice were housed two to five per cage in a specific pathogen-free mouse facility on a 12 h light/dark cycle, with ad libitum access to food and water. All procedures were performed in accordance with NIH guidelines and approval of the Baylor College of Medicine Institutional Animal Care and Use Committee. The Tg(APPswe/PS1dE9) (Tg; Jankowsky et al., 2004), IκBα+/− (Beg et al., 1995), C3aR−/− (Humbles et al., 2000), CR3 KO (Coxon et al., 1996), CamKIIα-TTA (TTA; Mayford et al., 1996), and tetO-APPswe/ind line 102 (APP; Jankowsky et al., 2005) mouse strains are available from The Jackson Laboratory. The GFAP-Cre transgenic mice (Bajenaru et al., 2002) were obtained from National Cancer Institute mouse repository. The IκBαfl/+ mice were obtained from Dr. Rudolf Rupec (University of Munich, Munich, Germany; Rupec et al., 2005). The C3aR−/− mice were backcrossed to C57BL/6J for five generations before use. All other lines were backcrossed to the C57BL/6J background for >10 generations. The double transgenic APP/TTA males on a C57BL/6J background were outcrossed to FVB/NJ females to generate F1 offspring for study. Genotyping was performed by PCR of tail DNA 1 week before weaning.
Mice with astroglial NF-κB/C3 upregulation [GFAP-Cre; IκBαfl/− or glial cell knock-out (GcKO)] have been described previously (Lian et al., 2015). The GcKO/Tg mice were generated by crossing the IκBαfl/+;Tg mice (obtained from Tg and IκBαfl/+ breeding) with GFAP-Cre; IκBα+/− mice (generated from GFAP-Cre and IκBα+/− breeding). These animals and their Tg controls were analyzed at 18 months. The APP/TTA mice and their TTA controls began C3aR antagonist (C3aRA) treatment at 7.25 months of age. Mice were injected intraperitoneally with 0.5% DMSO (vehicle) or C3aRA (1 mg/kg; SB290157, Calbiochem, catalog #559410) three times per week (Monday, Wednesday, and Friday) for 5 weeks. Only males were used for these experiments.
qRT-PCR.
cDNA was synthesized after reverse transcription of total RNA and analyzed as described previously (Yang et al., 2009). Briefly, total RNA was extracted from mouse hippocampi or from primary cells using the RNeasy mini kit (Qiagen, catalog #74106). Reverse transcription was performed using the Superscript III First Strand synthesis system (Invitrogen, catalog #18080-051). The qPCR recipe included 12.5 μl of 2× SYBR Green PCR Master Mix (Roche, catalog #04673484001), 1.5 μl each of sense and antisense primer at 5 μm, 5 μl of cDNA (diluted to 2 ng/μl), and 4.5 μl of H2O. The primer sequences were as follows: 5′- AAG CAT CAA CAC ACC CAA CA-3′ (C3 forward); 5′- CTT GAG CTC CAT TCG TGA CA-3′ (C3 reverse); 5′-AAT GTG TCC GTC GTG GAT CTG A-3′ (GAPDH forward); 5′-GATGCCTGCTTCACCACCTTCT-3′ (GAPDH reverse); 5′-CTCCGCTTTCATGTAGAGGAAG-3′ (PGK1 forward); 5′-GACATCTCCTAGTTTGGACAGTG-3′ [phosphoglycerate kinase 1 (PGK1) reverse]; 5′-CAACGAGCGGTTCCGATG-3′ [β-actin (ACTB) forward]; 5′-GCCACAGGATTCCATACCCA-3′ (ACTB reverse).
Primary microglial culture and phagocytosis and degradation assays.
Cortices and hippocampi were isolated from newborn pups in dissection medium (HBSS with 10 mm HEPES, 0.6% glucose, 1% v/v Pen/Strep) and cut into small pieces. Tissue was digested with 2.5% trypsin at 37°C for 15 min before trypsin inhibitor (1 mg/ml) was added. Tissue was centrifuged for 5 min at 1000 rpm centrifugation, triturated, and resuspended in culture media (DMEM supplemented with 10% fetal bovine serum). Cells were plated onto poly-d-lysine (PDL)-coated T-75 flasks at 50,000 cells/cm2 to generate mixed glial cultures. When confluent, microglia were purified by tapping the flasks against table and collecting the floating cells in media. These cells were then seeded at 50,000 cells/cm2 and cultured for another day in PDL-coated 12-well plates for mRNA assay or on coverslips for phagocytosis assay. For astrocyte culture, confluent mixed glial cultures were shaken at 220 rpm overnight at 37°C to remove unwanted cell types (microglia, oligodendrocytes, neurons, and fibroblasts), released with 0.5% trypsin in EDTA, and plated onto PDL-coated culture vessels.
Synthetic Aβ42 (Invitrogen, catalog #03-111) and reverse Aβ42 (rAβ42; Sigma, catalog #SCP0048) peptides were prepared following the protocol described previously (Stine et al., 2003). Briefly, lyophilized peptide was dissolved in hexafluoroisopropanol (HFIP) and incubated at RT for 2 h. HFIP was then dried down by SpeedVac, allowing the formation of Aβ films. The film was then resuspended in DMSO, sonicated in a water bath for 10 min, diluted in PBS, and incubated at 4°C for 24 h. Primary astroglial or microglial cultures were treated with 100 nm rAβ42 or Aβ42 for 20 h before they were harvested for qPCR analysis.
For phagocytosis assays using beads, aqueous green fluorescent latex beads of 1 μm diameter (Sigma, catalog #L1030) were preopsonized in FBS for 1 h at 37°C before dilution in DMEM. The final concentration for beads and FBS in DMEM were 0.01% (v/v) and 0.05% (v/v), respectively. For phagocytosis assay using fluorescent Aβ42, 6-carboxyfluorescein (FAM)-labeled Aβ42 peptides purchased from Anaspec (catalog #AS-23526-01) were dissolved in DMSO, diluted in DMEM to a final concentration of 500 nm, and incubated at 37°C for 1 h to promote aggregation. After treating microglial cells with 10 μg/ml C3 (Millipore, catalog #204885) for desired times (1, 24, or 48 h), phagocytosis assay was performed by replacing the C3 media with bead- or Aβ-containing DMEM, and cultures were incubated at 37°C for 1 h. For the treatment of C3aRA, DMSO or 10 μm C3aRA was added to primary microglia 1 h before vehicle or C3 treatment. Cultures were then washed thoroughly with ice-cold PBS for five times and fixed in 4% PFA. Cultures used for phagocytosis assay were prepared in duplicate and both were included in later staining, imaging, and quantification.
To measure the rate of Aβ42 degradation, primary microglia cells were plated in 24-well plates at 50,000 cells/cm2. One day after plating, 10 μg/ml of C3 or PBS in fresh media was added into the culture for 1 or 48 h. Aβ42 was added to the culture at 500 nm and incubated at 37°C for 1 h. Aβ42 was removed, and cells were washed with ice-cold PBS three times. Next, fresh media was added to cells and incubated at 37°C for 0, 1, 2 and 4 h. At each time point, cells were harvested with 0.25% Trypsin, centrifuged at 1500 rpm for 5 min, and lysed with RIPA buffer (25 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1% NP-40, 1% sodium deoxycholate, and 0.1% sodium dodecyl sulfate). Aβ42 concentration at each time point was measured using an Aβ42 ELISA kit (Invitrogen, KHB331).
Western blotting and Aβ ELISA.
Hippocampal and cortical tissues were homogenized in RIPA buffer and centrifuged at 14,000 rpm for 20 min. Supernatant was collected and quantified using a detergent compatible (DC) colorimetric protein assay (Bio-Rad). Lysates were boiled at 95°C for 7 min in sample buffer. Fifteen microgram protein samples were loaded onto 12% SDS-PAGE gels, transferred to nitrocellulose membranes (Bio-Rad) after running, and then blocked with 5% milk in PBS containing 0.1% Tween 20 (PBST). Membranes were probed with primary antibody (rabbit anti-APP Y188, Abcam), washed, and blotted with secondary antibodies (goat anti-rabbit, Millipore). The membranes were washed again with PBST, and the signal was developed with ECL solution (GE Healthcare Life Sciences).
For Aβ measurement, brain tissues as well as cell culture lysates were processed in RIPA buffer. Fifty microliters of concentration-determined samples were used for the detection of Aβ40 and Aβ42 with human Aβ40 and Aβ42 ELISA kits (Invitrogen, KHB3481 and KHB331) according to the manufacturer‘s instructions.
Histology and immunostaining.
Mice were perfused with 4% PFA, and the brains were post-fixed in 4% PFA overnight at 4°C followed by cryoprotection in 40% sucrose. Thirty micrometer coronal sections were cut with a microtome and stored at in cryoprotectant at −20°C. For each experiment, sections were collected randomly from at least three animals. Sections or fixed cultured microglia on coverslips were extensively washed in PBS, blocked with PBST containing 3% BSA and 2% donkey serum for 30 min, and then incubated in primary antibody diluted in blocking solution overnight at 4°C [mouse anti-GFAP (Millipore, catalog #MAB3402); rabbit anti-Iba1 (Wako, catalog #019-19741); mouse anti-Aβ42 (BioLegend, catalog #800702); rabbit anti-abeta42 (Cell Signaling, catalog #14974); rat anti-mouse C3aR (Hycult Biotech, catalog #HM1123); chicken anti-C3 (a kind gift from Dr. Scott Barnum, University of Alabama at Birmingham, Birmingham, AL); mouse anti-Lys-Asp-Glu-Leu (KDEL) (Stressgen, SPA-827); mouse anti-GM130 (BD Biosciences, 610822); rat anti-LAMP1 (BD Biosciences, 553792)]. After washing, sections or coverslips were incubated in secondary antibody for 1 h at room temperature and mounted in DAPI solution after final washing. Thioflavin S staining was performed before mounting by incubating sections in 0.002% Thioflavin S (Sigma, catalog #T1892) for 8 min followed by a 2 min rinse in 50% EtOH and a 5 min wash in PBS. DAB staining was performed using the Vectastain Elite ABC Kit (Vector Laboratories, catalog #PK-6200) following the manufacturer's instructions.
RNA in situ hybridization.
DNA primer probes used for subsequent synthesis of sense or antisense RNA were PCR amplified from mouse cDNA with primers for C3 and C3aR genes (Allen Brain Atlas) and carried T7 or SP6 RNA polymerase promoter sequences at the 5′ end: 5′-GCGTAATACGACTCACTATAGGGCAGGGGAGTATATTGAAGCCAG-3′ (C3 forward); 5′-GCGATTTAGGTGACACTATAGTCTATCTACTCCAGAGGCCAGC-3′ (C3 reverse); 5′-GCGTAATACGACTCACTATAGGGTGGCTTGTTCCTGTGCAA-3′ (C3aR forward); 5′-GCGATTTAGGTGACACTATAGACATCACAGCTTCCCCCA-3′ (C3aR reverse).
The in situ hybridization was performed as described by the core facility at Baylor College of Medicine (Yaylaoglu et al., 2005). After RNA labeling, sections were processed for immunofluorescence staining with anti-GFAP or anti-Iba1 antibodies as described above.
Image processing and quantification.
Fluorescent immunostained brain sections and microglial cultures were imaged using a Leica laser confocal microscope. Thioflavin S- and DAB-stained sections were scanned by SilverFast (LaserSoft Imaging) and then imaged by the EVOS FL Auto system. Images were processed by ImageJ, and background was subtracted by the software for fluorescence images before quantification.
For quantification of C3 fluorescence intensity in GFAP+ cells or of C3aR in Iba1+ cells, GFAP+ or Iba1+ cells were first chosen as a region of interest (ROI) by the ROI manager of ImageJ after threshold adjustment. The mean gray value of these ROIs in the C3 or C3aR channel was measured to indicate the fluorescence intensity of C3 or C3aR in the GFAP+ or Iba1+ cells.
For plaque load quantification, scanned images of Thioflavin S- and DAB-stained brain sections were adjusted for color threshold to allow the software to identify plaque regions. The hippocampal and cortical regions were identified according to the mouse brain atlas. The percentage of plaque-covered area in the total area of the cortex or hippocampus was measured and shown as plaque load. For quantification of gliosis, GFAP- or Iba1-stained area showing obvious shape of a cell body was considered as a GFAP+ or Iba1+ cell, and the number of GFAP+ or Iba1+ cells in the hippocampus or cortex was counted and compared between different animals.
For phagocytosis assays using beads, the total cell number in each image was counted by ImageJ after threshold adjustment and ROI manager selection of the DAPI channel. Based on the composite image of Iba1 and beads, cells having beads inside the cell body were manually counted using the Cell Counter ImageJ plugin. The number of bead-containing cells was divided by total cell number for each image to indicate the percentage of phagocytic cells. For phagocytosis assays using FAM-labeled Aβ42, the cell body was first identified as the ROI using the Iba1 channel. Once the ROIs were defined, the green channel showing FAM fluorescence was used to measure both the fluorescence intensity and percentage area of FAM inside each ROI.
Statistics.
All data are presented as mean ± SEM. Power analysis was performed using confident interval of α = 0.05. Outliers were identified using the Grubbs' test with α = 0.05. Pairwise comparisons were analyzed using a two-tailed Student's t test, whereas two-way ANOVA followed by Bonferroni post hoc analysis was used for multiple comparisons. Linear regression followed by F test was used for Aβ42 degradation assay. P values less than or equal to 0.05, 0.01, and 0.001 were considered significant.
Results
Astrocytes and microglia are the primary sources for C3 and C3aR, respectively
Our previous analysis indicated elevated C3 in APP/TTA transgenic mice and in Aβ-treated primary astroglial cultures (Lian et al., 2015). To directly investigate whether astrocytes are the primary cellular source of C3 expression in the brain, we performed C3 RNA in situ hybridization combined with immunostaining with an anti-GFAP antibody in 12-month-old wild-type (WT) mice. We found that the C3 mRNA detected by the antisense probe strongly colocalized with the astrocytic marker GFAP, whereas there was almost no signal using the sense probe (Fig. 1A). This result was corroborated by double immunostaining using anti-C3 and anti-GFAP antibodies, which also revealed high degree of colocalization of C3 protein in GFAP-positive cells (Fig. 1B). The specificity of the anti-C3 antibody was documented by the minimal staining in C3 knock-out (KO) brain sections. Similar RNA in situ hybridization for C3aR combined with immunostaining using the anti-Iba1 antibody showed strong expression of C3aR in Iba1-positive microglia (Fig. 1C), which was confirmed by double immunostaining using anti-C3aR and Iba1 antibodies (Fig. 1D). The data combined provide strong support that astrocytes and microglia are the primary cell types for C3 and C3aR expression in the brain, respectively.
Figure 1.

C3 and C3aR are specifically expressed in astrocytes and microglia, respectively. A, C3 RNA in situ hybridization combined with anti-GFAP antibody staining in 12-month-old wild-type mice. Sense, Sense probe for C3; antisense, antisense probe for C3. B, Representative anti-C3 and anti-GFAP double immunostaining in 3-month-old WT and C3 KO mice. C, C3aR RNA in situ hybridization combined with anti-Iba1 antibody staining in 12-month-old wild-type mice. Sense, Sense probe for C3aR; antisense, antisense probe for C3aR. D, Representative anti-C3aR and anti-Iba1 coimmunostaining in 3-month-old WT and C3aR KO mice. Arrowheads mark representative colocalized cells. Scale bars: 50 μm.
Next we moved to AD mouse models and performed double immunostaining for C3 and GFAP in brain sections from 8-month-old APP/TTA mice. Compared to littermate TTA controls, C3 intensity was substantially elevated in GFAP-positive astrocytes in APP/TTA mice (Fig. 2A, quantified in B). Similar results were obtained in the second AD model, APP/PS1, hereafter referred to as Tg, tested at 18 months of age (Fig. 2C,D). In contrast, costaining of C3 with the microglial marker Iba1 showed poor colocalization (Fig. 2E). Consistent with these in vivo findings, qPCR analysis of C3 mRNA levels in primary cultures revealed that basal C3 was significantly higher in astrocytes than in microglia, and that treatment with Aβ42 resulted in further C3 upregulation specifically in the astrocytes (Fig. 2F). Thus, astrocytes, but not microglia, are the major source of C3 under both basal and Aβ-induced conditions.
Figure 2.
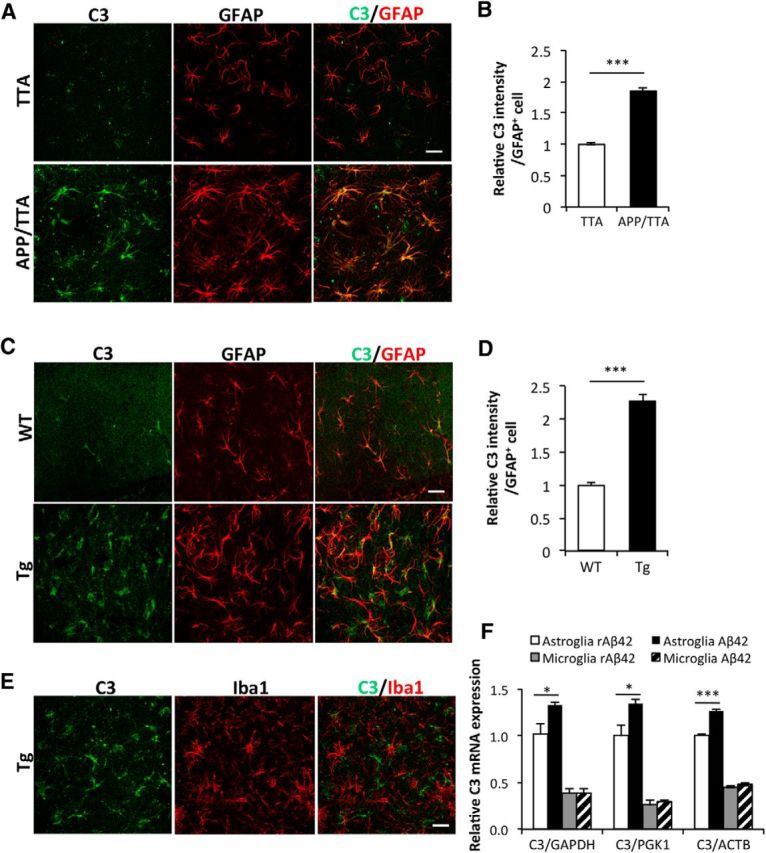
Astrocytic C3 upregulation in APP transgenic mice. A, C, Representative double immunostaining for C3 and astrocytic marker GFAP in the hippocampus of APP/TTA transgenic mice at 8 months of age (A) and APP/PS1 transgenic (Tg) animals at 18 months (C). Littermate TTA and WT mice were used as controls for bigenic APP/TTA and Tg mice, respectively. B, D, Quantification of C3 fluorescence intensity in GFAP+ cells in APP/TTA (B) or Tg (D) hippocampal regions. N = 126 (TTA), 145 (APP/TTA), 87 (WT), 88 (Tg) cells collected randomly from sections of three animals per genotype. E, Representative double immunostaining for C3 and microglial marker Iba1 in the hippocampus of Tg animals. F, qPCR measurement of C3 mRNA levels in WT primary astroglial and microglial cultures treated with 100 nm Aβ42 or reverse peptide (rAβ42). Three internal controls (GAPDH, PGK1, and ACTB) were used. N = 3 cultures per condition. Scale bars: 50 μm. *p ≤ 0.05; ***p ≤ 0.001 (B, D, Student's t test; F, two-way ANOVA followed by Bonferroni's post hoc analysis).
Astroglial NF-κB and complement activation worsens amyloid pathology
Our previous studies identified complement C3 as one of the downstream effectors of NF-κB signaling in astroglia (Lian et al., 2015). To assess whether astroglial NF-κB and complement activation plays a functional role in AD pathogenesis, we crossed the APP/PS1 Tg mice with a model that selectively overproduces NF-κB and C3 in astrocytes by eliminating IκBα from astrocytes using a GFAP-Cre mated with a floxed IκBα line, which we refer to as our GcKO. Loss of IκBα results in constitutive NF-κB activation and astrocytic upregulation of C3 release (Lian et al., 2012, 2015). As expected, C3 mRNA expression was elevated in the brains of GcKO/Tg mice compared to Tg controls (Fig. 3A). Remarkably, astrocytic NF-κB and C3 activation resulted in significantly greater plaque deposition in both the cortex and hippocampus of GcKO/Tg mice compared with Tg controls detected by Aβ antibody (Fig. 3B,C) and Thioflavin S (Fig. 3D,E) staining, and this was accompanied by significant elevation of both reactive astrogliosis and microgliosis (Fig. 3F,G). Together, the data suggest that increased NF-κB and C3 complement activation exacerbates amyloid pathology and promotes glial inflammation in the brain.
Figure 3.
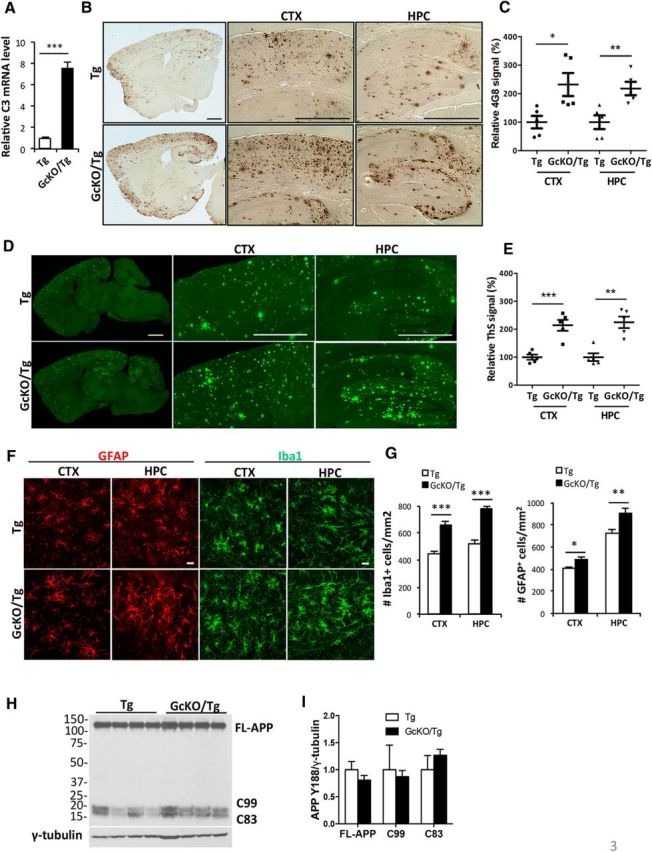
Astrocyte-specific NF-κB/C3 activation increases amyloid burden and exacerbates reactive gliosis in APP/PS1 transgenic (Tg) mice. A, qPCR measurement of C3 mRNA levels in Tg animals with astrocyte-specific IκBα deletion and NF-κB/C3 activation (GcKO/Tg) and Tg mice alone. N = 3 animals per genotype. B, Representative DAB immunostaining of amyloid plaques in Tg and GcKO/Tg animals using the rabbit anti-Aβ antibody. CTX, Cortex; HPC, hippocampus. C, Quantification of plaque load in the CTX and HPC of Tg and GcKO/Tg animals. N = 5 animals per genotype, 4–5 sections per mouse. D, Representative Thioflavin S (ThS) staining of amyloid plaques in the CTX and HPC of Tg and GcKO/Tg animals. E, Quantification of ThS-positive signals in the CTX and HPC of Tg and GcKO/Tg animals. N = 5 animals per genotype, 4–5 sections per mouse. F, Representative GFAP and Iba1 immunostaining in CTX and HPC of Tg and GcKO/Tg animals. G, Quantification of GFAP+ and Iba1+ cells in the CTX and HPC. N = 16 sections per genotype (GFAP), N = 13 sections per genotype (Iba1). H, I, Western blot analysis of FL-APP and its C-terminal fragments C83 and C99 using the APP Y188 antibody in Tg and GcKO/Tg animals (H) and its quantification (I, nonsignificant). Scale bars: B, D, 1 mm; F, 20 μm. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001 (two-tailed Student's t test). Filled circle, Tg CTX; filled square: GcKO/Tg CTX; filled triangle, Tg HPC; filled reverse triangle, GcKO/Tg HPC.
Heightened NF-κB-mediated C3 (C3a)–C3aR signaling compromises microglial phagocytosis
The worsening of amyloid pathology in GcKO/Tg mice suggests that either Aβ production was increased or its clearance was retarded. Using Aβ ELISA to measure peptide levels in 2-month-old predeposit GcKO/Tg mice and Tg controls, we found that NF-κB and accompanying complement activation did not affect steady-state levels of either Aβ40 or Aβ42 (data not shown). Furthermore, we examined the levels of full-length (FL)-APP and their C-terminal fragments (C99 and C83) by Western blotting and did not detect significant differences in any of the products, suggesting that APP expression and processing was not altered in GcKO/Tg mice compared to Tg controls (Fig. 3H,I). These results therefore pointed toward impaired Aβ clearance resulting from heightened astrocytic NF-κB and C3 signaling.
Microglia are known Aβ scavengers (Prokop et al., 2013; Fu et al., 2014), and the complement pathway has been shown to be required for their proper phagocytosis (Fu et al., 2012; Linnartz and Neumann, 2013). Since C3aR is abundantly expressed in microglia (Fig. 1C,D; Zhang et al., 2014), particularly under neuroinflammatory conditions (Davoust et al., 1999), we thought to test whether microglial phagocytic function might be compromised by aberrant complement signaling using primary microglial cultures. Consistent with our in vivo results, double immunostaining using anti-C3aR and Iba1 antibodies showed a high degree of colocalization between C3aR- and Iba1-positive cells in microglial cultures taken from wild-type mice. In contrast, the C3aR immunoreactivity was undetectable in cells derived from C3aR knock-out animals (Fig. 4A). We next treated the primary microglial cultures with either PBS or purified recombinant C3 protein and assessed the effect of C3 on fluorescent latex bead uptake (Fig. 4B). We observed an interesting bimodal response: short-term C3 treatment (1 h) promoted phagocytosis, whereas longer treatment (24 h) diminished bead uptake (Fig. 4C). Since multiple C3 cleavage products, including C3a, C3b, and iC3b, as well as additional downstream processing products have been shown to mediate Aβ phagocytosis (Wyss-Coray et al., 2002; Maier et al., 2008; Fu et al., 2012), we directly tested the effect of C3a in our system and observed similar results as C3 (Fig. 4D), thus supporting a functional role of C3a in C3-mediated response. Reduced bead uptake upon 24 h of C3 or C3a treatment was prevented by the addition of a C3aR antagonist, SB290157, that functions to compete with C3a for ligand binding (Ames et al., 2001; Fig. 4E,F), further strengthening the notion that the C3-induced decline in phagocytosis is mediated by C3a and microglial C3aR. As an additional support, we prepared primary microglial cultures from C3aR knock-out (C3aR KO) mice and littermate WT controls and repeated the phagocytosis experiments plus and minus C3. As expected, C3 treatment reduced phagocytosis in WT microglia. In contrast, microglia lacking C3aR displayed no response to C3 treatment at 24 h (Fig. 4G). Together, these findings suggest that prolonged C3/C3a–C3aR activation curtails microglial phagocytosis.
Figure 4.
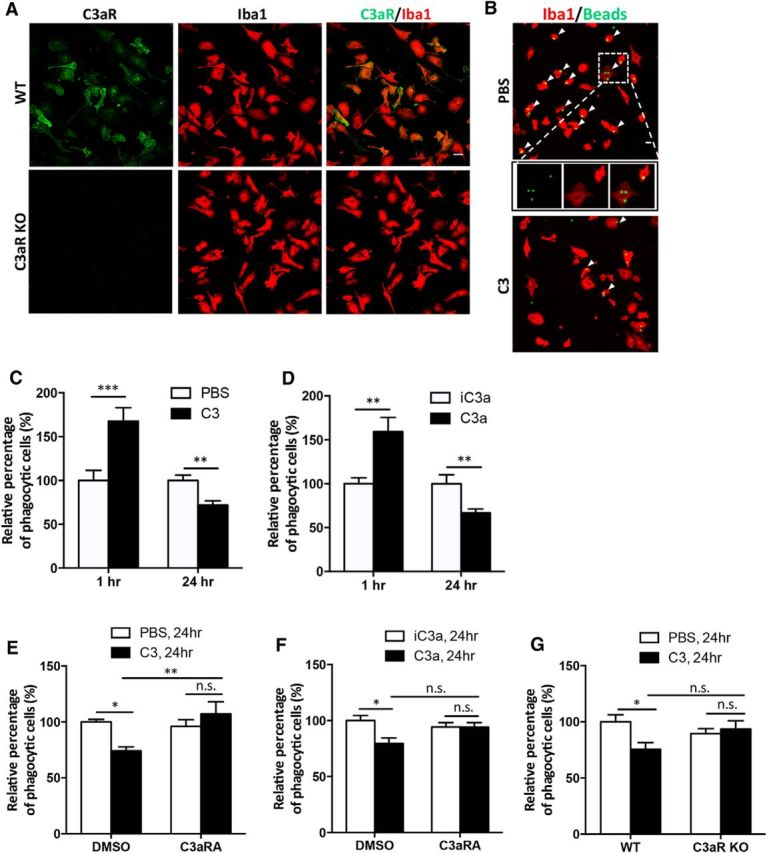
C3 and C3aR signaling modulates microglial phagocytosis in vitro. A, Double immunostaining for C3aR and Iba1 in WT and C3aR KO primary microglia. B, Representative images of fluorescent bead uptake in primary WT microglial cultures treated with PBS or 10 μg/ml C3 for 24 h. Microglia are labeled with an anti-Iba1 antibody in red, and beads are labeled with green fluorescence. Examples of phagocytic Iba1+ cells with internalized beads are marked by arrowheads, with one PBS-treated cell (inset) shown in higher-magnification views. C, Relative percentage of phagocytic microglia following 1 or 24 h of vehicle (PBS) or C3 (10 μg/ml) treatment. D, Relative percentage of phagocytic microglia following 1 or 24 h of control (iC3a) or C3a (100 nm) treatment. E, Relative percentage of phagocytic microglia after 24 h PBS or C3 treatment in the presence of 10 μm C3aRA or DMSO vehicle. F, Relative percentage of phagocytic microglia after 24 h iC3a or C3a treatment in the presence of 10 μm C3aRA or DMSO vehicle. G, Relative percentage of phagocytic cells in WT or C3aR KO microglia treated with PBS or C3 for 24 h. N = 12 random fields for each data point; each field contained >100 cells. Scale bars: 20 μm. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.01 (C, D, Student's t test; E–G, two-way ANOVA followed by Bonferroni's post hoc analysis; n.s., nonsignificant.
Having documented that C3/C3a–C3aR signaling could impact microglial phagocytosis of an artificial substrate, we next tested whether this pathway could reduce phagocytosis of disease-relevant Aβ aggregates. We applied FAM-labeled fluorescent Aβ42 aggregates to primary wild-type microglial cultures and measured Aβ uptake in the presence or absence of exogenous C3 (Fig. 5A–C). Similar to the latex bead experiments, C3 treatment resulted in higher intracellular Aβ fluorescence (Fig. 5B) and more Aβ-positive foci per Iba1-positive cell (Fig. 5C) at 1 h, followed by reversal of this effect after 48 h of C3 incubation. Reduced Aβ uptake upon 48 h of C3 treatment was blunted in microglia cultures derived from C3aR knock-out mice (Fig. 5D,E, C3aR KO, PBS vs C3, nonsignificant). In contrast, microglia cultured from mice deficient in another complement receptor, CR3 (CD11b/CD18), which interacts with iC3b, still responded to C3 (Fig. 5D,E, CR3 KO, PBS vs C3, p < 0.001), although the response appeared to be dampened compared to C3-treated wild-type controls (Fig. 5E, CR3 KO vs WT, C3, p < 0.05; Todd, 1996). Together, these results provide strong support for a major role of C3aR in mediating the C3/C3a effect on Aβ42 phagocytosis.
Figure 5.
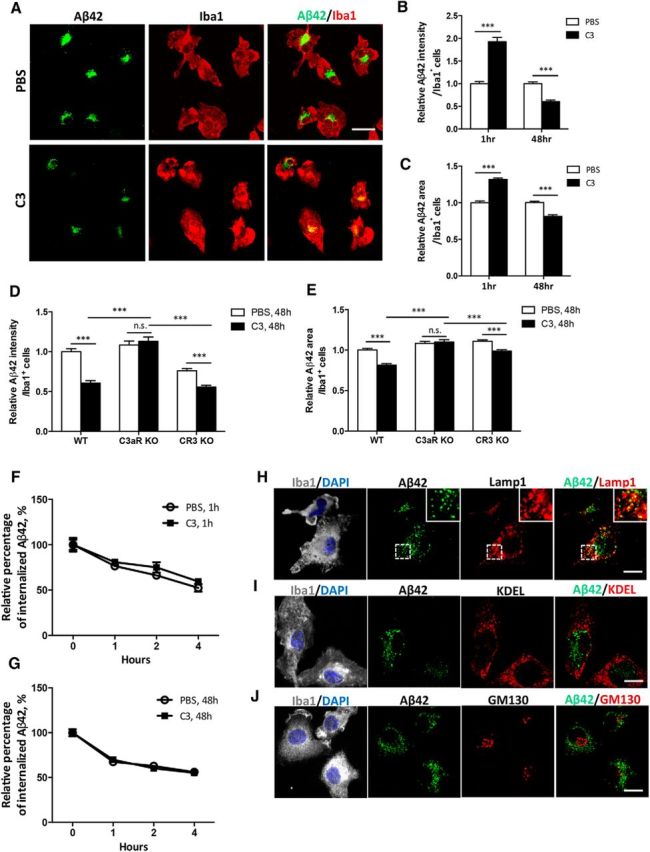
Complement activation mediates microglial phagocytosis of Aβ in vitro. A, Representative images of fluorescent FAM-labeled Aβ42 peptide uptake in primary wild-type microglial cultures treated with vehicle (PBS) or 10 μg/ml C3 for 48 h. Microglia are marked by Iba1 immunoreactivity in red; Aβ42 is labeled with green fluorescence. B, C, Quantification of relative Aβ42 fluorescence intensity per Iba1-positive cell (B) and percentage of Aβ42+ area within each cell (C) in microglial cultures treated with PBS or 10 μg/ml C3 for 1 h or 48 h. N = 165 cells (PBS, 1 h); N = 151 (C3, 1 h); N = 164 (PBS, 48 h); N = 158 (C3, 48 h). D, E, Quantification of relative Aβ42 fluorescence intensity per Iba1+ cell (D) and percentage of Aβ42+ area within each cell (E) in primary WT, C3aR KO, and CR3 KO microglia exposed to FAM-labeled Aβ42 after 48 h of PBS or C3 treatment. N = 164 cells (WT); N = 112 (C3aR KO); N = 154 (CR3 KO). F, G, Aβ42 degradation assay by application of the peptide to 1 h (F) or 48 h (G) PBS- or C3-treated microglial cultures and by measuring relative intracellular Aβ42 levels immediately after (0 h) and at 1, 2, and 4 h later. The experiments were performed in triplicate. H–J, Coimmunostaining of Iba1 and lysosome marker Lamp1 (H), ER marker KDEL (I), and cis-Golgi network marker GM130 (J) in primary WT microglia following 48 h C3 treatment and 1 h incubation with FAM-labeled Aβ. The Iba1/DAPI are merged z-stack images; all others are taken from a single z position to test colocalization. Insets in H are enlarged views of the bracketed areas within each image. Scale bars: A, 20 μm; H–J, 10 μm. ***p ≤ 0.001 (B, C, Student's t test; D, E, two-way ANOVA followed by Bonferroni's post hoc analysis; n.s., nonsignificant). F, G, Linear regression followed by F test (p > 0.5).
Reduced intracellular Aβ42 upon 48 h of C3 incubation could, in principle, be the result of reduced phagocytosis or elevated degradation. Since we obtained similar results using the degradation-resistant beads, we felt that it is unlikely due to altered clearance. However, to rule out this possibility, we performed Aβ degradation assay in wild-type microglia treated with PBS or C3 for 1 or 48 h and found no appreciable differences among the different groups (Fig. 5F,G). These results provide compelling evidence that the reduced intracellular Aβ signal in 48 h C3-treated microglia is due to impaired uptake, but not elevated degradation.
Consistent with the hypothesis that microglia-mediated Aβ clearance involves extracellular uptake followed by lysosomal degradation (Desjardins, 2003), we found strong colocalization of fluorescent Aβ42 with the lysosome marker Lamp1 (Fig. 5H). In contrast, immunostaining with markers for endoplasmic reticulum (KDEL; Fig. 5I) or Golgi complex (GM130; Fig. 5J) showed little Aβ localization in these compartments.
C3aR inhibition reduces amyloid pathology
The above in vitro studies support the idea that chronic C3–C3aR activation may impair Aβ uptake, which could be the underlying mechanism for the accelerated Aβ pathology in GcKO/Tg mice with astroglial NF-κB and complement activation. We thus examined whether C3aR is perturbed in these animals. qPCR analysis showed that, similar to that of C3, C3aR mRNA was indeed elevated in brain tissues of GcKO/Tg mice compared to their littermate Tg mice (Fig. 6A). Immunofluorescence examination revealed that in both Tg and GcKO/Tg mice, the C3aR signal was particularly prominent in Iba1-positive microglia surrounding Thioflavin S-labeled amyloid plaques, although the C3aR immunoreactivity was stronger in GcKO/Tg mice (Fig. 6B). This was verified by quantifying the C3aR intensity in Iba1-positive cells (Fig. 6C). Increased C3aR mRNA and immunoreactivity was also observed in the APP/TTA mice relative to the littermate TTA controls (Fig. 6D–F). These results suggest that Aβ accumulation induces microglial C3aR expression. Given that prolonged C3aR activation diminished microglial Aβ uptake in vitro, we reasoned that microglial C3aR upregulation in the vicinity of amyloid deposits may augment further accumulation by decreasing phagocytic clearance, and that this effect might be reversed by C3aR blockage. To test this idea, we treated 7-month-old APP/TTA mice and littermate TTA controls with the C3aR antagonist or the vehicle control DMSO and quantified the plaque burden 5 weeks later. Compared to DMSO-injected controls, APP/TTA mice treated with the C3aR antagonist showed a significant reduction in amyloid load across the forebrain (Fig. 6G, quantified in H). Quantification of RIPA-extractable Aβ40 and Aβ42 revealed that, although we failed to see changes in absolute Aβ values, we observed a significant decrease in the Aβ42/40 ratio, which is a reliable measure for Aβ pathology, in C3aRA-treated animals compared to the DMSO controls (Fig. 6I). The attenuation of the amyloid burden was accompanied by a decline in microgliosis (Fig. 6J,K).
Figure 6.
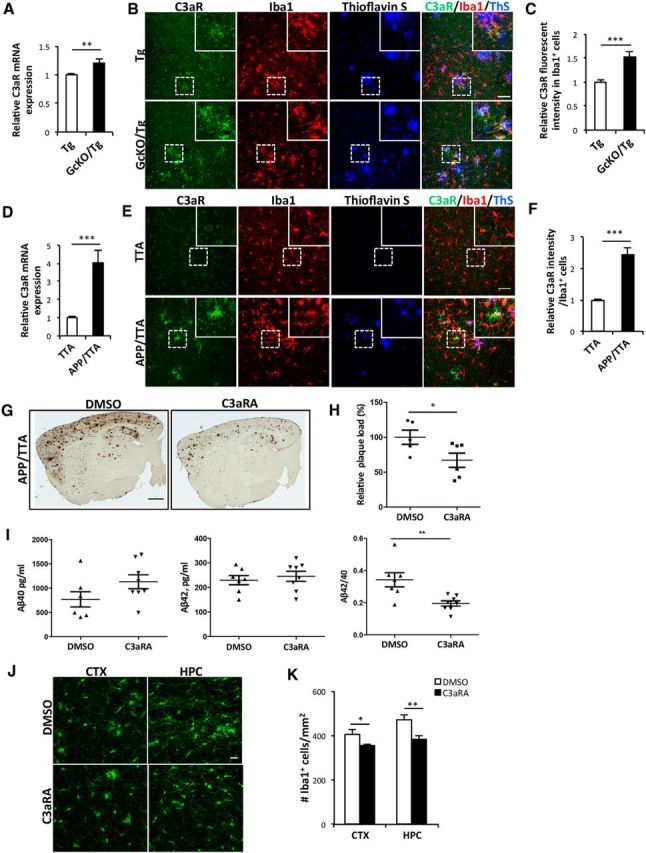
C3aR inhibition ameliorates amyloid pathology. A, qPCR measurement of C3aR mRNA in Tg and GcKO/Tg mouse brains. N = 6/group. B, Representative double immunostaining for C3aR and Iba1 with amyloid plaques labeled by Thioflavin S in cortical sections from 18-month-old Tg and GcKO/Tg mice. C, Quantification of relative C3aR immunofluorescence intensity in Iba1+ cells from Tg and GcKO/Tg mice brain sections. N = 40 cells (Tg); N = 35 (GcKO/Tg). Random sections were selected for analysis from three animals per genotype. D, qPCR measurement of C3aR mRNA in TTA and APP/TTA mouse brains. N = 6 animals (TTA); N = 7 (APP/TTA). E, Representative double immunostaining for C3aR and Iba1 with amyloid plaque labeled by Thioflavin S in cortical sections from 8-month-old TTA and APP/TTA mice. F, Quantification of relative C3aR immunofluorescence intensity in Iba1+ cells from TTA and APP/TTA brain sections. N = 35 cells (TTA); N = 28 (APP/TTA). Random sections were selected for analysis from three animals per genotype. G, Representative immunostaining of amyloid plaques in APP/TTA mice treated with vehicle (DMSO) or 1 mg/kg C3aRA. H, Quantification of plaque load in the forebrain of DMSO- or C3aRA-treated animals. N = 6 animals/genotype/treatment, 2–4 sections per mouse. I, Levels of RIPA-extracted Aβ40 and Aβ42 and the Aβ42/40 ratio in DMSO- or C3aRA-treated APP/TTA mice. N = 6 animals/genotype/treatment. J, Representative Iba1 immunostaining in the CTX and HPC of APP/TTA animals treated with DMSO or C3aRA. K, Number of Iba1+ cells in the CTX and HPC of APP/TTA animals treated with DMSO or C3aRA. N = 11 sections (CTX, APP/TTA, DMSO); N = 12 (CTX, APP/TTA, C3aRA); N = 12 (HPC, APP/TTA, DMSO); N = 16 (CTX, APP/TTA, C3aRA). Scale bars: B, E, 50 μm; G, 1 mm; J, 20 μm. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001 (Student's t test).
Discussion
In the current study, we expand on our past efforts to elucidate the role of NF-κB and complement activation in the pathogenesis of AD. Using a combination of genetic and pharmacologic tools, we identify a novel signaling pathway linking astroglial NF-κB and C3 with microglial C3aR to dynamically regulate microglial phagocytosis. We show that Aβ induces astrocytic C3 expression which, under chronic activation conditions and in a C3aR-dependent manner, attenuates microglial Aβ phagocytosis in vitro and exacerbates Aβ pathology in vivo. Our studies identify Aβ as an upstream stimulator of the NF-κB and complement activation and downstream effector of the complement-mediated phagocytic function, and establish an Aβ–C3/C3a–C3aR signaling cross talk among neuron, astrocyte, and microglia that is intimately involved in AD pathogenesis.
Elevated expression of complement protein and activation of complement signaling have been observed both in postmortem AD tissue and in APP transgenic mouse models (Rogers et al., 1992; Yasojima et al., 1999; Matsuoka et al., 2001; Reichwald et al., 2009). However, both neurons and glia have been reported to express complement proteins, leaving unclear which cell types mediate specific aspects of the complement response (Fischer et al., 1995; Shen et al., 1997; Walker et al., 1998; Lue et al., 2001). Using in situ hybridization and immunofluorescence staining, we show here that under physiological conditions, C3 is primarily expressed in astrocytes where its expression is further upregulated in APP/PS1 (Tg) and APP/TTA mice. This assessment is consistent with previous reports using Tg2576 and 3xTg animals (Zhou et al., 2008; Fonseca et al., 2011). However, in contrast to past reports describing microglial expression of complement proteins (Haga et al., 1996; Schäfer et al., 2000; Griffin et al., 2007; Rutar et al., 2014), we detected little C3 protein in microglia of APP transgenic mice. We further observed no upregulation of C3 mRNA following Aβ stimulation of purified microglia in vitro, despite the clear C3 response detected following TNFα stimulation (data not shown). These discrepancies with past reports may be due to differences in the preparation, duration, or dosage of Aβ exposure or differences in the sensitivity and specificity of the antibodies employed.
The C3aR is expressed by both neurons and glia (Davoust et al., 1999; Zhang et al., 2014), but its specific function in each cell type is not well understood. Previously, we found that neuronal activation of C3aR disrupts intracellular calcium dynamics, leading to aberrant dendritic morphology and neuronal excitability (Lian et al., 2015). Here we show that, in agreement with the published reports, microglia possess high levels of C3aR and their expression is particularly prominent in areas surrounding amyloid plaques in APP transgenic animals. The heightened C3/C3a–C3aR signaling may impair microglial function and dampen microglial Aβ clearance as prolonged complement activation impairs Aβ phagocytosis in vitro. We did not detect appreciable astrocytic C3aR expression in our AD mouse models even though such expression has been reported by others (Ischenko et al., 1998; Jauneau et al., 2006). Although the reason for this remains to be determined, it is interesting to note that transcriptional control of C3aR in microglia and astroglia has been shown to be distinctly regulated, which may result in variable expression patterns in different model systems (Martin et al., 2007). Nevertheless, cell-type specific knock-out mice are needed to definitively address the precise cellular expression of C3 and C3aR and their perspective contribution to disease pathogenesis.
Microglial uptake of both artificial substrates such as fluorescent latex beads and more realistic targets such as synthetic Aβ42 aggregates is accomplished through phagocytosis (Koenigsknecht and Landreth, 2004; Smith et al., 2010; Jones et al., 2013). The C3 cleavage products C3a, C3b, and iC3b and the subsequent processing products have been shown to mediate Aβ phagocytosis (Wyss-Coray et al., 2002; Maier et al., 2008; Fu et al., 2012). One mechanism by which phagocytic cells recognize and engulf target particles is through complement-mediated opsonization (Schafer et al., 2012; Linnartz and Neumann, 2013; Bodea et al., 2014). Not only does complement activation identify the target required for clearance, it also promotes microglial migration to the site by generating chemoattractant anaphylatoxin C3a from proteolytic cleavage of C3 (Zwirner et al., 1998; Gutzmer et al., 2006). Here we demonstrate that exposing primary microglia to exogenous C3 affects phagocytosis. This is primarily mediated by C3a and through microglial C3aR-dependent substrate uptake, but not degradation. Unexpectedly, we found that the microglial response was biphasic: acute C3/C3a treatment increased, whereas prolonged treatment of 1–2 d impaired, substrate uptake. Whether the differing outcomes are mediated by distinct molecular mechanisms or quantitative differences of the same signaling pathway remains to be established. The functional attenuation produced by extended complement activation is consistent with the idea that chronic neuroinflammation dampens the immune response (Frank-Cannon et al., 2009; Lian et al., 2012). In this regard, it is interesting to note that, besides the well-recognized proinflammatory function, C3a has been implicated to display anti-inflammatory responses, particularly under acute inflammatory conditions (Coulthard and Woodruff, 2015). Therefore, it is tempting to speculate that the anti- and proinflammatory activities of C3/C3a during acute and prolonged incubation, respectively, govern its contrasting effects on Aβ phagocytosis.
The effect of C3/C3a was abrogated by coadministration of C3aR antagonist and was absent in C3aR null microglia, indicating that C3aR is a key part of the C3/C3a response. It is important to note, however, phagocytosis is a complex cellular process involving multiple receptors and subcellular programs (Desjardins, 2003; Sierra et al., 2013; Fu et al., 2014). Of particular relevance, the iC3b receptor CR3 has also been shown to mediate microglial Aβ phagocytosis (Fu et al., 2012). We found that, different from those of the C3aR knock-out, microglia cultured from CR3 null mice still respond to C3 treatment, although the degree of the response is somewhat dampened compared to wild-type cultures. Our results overall support a potential activity of CR3 in this process, although the effect is not as prominent as that of C3aR. These in vitro results are corroborated with our mouse model studies in which we show that C3aR blockade leads to reduced Aβ pathology and neuroinflammation in APP/TTA mice. Intriguingly, we found that the C3aR antagonist resulted in a lower Aβ42/40 ratio but not absolute Aβ levels. The reason for this is not clear, but could be due to the specific RIPA-soluble pool of Aβ we captured, as has been seen by others (Chakrabarty et al., 2015). Nevertheless, the ratio of Aβ42/40 is viewed as a reliable readout to predict Aβ pathology (Wang et al., 2006), and our results as a whole suggest that C3aR inhibition positively affects Aβ dynamics in the mouse brain.
Although the overall impact of C3 on microglial phagocytosis of different substrates was similar, there were noticeable quantitative differences that may be informative. For example, C3 exposure reduced phagocytosis of beads within 24 h, but required twice as long to significantly impair uptake of Aβ. Conversely, 100% of cultured microglia internalized Aβ but only a fraction took up the latex beads. Similar nuances in substrate uptake were also reported by other groups (Koenigsknecht and Landreth, 2004; Smith et al., 2010). One possible explanation is that different membrane receptors may be involved in the phagocytosis of beads versus Aβ. Latex beads are opsonized in serum, and their uptake may be mediated through Fc receptors (Hishikawa et al., 1991; Kusner et al., 1999; Zhang et al., 2010). In contrast, Aβ42 phagocytosis does not require opsonization and may instead involve β1 integrin receptors (Bamberger et al., 2003; Koenigsknecht and Landreth, 2004). Differences in the capacity and kinetics of these receptors may well influence the efficiency and timing of microglial substrate uptake.
Therapeutic benefit from C3aR blockade has been reported in several disease models including CNS lupus (Jacob et al., 2010), multiple sclerosis (Boos et al., 2004), asthma (Ames et al., 2001; Banda et al., 2012), and ischemia/reperfusion injury (Ducruet et al., 2008). We too found that blocking C3aR was beneficial, as it restored learning and memory in amyloid-bearing APP transgenic mice (Lian et al., 2015). Here we reveal that treatment with the C3aR antagonist also reduced amyloid pathology and neuroinflammation in APP transgenic mice, and we provide in vitro evidence that this effect was mediated by restoring microglial phagocytosis. Collectively, data from our current and former studies suggest that the therapeutic efficacy of C3aR inhibition is likely due to its protection of both neuronal synaptic activity and microglial phagocytic function. Although the duration of this benefit remains to be seen and the extension to human patients remains to be shown, our findings provide cautious optimism for considering C3aR therapy in AD.
Footnotes
This work was supported in part by the RNA In Situ Hybridization Core facility of the Eunice Kennedy Shriver National Institute Of Child Health and Human Development (Grant U54HD083092) at Baylor College of Medicine, a Shared Instrumentation Grant from the NIH (1S10OD016167), and the R01s from the NIH (AG032051, AG020670, and NS076117 to H.Z.). We thank A. Cole for expert technical assistance and members of the Zheng laboratory for stimulating discussions. We thank Dr. Rupec (University of Munich, Munich, Germany) for providing the IκBα floxed mice.
The authors declare no competing financial interests.
References
- Ames RS, Lee D, Foley JJ, Jurewicz AJ, Tornetta MA, Bautsch W, Settmacher B, Klos A, Erhard KF, Cousins RD, Sulpizio AC, Hieble JP, McCafferty G, Ward KW, Adams JL, Bondinell WE, Underwood DC, Osborn RR, Badger AM, Sarau HM. Identification of a selective nonpeptide antagonist of the anaphylatoxin C3a receptor that demonstrates antiinflammatory activity in animal models. J Immunol. 2001;166:6341–6348. doi: 10.4049/jimmunol.166.10.6341. [DOI] [PubMed] [Google Scholar]
- Bajenaru ML, Zhu Y, Hedrick NM, Donahoe J, Parada LF, Gutmann DH. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol. 2002;22:5100–5113. doi: 10.1128/MCB.22.14.5100-5113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. 2003;23:2665–2674. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banda NK, Hyatt S, Antonioli AH, White JT, Glogowska M, Takahashi K, Merkel TJ, Stahl GL, Mueller-Ortiz S, Wetsel R, Arend WP, Holers VM. Role of C3a receptors, C5a receptors, and complement protein C6 deficiency in collagen antibody-induced arthritis in mice. J Immunol. 2012;188:1469–1478. doi: 10.4049/jimmunol.1102310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg AA, Sha WC, Bronson RT, Baltimore D. Constitutive NF-κB activation, enhanced granulopoiesis, and neonatal lethality in IκBα-deficient mice. Genes Dev. 1995;9:2736–2746. doi: 10.1101/gad.9.22.2736. [DOI] [PubMed] [Google Scholar]
- Benoit ME, Tenner AJ. Complement protein C1q-mediated neuroprotection is correlated with regulation of neuronal gene and microRNA expression. J Neurosci. 2011;31:3459–3469. doi: 10.1523/JNEUROSCI.3932-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodea LG, Wang Y, Linnartz-Gerlach B, Kopatz J, Sinkkonen L, Musgrove R, Kaoma T, Muller A, Vallar L, Di Monte DA, Balling R, Neumann H. Neurodegeneration by activation of the microglial complement-phagosome pathway. J Neurosci. 2014;34:8546–8556. doi: 10.1523/JNEUROSCI.5002-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boos L, Campbell IL, Ames R, Wetsel RA, Barnum SR. Deletion of the complement anaphylatoxin C3a receptor attenuates, whereas ectopic expression of C3a in the brain exacerbates, experimental autoimmune encephalomyelitis. J Immunol. 2004;173:4708–4714. doi: 10.4049/jimmunol.173.7.4708. [DOI] [PubMed] [Google Scholar]
- Bradt BM, Kolb WP, Cooper NR. Complement-dependent proinflammatory properties of the Alzheimer's disease beta-peptide. J Exp Med. 1998;188:431–438. doi: 10.1084/jem.188.3.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty P, Li A, Ceballos-Diaz C, Eddy JA, Funk CC, Moore B, DiNunno N, Rosario AM, Cruz PE, Verbeeck C, Sacino A, Nix S, Janus C, Price ND, Das P, Golde TE. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron. 2015;85:519–533. doi: 10.1016/j.neuron.2014.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulthard LG, Woodruff TM. Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. J Immuno. 2015;194:3542–3548. doi: 10.4049/jimmunol.1403068. [DOI] [PubMed] [Google Scholar]
- Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH, Arnaout MA, Mayadas TN. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/S1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- Crehan H, Holton P, Wray S, Pocock J, Guerreiro R, Hardy J. Complement receptor 1 (CR1) and Alzheimer's disease. Immunobiology. 2012;217:244–250. doi: 10.1016/j.imbio.2011.07.017. [DOI] [PubMed] [Google Scholar]
- Davoust N, Jones J, Stahel PF, Ames RS, Barnum SR. Receptor for the C3a anaphylatoxin is expressed by neurons and glial cells. Glia. 1999;26:201–211. doi: 10.1002/(SICI)1098-1136(199905)26:3%3C201::AID-GLIA2%3E3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Desjardins M. ER-mediated phagocytosis: a new membrane for new functions. Nat Rev Immunol. 2003;3:280–291. doi: 10.1038/nri1053. [DOI] [PubMed] [Google Scholar]
- Ducruet AF, Hassid BG, Mack WJ, Sosunov SA, Otten ML, Fusco DJ, Hickman ZL, Kim GH, Komotar RJ, Mocco J, Connolly ES. C3a receptor modulation of granulocyte infiltration after murine focal cerebral ischemia is reperfusion dependent. J Cereb Blood Flow Metab. 2008;28:1048–1058. doi: 10.1038/sj.jcbfm.9600608. [DOI] [PubMed] [Google Scholar]
- Fan R, Tenner AJ. Complement C1q expression induced by Abeta in rat hippocampal organotypic slice cultures. Exp Neurol. 2004;185:241–253. doi: 10.1016/j.expneurol.2003.09.023. [DOI] [PubMed] [Google Scholar]
- Fischer B, Schmoll H, Riederer P, Bauer J, Platt D, Popa-Wagner A. Complement C1q and C3 mRNA expression in the frontal cortex of Alzheimer's patients. J Mol Med (Berl) 1995;73:465–471. doi: 10.1007/BF00202265. [DOI] [PubMed] [Google Scholar]
- Fonseca MI, Chu SH, Berci AM, Benoit ME, Peters DG, Kimura Y, Tenner AJ. Contribution of complement activation pathways to neuropathology differs among mouse models of Alzheimer's disease. J Neuroinflammation. 2011;8:4. doi: 10.1186/1742-2094-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol Neurodegener. 2009;4:47. doi: 10.1186/1750-1326-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Liu B, Frost JL, Hong S, Jin M, Ostaszewski B, Shankar GM, Costantino IM, Carroll MC, Mayadas TN, Lemere CA. Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Abeta by microglia. Glia. 2012;60:993–1003. doi: 10.1002/glia.22331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu R, Shen Q, Xu P, Luo JJ, Tang Y. Phagocytosis of microglia in the central nervous system diseases. Mol Neurobiol. 2014;49:1422–1434. doi: 10.1007/s12035-013-8620-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin RS, Costigan M, Brenner GJ, Ma CH, Scholz J, Moss A, Allchorne AJ, Stahl GL, Woolf CJ. Complement induction in spinal cord microglia results in anaphylatoxin C5a-mediated pain hypersensitivity. J Neurosci. 2007;27:8699–8708. doi: 10.1523/JNEUROSCI.2018-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutzmer R, Köther B, Zwirner J, Dijkstra D, Purwar R, Wittmann M, Werfel T. Human plasmacytoid dendritic cells express receptors for anaphylatoxins C3a and C5a and are chemoattracted to C3a and C5a. J Invest Dermatol. 2006;126:2422–2429. doi: 10.1038/sj.jid.5700416. [DOI] [PubMed] [Google Scholar]
- Haga S, Aizawa T, Ishii T, Ikeda K. Complement gene expression in mouse microglia and astrocytes in culture: comparisons with mouse peritoneal macrophages. Neurosci Lett. 1996;216:191–194. doi: 10.1016/0304-3940(96)13040-8. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley K. Neuroinflammation in Alzheimer's disease: mechanisms, pathologic consequences, and potential for therapeutic manipulation. J Alzheimers Dis. 2010;21:1–14. doi: 10.3233/JAD-2010-1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishikawa T, Cheung JY, Yelamarty RV, Knutson DW. Calcium transients during Fc receptor-mediated and nonspecific phagocytosis by murine peritoneal macrophages. J Cell Biol. 1991;115:59–66. doi: 10.1083/jcb.115.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbles AA, Lu B, Nilsson CA, Lilly C, Israel E, Fujiwara Y, Gerard NP, Gerard C. A role for the C3a anaphylatoxin receptor in the effector phase of asthma. Nature. 2000;406:998–1001. doi: 10.1038/35023175. [DOI] [PubMed] [Google Scholar]
- Ischenko A, Sayah S, Patte C, Andreev S, Gasque P, Schouft MT, Vaudry H, Fontaine M. Expression of a functional anaphylatoxin C3a receptor by astrocytes. J Neurochem. 1998;71:2487–2496. doi: 10.1046/j.1471-4159.1998.71062487.x. [DOI] [PubMed] [Google Scholar]
- Jacob A, Bao L, Brorson J, Quigg RJ, Alexander JJ. C3aR inhibition reduces neurodegeneration in experimental lupus. Lupus. 2010;19:73–82. doi: 10.1177/0961203309348978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Gonzales V, Savonenko AV, Wen JC, Jenkins NA, Copeland NG, Younkin LH, Lester HA, Younkin SG, Borchelt DR. Persistent amyloidosis following suppression of Aβ production in a transgenic model of Alzheimer disease. PLoS Med. 2005;2:e355. doi: 10.1371/journal.pmed.0020355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauneau AC, Ischenko A, Chatagner A, Benard M, Chan P, Schouft MT, Patte C, Vaudry H, Fontaine M. Interleukin-1beta and anaphylatoxins exert a synergistic effect on NGF expression by astrocytes. J Neuroinflammation. 2006;3:8. doi: 10.1186/1742-2094-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RS, Minogue AM, Connor TJ, Lynch MA. Amyloid-beta-induced astrocytic phagocytosis is mediated by CD36, CD47 and RAGE. J Neuroimmune Pharmacol. 2013;8:301–311. doi: 10.1007/s11481-012-9427-3. [DOI] [PubMed] [Google Scholar]
- Koenigsknecht J, Landreth G. Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J Neurosci. 2004;24:9838–9846. doi: 10.1523/JNEUROSCI.2557-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusner DJ, Hall CF, Jackson S. Fc gamma receptor-mediated activation of phospholipase D regulates macrophage phagocytosis of IgG-opsonized particles. J Immunol. 1999;162:2266–2274. [PubMed] [Google Scholar]
- Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fiévet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- Lian H, Shim DJ, Gaddam SS, Rodriguez-Rivera J, Bitner BR, Pautler RG, Robertson CS, Zheng H. IκBα deficiency in brain leads to elevated basal neuroinflammation and attenuated response following traumatic brain injury: implications for functional recovery. Mol Neurodegener. 2012;7:47. doi: 10.1186/1750-1326-7-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, Shim DJ, Rodriguez-Rivera J, Taglialatela G, Jankowsky JL, Lu HC, Zheng H. NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer's disease. Neuron. 2015;85:101–115. doi: 10.1016/j.neuron.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnartz B, Neumann H. Microglial activatory (immunoreceptor tyrosine-based activation motif)- and inhibitory (immunoreceptor tyrosine-based inhibition motif)-signaling receptors for recognition of the neuronal glycocalyx. Glia. 2013;61:37–46. doi: 10.1002/glia.22359. [DOI] [PubMed] [Google Scholar]
- Loeffler DA, Camp DM, Bennett DA. Plaque complement activation and cognitive loss in Alzheimer's disease. J Neuroinflammation. 2008;5:9. doi: 10.1186/1742-2094-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue LF, Walker DG, Rogers J. Modeling microglial activation in Alzheimer's disease with human postmortem microglial cultures. Neurobiol Aging. 2001;22:945–956. doi: 10.1016/S0197-4580(01)00311-6. [DOI] [PubMed] [Google Scholar]
- Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA. Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci. 2008;28:6333–6341. doi: 10.1523/JNEUROSCI.0829-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CB, Ingersoll SA, Martin BK. Transcriptional control of the C3a receptor gene in glial cells: Dependence upon AP-1 but not Ets. Mol Immunol. 2007;44:703–712. doi: 10.1016/j.molimm.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Picciano M, Malester B, LaFrancois J, Zehr C, Daeschner JM, Olschowka JA, Fonseca MI, O'Banion MK, Tenner AJ, Lemere CA, Duff K. Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer's disease. Am J Pathol. 2001;158:1345–1354. doi: 10.1016/S0002-9440(10)64085-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- Orsini F, De Blasio D, Zangari R, Zanier ER, De Simoni MG. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front Cell Neurosci. 2014;8:380. doi: 10.3389/fncel.2014.00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips T, Robberecht W. Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 2011;10:253–263. doi: 10.1016/S1474-4422(11)70015-1. [DOI] [PubMed] [Google Scholar]
- Prokop S, Miller KR, Heppner FL. Microglia actions in Alzheimer's disease. Acta Neuropathol. 2013;126:461–477. doi: 10.1007/s00401-013-1182-x. [DOI] [PubMed] [Google Scholar]
- Reichwald J, Danner S, Wiederhold KH, Staufenbiel M. Expression of complement system components during aging and amyloid deposition in APP transgenic mice. J Neuroinflammation. 2009;6:35. doi: 10.1186/1742-2094-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, Civin WH, Brachova L, Bradt B, Ward P, Lieberburg I. Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1992;89:10016–10020. doi: 10.1073/pnas.89.21.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupec RA, Jundt F, Rebholz B, Eckelt B, Weindl G, Herzinger T, Flaig MJ, Moosmann S, Plewig G, Dörken B, Förster I, Huss R, Pfeffer K. Stroma-mediated dysregulation of myelopoiesis in mice lacking IκBα. Immunity. 2005;22:479–491. doi: 10.1016/j.immuni.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Rutar M, Valter K, Natoli R, Provis JM. Synthesis and propagation of complement C3 by microglia/monocytes in the aging retina. PLoS One. 2014;9:e93343. doi: 10.1371/journal.pone.0093343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer MK, Schwaeble WJ, Post C, Salvati P, Calabresi M, Sim RB, Petry F, Loos M, Weihe E. Complement C1q is dramatically up-regulated in brain microglia in response to transient global cerebral ischemia. J Immunol. 2000;164:5446–5452. doi: 10.4049/jimmunol.164.10.5446. [DOI] [PubMed] [Google Scholar]
- Shen Y, Li R, McGeer EG, McGeer PL. Neuronal expression of mRNAs for complement proteins of the classical pathway in Alzheimer brain. Brain Res. 1997;769:391–395. doi: 10.1016/S0006-8993(97)00850-0. [DOI] [PubMed] [Google Scholar]
- Shinjyo N, Ståhlberg A, Dragunow M, Pekny M, Pekna M. Complement-derived anaphylatoxin C3a regulates in vitro differentiation and migration of neural progenitor cells. Stem Cells. 2009;27:2824–2832. doi: 10.1002/stem.225. [DOI] [PubMed] [Google Scholar]
- Sierra A, Abiega O, Shahraz A, Neumann H. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci. 2013;7:6. doi: 10.3389/fncel.2013.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Gibbons HM, Dragunow M. Valproic acid enhances microglial phagocytosis of amyloid-β1–42. Neuroscience. 2010;169:505–515. doi: 10.1016/j.neuroscience.2010.04.041. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Stine WB, Jr, Dahlgren KN, Krafft GA, LaDu MJ. In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J Biol Chem. 2003;278:11612–11622. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- Tacnet-Delorme P, Chevallier S, Arlaud GJ. Beta-amyloid fibrils activate the C1 complex of complement under physiological conditions: evidence for a binding site for A beta on the C1q globular regions. J Immunol. 2001;167:6374–6381. doi: 10.4049/jimmunol.167.11.6374. [DOI] [PubMed] [Google Scholar]
- Todd RF., 3rd The continuing saga of complement receptor type 3 (CR3) J Clin Invest. 1996;98:1–2. doi: 10.1172/JCI118752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DG, Kim SU, McGeer PL. Expression of complement C4 and C9 genes by human astrocytes. Brain Res. 1998;809:31–38. doi: 10.1016/S0006-8993(98)00811-7. [DOI] [PubMed] [Google Scholar]
- Wang R, Wang B, He W, Zheng H. Wild-type presenilin 1 prectects against Alzheimer disease mutation-induced amyloid pathology. J Biol Chem. 2006;281:15330–15336. doi: 10.1074/jbc.M512574200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Hancock AM, Bradner J, Chung KA, Quinn JF, Peskind ER, Galasko D, Jankovic J, Zabetian CP, Kim HM, Leverenz JB, Montine TJ, Ginghina C, Edwards KL, Snapinn KW, Goldstein DS, Shi M, Zhang J. Complement 3 and factor h in human cerebrospinal fluid in Parkinson's disease, Alzheimer's disease, and multiple-system atrophy. Am J Pathol. 2011;178:1509–1516. doi: 10.1016/j.ajpath.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci U S A. 2002;99:10837–10842. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Wang Z, Wang B, Justice NJ, Zheng H. Amyloid precursor protein regulates Cav1.2 L-type calcium channel levels and function to influence GABAergic short-term plasticity. J Neurosci. 2009;29:15660–15668. doi: 10.1523/JNEUROSCI.4104-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasojima K, Schwab C, McGeer EG, McGeer PL. Up-regulated production and activation of the complement system in Alzheimer's disease brain. Am J Pathol. 1999;154:927–936. doi: 10.1016/S0002-9440(10)65340-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaylaoglu MB, Titmus A, Visel A, Alvarez-Bolado G, Thaller C, Eichele G. Comprehensive expression atlas of fibroblast growth factors and their receptors generated by a novel robotic in situ hybridization platform. Dev Dyn. 2005;234:371–386. doi: 10.1002/dvdy.20441. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Hoppe AD, Swanson JA. Coordination of Fc receptor signaling regulates cellular commitment to phagocytosis. Proc Natl Acad Sci U S A. 2010;107:19332–19337. doi: 10.1073/pnas.1008248107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Fonseca MI, Pisalyaput K, Tenner AJ. Complement C3 and C4 expression in C1q sufficient and deficient mouse models of Alzheimer's disease. J Neurochem. 2008;106:2080–2092. doi: 10.1111/j.1471-4159.2008.05558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009;9:729–740. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- Zwirner J, Werfel T, Wilken HC, Theile E, Götze O. Anaphylatoxin C3a but not C3a(desArg) is a chemotaxin for the mouse macrophage cell line J774. Eur J Immunol. 1998;28:1570–1577. doi: 10.1002/(SICI)1521-4141(199805)28:05<1570::AID-IMMU1570>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]