

Abstract
Two forms of NMDA receptor (NMDAR)-dependent long-term potentiation (LTP) at hippocampal CA1 synapses can be distinguished based on their sensitivity to inhibitors of protein kinase A (PKA). The PKA-dependent form requires multiple episodes of high-frequency stimulation (HFS) or theta burst stimuli (TBS) with a spacing between episodes in the order of minutes. To investigate the mechanism by which spaced episodes induce the PKA-dependent form of LTP, we have compared, in interleaved experiments, spaced (s) and compressed (c) TBS protocols in the rat CA1 synapses. We find that LTP induced by sTBS, but not that induced by cTBS, involves the insertion of calcium-permeable (CP) AMPARs, as assessed using pharmacological and electrophysiological criteria. Furthermore, a single TBS when paired with rolipram [4-(3-(cyclopentyloxy)-4-methoxyphenyl)pyrrolidin-2-one], to activate PKA, generates an LTP that also involves the insertion of CP-AMPARs. These data demonstrate that the involvement of CP-AMPARs in LTP is critically determined by the timing of the induction trigger and is associated specifically with the PKA-dependent form of LTP.
SIGNIFICANCE STATEMENT Long-term potentiation is a family of synaptic mechanisms that are believed to be important for learning and memory. Two of the most extensively studied forms are triggered by the synaptic activation of NMDA receptors and expressed by changes in AMPA receptor function. They can be distinguished on the basis of their requirement for activation of a protein kinase, PKA. We show that the PKA-dependent form also involves the transient insertion of calcium-permeable AMPA receptors. These results have implications for relating synaptic plasticity to learning and memory and suggest a specific linkage between PKA activation and the rapid synaptic insertion of calcium-permeable AMPA receptors during long-term potentiation.
Keywords: calcium-permeable AMPA receptor, hippocampus, long-term potentiation, NMDA receptor, PKA, rectification
Introduction
Long-term potentiation (LTP) and its counterpart long-term depression are the major forms of long-lasting synaptic plasticity in the vertebrate CNS. LTP was first described at perforant path synapses (Bliss and Gardner-Medwin, 1973; Bliss and Lomo, 1973) and has since been described in numerous excitatory pathways in the CNS. In particular, LTP has been studied extensively at the Schaffer collateral–commissural pathway (SCCP), in which, using the competitive NMDA receptor (NMDAR) antagonist 2-amino-5-phosphonopentanoate (AP-5; Davies et al., 1981), it was found that LTP is triggered by the synaptic activation of NMDARs (Collingridge et al., 1983). NMDAR–LTP is not a unitary phenomenon; rather, it can be divided into several mechanistically distinct components. After a period of high-frequency stimulation (HFS), there is often a decaying phase, known as short-term potentiation (STP), and a sustained phase that lasts hours in vitro and days in vivo (Abraham, 2003). The sustained phase of LTP has been subdivided based on whether it is sensitive or not to inhibitors of protein kinase A (PKA) and protein synthesis. The form of LTP that is insensitive to these inhibitors is commonly referred to as early LTP (E-LTP), whereas the form that is sensitive to these inhibitors is often called late-phase LTP (L-LTP; Huang et al., 1996). The PKA-sensitive form of LTP requires multiple episodes of HFS or theta burst stimulation (TBS) for its induction. When a single episode is used to induce LTP, the resulting potentiation is independent of both PKA and de novo protein synthesis. Interestingly, the induction of the PKA-sensitive form requires not only multiple episodes but for these episodes to be spaced in time, with an inter-episode interval in the order of minutes. If the episodes are compressed in time (with an inter-episode interval in the order of seconds), then only the PKA-independent form of LTP is induced (Woo et al., 2003; Kim et al., 2010; Park et al., 2014). This raises an important question as to why the timing of the stimuli matters.
In the present study, we tested the hypothesis that calcium-permeable (CP) AMPARs are a determinant factor in the generation of the PKA-dependent form of LTP. CP-AMPARs have been implicated in the induction of LTP at CA1 synapses under some (Jia et al., 1996; Plant et al., 2006) but not all (Adesnik and Nicoll, 2007) experimental conditions. When this occurs, it has been attributed to an increase in the expression of GluA2-lacking AMPARs, leading to a greater calcium permeability of AMPARs during and/or shortly after the HFS or TBS. In interleaved experiments, we have compared compressed TBS (cTBS) with spaced TBS (sTBS) protocols. We found that three different inhibitors of CP-AMPARs, IEM 1460 (N,N,H,-trimethyl-5-[(tricyclo[3.3.1.13,7]dec-1-ylmethyl)amino]-1-pentanaminiumbromide hydrobromide [IEM]), philanthotoxin 433 [(S)-N-[4-[[3-[(3-aminopropyl)amino]propyl]amino]butyl]-4-hydroxy-α-[(1-oxobutyl)amino]benzenepropanamide tris(trifluoroacetate) salt (PhTx)], and 1-naphthyl acetyl spermine (NASPM), were all without effect on LTP induced by a compressed protocol (hereafter referred to as cLTP). In contrast, all inhibitors substantially inhibited LTP induced by a spaced protocol (i.e., sLTP). Consistent with sLTP involving the insertion of CP-AMPARs, sTBS (but not cTBS) resulted in an increase in inward rectification of AMPAR-mediated synaptic transmission. We also found that inhibition of phosphodiesterase 4 (PDE4), using rolipram [4-(3-(cyclopentyloxy)-4-methoxyphenyl)pyrrolidin-2-one], resulted in an enhancement of LTP in response to a weak TBS (wTBS), that comprised a single episode of TBS. This rolipram-enhanced LTP was also sensitive to IEM and was associated with an increase in inward rectification. Collectively, these data suggest that the first TBS episode induces LTP via a mechanism that is independent of PKA and CP-AMPARs (i.e., cLTP), but, at the same time, this TBS activates PKA to prime for sLTP. If a second TBS is delivered within a critical time window, then a PKA-dependent form of LTP (sLTP) is induced and this process involves the transient insertion of CP-AMPARs.
Materials and Methods
Hippocampal slice preparation.
Transverse hippocampal slices (400 μm) were prepared from male Sprague Dawley rats (3–12 weeks of age). Animals were anesthetized with isoflurane and killed by decapitation in accordance with Korean and United Kingdom animal legislation. Hippocampi were isolated rapidly from the brain and sliced using a vibratome (Leica). The CA3 region was removed, and slices were transferred to an incubation chamber with artificial CSF (ACSF) saturated with 95% O2 and 5% CO2. ACSF contained the following (mm): 124 NaCl, 3 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 MgSO4, 10 glucose, and 2 CaCl2. Slices were allowed to recover at 32–34°C for 30 min and then maintained at 26–28°C for a minimum of 1 h before recordings were made.
Extracellular electrophysiology.
The extracellular recording was performed in an interface chamber (Campden Instruments) maintained at 32°C and perfused continuously at 2–3 ml/min with ACSF. Standard extracellular recordings were performed in the CA1 region of hippocampal slices, as described by Choi et al., 2014, to measure the slope of evoked field EPSPs (fEPSPs). Recordings were monitored and analyzed using WinLTP (Anderson and Collingridge, 2007). Each experiment was conducted on slices from separate animals, so the n value indicates both the number of slices and animals used. Two independent SCCPs were stimulated alternatively, each at a frequency of 0.033 Hz (or 0.1 Hz for data shown in Fig. 4). After a stable baseline of at least 20 min, LTP was induced using TBS delivered at basal stimulus intensity. An episode of TBS comprised five bursts at 5 Hz, with each burst composed of five pulses at 100 Hz (Fig. 1A). For cLTP induction, a train of three TBS episodes was given with an interval of 10 s. For sLTP induction, a train of three TBS episodes was given with an interval between 2 min and 1 h. Representative sample traces are an average of five consecutive responses, collected from typical experiments (stimulus artifacts were blanked for clarity).
Figure 4.
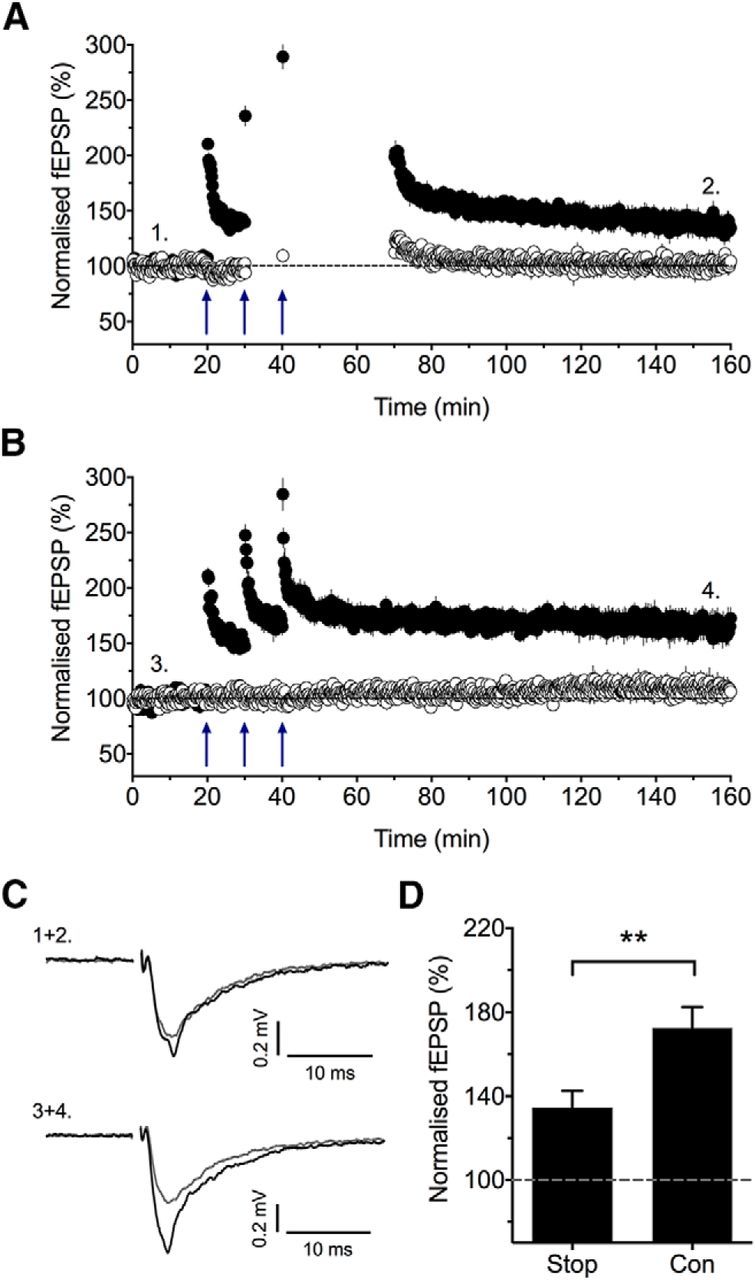
Stopping stimulation after the delivery of TBS reduces the magnitude of LTP. A, The effects of stopping stimulation after the second and third episodes of TBS on sLTP (n = 9). Initial test responses were collected after the delivery of the second and third TBS, so as to estimate the level of sLTP induction. B, Interleaved control experiments (n = 7). C, Example traces from representative experiments. D, A comparison of the levels of LTP, quantified 2 h after the delivery of the third TBS episode, for the data presented in A (Stop) and B (Con). **p < 0.01 versus control.
Figure 1.
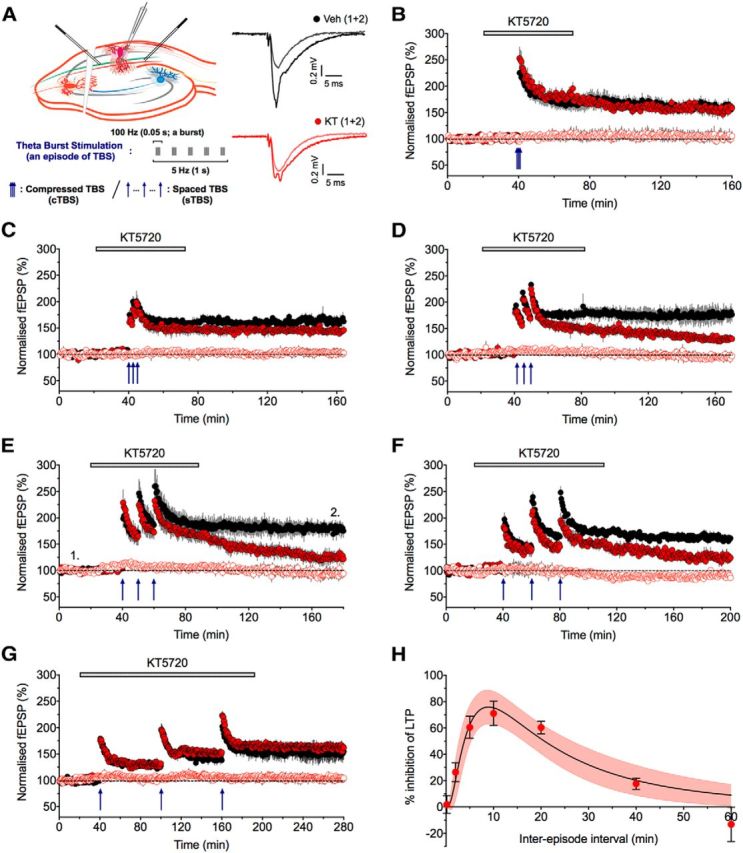
Defining the PKA dependence of LTP. A, A schematic of the protocol for studying LTP. Two inputs were stimulated alternately and TBS delivered to one input with the second input serving as a control for stability. Traces are the averages of five successive records of baseline and LTP, obtained at the times indicated by numbers in E for a vehicle (Veh) control (black) and a KT experiment (red). B, The lack of effect of KT on LTP using a cTBS protocol (inter-episode interval of 10 s). Data are pooled (mean ± SEM from 5 experiments) and illustrate the effects of treated (colored) and interleaved controls (n = 8, black) for the test (filled symbols) and control (open symbols) input. The timing of TBS is shown by the blue arrows. KT (1 μm) was applied for the duration indicated by the gray bar. C–G, The effects of KT on a sTBS with an inter-episode interval of 2 min (C; n = 5 and 3 for the KT and vehicle experiment, respectively), 5 min (D; 4 and 3), 10 min (E; 5 and 5), 20 min (F; 4 and 6), and 60 min (G; 4 and 3). H, The time course of inhibition (percentage) of LTP by KT. Each value was calculated by comparing the level of LTP in KT with its interleaved control experiment. The line is a log Gaussian curve with 95% confidence intervals (shaded).
Simultaneous whole-cell and field recording.
In some experiments, simultaneous whole-cell and field recordings were obtained to monitor EPSCs and fEPSPs under identical conditions. Peak amplitude (picoamperes) and initial slope (volts per seconds) of EPSCs and fEPSPs were measured using WinLTP (Anderson and Collingridge, 2007). Recording chamber was maintained at 32°C, and CA1 pyramidal cells were visualized with infrared differential interference contrast optics (Hamamatsu Photonics). ACSF contained 50 μm picrotoxin and 20 μm (+)-bicuculline. The whole-cell solution comprised the following (in mm): 8 NaCl, 130 CsMeSO3, 10 HEPES, 0.5 EGTA, 4 Mg-ATP, 0.3 Na3-GTP, 5 QX-314, and 0.1 spermine. The pH was adjusted to 7.2–7.3 with CsOH, and osmolarity was to 285–290 mOsm. Two independent SCCPs were stimulated alternatively, each at a frequency of 0.1 Hz. Borosilicate glass pipettes were used with a resistance of 4–6 MΩ for whole-cell recordings, and experiments were only accepted for analysis if series resistance values were <25 MΩ and varied by <20% during the course of experiment. Cells were clamped at a holding potential of −70 mV. Whole-cell patch-clamp recordings were initiated following collection of at least 10 min stable baseline of extracellular recordings. LTP was obtained within 10 min of whole cell (to prevent “washout” of LTP) under current-clamp conditions.
Rectification index measurement.
The rectification index (RI) of AMPAR EPSCs was measured 10 min after LTP induction. Currents through AMPARs were isolated pharmacologically using a combination of a competitive NMDAR antagonist (d-AP-5; 100 μm) plus a glycine-site antagonist [l-689,560 (trans-2-carboxy-5,7-dichloro-4-phenylaminocarbonylamino-1,2,3,4-tetrahydroquinoline); 5 μm]. The neuron was immediately depolarized to 40 mV for 100 s and then to 0 mV for 50 s, and consecutive responses obtained at these holding potentials were averaged. The index was calculated by taking the ratio of the slopes from 0 to 40 mV and −70 to 0 mV. RIs were compared between LTP and control inputs within each neuron.
Compounds.
Drugs were prepared as frozen stock solutions (stored below −20°C) and dissolved into ACSF at least 20 min before their bath application. Compounds used were as follows: IEM 1460, NASPM, L-689,560 (Tocris Bioscience), rolipram (Abcam), KT5720 [(9R,10S,12S)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid, hexyl ester (KT)], d-AP-5 (Tocris, Abcam, or Hello Bio), and philanthotoxin 433 (Sigma).
Analysis.
All treatment groups were interleaved with control (i.e., vehicle) experiments. Data were normalized to the baseline preceding drug application and are presented as mean ± SEM. Statistical significance was assessed using either Student's t test or one-way ANOVA with post hoc Bonferroni's correction as appropriate; the level of significance is denoted as follows: *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
PKA inhibitors inhibit LTP induced by sTBS but not cTBS
Previous work has shown that the sensitivity of LTP to inhibitors of PKA is dependent on the induction parameters. A single episode (Huang and Kandel, 1994; Bortolotto and Collingridge, 2000) or multiple episodes that are compressed in time (Woo et al., 2003; Kim et al., 2010; Park et al., 2014) induce a PKA-independent form, whereas multiple episodes that are spaced in time induce a PKA-dependent component of LTP (Huang and Kandel, 1994; Woo et al., 2003; Kim et al., 2010; Park et al., 2014). Before investigating the underlying mechanism, we wanted to define more precisely the timing requirements for the induction of sLTP (Fig. 1). We performed two-input experiments and delivered 75 stimuli to one input (test input) leaving the other unconditioned (control input), to ensure stability of the recordings. In each case, the stimuli were delivered as three episodes, with each episode comprising five bursts (of five shocks at 100 Hz, with an interburst interval of 200 ms). The only variable was the inter-episode interval that was varied between 10 s and 1 h (Fig. 1A). We probed for the involvement of PKA using the extensively studied inhibitor KT (1 μm). We found that KT has no effect on LTP induced with an inter-episode interval of 10 s (Fig. 1B) or 1 h (Fig. 1G) but inhibited LTP induced with intermediate intervals of 2 min (Fig. 1C), 5 min (Fig. 1D), 10 min (Fig. 1E), 20 min (Fig. 1F), and 40 min (data not shown). For example, the levels of LTP, expressed as percentage baseline and quantified 2 h after the third TBS episode, using an inter-episode interval of 20 min were 161 ± 4% (n = 6) and 124 ± 7% (n = 4, p = 0.004, unpaired Student's t test) for the vehicle and KT groups, respectively. To determine the time course, we measured the level of inhibition at each time point, by comparing the level of LTP in the presence of KT with the corresponding interleaved control group. These results are presented graphically in Figure 1H.
CP-AMPARs are important for sLTP
The present observations can be explained by the first TBS priming for, and the subsequent TBS being involved in inducing, the PKA-sensitive form of LTP. In which case, an important question is how does this priming work? We wondered whether this priming might recruit CP-AMPARs and whether these are required for sLTP. We based this idea on the findings that PKA is known to recruit GluA1-containing AMPARs to the plasma membrane (Roche et al., 1996; Oh et al., 2006; Man et al., 2007) and because inhibitors of CP-AMPARs have been reported to inhibit LTP at these synapses under certain (Jia et al., 1996; Plant et al., 2006) but not all circumstances (Adesnik and Nicoll, 2007). To test this idea, we compared the ability of the CP-AMPAR inhibitor IEM (30 μm) to inhibit LTP induced by cTBS or sTBS.
IEM had no effect on cLTP (Fig. 2A) but substantially inhibited sLTP (Fig. 2F). The levels of LTP, expressed as percentage baseline and quantified 2 h after the third TBS episode for the IEM and vehicle groups, were 164 ± 10% (n = 6) versus 160 ± 7% (n = 8, p = 0.457, unpaired Student's t test) for the cTBS and 123 ± 4% (n = 4) vs 158 ± 5% (n = 6, p = 0.001, unpaired Student's t test) for the sTBS with an inter-episode interval of 20 min. In a previous study, in which CP-AMPARs were found to be involved in LTP, there was a time window after the induction of LTP in which they were involved (Plant et al., 2006). Therefore, we applied IEM at different times during and after the sTBS protocol. When IEM was applied immediately after the first TBS (Fig. 2B), the second TBS (Fig. 2C) or the third TBS (Fig. 2D) it was able to inhibit LTP in an application timing-dependent manner. However, when it was applied 1 h after the last TBS, it was ineffective (Fig. 2E). These data, compared in Figure 2F, show that the IEM-sensitive process extends beyond the TBS but is fairly transient in nature.
Figure 2.

A role for CP-AMPARs in LTP. A, The lack of effect of IEM (30 μm) on LTP using the cTBS protocol (n = 6 and 8 for the IEM and vehicle groups, respectively). B–F, The effects of IEM on LTP induced with sTBS, with an inter-episode interval of 20 min. IEM was applied immediately after the first TBS (B; n = 7), second TBS (C; n = 6), third TBS (D; n = 10), or 1 h after the third TBS (E; n = 5). A single set of interleaved vehicle controls (n = 21) are shown. F, Quantification of the effects of IEM on LTP. The histograms plot the level of inhibition (percentage) of LTP, measured 2 h after the last TBS, for the conditions illustrated in A–E. *p < 0.05, **p < 0.01, and ***p < 0.001 versus control.
Although IEM is used extensively as an inhibitor of CP-AMPARs, there is the possibility of off-target effects. Therefore, we tested two additional inhibitors, PhTx (5–10 μm) and NASPM (30 μm), applying them immediately after the first TBS. Consistent with an action on CP-AMPARs, PhTx had no effect on cLTP (Fig. 3A) but inhibited sLTP (Fig. 3B). Similarly, NASPM had no effect on cLTP (Fig. 3C) but inhibited sLTP (Fig. 3D).
Figure 3.

Additional evidence for a role of CP-AMPARs in LTP. A, The lack of effect of PhTx (5–10 μm) on LTP using a cTBS protocol (inter-episode interval of 10 s, n = 5). B, Inhibitory effect of PhTx on LTP induced by sTBS protocol (inter-episode interval of 20 min, n = 8). C, The lack of effect of NASPM (30 μm) on LTP using a cTBS protocol (inter-episode interval of 10 s, n = 6). D, Inhibitory effect of NASPM of LTP induced by sTBS protocol (inter-episode interval of 20 min, n = 7).
sLTP requires low-frequency stimulation after the delivery of TBS
It has been reported (Plant et al., 2006) but disputed (Adesnik and Nicoll, 2007) that CP-AMPARs are activated by the test pulses delivered after the induction trigger and that this is required for the expression of LTP. To test whether this is the case for sLTP, we compared, in interleaved experiments, the effects of stopping stimulation after the TBS (Fig. 4A) to the standard condition of continuous low-frequency stimulation (Fig. 4B). In these experiments, we stimulated as usual after the delivery of the first TBS to assess the level of cLTP. We also collected a test response after the delivery of the second and third TBS to estimate the level of sLTP induction. However, for the remainder of the time, until 30 min after the third TBS, we stopped the test stimuli on both inputs. The level of LTP, quantified 2 h after the third TBS, was significantly less than that induced in experiments in which test stimuli were delivered continuously [135 ± 8% (n = 9) vs 173 ± 10% (n = 7), respectively, p = 0.01; Fig. 4D]. These observations are consistent with the scenario in which test stimuli given after the initial induction trigger are required for the full expression of sLTP.
Rolipram facilitates LTP via a mechanism involving CP-AMPARs
We have assumed that the priming effect of the first TBS involves the activation of PKA based on the sensitivity of the LTP induced by the subsequent TBS to KT. To test this in another way, we activated PKA using rolipram to inhibit cAMP hydrolysis by PDE4 and delivered a single episode of TBS (wTBS). Under control conditions, this induced a relatively small LTP (122 ± 4% of baseline, n = 4; Fig. 5A), and, in the presence of rolipram (0.1 μm), the LTP was much larger (157 ± 5%, n = 5, p = 0.002, one-way ANOVA with Bonferroni's correction for vehicle vs rolipram; Fig. 5B). IEM had no effect on the weak LTP induced under control conditions (124 ± 7%, n = 4; Fig. 5C) but substantially inhibited the rolipram-enhanced LTP (128 ± 5%, n = 6; Fig. 5D), to a level that was not significantly different from the LTP recorded under control conditions (p > 0.999, one-way ANOVA with Bonferroni's correction). These data show that, although the LTP induced by a wTBS is independent of CP-AMPARs, the additional LTP induced by a rolipram-primed TBS is primarily, if not exclusively, mediated by CP-AMPARs.
Figure 5.

Rolipram enhances LTP via the insertion of CP-AMPARs. A, The effects of a wTBS. B, Enhancement of LTP induced by a wTBS in the presence of rolipram (0.1 μm). C, The lack of effect of IEM on LTP induced by a wTBS. D, Inhibition of the rolipram-enhanced LTP by IEM. In B–D, the vehicle dataset is replotted for illustrative purposes. E, Traces from representative experiments to illustrate rolipram-enhanced LTP (red) and the effects of rolipram plus IEM (purple), which were obtained at the times indicated by numbers during baseline and 1 h after the delivery of wTBS. F, Quantification of the effects measured 1 h after LTP induction. **p < 0.01 versus vehicle. Rol, Rolipram; Veh, vehicle.
sLTP is associated with an increase in inward rectification of AMPARs
So far, the evidence for the insertion of CP-AMPARs has been based on sensitivity to polyamine inhibitors. Because CP-AMPARs have an inwardly rectifying current–voltage relationship, we tested whether LTP was associated with any alteration in rectification properties. To do this, we made simultaneous field potential recordings and whole-cell patch-clamp recordings and compared the rectification of pharmacologically isolated AMPAR-mediated EPSCs between potentiated and control inputs. As shown in Fig. 6, cTBS resulted in input-specific LTP that was similar in magnitude when the fEPSP (Fig. 6A1) and EPSC (Fig. 6A2) recordings were compared. Immediately after delivering the cTBS, NMDARs were blocked by applying d-AP-5 (100 μm) plus L-689,560 (5 μm), and, 10 min after TBS, the rectification of AMPARs was estimated by recording AMPAR-mediated EPSCs at three holding potentials (−70, 0, and 40 mV). The RI was not significantly different between the two inputs (the respective values for test and control inputs were 0.81 ± 0.03 and 0.83 ± 0.03; p = 0.240, n = 21 neurons, 10 animals, paired Student's t test; Fig. 6A3). Because of washout of LTP, it was not possible to estimate the RI for sLTP in exactly the same way. Instead, two TBS were delivered to ensure that LTP was induced within the neuronal population (Fig. 6B1) and a whole-cell recording obtained shortly after the second TBS (Fig. 6B2). Thereafter, the protocol was the same. In contrast to cLTP, the sLTP was associated with a small, but highly significant, change in the RI (0.78 ± 0.03 and 0.84 ± 0.03, n = 23, 11 animals, p = 0.003, paired Student's t test), which is consistent with the insertion of CP-AMPARs (Fig. 6B3).
Figure 6.

Alterations in AMPAR rectification associated with LTP. A1, A representative example of LTP recorded extracellularly induced by a cTBS. A2, The simultaneous whole-cell recording from a neuron in the same region of the slice. d-AP-5 (100 μm) plus L-689,560 (5 μm) were applied immediately after the delivery of TBS, as indicated by the gray bar. After 10 min, the holding current was varied to obtain an RI. A3, The traces are averages of five successive EPSCs recorded at 40, 0, and −70 mV for the LTP pathway (top), the control pathway (middle), and the two inputs normalized at 40 mV and superimposed (bottom). Note the absence of any alteration. The histogram plots the RI calculated for 21 neurons from 10 animals. Data from the individual neurons are shown in the plot to the right. Dotted lines connect the RI measurements for the control and tetanized inputs for each neuron. B1–B3, Equivalent records for the LTP induced by sTBS. Note that the whole-cell recording (WC) was obtained after the second TBS. This was necessary because of the lability of LTP washout and to enable the RI measurements to be made after a similar length of whole-cell dialysis. Note that there is a small but discernible alteration in AMPAR rectification (quantification of 23 neurons from 11 animals, **p < 0.01, paired Student's t test). Con, Control.
Rolipram-enhanced LTP is associated with an alteration in rectification of AMPARs
Last, we determined whether the rolipram-enhanced LTP is also associated with an alteration in AMPAR rectification. As illustrated in Fig. 7A, a wTBS resulted in a modest, input-specific LTP that was of similar magnitude in the field (Fig. 7A1) and patch-clamp (Fig. 7A2) recordings. The rectification was assessed 10 min after the TBS, and again there was no difference between the potentiated (0.87 ± 0.03, n = 20, 10 animals) and control (0.87 ± 0.02, p = 0.823, paired Student's t test; Fig. 7A3) inputs. In interleaved slices treated with rolipram, there was a larger LTP that was similar between the field (Fig. 7B1) and patch-clamp (Fig. 7B2) recordings, and this was associated with a small, but highly significant, change in the RI of AMPARs (0.83 ± 0.02 vs 0.89 ± 0.02, n = 24, 10 animals, p = 0.004, paired Student's t test; Fig. 7B3), again consistent with the insertion of CP-AMPARs.
Figure 7.

Alterations in AMPAR rectification associated with rolipram-enhanced LTP. A1, Extracellular recordings were performed as described in Figure 6 except that wTBS was used for LTP induction. A2, The simultaneous whole-cell recording from a neuron in the same region of the slice. After 10 min, the holding current was varied to obtain the RI. A3, The traces are averages of five successive EPSCs recorded at 40, 0, and −70 mV for the LTP pathway (top), the control pathway (middle), and the two inputs normalized at 40 mV and superimposed (bottom). Note the absence of any alteration. The histogram plots the RI calculated for 20 neurons from 10 animals. Individual data points are shown in the plot on the right. B1–B3, Equivalent records for the LTP induced by wTBS in the presence of rolipram. Note that there is a small but discernible alteration in AMPAR rectification (quantification of 24 neurons from 10 animals, **p < 0.01, paired Student's t test). Con, Control.
Discussion
In the present study, we have found that the synaptic recruitment of CP-AMPARs is required for the generation of the PKA-dependent form of LTP that is induced using an sTBS protocol. We base this conclusion on the effects of three distinct antagonists of these receptors on sLTP and the finding that there is a small, but highly significant, increase in AMPAR inward rectification. Interestingly, CP-AMPARs are specifically involved in sLTP because, in interleaved experiments, the same blockers had no effect whatsoever on cLTP and there was no change in AMPAR rectification.
LTP can be divided into mechanistically distinct forms
LTP at CA3–CA1 synapses comprises a family of different, mechanistically distinct, forms of potentiation. In the present study, we have observed three separate forms of NMDAR-dependent synaptic potentiation. One form, STP, was not specifically studied in the present investigation. The other two forms of potentiation are both able to persist for many hours in vitro, but they can be distinguished on the basis of their sensitivity to inhibitors of PKA. In response to a cTBS, we found that the LTP was completely insensitive to inhibitors of PKA and protein synthesis (data not shown). This form of LTP, termed here as cLTP, is equivalent to that which has been referred to previously as E-LTP, a termed coined because, in some studies, the LTP decayed back to baseline within ∼1 h. In contrast, in response to an sTBS, we observed an LTP that was sensitive to inhibitors of PKA and protein synthesis (data not shown). This form of LTP, termed here as sLTP, is equivalent to that commonly referred to as L-LTP. Because we observe that the PKA-independent form of LTP can last for many hours and that the PKA-dependent form can be induced rapidly (Bortolotto et al., 2000; Park et al., 2014), we have used the operational terms (cLTP and sLTP) rather than descriptors “early” and “late” to avoid any ambiguity.
We defined the PKA-sensitive component using KT. Although KT may have off-target effects, the sensitivity of sLTP to a variety of different PKA inhibitors and to genetic deletion of PKA subunits clearly defines sLTP as dependent on PKA (Frey et al., 1993; Qi et al., 1996; Woo et al., 2003). Additional support for this is provided by the observation that a similar KT-sensitive component of LTP was induced when a wTBS was delivered in the presence of rolipram, which enhances the activation of PKA by inhibiting the breakdown of cAMP (Barad et al., 1998).
Interestingly, the magnitude of cLTP and sLTP, induced by the same number (i.e., 75) of stimuli, were similar. This suggests that, when the PKA-dependent from of LTP is induced, there is suppression of the PKA-independent form (which would otherwise be additive). It seems likely that the small LTP induced by sTBS in the presence of a PKA or CP-AMPAR inhibitor is the residual PKA-independent form, although the possibility it represents an incompletely inhibited PKA-dependent form cannot be excluded. Additional work will be required to address this issue.
A role for CP-AMPARs in the induction of the PKA-sensitive form of LTP
We have found that sLTP is highly sensitive to inhibitors of CP-AMPARs and is associated with an increase in AMPAR rectification. In contrast, cLTP was insensitive to these inhibitors and was not associated with any change in AMPAR rectification. These findings may reconcile a previous controversy over the role of CP-AMPARs in the induction of LTP at these synapses. Retrospectively, one may conclude that the study that reported an involvement of CP-AMPARs (Plant et al., 2006) studied predominantly the PKA-dependent form of LTP, whereas that which saw no evidence for a role (Adesnik and Nicoll, 2007) studied the PKA-independent form. Superficially, these studies were similar, but there are many unknown variables that could account for the differences between studies. In this context, it is interesting to note that acute stress recruits CP-AMPARs, and these are able to magnify LTP induced by compressed induction protocols (Whitehead et al., 2013). Therefore, the level of stress experienced by the animals before the preparation of slices may have been an important factor.
Based on our experiments, it is not possible to determine whether the CP-AMPARs responsible for sLTP are GluA2-lacking or contain unedited GluA2(Q) subunits, because both are sensitive to inhibitors, such as IEM (Schlesinger et al., 2005). However, the majority of GluA2 subunits undergo mRNA editing to the GluA2(R) form (Sommer et al., 1991), and so it is highly likely that the CP-AMPARs are GluA2 lacking.
It is possible to estimate the proportion of the synaptic current that is mediated by CP-AMPARs after the induction of sLTP. In GluA2 knock-out mice, in which all the AMPARs are of the CP variety, the RI, estimated using outside-out patches, is 0.16, whereas the value in wild-type mice is 0.83 (Lu et al., 2009). Assuming that this equates to 100 and 0% CP-AMPARs, respectively, then in the present study, the RI change would be explained by the newly inserted CP-AMPARs comprising ∼10% of the total synaptic current. The acute effect of the polyamine inhibitors, assessed 10–20 min after the third TBS, are consistent with this level of contribution of CP-AMPARs to the total AMPA receptor-mediated synaptic response. However, these newly inserted CP-AMPARs seem to be required for the stabilization of sLTP, such that, if they are blocked during the critical time window, LTP decays toward baseline. In additional support of this role of CP-AMPARs, which outlasts the initial induction trigger, we found that stopping stimulation after TBS resulted in a smaller magnitude of LTP. This observation is consistent with the notion that the CP-AMPARs are activated by low-frequency transmission over a period of tens of minutes after the TBS or HFS (Plant et al., 2006) and that this is required for the stabilization of sLTP. How the activation of CP-AMPARs is able to selectively trigger sLTP under these conditions remains to be determined. Interestingly, in GluA2 knock-out mice, it has been shown that the CP-AMPAR-dependent form of LTP requires activation of phosphatidylinositol 3-kinase and MAPKs (Asrar et al., 2009). Additional work is required to assess the role of these kinases in the induction of sLTP.
The requirement for the activation of CP-AMPARs was only transient because, 1 h after the last TBS, IEM had no effect on synaptic transmission. This suggests that CP-AMPARs, although necessary for the induction of sLTP and contributing to the initial expression of the sLTP, are not involved in the later expression of sLTP. As suggested previously (Plant et al., 2006), GluA2-lacking AMPARs are presumably replaced by GluA2-containing AMPARs over time.
On the mechanisms underlying two forms of LTP
We propose that the first episode of TBS (or HFS) does two distinct things. First, it induces cLTP via a process that does not require PKA. Rather it may involve, for example, CaMKII-mediated alterations in AMPAR function and/or number (Derkach et al., 1999; Poncer et al., 2002; Kristensen et al., 2011). Second, and concurrently, it primes for sLTP. The priming involves the activation of PKA, presumably via a pathway involving Ca2+-sensitive adenylyl cyclase to generate cAMP, which leads to the rapid membrane insertion of GluA2-lacking AMPARs. These may be GluA1 homomers or GluA1/3 heteromers and could involve the PKA-dependent phosphorylation of GluA1 at Ser845 (Roche et al., 1996; Oh et al., 2006; Man et al., 2007). Such a phosphorylation has been shown to lead to the insertion of CP-AMPARs to the plasma membrane (He et al., 2009; Clem and Huganir, 2010). Although it has been shown that LTP is unaffected in GluA1S845A mice, the timing of the stimuli was such that only cLTP would have been expected (Lee et al., 2010). However, if the first priming stimulus causes membrane insertion of CP-AMPARs, then why do inhibitors of CP-AMPARs not affect cLTP? The simplest explanation is that PKA drives CP-AMPARs into the plasma membrane but not into the synapse. Rather, this requires a subsequent TBS (or HFS), with a time interval of at least a couple of minutes, to drive the CP-AMPARs into the synapse. Consistent with this model, it has been shown that increasing cAMP, with for example forskolin, requires NMDAR activation to facilitate synaptic transmission (Otmakhov et al., 2004). Therefore, this predicts that, to induce sLTP, NMDARs are required to, first, activate PKA during the priming stimulus to drive AMPARs into the plasma membrane at extra synaptic sites and, second, during the subsequent TBS, to drive these CP-AMPARs into the synapse. Because they are also required to induce STP (Volianskis et al., 2013) and cLTP, NMDARs have multiple actions during the induction of LTP, a scenario far more complex than envisaged originally (Collingridge et al., 1983; Bliss and Collingridge, 1993).
Footnotes
This work was supported by the Medical Research Council (G.L.C.), ERC Grants RD1819 and RQ8898 (G.L.C.), the Canadian Institute for Health Research (CIHR) Michael Smith Chair in Neurosciences and Mental Health (M.Z.), Canada Research Chair (M.Z.), CIHR Operating Grant MOP-124807 (M.Z.), Natural Sciences and Engineering Research Council of Canada Discovery Grant RGPIN 402555 (M.Z.), the Azrieli Neurodevelopmental Research Program and Brain Canada (M.Z.), and the National Honor Scientist Program of the National Research Foundation funded by the Korea government (B.-K.K.).
The authors declare no competing financial interests.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License Creative Commons Attribution 4.0 International, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Abraham WC. How long will long-term potentiation last? Philos Trans R Soc Lond B Biol Sci. 2003;358:735–744. doi: 10.1098/rstb.2002.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adesnik H, Nicoll RA. Conservation of glutamate receptor 2-containing AMPA receptors during long-term potentiation. J Neurosci. 2007;27:4598–4602. doi: 10.1523/JNEUROSCI.0325-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson WW, Collingridge GL. Capabilities of the WinLTP data acquisition program extending beyond basic LTP experimental functions. J Neurosci Methods. 2007;162:346–356. doi: 10.1016/j.jneumeth.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Asrar S, Zhou Z, Ren W, Jia Z. Ca2+ permeable AMPA receptor induced long-term potentiation requires PI3/MAP kinases but not Ca/CaM-dependent kinase II. PLoS One. 2009;4:e4339. doi: 10.1371/journal.pone.0004339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barad M, Bourtchouladze R, Winder DG, Golan H, Kandel E. Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-lasting long-term potentiation and improves memory. Proc Natl Acad Sci U S A. 1998;95:15020–15025. doi: 10.1073/pnas.95.25.15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Gardner-Medwin AR. Long-lasting potentiation of synaptic transmission in the dentate area of the unanaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:357–374. doi: 10.1113/jphysiol.1973.sp010274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bortolotto ZA, Collingridge GL. A role for protein kinase C in a form of metaplasticity that regulates the induction of long-term potentiation at CA1 synapses of the adult rat hippocampus. Eur J Neurosci. 2000;12:4055–4062. doi: 10.1046/j.1460-9568.2000.00291.x. [DOI] [PubMed] [Google Scholar]
- Choi JH, Park P, Baek GC, Sim SE, Kang SJ, Lee Y, Ahn SH, Lim CS, Lee YS, Collingridge GL, Kaang BK. Effects of PI3Kγ overexpression in the hippocampus on synaptic plasticity and spatial learning. Mol Brain. 2014;7:78. doi: 10.1186/s13041-014-0078-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RL, Huganir RL. Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science. 2010;330:1108–1112. doi: 10.1126/science.1195298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, McLennan H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol. 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J, Francis AA, Jones AW, Watkins JC. 2-amino-5-phosphonovalerate (2APV), a potent and selective antagonist of amino acid-induced and synaptic excitation. Neurosci Lett. 1981;21:77–81. doi: 10.1016/0304-3940(81)90061-6. [DOI] [PubMed] [Google Scholar]
- Derkach V, Barria A, Soderling TR. Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci U S A. 1999;96:3269–3274. doi: 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- He K, Song L, Cummings LW, Goldman J, Huganir RL, Lee HK. Stabilization of Ca2+-permeable AMPA receptors at perisynaptic sites by GluR1-S845 phosphorylation. Proc Natl Acad Sci U S A. 2009;106:20033–20038. doi: 10.1073/pnas.0910338106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learn Mem. 1994;1:74–82. doi: 10.1101/lm.1.1.74. [DOI] [PubMed] [Google Scholar]
- Huang YY, Nguyen PV, Abel T, Kandel ER. Long-lasting forms of synaptic potentiation in the mammalian hippocampus. Learn Mem. 1996;3:74–85. doi: 10.1101/lm.3.2-3.74. [DOI] [PubMed] [Google Scholar]
- Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna FA, Velumian A, MacDonald J, Carlen P, Abramow-Newerly W, Roder J. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron. 1996;17:945–956. doi: 10.1016/S0896-6273(00)80225-1. [DOI] [PubMed] [Google Scholar]
- Kim M, Huang T, Abel T, Blackwell KT. Temporal sensitivity of protein kinase A activation in late-phase long term potentiation. PLoS Comput Biol. 2010;6:e1000691. doi: 10.1371/journal.pcbi.1000691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen AS, Jenkins MA, Banke TG, Schousboe A, Makino Y, Johnson RC, Huganir R, Traynelis SF. Mechanism of Ca2+/calmodulin-dependent kinase II regulation of AMPA receptor gating. Nat Neurosci. 2011;14:727–735. doi: 10.1038/nn.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, He K, Song L, Huganir RL. Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol. 2010;103:479–489. doi: 10.1152/jn.00835.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron. 2009;62:254–268. doi: 10.1016/j.neuron.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man HY, Sekine-Aizawa Y, Huganir RL. Regulation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc Natl Acad Sci U S A. 2007;104:3579–3584. doi: 10.1073/pnas.0611698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, Soderling TR. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem. 2006;281:752–758. doi: 10.1074/jbc.M509677200. [DOI] [PubMed] [Google Scholar]
- Otmakhov N, Khibnik L, Otmakhova N, Carpenter S, Riahi S, Asrican B, Lisman J. Forskolin-induced LTP in the CA1 hippocampal region is NMDA receptor dependent. J Neurophysiol. 2004;91:1955–1962. doi: 10.1152/jn.00941.2003. [DOI] [PubMed] [Google Scholar]
- Park P, Volianskis A, Sanderson TM, Bortolotto ZA, Jane DE, Zhuo M, Kaang BK, Collingridge GL. NMDA receptor-dependent long-term potentiation comprises a family of temporally overlapping forms of synaptic plasticity that are induced by different patterns of stimulation. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130131. doi: 10.1098/rstb.2013.0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci. 2006;9:602–604. doi: 10.1038/nn1678. [DOI] [PubMed] [Google Scholar]
- Poncer JC, Esteban JA, Malinow R. Multiple mechanisms for the potentiation of AMPA receptor-mediated transmission by α-Ca2+/calmodulin-dependent protein kinase II. J Neurosci. 2002;22:4406–4411. doi: 10.1523/JNEUROSCI.22-11-04406.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi M, Zhuo M, Skålhegg BS, Brandon EP, Kandel ER, McKnight GS, Idzerda RL. Impaired hippocampal plasticity in mice lacking the Cβ1 catalytic subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1996;93:1571–1576. doi: 10.1073/pnas.93.4.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/S0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- Schlesinger F, Tammena D, Krampfl K, Bufler J. Two mechanisms of action of the adamantane derivative IEM-1460 at human AMPA-type glutamate receptors. Br J Pharmacol. 2005;145:656–663. doi: 10.1038/sj.bjp.0706233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer B, Köhler M, Sprengel R, Seeburg PH. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell. 1991;67:11–19. doi: 10.1016/0092-8674(91)90568-J. [DOI] [PubMed] [Google Scholar]
- Volianskis A, Bannister N, Collett VJ, Irvine MW, Monaghan DT, Fitzjohn SM, Jensen MS, Jane DE, Collingridge GL. Different NMDA receptor subtypes mediate induction of long-term potentiation and two forms of short-term potentiation at CA1 synapses in rat hippocampus in vitro. J Physiol. 2013;591:955–972. doi: 10.1113/jphysiol.2012.247296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead G, Jo J, Hogg EL, Piers T, Kim DH, Seaton G, Seok H, Bru-Mercier G, Son GH, Regan P, Hildebrandt L, Waite E, Kim BC, Kerrigan TL, Kim K, Whitcomb DJ, Collingridge GL, Lightman SL, Cho K. Acute stress causes rapid synaptic insertion of Ca2+-permeable AMPA receptors to facilitate long-term potentiation in the hippocampus. Brain. 2013;136:3753–3765. doi: 10.1093/brain/awt293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo NH, Duffy SN, Abel T, Nguyen PV. Temporal spacing of synaptic stimulation critically modulates the dependence of LTP on cyclic AMP-dependent protein kinase. Hippocampus. 2003;13:293–300. doi: 10.1002/hipo.10086. [DOI] [PubMed] [Google Scholar]