

Abstract
The homozygous loss of the survival motor neuron 1 (SMN1) gene is the primary cause of spinal muscular atrophy (SMA), a neuromuscular degenerative disease. A genetically similar gene, SMN2, which is not functionally equivalent in all SMA patients, modifies the clinical SMA phenotypes. We analyzed the methylation levels of 4 CpG islands (CGIs) in SMN2 in 35 Chinese children with SMA by MassARRAY. We found that three CpG units located in CGI 1 (nucleotides (nt) −871, −735) and CGI 4 (nt +999) are significantly hypomethylated in SMA type III compared with type I or II children after receiving Bonferroni correction. In addition to the differentially methylated CpG unit of nt −871, the methylation level of the nt −290/−288/−285 unit was negatively correlated with the expression of SMN2 full-length transcripts (SMN2-fl). In addition, the methylation level at nt +938 was inversely proportional to the ratio of SMN2-fl and lacking exon 7 transcripts (SMN2-(7, fl/(7), and was not associated with the SMN2 transcript levels. Thus, we can conclude that SMN2 methylation may regulate the SMA disease phenotype by modulating its transcription.
Keywords: CpG island, Methylation, Survival motor neuron 2 (SMN2), Spinal muscular atrophy
1. Introduction
Degeneration of motor neurons in the anterior horn of the spinal cord causes progressive muscle weakness and atrophy in the autosomal recessive disorder proximal spinal muscular atrophy (SMA). The disease forms are categorized by order of increasing age and decreasing clinical severity as follows: childhood onset of SMA type I (MIM #253300) SMA type II (MIM #253550) SMA type III (MIM #253400) and adult onset of SMA type IV (MIM #271150). SMA is caused by deletions or mutations in the survival motor neuron 1 (SMN1) gene on chromosome 5q. The copy gene SMN2 varying in a nucleotide in exon 7 modifies the SMA clinical symptoms. For example patients with deleted or altered SMN1 often have increased copies of SMN2 which contributes to SMA phenotypes. The SMN2 copy number inversely correlates with the SMA severity (Prior et al. 2009). However siblings with the identical SMN1 genetics and SMN2 copies show marked clinical discrepancies indicating that epigenetic modification may control the individual variations in the SMN2 function (Bernal et al. 2011). In fact SMA type I and III patients have two differentially methylated CpG sites (CpGs) which co-localize within the first transcriptional start site of SMN2 correlating with the disease severity (Hauke et al. 2009). Thus DNA methylation regulates gene expression by altering the transcriptional activity and is responsible for non-equivalent SMN2 functions. However the role of SMN2 DNA methylation in the SMA progression particularly in young patients still needs further elucidation.
Therefore in this study we analyzed the DNA methylation levels of SMN2 in 35 children with SMA and evaluated the correlation between the methylation modification and childhood onset of SMA progression. This is a second study being conducted after Hauke et al. (2009) completed their study on SMN2 promoter methylation in connection to SMA severity.
2. Materials and methods
2.1. Subjects
Thirty-five Chinese patients with SMA determined by homozygous exon 7 deletion in SMN1 were enrolled with the Department of Medical Genetics in the Capital Institute of Pediatrics (Beijing China). Informed consent was obtained from the minors’ parents and the Ethical Committee of the Capital Institute of Pediatrics approved the study protocol.
2.2. DNA extraction and SMN copy number
Genomic DNA was extracted from peripheral blood samples by proteinase K digestion and phenol/chloroform extraction. The SMN1 and SMN2 copy numbers were determined by multiplex ligation-dependent probe amplification (MLPA) (MRC-Holland Amsterdam the Netherlands) according to the manufacturer’s instructions.
2.3. Bisulfite conversion and SMN2 DNA methylation analyses
For epigenetic analyses, unmethylated cytosines were bisulfite converted, using EpiTect Bisulfite Kit (Qiagen, Hilden, Germany), following the manufacturer′s protocol. Four polymerase chain reaction (PCR) fragments were cloned into T1 vectors, using a pEASYTM-T1 Cloning Kit (TransGen Biotech, Beijing, China). Subsequently, ten positive clones were sequenced by ABI 3730 XL (Life Technologies, Carlsbad, USA) to evaluate the methylation status of four CpG islands (CGIs) in SMN2
Quantitative DNA methylation analysis was performed using a MassARRAY platform (Sequenom, San Diego, CA, USA). Four CGIs were amplified by nested PCR using AmpliTag Gold DNA polymerase (Life Technologies, Carlsbad, USA) and previously described primers with some modifications (Hauke et al., 2009). Each reverse primer for the inner PCR contained a T7 promoter sequence for in vitro transcription. The PCR conditions were as follows: 95 °C for 5 min, followed by 40 amplification cycles (95 °C for 15 s, 52−60 °C for 45 s, 72 °C for 60 s) and then a final extension for 5 min at 72 °C. Following dephosphorylation and in vitro transcription, the samples were purified through a Clean Resin (Sequenom, San Diego, CA, USA), according to the manufacturer’s instructions. EpiTyper software (Sequenom, San Diego, CA, USA) analyzed and generated methylation data on the CpG-positive fragments. The primers used for bisulfite sequencing and MassARRAY are shown in Tables S1 and S2.
2.4. RNA extraction and SMN2 expression level
Total RNA was extracted from the peripheral blood using an RNAsimple Total RNA Kit (Tiangen, Beijing, China) and reverse-transcribed by an Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen, Carlsbad, CA, USA). Then, real-time PCR determined the transcript levels using the following primer pairs: (1) full-length SMN2 (SMN2-fl), 5'-TAC ATG AGT GGC TAT CAT ACT GGC TA-3' and 5'-AAT GTG AGC ACC TTC CTT CTT TT-3'; (2) truncated SMN2 lacking exon 7 (SMN2-(7), 5'-CAT GGT ACA TGA GTG GCT ATC ATA C-3' and 5'-AGT GGT GTC ATT TAG TGC TGC TCT A-3'. TaqMan MGB probes, 5'-FAM-TAT GGG TTT TAG ACA AAA-MGB-NFQ-3' and 5'-FAM-CTA TTA TAT GGA AAT GCT GG-MGB-NFQ-3', were used to specifically detect SMN2-fl transcripts and SMN2-(7 transcripts, respectively. Overall, the SMN2 transcript levels were calculated as the sum of the SMN2-fl and SMN2-(7 transcripts. Endogenous reference primers and previously described TaqMan probes (Tiziano et al., 2010) were used for the internal control, glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The construction of a standard curve and thermal cycling conditions were adapted from Qu et al. (2012). The quantities of SMN2-fl transcript, SMN2-(7 transcript, and GAPDH were verified against the appropriate standard curve. All samples were analyzed in triplicate.
2.5. Statistical analysis
A Kolmogorov-Smirnov test was used to assess the normal distribution for all variables. The methylation levels of SMN2 CGIs and units in different groups were compared using an analysis of variance (ANOVA) or Brown-Forsythe test when appropriate, and followed by a least significant difference (LSD) or Tamhane’s T2 post-hoc comparison test. The correlation between the CpG methylation and SMN2 transcript levels was determined by Spearman correlation analysis. Statistical analyses were performed using SPSS 19.0 for Windows (SPSS Inc., Chicago, IL, USA). P<0.05 was considered to be statistically significant. However, after Bonferroni correction, the statistically significant P-value should be adjusted based on the number of comparing units.
3. Results
3.1. Subjects’ characteristics
Thirty-five SMN1-deleted patients were all carrying three copies of SMN2, including 12 type I, 15 type II, and 8 type III samples. The ages of these patients ranged from 3 months to 5.5 years (mean, (1.4±1.3) years) and the male to female ratio was 19:16 (Table 1). Among them, 3 of type I, 13 of type II, and 8 of type III patients were evaluated for the related association between SMN2 methylation and transcript levels. The ages ranged from 3 months to 5.5 years (mean, (1.8±1.4) years) and the male to female ratio was 12:12.
Table 1.
Characteristics of SMA children and SMN2 methylation according to their clinical types
| Type | Age (year) | Gender male/female | Methylation (%) |
|||
| CGI 1 | CGI 2 | CGI 3 | CGI 4 | |||
| SMA I (n=12) | 0.6±0.5 | 8/4 | 76.7±8.6 | 59.9±5.0 | 13.5±4.6 | 70.0±3.8 |
| SMA II (n=15) | 1.3±1.1 | 7/8 | 79.6±3.9 | 58.6±3.5 | 13.1±3.1 | 73.4±6.3 |
| SMA III (n=8) | 2.9±1.3 | 4/4 | 77.8±3.1 | 56.8±2.8 | 12.8±1.7 | 68.0±1.7 |
|
| ||||||
| P-value | 0.000 | 0.558 | 0.453 | 0.236 | 0.916 | 0.032 |
| Adjusted P-value | 0.474 | 0.098 | 0.824 | 0.014 | ||
Results are shown as mean±standard deviation (SD) unless otherwise stated. Statistically significant results are shown in bold. The P-value was obtained by performing an ANOVA test or chi-square test when appropriate. The adjusted P-value was obtained by an analysis of covariance and age was considered as the co-variable
3.2. SMN2 CGIs methylation pattern
Putative CGIs within the genomic region, 3000 nucleotides (nt) upstream and downstream of the translational SMN2 start site, were predicted through a CGI finder algorithm (http://www.EBI.ac.uk/emboss). For the four CGIs, 1–4 extended from nt −896 to −645, nt −469 to −247, nt −151 to +295, and nt +844 to +1146, respectively.
To identify the SMN2 CGIs methylation in Chinese SMA children, the bisulfite-treated DNA derived from the peripheral blood samples of three SMN1-deleted subjects with SMA types I, II, and III (SMN2 copy number=3) were examined by cloning and sequencing. The results demonstrated that the hypermethylations of CGIs 1 and 4 (mean, (98.67±1.15)% and (89.33±3.06)%, respectively) were independent of the SMA type. CGI 2 had intermediate methylation ((47.33±2.89)%), but CGI 3 was hypomethylated ((0.67±0.29)%) (Fig. 1).
Fig. 1.

Methylation analysis of four SMN2 CGIs by bisulfite treatment, cloning, and sequencing
Three DNA samples were isolated from the peripheral blood of the SMA I, II, and III patients. Ten independent clones for each CGI of each patient were analyzed for methylation levels. Full circles represent methylated CpGs, while empty circles correspond to unmethylation
3.3. SMN2 CGIs methylation correlation to SMA type, age, and gender
We subsequently employed a MassARRAY to examine the degree of methylation in SMN2 CGIs from the 35 SMA children. The few closely located CpG dinucleotides were measured as one unit. Methylation was measured at 9, 8, 26, and 12 CpG units in CGIs 1, 2, 3, and 4, respectively. Consistent with the results of cloning and sequencing, the MassARRAY revealed the same methylation pattern: CGIs 1 and 4 were hypermethylated and CGI 3 was hypomethylated, while CGI 2 was at the intermediate level of the methylation (Fig. 2). There were no differences in methylation status from CGI 1 to CGI 3 by the SMA type. However, the status of CGI 4 was still different among the SMA types after consideration of the patients’ ages. The patients with SMA type II had significantly higher CGI 4 methylation than the SMA type III patients (P=0.017; Table 1). Gender did not influence the methylation status in any SMA patients (data not shown, P>0.05).
Fig. 2.

Methylation analysis of four SMN2 CGIs by MassARRAY
Black triangles represent mean methylation levels of each CpG unit within each SMN2 CGI in the SMA patients. The mean methylation levels of each SMN2 CGI are indicated by a horizontal line: (a) CGI 1: 76.7%, CGI 2: 59.9%, CGI 3: 13.5%, CGI 4: 70.0%; (b) CGI 1: 79.6%, CGI 2: 58.6%, CGI 3: 13.1%, CGI 4: 73.4%; (c) CGI 1: 77.8%, CGI 2: 56.8%, CGI 3: 12.8%, CGI 4: 68.0%
In this current study, thirteen CpG units of SMN2 CGIs, including 19 CpGs (1–4 CpGs per unit) were detected that significantly varied in their methylation levels based on SMA classification (Table 2). Among these, eight CpG units located in CGI 1 (nt −871, −735), CGI 2 (nt −290/−288/−285), and CGI 4 (nt +890/+894/+898/+900, +938, +988, +999, +1064) had lower methylation levels in SMA type III compared with type I or II patients (Fig. 3). After Bonferroni correction, there were three CpG units (nt −871, −735, and +999) that still remained differently methylated (P<0.001).
Table 2.
Differentially methylated units in SMN2 among SMA types
| Methylation unit (dinucleotide position, nt) | Methylation level (%) |
P-value | ||
| SMA I (n=12) | SMA II (n=15) | SMA III (n=8) | ||
| CGI 1_2 (−871) | 94.1±5.3 | 85.7±5.6 | 80.3±4.4 | 0.000 |
| CGI 1_8 (−735) | 93.7±5.1 | 86.6±7.7 | 82.0±4.4 | 0.001 |
| CGI 2_9 (−290/−288/−285) | 54.2±5.7 | 50.5±5.6 | 48.3±3.4 | 0.046 |
| CGI 3A_7 (−81) | 3.2±3.3 | 6.8±3.4 | 2.3±1.7 | 0.003 |
| CGI 3B_5 (+160) | 1.3±1.1 | 2.2±1.2 | 1.1±0.9 | 0.046 |
| CGI 3B_10 (+213) | 8.0±1.6 | 9.4±2.5 | 7.0±1.6 | 0.032 |
| CGI 3B_11 (+228/+237) | 8.1±3.5 | 11.6±2.6 | 8.5±2.3 | 0.008 |
| CGI 4_2 (+855) | 21.5±8.6 | 53.5±18.2 | 39.3±8.6 | 0.000* |
| CGI 4_2 (+890/+894/+898/+900) | 83.1±6.5 | 79.4±4.0 | 76.4±1.4 | 0.010* |
| CGI 4_3 (+938) | 69.6±4.1 | 73.7±9.9 | 63.4±5.1 | 0.006* |
| CGI 4_6 (+988) | 49.6±6.2 | 50.1±11.6 | 36.5±7.3 | 0.004 |
| CGI 4_7 (+999) | 78.1±4.2 | 75.7±5.9 | 62.2±4.3 | 0.000 |
| CGI 4_10 (+1064) | 83.5±7.3 | 81.9±10.8 | 69.6±6.4 | 0.003 |
Results are shown as mean±SD. P-values were obtained using an ANOVA test or a Brown-Forsythe test (*)
Fig. 3.
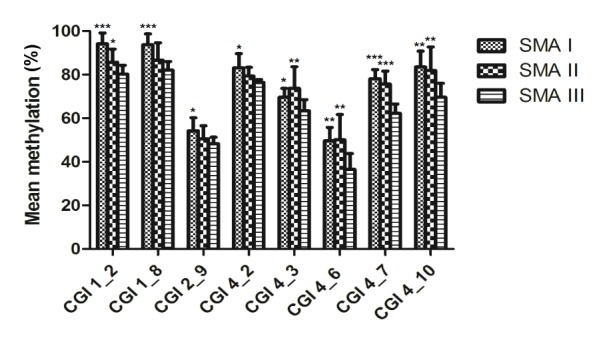
Comparison of SMN2 methylation levels by SMA types
Eight CpG units had different methylation frequencies in type I or II compared with type III SMA patients (* P<0.05, ** P<0.01, and *** P<0.001 significantly differ). The bar graph depicts the mean methylation levels (±SD) of each CpG unit within the respective CGIs and each CpG unit includes one to four CpGs. The CpG unit corresponding dinucleotide position is as follows: CGI 1_2: nt −871, CGI 1_8: nt −735, CGI 2_9:nt −290/−288/−285, CGI 4_2: nt +890, CGI 4_3: nt +938, CGI 4_6: nt +988, CGI 4_7: nt +999, and CGI 4_10: nt +1064
3.4. SMN2 CGIs methylation frequencies and transcript levels
To determine whether the methylation status of SMN2 affects its transcript level, we evaluated SMN2-fl and SMN2-(7 transcripts in 24 SMN1-deleted types I, II, and III SMA patients carrying three SMN2 copies (Table 3).
Table 3.
Correlation between differentially methylated units and SMN2 transcription
| Transcription | nt −871 |
nt −290/−288/−285 | nt +938 |
|||
| r | P-value | r | P-value | r | P-value | |
| SMN2-fl | −0.550 | 0.007 | −0.500 | 0.015 | −0.383 | 0.071 |
| SMN2-Δ7 | −0.512 | 0.012 | −0.141 | 0.522 | −0.024 | 0.913 |
| SMN2-fl+ SMN2-Δ7 | −5.990 | 0.003 | −0.355 | 0.097 | −0.109 | 0.621 |
| fl/Δ7 | 0.077 | 0.726 | −0.206 | 0.346 | −0.515 | 0.012 |
Spearman correlation analysis was used to evaluate the correlation between differentially methylated units and SMN2 transcription
Correlation analysis revealed that there was no association between SMN2 CGIs methylation levels and total SMN2 transcript levels (SMN2-fl+SMN2-(7) (Crawford et al., 2012). However, 3/8 differentially methylated CpG units correlated with different SMN2 transcripts after applying a Bonferroni correction (P<0.017). The CGI 1_2 (nt −871) methylation frequency was inversely proportional to the SMN2-fl, SMN2-(7, and total SMN2 transcript levels (r=−0.550, P=0.007; r=−0.512, P=0.012; r=−5.990, P=0.003). Meanwhile, CGI 2_9 (nt −290/−288/−285) and CGI 4_3 (nt +938) inversely correlated with SMN2-fl (r=−0.500, P=0.015) and the ratio reflecting SMN2 exon 7 inclusion (fl/(7, r=−0.515, P=0.012), respectively.
4. Discussion
Despite identifying the causative gene for SMA, the contribution of a significantly homologous gene, SMN2 complicates the molecular interactions leading to SMA phenotypes. Both the SMN2 copy number and epigenetic modification underly the SMA clinical symptoms. Methylation levels of seven CpGs upstream and downstream to the SMN2 translational start site significantly varied between the types I and III SMA patients. And CpGs at position nt −296 and −290 were more hypermethylated in SMA type I than type III patients. Moreover, their methylation levels negatively correlated with the activity of the first transcriptional start site of SMN2 (Hauke et al., 2009). These findings suggest that SMN2 methylation may modify the predicted SMA phenotypes. Because DNA methylation is affected by an individual’s genetic makeup, age, gender, and lifestyle (Murrell et al., 2005; Schumacher and Petronis, 2006; Barrès et al., 2012), we analyzed the SMN2 methylation status upstream and downstream of the translational start site in 35 Chinese SMA children.
The aim of this study was to clarify whether DNA methylation affects SMA clinicopathology by altering the expression levels of SMN2. In accordance to a previous report (Hauke et al., 2009), four CGIs predicted the upstream and downstream of the SMN2 translational start site, which had different methylation patterns, wherein the CGIs 1 and 4 were hypermethylated, CGI 2 was moderately methylated and CGI 3 was hypomethylated. These results suggest evolutionary conservation of the methylation pattern of CGIs proximal to the SMN2 translational start site, which is independent of the tissue specificity and race, and has negligible inter-individual variability. It is obvious that the age distributions in SMA types are significantly different due to the criteria for SMA clinical classification. In our study, with or without consideration of patients’ ages, the statistical results for the average methylation levels from CGIs 1–4 were the same. Additionally, we also found that gender did not affect the methylation levels of any CGIs. Taken together, our data suggested that neither age nor gender might affect CGI methylation levels. Moreover, our study showed that although the average methylation levels of CGI 4 in children with type III SMA were significantly lower than those in children with type II SMA (P=0.017), this difference was no longer significant after applying the Bonferroni correction. Therefore, it should be further confirmed in a larger sample study.
In this study, 13 differentially methylated units were detected in the four CGIs in the SMN2 gene, and 8/13 CpG units associated with the disease severity. For example, the methylation levels in type I SMA were significantly higher than those in type III SMA (P<0.05). In comparison to that reported by Hauke et al. (2009), seven differentially methylated sites (nt −871, −695, −296, −290, +855, +988, and +1103), our data were consistent for nt −871, −290, and +988 results; however, the methylation levels at nt −695, −296, +855, and +1103 had insignificant differences. Moreover, our data demonstrated significantly higher methylation at nt −735, +890/+894/+898/+900, +938, +999, and +1064 in children with type I SMA than that in children with type III SMA. These results suggest that race may affect the methylation at specific loci despite conservation of methylation of each CGI in the SMN2 gene.
To determine the influence of SMN2 methylation on its transcription, we evaluated the correlation between SMN2 methylation and transcription in 24 children with types I–III SMA. The methylation level of individual CGI did not correlate with total SMN2 transcription levels. However, the correlation analysis of levels of differentially methylated units and SMN2 transcriptional levels indicated that at nt −871 and nt −290/−288/−285, the methylation levels negatively correlate with SMN2-fl transcript levels. Jan Hauke et al. (2009) indicated that the differentially methylated sites at nt −296 and nt −290 correlated with the SMA severity. However, in our study, we did not find differences in the methylation level at nt −296 among the different SMA types. Moreover, based on the other two adjacent sites along with nt −290 in the same differentially methylated unit (nt −290/−288/−285), it is suggested that the variations in methylation of this unit may not be very significantly caused by nt −290 differential methylation (0.001<P<0.05). Thus, the methylation levels at nt −871 and −290 most likely affect SMN2 transcription. Interestingly, methylation of nt +938, an intragenic methylation site located in SMN2 CGI 4, inversely correlated with the ratio of fl/Δ7, but not SMN2 transcript levels. Intragenic methylation is associated with alternative gene splicing (Jones, 2012; Maunakea et al., 2013), and this data suggest that hypermethylation at nt +938 might decrease the ratio of fl/Δ7 by promoting the alternative splicing of exon 7. However, we need to perform further experiments to confirm the underlying mechanism.
Despite the association of differentially methylated CpG units with SMA severity, the exact pathogenesis needs to be elucidated, and will be our focus in the future. For example, the methylation of other genes, such as CHML, ARHGAP22, CYTSB, CDK2AP1, and SLC23A2, also contributes to the SMA phenotype modification (Zheleznyakova et al., 2013). SMA clinicopathology may also be affected by other epigenetic modifications such as acetylation. In fact, histone deacetylase (HDAC) inhibitors can increase the in vitro transcript levels of full-length SMN2 proteins, which in turn affect SMA severity in animal models (Chang et al., 2001; Andreassi et al., 2004; Hahnen et al., 2006). In-depth studies of these modifying factors will improve our understanding of SMA pathogenesis, and allow for the development of effective therapeutic regimens.
In conclusion, our studies indicate that SMN2 methylation, particularly at nt −871 and −290, regulates SMA clinical phenotypes by possibly regulating SMN2 transcription. Moreover, nt +938 potentially affects SMN2 transcription by regulating the alternative splicing, which will be studied extensively in the near future.
Acknowledgements
We would like to thank the patients and their parents for their collaborations in this investigation. We would also like to thank Editage for providing editorial assistance.
List of electronic supplementary materials
Table S1 Primers for SMN2 CGIs cloning sequencing
Table S2 Primers for SMN2 nested PCR
Footnotes
Project supported by the National Natural Science Foundation of China (Nos. 81050034 and 81500979), the Research Foundation of the Capital Institute of Pediatrics (No. Fangxiang-2014-01), and the Beijing Talents Fund (No. 2014000021469G228)
Electronic supplementary materials: The online version of this article (http://dx.doi.org/10.1631/jzus.B1500072) contains supplementary materials, which are available to authorized users
Compliance with ethics guidelines: Yan-yan CAO, Yu-jin QU, Sheng-xi HE, Yan LI, Jin-li BAI, Yu-wei JIN, Hong WANG, and Fang SONG declare that they have no conflict of interest.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2008 (5). Informed consent was obtained from all patients for being included in the study.
References
- 1.Andreassi C, Angelozzi C, Tiziano FD, et al. Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur J Hum Genet. 2004;12(1):59–65. doi: 10.1038/sj.ejhg.5201102. [DOI] [PubMed] [Google Scholar]
- 2.Barrès R, Yan J, Egan B, et al. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012;15(3):405–411. doi: 10.1016/j.cmet.2012.01.001. (Available from: http://dx.doi.org/10.1016/j.cmet.2012.01.001) [DOI] [PubMed] [Google Scholar]
- 3.Bernal S, Also-Rallo E, Martínez-Hernández R, et al. Plastin 3 expression in discordant spinal muscular atrophy (SMA) siblings. Neuromuscul Disord. 2011;21(6):413–419. doi: 10.1016/j.nmd.2011.03.009. (Available from: http://dx.doi.org/10.1016/j.nmd.2011.03.009) [DOI] [PubMed] [Google Scholar]
- 4.Chang JG, Hsieh-Li HM, Jong YJ, et al. Treatment of spinal muscular atrophy by sodium butyrate. PNAS. 2001;98(17):9808–9813. doi: 10.1073/pnas.171105098. (Available from: http://dx.doi.org/10.1073/pnas.171105098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crawford TO, Paushkin SV, Kobayashi DT, et al. Pilot Study of Biomarkers for Spinal Muscular Atrophy Trial Group. Evaluation of SMN protein transcript and copy number in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. PLoS ONE. 2012;7(4):e33572. doi: 10.1371/journal.pone.0033572. (Available from: http://dx.doi.org/10.1371/journal.pone.0033572) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hahnen E, Eyüpoglu IY, Brichta L, et al. In vitro and ex vivo evaluation of second-generation histone deacetylase inhibitors for the treatment of spinal muscular atrophy. J Neurochem. 2006;98(1):193–202. doi: 10.1111/j.1471-4159.2006.03868.x. (Available from: http://dx.doi.org/10.1111/j.1471-4159.2006.03868.x) [DOI] [PubMed] [Google Scholar]
- 7.Hauke J, Riessland M, Lunke S, et al. Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Hum Mol Genet. 2009;18(2):304–317. doi: 10.1093/hmg/ddn357. (Available from: http://dx.doi.org/10.1093/hmg/ddn357) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492. doi: 10.1038/nrg3230. (Available from: http://dx.doi.org/10.1038/nrg3230) [DOI] [PubMed] [Google Scholar]
- 9.Maunakea AK, Chepelev I, Cui K, et al. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013;23(11):1256–1269. doi: 10.1038/cr.2013.110. (Available from: http://dx.doi.org/10.1038/cr.2013.110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murrell A, Rakyan VK, Beck S. From genome to epigenome. Hum Mol Genet. 2005;14(Suppl. 1):R3–R10. doi: 10.1093/hmg/ddi110. (Available from: http://dx.doi.org/10.1093/hmg/ddi110) [DOI] [PubMed] [Google Scholar]
- 11.Prior TW, Krainer AR, Hua Y, et al. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet. 2009;85(3):408–413. doi: 10.1016/j.ajhg.2009.08.002. (Available from: http://dx.doi.org/10.1016/j.ajhg.2009.08.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qu YJ, Du J, Li EZ, et al. Subtle mutations in the SMN1 gene in Chinese patients with SMA: p.Arg288Met mutation causing SMN1 transcript exclusion of exon7. BMC Med Genet. 2012;13:86. doi: 10.1186/1471-2350-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schumacher A, Petronis A. Epigenetics of complex diseases: from general theory to laboratory experiments. Curr Top Microbiol Immunol. 2006;310:81–115. doi: 10.1007/3-540-31181-5_6. [DOI] [PubMed] [Google Scholar]
- 14.Tiziano FD, Pinto AM, Fiori S, et al. SMN transcript levels in leukocytes of SMA patients determined by absolute real-time PCR. Eur J Hum Genet. 2010;18(1):52–58. doi: 10.1038/ejhg.2009.116. (Available from: http://dx.doi.org/10.1038/ejhg.2009.116) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheleznyakova GY, Voisin S, Kiselev AV, et al. Genome-wide analysis shows association of epigenetic changes in regulators of Rab and Rho GTPases with spinal muscular atrophy severity. Eur J Hum Genet. 2013;21(9):988–993. doi: 10.1038/ejhg.2012.293. (Available from: http://dx.doi.org/10.1038/ejhg.2012.293) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primers for SMN2 CGIs cloning sequencing
Table S2 Primers for SMN2 nested PCR