

Abstract
Background
Sorghum is an important C4 crop which relies on applied Nitrogen fertilizers (N) for optimal yields, of which substantial amounts are lost into the atmosphere. Understanding the genetic variation of sorghum in response to limited nitrogen supply is important for elucidating the underlying genetic mechanisms of nitrogen utilization.
Results
A bi-parental mapping population consisting of 131 recombinant inbred lines (RILs) was used to map quantitative trait loci (QTLs) influencing different agronomic traits evaluated under normal N (100 kg.ha−1 fertilizer) and low N (0 kg.ha−1 fertilizer) conditions. A linkage map spanning 1614 cM was developed using 642 polymorphic single nucleotide polymorphisms (SNPs) detected in the population using Genotyping-By-Sequencing (GBS) technology. Composite interval mapping detected a total of 38 QTLs for 11 agronomic traits tested under different nitrogen levels. The phenotypic variation explained by individual QTL ranged from 6.2 to 50.8 %. Illumina RNA sequencing data generated on seedling root tissues revealed 726 differentially expressed gene (DEG) transcripts between parents, of which 108 were mapped close to the QTL regions.
Conclusions
Co-localized regions affecting multiple traits were detected on chromosomes 1, 5, 6, 7 and 9. These potentially pleiotropic regions were coincident with the genomic regions of cloned QTLs, including genes associated with flowering time, Ma3 on chromosome 1 and Ma1 on chromosome 6, gene associated with plant height, Dw2 on chromosome 6. In these regions, RNA sequencing data showed differential expression of transcripts related to nitrogen metabolism (Ferredoxin-nitrate reductase), glycolysis (Phosphofructo-2-kinase), seed storage proteins, plant hormone metabolism and membrane transport. The differentially expressed transcripts underlying the pleiotropic QTL regions could be potential targets for improving sorghum performance under limited N fertilizer through marker assisted selection.
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-015-0696-x) contains supplementary material, which is available to authorized users.
Keywords: Sorghum, Agronomic traits, Differentially expressed gene transcripts, Genotyping-by-sequencing, Nitrogen fertilizer, QTL mapping, Illumina RNA-seq
Background
Sorghum (Sorghum bicolor (L.) Moench) is the fifth most cultivated cereal crop worldwide (http://www.fao.org/3/a-ax443e.pdf) and also an important source of fodder, fiber and biofuel [1]. Sorghum performs C4 photosynthesis like maize and sugarcane, and uses Nitrogen, CO2 and water more efficiently than maize and most C3 plants [2]. Sorghum is an important model for genome analysis among the C4 grasses because its genome is relatively small (~818 Mbp) [3], and the cultivated species is diploid (2n = 20). Due to its deep root system, sorghum is drought tolerant and is preferentially grown in water-limited environments [4]. Despite being a C4 crop, sorghum still relies on applied fertilizer to achieve maximal yields. Nitrogen (N) is the macronutrient which is often limiting sorghum production. N is the most abundantly absorbed mineral nutrient by plant roots [5] and 75 % of the leaf N is allocated to the chloroplasts [6]. As nitrogen is an essential part of many biomolecules, it comprises 1.5 to 2 % of plant dry matter and 16 % of the total plant protein [7].
N fertilizer application is expected to rise approximately three-fold in the next 40 years [8]. In general, plants absorb less than half of the applied fertilizer [7]. Both phosphorus and potassium are immobile nutrients in the soil and are generally not vulnerable for leaching. However, nitrogen is a mobile nutrient and when present in excess, it is released in to the atmosphere through volatilization or lost through leaching and ground water runoff, of which both have adverse environmental effects [8]. Excess N fertilizer application is a major economic cost to farmers, and also leads to acidification of soils [9]. Because of their potential positive effects on improving economic returns and limiting global climate change, lowering fertilizer input and breeding plants with better nitrogen use efficiency (NUE) are two major goals of research in plant nutrition [10]. As a function of multiple interacting genetic and environmental factors, the molecular basis of NUE is complex. NUE is defined as the grain yield [11] or fresh/dry matter produced [8] per unit of available N in the soil. Uptake of N from the soil involves a variety of transporters, and a number of enzymes for assimilation and transfer of the absorbed N into amino acids and other compounds [12]. However, little is known about how these processes are regulated especially under different N conditions.
QTL analysis, based on high density linkage maps, is a powerful tool for dissecting the genetic basis underlying complex traits [13]. QTL mapping studies have been conducted under different N conditions for NUE and other agronomic traits in maize [14], Arabidopsis [15], and rice [16, 17]. QTLs associated with low-nitrogen tolerance were detected in rice [18] and barley [19] for different traits, at the seedling stage. In barley, Mickelson et al. [20] mapped a QTL for grain protein concentration, which is homologous to a durum wheat grain protein QTL mapped by Joppa et al. [21]. QTLs for NUE and enzymes involved in nitrogen metabolism were reported in wheat [22] and QTLs for glutamine synthetase (GS) activity were co-localized with those for grain N [23] and confirmed in another population [24]. In wheat, Quraishi et al. [25] identified 11 major regions controlling NUE, which co-localized with key developmental genes such as Ppd (photoperiod sensitivity), Vrn (vernalization) and Rht (reduced height). However, there are no previous QTL mapping reports for agronomic traits tested under different nitrogen levels in sorghum. Significant genotypic differences for N utilization efficiency have been documented in sorghum [26, 27]. N utilization of genotypes varied with different nitrogen sources, nitrogen amounts and other environmental conditions [28]. Thus, there is good reason to believe that improvements in N utilization efficiency in sorghum can be achieved using genetic approaches.
Different kinds of DNA based low-throughput marker systems such as restriction fragment length polymorphism (RFLP), amplified fragment length polymorphism (AFLP), and simple sequence repeat (SSR) markers have been developed and used to investigate the variants and quantitative trait loci (QTLs) controlling >150 traits in sorghum. AFLPs, SSRs and RFLPs were used for generating the dense linkage maps [29]. Diversity Array Technology was evolved [30] as a cost effective hybridization-based alternative to the gel-based marker technologies, which offers a multiplexed genotyping independent of sequence information. DArT markers were developed for sorghum and used for genotyping a diverse set of sorghum lines and a bi-parental mapping population [31]. With the availability of sorghum whole genome sequence [32], Mace et al. [4] generated a single, reference consensus map by integrating six independent sorghum genetic maps containing 2029 unique loci consists of SSRs, AFLPs, and DArT markers. Using this as a framework map, Mace and Jordon et al. [33] mapped 35 major effect genes commonly observed in segregating mapping populations onto a common reference map to enable sorghum researchers link the information of QTLs and select the major genes. Furthermore, Mace et al. [34] projected 771 QTL relating to 161 unique traits from 44 studies onto the sorghum consensus map, which is useful for development of efficient marker-assisted breeding strategies. With the advent of high-throughput DNA sequencing technologies, it became possible to re-sequence genomes and detect single nucleotide polymorphisms (SNPs) which can be used for rapid genotyping [35]. Zou et al. [36] developed a linkage map based on SNPs generated from whole-genome re-sequencing by the Illumina Genome Analyzer IIx as described by Huang et al. [37] and used it for detecting QTLs for important agronomic traits under contrasting photoperiods in sorghum. However, it remains costly to employ whole-genome sequencing to evaluate multiple individuals in mapping populations. Next generation sequencing of a reduced representation genomic library, where fewer sequence reads are needed to obtain meaningful information compared to whole genome sequencing, is a convenient approach for capturing genetic variation. Genotyping-by-sequencing (GBS) is an efficient strategy for constructing multiplexed reduced representation library [38]. This technique has successfully been applied to generate high-density genetic maps and QTL mapping in several plant species [39].
In this study, we used SNPs generated from GBS technology to develop a linkage map and which then used to map QTLs for different agronomic traits in RIL population of sorghum. This process of QTL detection enabled us to link variation at the trait level to the variation at sequence level. However, a QTL may contain tens to hundreds of genes, figuring out the genes responsible for trait variation is a major challenge. With the advancement of sequencing technology, transcriptome comparisons were made between different sorghum genotypes at different tissue levels and at different growing conditions [40–44]. In addition, Morokoshi et al. [44] compiled all these datasets and developed a transcriptome database for sorghum which will be useful to researchers for transcriptome comparisons. The desire to identify the underlying genes responsible for trait variation in QTL regions has been increasing and to this end, we used previously generated high throughput Illumina-based RNA sequencing data [43] to identify differentially expressed gene transcripts in QTL regions. By further evaluation, the resulting candidate genes could be potential targets for improving N-stress tolerance and nitrogen utilization of sorghum and related crops.
Methods
Plant material
A mapping population derived from a cross between the inbred lines CK60 and China17 was used in this study. CK60, a public sorghum line, which is short, photoperiod-sensitive, late-maturing U.S. sorghum line and an inefficient N user. China17, a photoperiod-insensitive Chinese sorghum line was provided by Dr. Jerry Maranville (University of Nebraska, Lincoln, USA), uses nitrogen more efficiently than CK60 and has higher assimilation efficiency indices at both low and high soil nitrogen levels [45]. China17 retains higher phosphoenolpyruvate carboxylase (PEPcase) activity than CK60 when grown under low N conditions [45]. The seedlings of China17 had greater root and shoot mass than CK60 under both low N and normal N conditions [43]. Each of the 131 RILs was derived from a single F2 plant following a single seed descent method until the F7 generation.
Experimental design
The F7 RILs and the two parents (CK60 and China17) were evaluated in an alpha lattice incomplete block design under two N levels with two independent replicates each for two years (2011 and 2012). The two N treatments were low N (LN, 0 kg.ha−1 fertilizer) and normal N (NN, 100 kg.ha−1 anhydrous ammonia fertilizer). The preceding crops were soybean in the NN field and oats or maize in the LN filed. The LN field had not received nitrogen fertilizer since 1986. The soil testing was done by collecting soil samples from 0 to 12 in. and 12–24 in. randomly across the NN and LN fields and results were described in Additional file 1. Single-row plots measuring five meters long at 0.75 m row spacing were sown at a density of 50 seeds for each RIL and parents. All entries were planted on the same day in conventionally tilled plots and maintained under rain fed conditions.
Phenotyping of important agronomic traits
Three plants were randomly selected for each genotype for phenotypic evaluation of eleven agronomic traits. The measured phenotypes include leaf chlorophyll content at three different stages of plant growth: before flowering (vegetative stage, Chl1), during flowering (Chl2) and at maturity (Chl3); plant height (PH, from base of the plant to tip of the head, in centimeters); and days to anthesis (AD, no. of days from planting to 50 % anthesis). Stover moisture contents (MC1) and head moisture contents (MC2) were calculated as the percent difference between wet and dry weights. Total biomass yield (BY, t.ha−1), grain yield (GY, t.ha−1), 1000 seed weight in grams (Test weight, TW) and grain-to-stover ratio (GS, %) were calculated and recorded from NN and LN fields. Haussmann et al. [46] described that the upper six leaves are a good source for measuring the greenness of leaves since they are photosynthetically active at anthesis and contribute nutrients to the grain [47]. In this study, chlorophyll contents were measured in the 3rd leaf from the top using a portable chlorophyll meter model SPAD-502 (Minolta, Japan). In summary, the phenotypes were classified into three groups, chlorophyll contents (Chl1, Chl2, and Chl3), morphological traits (PH, AD, MC1, and MC2), and yield-related traits (BY, GY, TW and GS).
Statistical analysis
The statistical model adopted for the alpha lattice incomplete block design in each N condition was Yijk = μ + gi + rj + bk(j) + eij. Yijk is the response of ith genotype in kth bock of jth replication, μ is the grand mean, gi is the genotype or line effect, rj is the replication effect, bk(j) is the random block k (k = 1…n) effect within replicate with bk(j) ~ N(0, σ2b) and eij is the residual term with ~ N(0, σ2e). Analysis of variance (ANOVA) for eleven traits was performed for each individual environment using the PROC MIXED procedure [48] of SAS version 9.2 (SAS Institute, 2008) where the genotype was considered as fixed, replications and blocks as random effects. The phenotypic data, from both seasons (2011 and 2012), were pooled to obtain single trait values for each family under NN and LN [13]. ANOVA was performed on pooled data by considering that genotype effect is fixed and environments (years), replication within environments, blocks within environments, and genotype by environment (GxE) interaction effects are random. Narrow-sense heritability with standard error was estimated using the PROC MIXED procedure of SAS version 9.2. For the heritability estimates, parental lines data were excluded, and estimates followed a method described by Holland et al. [49]. Pearson’s correlation coefficients between traits were calculated for the least square genotype means using the PROC CORR procedure of SAS. The RIL trait data were subjected to normality test using PROC UNIVARIATE to determine its suitability for QTL analysis.
High-throughput Genotyping and Linkage map construction
Total genomic DNA of the RILs and their parents were isolated from leaf tissues using a DNeasy Plant Mini Kit (Qiagen). DNA (500 ng) from each sample was digested with ApeKI (New England Bio-labs, Ipswich, MA), a type II restriction endonuclease that recognizes a degenerate 5 bp sequence (5’-GCWGC) and creates 5’ overhangs. Adapters with specific barcodes [38] were then ligated to the overhanging sequences using T4 ligase. A set of 96 DNA samples, each sample with a different barcode adapter, were combined and purified (Quick PCR Purification Kit; Qiagen, Valencia, CA) according to the manufacturer’s instructions. DNA fragments containing ligated adapters were amplified with primers containing complementary sequences for each adapter. PCR products were then purified and diluted for sequencing [38]. Single-end, 100 bp reads were collected for one 48- or 96-plex library per flow cell channel on a Genome Analyzer IIx (GAIIx; Illumina, Inc., San Diego, CA) [50] at Cornell University, USA.
Raw reads obtained from GAIIx were filtered [38] and aligned to the sorghum reference genome version 1.4 [32]. The genotypes of the population were determined based on the procedure described by Elshire et al. [38]. The biallelic SNP markers were checked for polymorphism between the parents. Prior to map construction, all polymorphic SNPs were checked by the chi-square (χ2) test for the goodness of fit against a 1:1 segregation ratio at the 0.05 probability level. SNPs with >70 % missing data were removed from data set. A total of 668 SNPs were selected and used for constructing linkage maps using Mapmaker/EXP 3.0 along with IciMapping (Inclusive composite interval mapping) V3.2 [51]. The genetic distance (cM) was calculated using the Kosambi mapping function.
QTL analysis
The composite interval mapping method of WinQTLcart2.5 [52] was used for QTL detection. QTL analysis was performed based on averaged mean values of each trait across two NN and two LN environments respectively. The walking speed chosen for all traits was 1 cM. Cofactors were determined using the forward and backward step-wise regression method with a probability in and out of 0.1 and a window size of 10 cM. A thousand-permutation test was applied to each data set to decide the LOD (logarithm of odds) thresholds (P ≤ 0.05) to determine significance of identified QTLs [53]. A 2-LOD support interval was calculated for each QTL to obtain a 95 % confidence interval. Adjacent QTLs on the same chromosome for the same trait were considered different when the support intervals were non-overlapping. The contribution rate (R2) was estimated as the percentage of variance explained by each QTL in proportion to the total phenotypic variance. The additive effect of a putative QTL was estimated by half the difference between two homozygous classes. QTLs were named according to McCouch et al. [54] and alphabetical order was used for QTLs on the same chromosome. QTLs with a positive or negative additive effect for a trait imply that the increase in the phenotypic value of the trait is contributed by alleles from CK60 or China17.
Detection of differentially expressed gene transcripts in the QTL intervals
In an earlier study [43], we detected several common DEG transcripts between the transcriptomes of seven sorghum genotypes (four low-N tolerant and three low-N sensitive) using Illumina RNA sequencing. Transcriptomes were prepared from root tissues of 3 week old seedlings grown under N-stress from four N-stress tolerant (China17, San Chi San, KS78 and high NUE bulk) and three sensitive (CK60, BTx623 and low NUE bulk) genotypes. In the present study, we used the RNA-seq data generated earlier in order to check the differential expression of gene transcripts between CK60 and China17 in the QTL regions. Pair-wise comparison was made between the transcriptomes of CK60 and China17 to detect DEG transcripts. The cutoff of log2-fold value >1 (2-fold absolute value) and adjusted P-value <0.001 (FDR) were used for determining significant DEG transcripts.
Results
Statistical analysis of phenotypic data
Mean values of 11 traits measured for parents (CK60, and China17) and the RIL population under NN and LN environments are given in Tables 1 and 2, respectively. The mean chlorophyll content was higher at flowering than at vegetative and mature stages under both N-conditions. CK60 retained more chlorophyll at all stages compared to China17 and the mean chlorophyll content of the RIL population was lower under LN compared to NN conditions. The plant height of CK60 was reduced by 23 cm, while that of China17 remained the same under LN compared to NN. Days to anthesis for the two parental lines were also significantly affected by N-condition, and LN delayed flowering in both parents. Compared to China17, the flowering was delayed more in CK60 under both N-levels. The biomass yield of CK60 was lower than China17 in both N conditions. The grain yield was also significantly different between the two parents; CK60 had lower grain yield under the two N-conditions. The average values of biomass and grain yield for the RILs were greatly reduced from NN to LN conditions, respectively. Similarly, the test weight of China17 was higher than CK60 under both N-conditions. The grain/stover ratio of China17 was decreased almost half, while no significant change was observed for CK60 under LN compared to NN. In contrast, the stover and head moisture contents of CK60 were higher than China17 under both N-conditions. The average of grain/stover ratio and stover moisture contents of the RILs remained the same under both N conditions but the average of head moisture content in the RIL population was increased under LN conditions.
Table 1.
Descriptive statistics, h 2, and mean squares of ANOVA results for the traits measured across two normal-N conditions in CK60 x China17 RIL population
| Category | Source of variation | Df | Chl1 | Chl2 | Chl3 | PH | AD | MC1 | MC2 | BY | GY | TW | GS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Descriptive statistics | CK60 | 49.8 | 55.6 | 53.6 | 99 | 71.5 | 68.6 | 24.8 | 7.69 | 2.89 | 20.3 | 0.52 | |
| China17 | 46.6 | 52.7 | 48.3 | 150 | 66.3 | 65 | 19.5 | 14.6 | 6.25 | 31.6 | 0.95 | ||
| RIL Mean | 47.8 | 53.3 | 47.2 | 161.3 | 67 | 66.5 | 19.4 | 11.2 | 3.39 | 23.6 | 0.47 | ||
| Std | 4.09 | 3.72 | 5.85 | 35 | 4.2 | 3.32 | 6.34 | 3.99 | 1.49 | 3.15 | 0.16 | ||
| Min | 38.7 | 38.2 | 32.3 | 70 | 55.1 | 52.8 | 8.16 | 3.09 | 0.4 | 14.4 | 0.05 | ||
| Max | 58.4 | 62.2 | 62.5 | 236.5 | 85.9 | 76.1 | 46.8 | 24.2 | 9.04 | 29.9 | 0.88 | ||
| h2 (%) | 71 | 56 | 51 | 64 | 61 | 40 | 53 | 62 | 55 | 64 | 39 | ||
| SE (%) | 6 | 9 | 10 | 7 | 8 | 12 | 9 | 0.8 | 9 | 7 | 12 | ||
| ANOVA | Env | 1 | 626.9 | 4276*** | 17016*** | 40478 | 11333*** | 347.6 | 4500 | 271.6 | 32.8* | 946.9 | 0.07 |
| Rep(Env) | 2 | 89.1* | 9.93 | 57.8 | 10976** | 55.6* | 191.1** | 2501*** | 34.6 | 2.57 | 1483*** | 0.02 | |
| Blk(Env*Rep) | 44 | 12.5 | 12.0* | 40.4*** | 429* | 11.3 | 14.8 | 31.7 | 13.1 | 2.43* | 5.69 | 0.02 | |
| Line | 130 | 50.9*** | 41.9*** | 105.3*** | 4037*** | 49.6*** | 34.3** | 123.9*** | 49.4*** | 6.8*** | 28.0*** | 0.08** | |
| Env*Line | 104 | 15.6** | 18.1*** | 58.2*** | 1634*** | 20.5** | 21.8** | 59.3*** | 19.6*** | 3.2*** | 10.5*** | 0.048*** | |
| Residual | 190 | 9.74 | 8.06 | 16.7 | 290 | 12.8 | 12.7 | 26.7 | 10.5 | 1.54 | 5.2 | 0.02 |
Df, degrees of freedom; chlorophyll contents at vegetative stage (Chl1), at anthesis (Chl2), and at maturity (Chl3); PH, plant height (cm)
AD, days to anthesis; MC1, % stover moisture content; MC2, % head moisture content; BY, biomass yield (t.ha−1); GY, grain yield (t.ha−1)
TW, test weight (g); GS, grain/stover ratio (%). Std, standard deviation; h2 (%), narrow sense heritability; SE (%), standard error %; ***P < 0.0001; **P < 0.01; *P < 0.05
Table 2.
Descriptive statistics, h2, and mean squares of ANOVA results for the traits measured across two low-N conditions in CK60 x China17 RIL population
| Category | Source of variation | Df | Chl1 | Chl2 | Chl3 | PH | AD | MC1 | MC2 | BY | GY | TW | GS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Descriptive statistics | CK60 | 31.8 | 39.5 | 40.2 | 76.3 | 90.5 | 68.6 | 34.4 | 3.75 | 1.21 | 20.2 | 0.49 | |
| China17 | 32.7 | 33.9 | 28.7 | 153.1 | 77.1 | 60.7 | 23.8 | 6.83 | 2.72 | 28.3 | 0.46 | ||
| RIL Mean | 33.3 | 36.8 | 31.8 | 131.7 | 82.6 | 66.2 | 27.4 | 6.43 | 1.86 | 20.3 | 0.42 | ||
| Std Dev | 3 | 3.9 | 5.4 | 38.7 | 7.8 | 3.2 | 9 | 2.1 | 0.79 | 3.3 | 0.14 | ||
| Min | 27.3 | 25.2 | 12.3 | 55.9 | 66.7 | 55.5 | 13.4 | 2.91 | 0.06 | 12.1 | 0.01 | ||
| Max | 40.2 | 48.1 | 46.2 | 214 | 108.2 | 74.6 | 57.8 | 13.2 | 5.02 | 27.8 | 0.96 | ||
| h2 (%) | 59 | 43 | 50 | 80 | 75 | 71.6 | 76 | 48 | 47 | 75 | 32 | ||
| SE (%) | 8 | 12 | 10 | 4 | 5 | 6 | 5 | 10 | 10 | 5 | 14 | ||
| ANOVA | Env | 1 | 360.5 | 16104*** | 24768** | 4740.9 | 54521*** | 186 | 264 | 435.9* | 163*** | 368.1 | 6.32* |
| Rep(Env) | 2 | 87.7* | 23.3 | 131.7* | 771.4 | 48.85 | 129*** | 670.5** | 21.4 | 0.16 | 1779*** | 0.22** | |
| Blk(Env*Rep) | 44 | 16.6* | 19.1 | 18.66 | 412.3** | 36.67 | 7.8 | 35 | 5.9 | 0.69 | 6.14 | 0.01 | |
| Line | 130 | 27.2*** | 45.6** | 87.7** | 4475*** | 167.4*** | 31.7*** | 238.0*** | 15.0** | 1.97** | 32.7*** | 0.05* | |
| Env*Line | 104 | 12.1 | 27.3*** | 44.03*** | 1001*** | 46.8* | 9.9*** | 66.2*** | 8.87** | 1.17** | 9.48** | 0.03*** | |
| Residual | 190 | 10.7 | 13.9 | 15.13 | 189.9 | 31.78 | 6.58 | 34.2 | 5.5 | 0.75 | 5.67 | 0.02 |
Df, degrees of freedom; chlorophyll contents at vegetative stage (Chl1), at anthesis (Chl2), and at maturity (Chl3); PH, plant height (cm); AD, days to anthesis; MC1, % stover moisture content; MC2, % head moisture content; BY, biomass yield (t.ha−1); GY, grain yield (t.ha−1); TW, test weight (g); GS, grain/stover ratio (%). Std, standard deviation; h2 (%), narrow sense heritability; SE (%), standard error %; ***P < 0.0001; **P < 0.01; *P < 0.05
The narrow sense heritability (h2) was estimated for each trait measured under both N conditions (Tables 1 and 2). Under NN, the heritability estimates of the 11 traits ranged from 39 to 71 %. Chlorophyll at the vegetative stage had the highest h2 value followed by plant height and test weight. Grain/stover ratio had the lowest heritability estimate. Under LN, h2 values ranged from 32 to 80 %. Plant height had the highest h2 values and grain/stover ratio had the lowest h2 value. ANOVA showed significant phenotypic variation for all the traits among RILs (Tables 1 and 2). GxE interaction was mainly associated with differences in magnitude of effects between years. Therefore, phenotypic data from 2011 and 2012 seasons were averaged separately for NN and LN conditions. GxE interactions were significant for all the traits except chlorophyll at the vegetative stage across two LN environments. Genotype variance was greater than GxE interaction variance for all traits across NN and LN environments (Tables 1 and 2).
Correlation of the traits
The focus of this work was evaluation of the genetic control of traits under NN and LN conditions in sorghum. Correlation coefficients based on the line means among three chlorophyll contents, yield-related traits and other morphological traits showed that most of the traits tested under the contrasting N conditions were significantly correlated (P < 0.05) (Table 3). Interestingly, leaf chlorophyll contents measured at three different stages of plant growth were negatively correlated with most of the yield-related (biomass yield, grain yield and test weight) and morphological traits (plant height, days to anthesis and head moisture content) in both N-conditions (Table 3). Under NN conditions, significant positive correlations were observed between chlorophylls and stover moisture content (P < 0.01). In addition, plant height had significant positive correlation with biomass and grain yield in both N conditions. Highest positive correlation was observed between biomass and grain yield in both NN and LN environments. Days to anthesis was positively correlated with stover and head moisture contents under both N conditions. Grain/stover ratio was not significantly correlated with many traits, but it had significant positive correlation with grain yield.
Table 3.
Correlation coefficient of the traits investigated
| Chl1 | Chl2 | Chl3 | PH | AD | MC1 | MC2 | BY | GY | GS | TW | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Chl1 | 0.76*** | 0.65*** | −0.35*** | −0.17* | 0.134 | −0.065 | −0.069 | 0.066 | 0.144 | −0.065 | |
| Chl2 | 0.77*** | 0.73*** | −0.38*** | −0.36*** | 0.08 | −0.23** | −0.18* | 0.085 | 0.30** | −0.016 | |
| Chl3 | 0.57*** | 0.58*** | −0.51*** | 0.077 | 0.22* | 0.163 | −0.162 | −0.016 | 0.118 | −0.24** | |
| PH | −0.62*** | −0.51*** | −0.52*** | −0.16 | −0.22* | −0.40*** | 0.54*** | 0.39*** | 0.022 | 0.43*** | |
| AD | −0.26** | −0.30** | 0.15 | 0.13 | 0.34*** | 0.76*** | 0.003 | −0.19* | −0.28** | −0.47*** | |
| MC1 | 0.28** | 0.16 | 0.38*** | −0.40*** | 0.19* | 0.39*** | −0.046 | −0.097 | 0.037 | −0.21* | |
| MC2 | −0.003 | −0.21* | 0.27** | −0.078 | 0.51*** | 0.38*** | −0.04 | −0.35*** | −0.45*** | −0.49*** | |
| BY | −0.64*** | −0.56*** | −0.37*** | 0.63*** | 0.25** | −0.24** | 0.12 | 0.75*** | 0.033 | 0.38*** | |
| GY | −0.56*** | −0.36*** | −0.35*** | 0.50*** | 0.142 | −0.31** | −0.27** | 0.73*** | 0.60*** | 0.40*** | |
| GS | −0.078 | 0.04 | −0.1 | 0.043 | −0.132 | −0.151 | −0.53*** | −0.094 | 0.52*** | 0.26** | |
| TW | −0.23** | 0.02 | −0.166 | 0.20* | −0.1 | −0.24** | −0.21* | 0.23** | 0.31** | 0.064 |
The numbers below the diagonal are correlation coefficients under normal N environments and numbers above the is diagonal are correlation coefficients under low N environments. Chlorophyll contents at vegetative stage (Chl1), at anthesis (Chl2), and at maturity (Chl3); PH, plant height (cm); AD, days to anthesis; MC1, % stover moisture content; MC2, % head moisture content; BY, biomass yield (t.ha−1); GY, grain yield (t.ha−1); TW, test weight (g); GS, grain/stover ratio (%). ***P < 0.0001; **P < 0.01; *P < 0.05
Linkage mapping and QTL analysis
Polymorphic SNP markers between CK60 and China17 were identified by the GBS pipeline. A linkage map was developed with 642 polymorphic SNPs (Additional file 2) with an average inter marker distance of 2.55 cM. The resulting linkage map comprised of 10 linkage groups and map spanning a total length of 1641 cM. Composite interval mapping detected a total of 38 QTLs for 11 traits analyzed across NN and LN environments. No significant QTLs were detected on chromosomes 2, 3, 4 and 10 (not shown in Fig. 1). The number of QTLs per trait ranged from one to four, and is listed in Tables 4 and 5 and shown in Fig. 1. Across two NN conditions, four QTLs for chlorophyll contents were detected including one QTL each for chlorophyll at vegetative and flowering stage, and two QTLs for chlorophyll at maturity explaining phenotypic variation range from 7.1 to 50.8 % (Table 4). Six QTLs were identified for four morphological traits including one major QTL for days to anthesis on chromosome 1, for which the CK60 allele delayed flowering by 3.6 days. Two QTLs each for stover and head moisture contents were detected under NN conditions. For all these QTLs, the CK60 allele contributed to increase the chlorophyll contents and the moisture contents. In contrast, the China17 allele contributed to an increase in the plant height by 39.8 cm for the QTL detected on chromosome 9. Similarly, we detected eight significant QTLs for yield-related traits. Of the eight detected, two QTLs are for biomass yield, three for grain yield, one for test weight and two for grain/stover ratio. For the two QTLs detected for biomass yield, China17 allele increased the biomass yield by 1.8 t.ha−1. For grain yield, CK60 allele increased grain yield by 0.5 t.ha−1 for the two QTLs on chromosome 1 and China17 allele increased grain yield for the other QTL on chromosome 9. CK60 allele responsible for an increase in the test weight of seeds for the major QTL detected on chromosome 5 for test weight. In contrast, the China17 allele increased the grain/stover ratio for two QTLs.
Fig. 1.
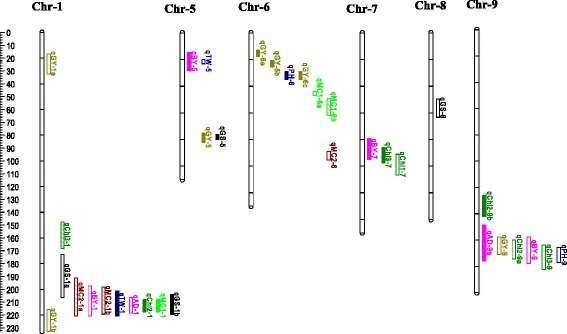
QTLs mapped to the linkage groups for 11 agronomically important traits across two normal N and two low-N conditions. Chr, indicate chromosome. Chlorophyll contents at vegetative stage (Chl1), at anthesis (Chl2), and at maturity (Chl3); plant height (PH, cm), days to anthesis (AD, days), stover moisture content (MC1,%), head moisture content (MC2,,%), biomass yield (BY, t.ha−1), grain yield (GY, t.ha−1), test weight (TW, g), and grain/stover ratio (GS, %); each trait was shown with different color; open bars indicates QTLs detected under NN, closed bars indicates QTLs detected under LN and open bar with strikes indicates QTLs detected consistently across environments. Supported intervals for each QTL are indicated by the length of vertical bars. Chr doesn’t contain QTLs not shown here. Left side scale is in cM
Table 4.
QTLs detected for 11 traits using the SNP linkage map across two normal N conditions
| Trait | QTL | Chr | Position (cM) | Flanking marker | Interval (cM)a | LOD score | Additiveb | R2 (%)c |
|---|---|---|---|---|---|---|---|---|
| Chl-1 | qChl1-7 | 7 | 97.8 | S7_60490830 - S7_60947414 | 91.1–106.6 | 2.76 | 1.18 | 7.1 |
| Chl-2 | qChl2-9a | 9 | 167.4 | S9_45363122 - S9_58417131 | 159.9–174.4 | 10.2 | 3.20 | 50.8 |
| Chl-3 | qChl3-1 | 1 | 157.6 | S1_50614823-S1_50837764 | 147.6–168.1 | 3.72 | 2.77 | 8.10 |
| qChl3-9 | 9 | 169.4 | S9_45363122 - S9_58417132 | 163.6–182.6 | 2.64 | 2.13 | 11.8 | |
| PH | qPH-9 | 9 | 171.4 | S9_45363122 - S9_58417133 | 165.8–177.2 | 4.33 | −39.8 | 44.2 |
| AD | qAD-1 | 1 | 213.9 | S1_61836509 – S1_62490042 | 208.7–218.3 | 5.15 | 3.6 | 16 |
| MC1 | qMC1-6a | 6 | 45.6 | S6_48858797 - S6_49609588 | 44–46.8 | 13.5 | 2.39 | 29.1 |
| qMC1-6b | 6 | 54.6 | S6_52982294 - S6_54274803 | 49.3–61.9 | 4.84 | 1.47 | 16.2 | |
| MC2 | qMC2-1 | 1 | 203 | S1_55726325 - S1_57821154 | 191.3–210.9 | 4.57 | 2.70 | 15.3 |
| qMC2-6 | 6 | 91.6 | S6_57001245 - S6_57540748 | 88.8–95.5 | 3.19 | 1.96 | 8.56 | |
| BY | qBY-1 | 1 | 204 | S1_55726325 - S1_57821154 | 197.3–220.9 | 2.60 | −1.82 | 10.8 |
| qBY-9 | 9 | 167.4 | S9_45363122 - S9_58417134 | 157.6–177.7 | 5.34 | −2.41 | 33.8 | |
| GY | qGY-1a | 1 | 28.3 | S1_2983876 - S1_3356469 | 16.7–32.3 | 3.84 | 0.52 | 9.84 |
| qGY-1b | 1 | 223 | S1_64266923 - S1_71768492 | 215.9–232.7 | 3.59 | 0.92 | 17.3 | |
| qGY-9 | 9 | 161.4 | S9_45363122 - S9_58417134 | 157.6–170.9 | 4.82 | −0.67 | 17.6 | |
| TW | qTW-5 | 5 | 22 | S5_44956096 - S5_45759643 | 20–23.1 | 4.51 | 1.72 | 15.0 |
| GS | qGS-1a | 1 | 190 | S1_54743129 - S1_54776428 | 173.1–206.4 | 3.30 | −0.06 | 11.3 |
| qGS-8 | 8 | 56.5 | S8_398073 - S8_5494183 | 49.6–63.3 | 3.84 | −0.09 | 17.6 |
Chlorophyll contents at vegetative stage (Chl1), at anthesis (Chl2), and at maturity (Chl3); PH, plant height (cm)
AD, days to anthesis; MC1, % stover moisture content; MC2, % head moisture content; BY, biomass yield (t.ha−1)
GY, grain yield (t.ha−1); TW, test weight (g); GS, grain/stover ratio (%). a2.0-LOD drop support interval of the QTL; bAdditive effect: positive values of the additive effect indicate that alleles from CK60 were in the direction of increasing the trait score and vice versa; c Percentage of phenotypic variation explained by the QTL. The SNP underlined is the corresponding SNP of QTL
Table 5.
QTLs detected for 11 traits using the SNP linkage map across two low-N conditions
| Trait | QTL | Chr | Position (cM) | Flanking marker | Interval (cM)a | LOD score | Additiveb | R2 (%)c |
|---|---|---|---|---|---|---|---|---|
| Chl-2 | qChl2-1 | 1 | 211 | S1_61786623 - S1_61836509 | 208.1–217.7 | 3.28 | −1.26 | 8.69 |
| qChl2-9b | 9 | 136 | S9_55230722 - S9_56646280 | 126.1–142.2 | 3.07 | 1.25 | 8.49 | |
| Chl-3 | qChl3-1 | 1 | 157.8 | S1_50614823 - S1_50837764 | 150.6–178.1 | 3.29 | 1.79 | 10.1 |
| qChl3-7 | 7 | 87.7 | S7_57772979 - S7_60426792 | 85.9–97.8 | 4.57 | 2.71 | 14.2 | |
| PH | qPH-6 | 6 | 30.8 | S6_41970042 - S6_43222258 | 28.9–34.8 | 5.30 | −16.4 | 13.2 |
| AD | qAD-1 | 1 | 210.8 | S1_61709596- S1_61786623 | 206.5–218.1 | 5.15 | 3.63 | 16.7 |
| qAD-9a | 9 | 159.4 | S9_45363122 - S9_58417131 | 148.8–176 | 3.29 | −3.00 | 16.4 | |
| MC1 | qMC1-1 | 1 | 211 | S1_61786623 - S1_61836509 | 208.1–217.7 | 3.14 | 1.44 | 10.0 |
| qMC1-6b | 6 | 54.6 | S6_52982294 - S6_54274803 | 49.3–61.9 | 2.69 | 1.14 | 6.97 | |
| MC2 | qMC2-1a | 1 | 203 | S1_55726325 - S1_57821154 | 201.3–220.9 | 4.74 | 4.74 | 16.4 |
| qMC2-1b | 1 | 209.4 | S1_59080688- S1_59595161 | 198.2–219.4 | 8.10 | 4.44 | 20.3 | |
| BY | qBY-5 | 5 | 20 | S5_44956096 - S5_45759643 | 14.5–28.4 | 2.95 | 0.72 | 9.04 |
| qBY-7 | 7 | 85.9 | S7_57557894 - S7_57772979 | 79.3–95.1 | 3.86 | −1.01 | 12.5 | |
| GY | qGY-5 | 5 | 78.6 | S5_59373257 - S5_59373316 | 75.3–82.2 | 3.22 | 0.37 | 10.8 |
| qGY-6a | 6 | 15.8 | S6_3799293 - S6_8141493 | 12.7–17.8 | 3.06 | −0.25 | 9.83 | |
| qGY-6b | 6 | 22.3 | S6_13884102 - S6_37768125 | 20.6–25.6 | 3.03 | −0.38 | 11.0 | |
| qGY-6c | 6 | 30.8 | S6_41970042 - S6_43222258 | 28.9–34.8 | 2.73 | −0.23 | 8.00 | |
| TW | qTW-1 | 1 | 209.4 | S1_59595161- S1_61709496 | 201.3–220.9 | 5.79 | −1.79 | 17.9 |
| GS | qGS-1b | 1 | 212.7 | S1_61786623 - S1_61836509 | 204.1–219.7 | 4.67 | −0.08 | 14.4 |
| qGS-5 | 5 | 78.6 | S5_59373257 - S5_59373316 | 76.2–80.1 | 5.30 | 0.07 | 14.6 |
Chlorophyll contents at vegetative stage (Chl1), at anthesis (Chl2), and at maturity (Chl3); PH, plant height (cm); AD, days to anthesis; MC1, % stover moisture content; MC2, % head moisture content; BY, biomass yield (t.ha−1); GY, grain yield (t.ha−1); TW, test weight (g); GS, grain/stover ratio (%). a2.0-LOD drop support interval of the QTL; bAdditive effect: positive values of the additive effect indicate that alleles from Ck60 were in the direction of increasing the trait score and vice versa; c Percentage of phenotypic variation explained by the QTL. The SNP underlined is the corresponding SNP of QTL
Under LN conditions, 20 QTLs were found to be significant for 11 traits studied (Table 5, Fig. 1). We detected four QTLs for chlorophyll content including two each for chlorophyll at flowering and maturity. No significant QTLs were detected for chlorophyll content at the vegetative stage. For these QTLs, the China17 allele increased the chlorophyll content at flowering for the QTL on chromosome 1 and the CK60 alleles increased the chlorophyll contents for the other QTLs. We detected seven significant QTLs for morphological traits. One major QTL explaining 13.2 % of the phenotypic variation was associated with plant height with the allele from China17 increasing plant height by 16.4 cm. Two QTLs were detected for days to anthesis. The CK60 allele associated with the QTL on chromosome 1 delayed heading by 3.6 d, while the China17 allele, associated with the QTL on chromosome 9, delayed heading by 3 d. Two QTLs for stover moisture content and head moisture content were identified with presence of the CK60 alleles resulting in increasing the moisture contents. Nine significant QTLs were found for yield-related traits under LN conditions. Two QTLs were detected for biomass yield, of which the China17 allele contributed for increased biomass yield by 1.0 t.ha−1 for QTL on chromosome 5, while the CK60 allele increased biomass yield at other QTL. Four QTLs were identified for grain yield, of which the CK60 allele increased the grain yield for one QTL on chromosome 5 and China17 alleles improved the grain yield for all other QTLs. One significant QTL explaining 17.9 % of the phenotypic variation was detected for test weight on chromosome 1 with the China17 allele increasing test weight by 1.8 g. Two QTLs were found for grain/stover ratio on chromosomes 1 and 5. The China17 allele contributed to an increase the grain/stover ratio for QTL on chromosome 1 while the CK60 allele was responsible for increasing the grain/stover ratio at the other QTL on chromosome 5. The additive effect of a single QTL could explain 7 to 20.3 % of the total phenotypic variation.
Differential expression of gene transcripts in the QTL regions
The previously generated Illumina RNA-sequencing data [43] was used to determine the variations in transcript abundance between nitrogen use inefficient (CK60) and efficient (China17) genotypes of sorghum. False discovery rate (FDR) ≤ 0.001 and the absolute value of |log2 -Ratio| ≥ 1 were used as thresholds to judge the significance of differences in transcript abundance of the same gene between two genotypes. Pair-wise comparison of the transcriptomes of CK60 and China17 seedling root tissues grown under N-stress revealed a total of 726 DEGs detected using v1.4 sorghum genome (Additional file 3). The sequences of all these DEGs compared to v2.1 sorghum genome and respective gene IDs were listed in Additional file 3. In addition, compared the sequences of polymorphic SNPs between CK60 and China17 to the sequences of DEG transcripts, and differential expression levels were listed in Additional file 2.
Out of 726 DGE transcripts observed between CK60 and China17 (Additional file 3), 108 DEGs were located in the vicinity of the QTL confidence intervals on chromosome 1, 6, 7, 8, and 9 (Additional file 3) and some of those were listed in Table 6. The QTL interval on chromosome 1 has 40 DEGs and chromosome 9 has 28 DEGs. Gene transcripts related to nitrogen metabolism (Ferredoxin-nitrate reductase), glycolysis (Phosphofructo-2-kinase), seed storage proteins, plant hormone metabolism (Gibberellin receptor GID1L2, Auxin response factor 2) were differentially expressed between CK60 and China17. The majority of these gene transcripts were expressed higher in CK60 than China17 under N-stress conditions in the seedling stage. For example, transcripts of Frigida, Auxin response factor 2 and translation elongation factor expressed six-fold higher in CK60 than China17. In contrast, magnesium transporter6, HSP21 and senescence associated protein were expressed higher in China17. A ferredoxin-nitrite reductase gene transcript which had higher expression in China17, coincided with the pleiotropic QTL region on chromosome 9.
Table 6.
Differential expression of gene transcripts associated with QTLs detected using RNA-seq
| Gene id (v1.4) | Chr | Start (bp) | logFC | Annoatation | Low N QTLs | Normal N QTLs |
|---|---|---|---|---|---|---|
| Sb01g032875 | 1 | 55828932 | 6.3 | Translation elongation factor EF1B | qMC2-1a | qMC2-1, qBY-1 |
| Sb01g032880 | 1 | 55840286 | 4 | SPX domain-3 | qMC2-1a | qMC2-1, qBY-1 |
| Sb01g032920 | 1 | 55885605 | 6.84 | Frigida putative expressed | qMC2-1a | qMC2-1, qBY-1 |
| Sb01g033010 | 1 | 56047918 | 9.1 | Retrotransposon protein, | qMC2-1a | qMC2-1, qBY-1 |
| Sb01g033090 | 1 | 56202769 | 3.98 | Mannose-binding lectin superfamily | qMC2-1a | qMC2-1, qBY-1 |
| Sb01g033360 | 1 | 56595053 | −5.2 | Acetoacetyl-CoA thiolase 2 | qMC2-1a | qMC2-1, qBY-1 |
| Sb01g033410 | 1 | 56731990 | 4.27 | Cation/carnitine transporter 3 | qMC2-1a | qMC2-1, qBY-1 |
| Sb01g033620 | 1 | 56944250 | 2.77 | Metacaspase 1 | qMC2-1a | qMC2-1, qBY-1 |
| Sb01g034190 | 1 | 57636134 | 2.49 | O-Glycosyl hydrolases | qMC2-1a | qMC2-1, qBY-1 |
| Sb01g035910 | 1 | 59529076 | 9.28 | Glutathione S-transferase | qMC2-1b | |
| Sb01g036330 | 1 | 59936853 | −3.38 | Ribosomal protein L16p/L10e family | qTW1 | |
| Sb01g036790 | 1 | 60395728 | 2.56 | Late embryogenesis abundant protein 1 | qTW1 | |
| Sb01g037480 | 1 | 61031332 | 4.1 | Nicotianamine synthase 4 | qTW1 | |
| Sb01g038720 | 1 | 62214256 | −7.69 | LHT1 lysine histidine transporter 1 | qAD-1 | |
| Sb01g041180 | 1 | 64497962 | −3.32 | HSP21 Heat shock protein 21 | qGY-1b | |
| Sb01g041350 | 1 | 64653444 | 2.63 | Subtilisin-like serine protease 2 | qGY-1b | |
| Sb01g041390 | 1 | 64692370 | −2.39 | Senescence associated protein | qGY-1b | |
| Sb01g041640 | 1 | 64933485 | 2.04 | Oxidoreductase superfamily protein | qGY-1b | |
| Sb01g041810 | 1 | 65045823 | 3.75 | STRUBBELIG-RECEPTOR FAMILY 7 | qGY-1b | |
| Sb01g042500 | 1 | 65775142 | 1.99 | Caleosin-related family protein | qGY-1b | |
| Sb01g042530 | 1 | 65792345 | 2.89 | MA3 domain-containing protein | qGY-1b | |
| Sb01g044230 | 1 | 67360823 | 2.26 | Polyamine oxidase 1 | qGY-1b | |
| Sb01g044810 | 1 | 67970813 | 3.04 | MADS-box transcription factor family | qGY-1b | |
| Sb01g045620 | 1 | 68676107 | 2.29 | Lectin protein kinase family protein | qGY-1b | |
| Sb01g047250 | 1 | 70350087 | 2.412 | Leucine-rich repeat transmembrane protein kinase | qGY-1b | |
| Sb01g047550 | 1 | 70645925 | 2.215 | Tetratricopeptide repeat (TPR)-like superfamily protein | qGY-1b | |
| Sb01g047650 | 1 | 70741548 | −3.156 | CCT motif family protein | qGY-1b | |
| Sb01g047780 | 1 | 70919577 | −8.348 | Magnesium transporter 6 | qGY-1b | |
| Sb01g048000 | 1 | 71075448 | 1.861 | Glutathione S-transferase | qGY-1b | |
| Sb01g048030 | 1 | 71101357 | 3.126 | Cytochrome P450, family 78, | qGY-1b | |
| Sb01g048100 | 1 | 71159986 | −4.194 | LYM2 lysm domain GPI-anchored protein 2 precursor | qGY-1b | |
| Sb01g048640 | 1 | 71596638 | 6.418 | Leucine-rich repeat family protein | qGY-1b | |
| Sb06g002090 | 6 | 3921155 | 3.276 | F-box/RNI-like/FBD-like domains-containing protein | qGY-6a | |
| Sb06g002180 | 6 | 4089379 | −2.511 | UDP-glucosyltransferase | qGY-6a | |
| Sb06g003180 | 6 | 6636307 | 3.018 | CAP (Cysteine-rich secretory proteins | qGY-6a | |
| Sb06g005420 | 6 | 13588101 | 6.889 | expressed protein | ||
| Sb06g006920 | 6 | 16373350 | −2.546 | purple acid phosphatase 27 | qGY-6b | |
| Sb06g008990 | 6 | 26359064 | 2.375 | Oxidoreductase, zinc-binding dehydrogenase family protein | qGY-6b | |
| Sb06g010870 | 6 | 30379011 | 3.616 | Cytochrome P450, family 71 | qGY-6b | |
| Sb06g011767 | 6 | 32144416 | 6.478 | auxin response factor 2 | qGY-6b | |
| Sb06g011770 | 6 | 32145882 | 6.844 | C2H2-like zinc finger protein | qGY-6b | |
| Sb06g012040 | 6 | 32755618 | 2.708 | Minichromosome maintenance (MCM2/3/5) family protein | qGY-6b | |
| Sb06g012280 | 6 | 33774679 | 2.815 | UDP-Glycosyltransferase superfamily | qGY-6b | |
| Sb06g012290 | 6 | 33921889 | 4.708 | Galactose oxidase/kelch repeat superfamily protein | qGY-6b | |
| Sb06g014250 | 6 | 39313831 | 4.128 | multidrug resistance-associated protein 9 | qGY-6b | |
| Sb06g014400 | 6 | 39867816 | −3.085 | HSP70 Heat shock protein 70 | ||
| Sb06g015520 | 6 | 43082617 | 3.501 | B-block binding subunit of TFIIIC | qPH-6, qGY-6c | |
| Sb06g016160 | 6 | 44576681 | 2.202 | seed storage 2S albumin superfamily | qPH-6, qGY-6c | |
| Sb06g016230 | 6 | 44708205 | 2.164 | Late embryogenesis abundant hydroxyproline-rich glycoprotein | qPH-6, qGY-6c | |
| Sb06g016260 | 6 | 44735351 | 2.5 | Aluminium activated malate transporter | qPH-6, qGY-6c | |
| Sb06g019470 | 6 | 49032854 | 1.3 | Copper transport protein family | qMC1-6a | |
| Sb06g019600 | 6 | 49168975 | 4.279 | Cytochrome P450 superfamily protein | qMC1-6a | |
| Sb06g019610 | 6 | 49174004 | 2.452 | phosphofructokinase 2 | qMC1-6a | |
| Sb06g024400 | 6 | 53535970 | 1.683 | NUDIX family, domain containing protein | qMC1-6b | qMC1-6b |
| Sb06g024590 | 6 | 53686490 | 1.958 | tonoplast intrinsic protein | qMC1-6b | qMC1-6b |
| Sb06g024650 | 6 | 53730735 | 4.367 | expansin B2 | qMC1-6b | qMC1-6b |
| Sb06g025220 | 6 | 54190773 | −2.114 | calcium-dependent protein kinase 29 | qMC1-6b | qMC1-6b |
| Sb06g025250 | 6 | 54207289 | 2.782 | Prolyl oligopeptidase family protein | qMC1-6b | qMC1-6b |
| Sb06g025330 | 6 | 54262137 | 2.362 | expressed protein | qMC1-6b | qMC1-6b |
| Sb06g028210 | 6 | 57045616 | 2.252 | Terpenoid cyclases/Protein prenyltransferases superfamily protein | qMC2-6 | |
| Sb06g028480 | 6 | 57260405 | 2.019 | unknown | qMC2-6 | |
| Sb06g028760 | 6 | 57496434 | 1.729 | Leucine-rich receptor-like protein kinase | qMC2-6 | |
| Sb07g022670 | 7 | 57311597 | 2.956 | Glutamate decarboxylase | qChl3-7, qBY-7 | |
| Sb07g022800 | 7 | 57514740 | 2.625 | aspartyl protease family protein | qChl3-7, qBY-7 | |
| Sb07g023140 | 7 | 57977647 | 4.794 | Gibberellin receptor GID1L2 | qChl3-7, qBY-7 | |
| Sb07g023220 | 7 | 58087984 | −3.707 | phospholipase A | qChl3-7 | |
| Sb07g023300 | 7 | 58178273 | 2.495 | expressed protein | qChl3-7 | |
| Sb07g023770 | 7 | 58722654 | 11.097 | rotamase | qChl3-7 | |
| Sb07g024200 | 7 | 59189842 | −7.492 | Ribosomal protein L1p/L10e family | qChl3-7 | |
| Sb07g025190 | 7 | 60212195 | 4.048 | MATE efflux family protein | qChl3-7 | |
| Sb07g025240 | 7 | 60280418 | −2.248 | hydroxymethylglutaryl-CoA synthase | qChl3-7 | |
| Sb07g025843 | 7 | 60959427 | 2.544 | ethylene-responsive transcription factor ERF114, putative, expressed | qChl1-7 | |
| Sb08g000550 | 8 | 482761 | −2.111 | ferritin-1, chloroplast precursor | qGS-8 | |
| Sb08g002590 | 8 | 2673615 | −2.004 | WRKY DNA-binding protein 55 | qGS-8 | |
| Sb08g002660 | 8 | 2779571 | 2.726 | Protease inhibitor/seed storage/LTP | qGS-8 | |
| Sb08g003170 | 8 | 3513569 | 3.001 | Chalcone and stilbene synthase family | qGS-8 | |
| Sb08g003820 | 8 | 4423015 | −3.008 | zinc finger superfamily protein | qGS-8 | |
| Sb08g004500 | 8 | 5410066 | −2.127 | fructose-bisphosphate aldolase 2 | qGS-8 | |
| Sb09g018440 | 9 | 46098300 | 6.288 | methyl esterase 3 | qAD-9a | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g020000 | 9 | 49032969 | 8.222 | inosine-uridine preferring nucleoside hydrolase family protein | qAD-9a | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g020240 | 9 | 49471823 | 3.041 | Major facilitator superfamily protein | qAD-9a | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g021016 | 9 | 50446536 | 3.241 | ethylene-responsive transcription factor | qAD-9a | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g021250 | 9 | 50714173 | 2.158 | alpha/beta-Hydrolases superfamily protein | qAD-9a | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g021490 | 9 | 50944384 | 4.533 | Subtilase family protein | qAD-9a | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g021720 | 9 | 51194456 | −1.869 | histone deacetylase 8 | qAD-9a | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g022390 | 9 | 52044973 | 8.308 | Ribosomal protein | qAD-9a | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g023150 | 9 | 52794183 | 2.46 | ribonuclease P family protein | qAD-9a | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g023320 | 9 | 52948577 | 6.593 | Major facilitator superfamily protein | qAD-9a | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g024840 | 9 | 54319624 | −2.168 | ferredoxin--nitrite reductase | qAD-9a | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g025530 | 9 | 55006797 | 2.719 | O-methyltransferase family protein | qAD-9a | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g025540 | 9 | 55018768 | 2.155 | O-methyltransferase family protein | qAD-9a | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g025730 | 9 | 55141225 | 3.406 | non-symbiotic hemoglobin 2 | qAD-9a | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g025900 | 9 | 55284480 | −2.24 | HSP101 Heat shock protein 101 | qAD-9a, qChl2-9b | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g026590 | 9 | 55803666 | 1.86 | RING/U-box superfamily protein | qAD-9a, qChl2-9b | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g027380 | 9 | 56449825 | −2.508 | serine/threonine-protein kinase SNT7, chloroplast precursor | qAD-9a, qChl2-9b | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g027470 | 9 | 56561299 | 7.612 | Disease resistance protein | qAD-9a, qChl2-9b | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g027590 | 9 | 56662520 | 2.783 | seed storage 2S albumin superfamily | qAD-9a, qChl2-9b | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g028890 | 9 | 57684814 | 2.326 | Iron-sulfur cluster, SufE/NifU family protein | qAD-9a | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g028960 | 9 | 57721281 | 3.573 | ribosomal protein L13 | qAD-9a | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
| Sb09g029540 | 9 | 58186320 | 5.903 | AMP-dependent synthetase | qAD-9a | qGY-9, qChl2-9a, qBY-9, qChl3-9, qPH-9 |
Chr, chromosome number; log2 ratio; number of folds the gene transcript is differentially expressed in RNA-seq. Log2 ratio >0 indicates, positive values indicates gene transcript expressed high in CK60. ns, indicate the transcript is not differentially expressed between CK60 and china17
Discussion
Trait variation in the mapping population under different N regimes
The RILs showed transgressive segregation for all the traits measured and in most cases, the mean value of the traits was intermediate between the parental lines, CK60 and China17 (Tables 1 and 2), suggesting a polygenic inheritance of the traits. Transgressive segregation can be caused by both parental lines contributing favorable or unfavorable alleles for a particular trait and is common in inbred populations [55]. In both N conditions, the genetic variance was greater than genotype by environment interaction variance for all the traits (Tables 1 and 2). This finding is in agreement with earlier studies [56]. The more marked contribution of genetic variance to trait determination suggests the opportunity for more robust detection of QTLs that govern nitrogen use efficiency [14]. Here, for both parental lines and RILs marked reductions were observed in mean values for chlorophyll contents measured at three different stages, plant height, biomass and grain yield traits grown under LN compared to NN. In maize, a 38 % reduction in grain yield was observed in plants grown under low-N compared to high-N conditions [14]. This decrease was caused by a significant reduction in kernel number, but has little effect on kernel size. Kernel number is very susceptible to N-stress because ovules are susceptible to abortion soon after fertilization [57], a possible result of limitation in supply of photosynthetic products [58].
Comparison of QTL regions under contrasting N environments
In this study, a total of 38 QTLs were identified using a SNP based genetic map in the RIL mapping population tested under two different nitrogen levels. However, almost half of these QTLs were detected under one N level, indicating that these traits were controlled by different genes under different N conditions. Major QTLs detected across two normal and two low-N environments were considered as consistent across environments. However, five QTLs for four morphological traits were detected consistently under both N conditions. These included, one QTL each for chlorophyll at maturity, day to anthesis and stover moisture content and two QTLs for head moisture content. For all these QTLs, the CK60 alleles increased chlorophyll content, delayed flowering, and increased stover and head moisture contents under NN and LN. This indicates that these traits shared a similar genetic basis under different N conditions.
Co-localization of QTLs between traits and associated differentially expressed gene transcripts
Co-localization may suggest pleiotropy whereby a genomic region contains genes that affect a number of traits [59]. In this study, co-localized QTLs affecting different traits were detected on chromosomes 1, 5, 6, 7, and 9 (Fig. 1). For example, the support intervals of ten QTLs explaining 8.1 to 20.3 % of phenotypic variation for eight traits were overlapping in the distal end of chromosome 1. Of the ten QTLs detected, two QTLs are for grain moisture content, one QTL each for test weight, chlorophyll content at anthesis, stover moisture content and grain/stover ratio detected under LN conditions, biomass yield under NN and for days to anthesis detected under NN and LN conditions. An additive effect from CK60 increased days to anthesis (delayed flowering), stover and head moisture content and grain yield. These traits were highly correlated (Table 3) and the correlations resulted in co-localization. Within this co-localized region, QTLs for green leaf area at maturity [60], days to anthesis [60, 61] fresh panicle weight, plant height [59, 62], and panicle architecture [63] were reported earlier. Stay green QTLs and the Ma3 gene encoding phytochrome B, which is involved in photoperiod sensitivity [64], were also reported in this region.
In this co-localized region containing ten QTLs, RNA-seq detected 19 differentially expressed gene transcripts between CK60 and China17, of which only six DEGs had higher expression in China17 (Table 6). Some of these DEGs including SPX domain-3, Frigida, late embryogenesis abundant protein 1 (LEA) were expressed higher in CK60, and lysine histidine transporter 1 (LHT1) had higher expression in China17. An SPX domain gene-3 was reported to be up-regulated and plays an important role in plant adaptation to phosphate starvation [65]. This region containing a major QTL for days to anthesis, was detected under both N conditions explaining 16 % of phenotypic variation. The CK60 allele contributed to flowering delay by three days. This region contained the flowering time gene transcript, Frigida, Which showed more abundant expression in CK60. It was reported earlier that ethylene insensitive 3-Like 1 (EIL-1), key regulator of ethylene biosynthesis, underlies the QTL cluster for days to anthesis, and green leaf area at maturity [60]. However, this gene is not differentially expressed in the root tissues of young seedlings in our RNA-seq analysis (not listed in Table 6). Together, these data suggest that high expression levels of the Frigida gene may contribute to the delayed flowering in CK60, but this is not the only gene influencing this phenotype. Similarly another DEG transcript, LEA had two-fold higher expression in CK60 under N-stress condition. Transgenic expression of a barley LEA protein in rice resulted in increased growth rate of transgenic plants than non-transformed plants under stress conditions [66]. Thus, LEA proteins play an important role in protection of plants under stress, a potential tool for genetic improvement towards stress tolerance. In contrast, a DEG transcript encoding high affinity amino acid transporter, lysine histidine transporter (LHT1), was massively expressed in China17 compared CK60 (Table 6). It was reported that being expressed in the root, LHT1 is responsible for uptake of amino acids from soil into root tissue [67], and distributes from roots to shoots through xylem [68] for further metabolism especially under N-stress conditions. The amino acid uptake, and thus nitrogen use efficiency could be higher with increased LHT1 expression under limited inorganic N supply.
A QTL for grain yield is located on distal end of chromosome 1. In this region QTLs for kernel weight [69], maturity [60], number of kernels/panicle and panicle length [70] and panicle architecture [71] were reported earlier. In this region, our RNA seq data detected 20 DEG transcripts including caleosin-related (Ca+2 binding) protein, a MADS-box transcription factor, polyamine oxidase 1 were expressed higher in CK60. Gene transcripts for magnesium transporter 6, a heat shock protein (HSP21) and senescence associated protein were more abundant in China17 (Table 6). Polyamines (PAs) and ethylene are endogenous plant growth regulators mediating many physiological processes such as growth, senescence, and responses to environmental stresses [72]. High levels of PAs were reported to be associated with higher kernel set and better seed development in maize [73] and increased grain-filling rates in rice [74]. On chromosome 5, QTLs for biomass yield detected under LN and test weight under NN are co-localized (Fig. 1). For these QTLs, the positive allele from China17 increased biomass yield by 1.0 t.ha−1 under LN conditions. In this co-localized region, QTLs for stay green [75, 76], fresh panicle weight and plant height [62] were detected earlier. In this region, RNA seq didn’t detect any significant DEG transcripts between Ck60 and China17.
On chromosome 6, co-localization was observed between major QTLs for plant height and grain yield under LN conditions. For these QTLs, the positive allele from China17 increased plant height by 16.4 cm as well as grain yield. In this region, QTLs for culm height and kernel weight [61], maturity and total dry matter [59], panicle architecture [63] and a major photoperiod sensitivity locus, Ma1 [77, 78] were reported earlier. Also, a major QTL for plant height, QPhe-sbi06-1, conditioned by the Dw2 gene was detected earlier by [60], and showed pleiotropic effects on panicle length, yield, and seed weight [79]. Transcriptome comparison showed that a Dw2 transcript encoding a multidrug resistance-associated protein 9 homolog showed higher expression levels in CK60, which may be involved in regulating plant height under N-stress in the seedlings (Table 6). In addition, RNA-seq found several differentially abundant gene transcripts in this co-localized region, including auxin response factor 2, seed storage 2S albumin, aluminum activated malate transporter, copper transporter and phosphofructokinase 2, all of which were expressed higher in CK60 and HSP70 was expressed higher in China17. Phosphofructo-2-kinase is the principle enzyme regulating the entry of metabolites into glycolysis [80] through conversion of fructose-6-phosphate to fructose-1,6-bisphosphate. This results in an increase of hexose phosphate, supplying more energy and substrates that are necessary for strong seedling development. It would be of interest to see whether differential expression of these transcripts holds true with the adult tissues and use them in marker assisted selection to regulate the pleotropic regions under LN conditions.
On chromosome 7, QTLs for biomass yield, chlorophyll content at vegetative and maturity were co-localized. For these QTLs, the positive allele from China17 increased biomass yield by 1.0 t.ha−1 under LN conditions. In this region, QTLs for fresh total biomass yield and dry total biomass yield was reported by Murray et al. [81]. In this co-localized region, a major plant height gene, Dw3 (Sb07g0232730), is located. Dw3 encodes a phosphoglycoprotein auxin efflux carrier orthologous to PGP1 in Arabidopsis [82]. QTL for panicle architecture [61, 69], total biomass yield t.ha−1 [81] and plant height [60] were reported earlier. In this region, RNA seq detected 12 DEG’s between CK60 and China 17 (Table 6). Glutamate decarboxylase, gibberellin receptor GID1L2 and ethylene responsive transcription factor ERF114 were expressed higher in CK60 and ribosomal protein L1p/L10e was abundant in China17. Glutamate decarboxylase (GAD1) was reported to be expressed in roots and catalyze the synthesis of γ-aminobutyric acid (GABA) under heat stress, disruption of GAD1 gene prevented accumulation of GABA in roots in response to heat stress [83].
A co-localized region at the distal end of the chromosome 9 contains QTLs for chlorophyll at flowering and days to anthesis across two LN and chlorophyll at maturity, plant height, biomass and grain yield traits across two NN. This clustering of QTLs is supported by the negative correlation observed between the chlorophyll contents at flowering and maturity, morphological and yield-related traits. In this region, alleles from China17 increased plant height, biomass and grain yield but caused negative effects on chlorophyll content at flowering and maturity. QTLs for stay green [76, 84], total seed weight [63], plant height [62], maturity [61, 78] were reported previously in this region. Moreover, a QTL interval for plant height (Sb-HT9.1) was fine mapped to ~100 kb region through association mapping [85], Dw3 and Sb-HT9.1 were consistently identified as two of the most important plant height loci in crosses between tall and dwarf sorghum [69, 78]. Our RNA-seq data showed that this region contains 28 DEG transcripts including those encoding ferredoxin-nitrite reductase (FNR), chloroplast localized serine/threonine-protein kinase, and a SufE/NifU family protein. FNR gene transcripts were highly expressed in China17 root tissues compared to CK60. In general nitrate is absorbed from soil, reduced to nitrite and then to ammonia by FNR in the plastids of root cells. The ammonia produced is incorporated into amino acids via the glutamine synthetase-glutamate synthase (GS-GOGAT) pathway. This region of chromosome 9 harbors the highly expressed gene encoding NADH-GOGAT and a glutamine-rich protein. However, these genes are not differentially expressed between the root tissues of CK60 and China17 according to RNA-seq data. Further, it would be important to check whether the expression levels of NADH-GOGAT between China17 and CK60 are changed in the shoots because most of the nitrogen assimilation takes place in shoots rather than root tissues. Transgenic over-expression of NADH-GOGAT in rice resulted in an increase in grain weight, indicating that NADH-GOGAT is indeed a key enzyme in nitrogen utilization and grain filling in rice [86]. In wheat, Quraishi et al. [25] validated the NUE QTL on chromosome-3B, and proposed that a GOGAT gene is conserved structurally and functionally at orthologous positions in rice, sorghum and maize genomes and that this gene likely contributes significantly to NUE in wheat and other cereals. It will be of interest to determine if breeding that allows for higher expression of FNR and GOGAT can increase biomass and grain yield by increasing nitrate assimilation and ammonium production.
Conclusion
QTLs detected for the different agronomic traits in the same genomic regions were consistent with previous QTL mapping studies conducted in diverse genetic and environmental backgrounds in sorghum. RNA-seq analyses detected differential expression of gene transcripts in the pleiotropic QTLs related to nitrogen uptake and metabolism and their expression levels were influenced by the availability of nitrogen. These potential DEG transcripts can possibly be used for improving sorghum performance through marker-assisted selection (MAS) strategies under N-stress conditions by further validation in other mapping populations. The markers and genes reported in this study will have applications in QTL mapping studies, diversity studies, and association mapping studies in sorghum and other members of the Poaceae family collectively aimed at improving nitrogen utilization.
Availability of supporting data
Supporting data are included as additional files
We deposited the RNA-seq data in Gene Expression Omnibus (http://www.ncbi.nm.nih.gov/geo/query/acc.cgi?acc=GSE54705) and it was mentioned in Gelli et al. 2014, BMC Genomics v15.
Acknowledgements
This study was supported by Plant Feedstock Genomics for Bioenergy #DE-Sc0002259 and The United Sorghum Check off Program # R0002-10. We thank Mei Chen and Jean Jack Reithoven of the University of Nebraska Genomics Core Facility for RNA-sequencing and Dr. Yongchao Dou for assisting with RNA-seq data analysis. We thank Tejindar Kumar Mall and Kanokwan for assisting in field data collection and Anji Reddy Konda for extensive help in experimental layout, field data collection, and critical review of the manuscript.
Abbreviations
- RILs
Recombinant inbred lines
- QTLs
Quantitative trait loci
- SNPs
Single nucleotide polymorphisms
- GBS
Genotyping-By-Sequencing
- DEG
Differentially expressed gene
- NUE
Nitrogen use efficiency
- GS
Glutamine synthetase
- Ppd
Photoperiod sensitivity
- Vrn
Vernalization
- Rht
Reduced height
- PEPcase
Phosphoenolpyruvate carboxylase
- LN
Low Nitrogen
- NN
Normal Nitrogen
- Chl1
Chlorophyll content at vegetative stage
- Chl2
Chlorophyll content at anthesis
- Chl3
Chlorophyll content at maturity
- PH
Plant height (cm)
- AD
Days to anthesis (days)
- MC1
Stover moisture content (%)
- MC2
Head moisture content (%)
- BY
Biomass yield (t.ha−1)
- GY
Grain yield (t.ha−1)
- TW
Test weight (g)
- GS
Grain/stover ratio (%)
- ANOVA
Analysis of variance
- IciMapping
Inclusive composite interval mapping
- LOD
Logarithm of odds
- h2
Narrow sense heritability
- FDR
False discovery rate
- HSP
Heat shock protein
- PAs
Polyamines
- EIL
1-ethylene insensitive 3-Like-1
- FNR
Ferredoxin-nitrite reductase
- GOGAT
Glutamate synthase
Additional files
Basic parameters showing soil properties at two N levels across years. (xls 22.0 kb)
Genetic distribution of SNPs discovered using genotyping-by-sequencing (GBS) in CK60 x China17 population. (xlsx 41.1 kb)
The list of differentially expressed genes identified between CK60 and China17 using RNA-seq. (xls 169 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MG designed the study, collected genotypic and phenotypic data, analyzed data for linkage map, QTL analysis, designed and executed Illumina RNA sequencing experiment, interpreted data, drafted and revised the manuscript, SM performed GBS for SNP discovery, CZ and KL for bioinformatics support; DH designed and supervised the RNA-seq study and critically reviewed the manuscript; ID coordinated the project, developed the RIL population and critically reviewed the manuscript; TC and DW are Co-PI’s on the DOE grant and both contributed to the phenotyping of the RIL population. All the authors read and approved the final manuscript.
Contributor Information
Malleswari Gelli, Email: malleswari.gelli@gmail.com.
Sharon E. Mitchell, Email: sem30@cornell.edu
Kan Liu, Email: kliu7@unl.edu.
Thomas E. Clemente, Email: tclemente1@unl.edu
Donald P. Weeks, Email: donald.weeks@unl.edu
Chi Zhang, Email: czhang5@unl.edu.
David R. Holding, Email: dholding2@unl.edu
Ismail M. Dweikat, Phone: (402)472-5328, Email: idweikat2@unl.edu
References
- 1.Doggett H. Sorghum. 2. New York: Wiley; 1988. [Google Scholar]
- 2.Paterson AH. Genomics of sorghum (A Review) 2008. Int J Plant Genomics. 2008;362451. doi:10.1155/2008/362451. [DOI] [PMC free article] [PubMed]
- 3.Price HJ, Dillon SL, Hodnett G, Rooney WL, Ross L, Johnston JS. Genome evolution in the genus Sorghum (Poaceae) Ann Bot. 2005;95:219–227. doi: 10.1093/aob/mci015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mace ES, Rami J, Bouchet S, Klein PE, Klein RR, Kilian A, et al. A consensus genetic map of sorghum that integrates multiple component maps and high-throughput diversity array technology (DArT) markers. BMC Plant Biol. 2009;9:13. doi: 10.1186/1471-2229-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marschner H. Mineral nutrition of higher plants. 2. London: Academic; 1995. [Google Scholar]
- 6.Hak R, Rinderle-Zimmer U, Lichtenthaler HK, Natr L. Chlorophyll a fluorescence signatures of nitrogen deficient barley leaves. Photosynthetica. 1993;28:151–159. [Google Scholar]
- 7.Frink CR, Waggoner PE, Ausubel JH. Nitrogen fertilizer: retrospect and prospect. Proc Natl Acad Sci U S A. 1999;96:1175–1180. doi: 10.1073/pnas.96.4.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Good AG, Shrawat AK, Muench DG. Can less yield more? Is reducing nutrient input into the environment compatible with maintaining crop production? Trends Plant Sci. 2004;9:597–605. doi: 10.1016/j.tplants.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 9.Guo JH, Liu XJ, Zhang Y, Shen JL, Han WX, Zhang WF, et al. Significant acidification in major Chinese croplands. Science. 2010;327:1008–1010. doi: 10.1126/science.1182570. [DOI] [PubMed] [Google Scholar]
- 10.Hirel B, Le Gouis JL, Ney B, Gallais A. The challenge of improving nitrogen use efficiency in crop plants: towards a more central role for genetic variability and quantitative genetics within integrated approaches. J Exp Bot. 2007;58:2369–2387. doi: 10.1093/jxb/erm097. [DOI] [PubMed] [Google Scholar]
- 11.Moll RH, Kamprath EJ, Jackson WA. Analysis and interpretation of factors which contribute to efficiency of nitrogen utilization. Agron J. 1982;74:562–564. doi: 10.2134/agronj1982.00021962007400030037x. [DOI] [Google Scholar]
- 12.Hirel B, Lea PJ. Ammonia assimilation. In: Lea PJ, Morot-Gaudry J-F, editors. Plant nitrogen. Berlin Heidelberg New York: Springer; 2001. pp. 79–99. [Google Scholar]
- 13.Agrama HAS, Zakaria AG, Said FB, Tuinstra M. Identification of quantitative trait loci for nitrogen use efficiency in maize. Mol Breed. 1999;5:187–195. doi: 10.1023/A:1009669507144. [DOI] [Google Scholar]
- 14.Gallais A, Hirel B. An approach to the genetics of nitrogen use efficiency in maize. J Exp Bot. 2004;55:295–306. doi: 10.1093/jxb/erh006. [DOI] [PubMed] [Google Scholar]
- 15.Loudet O, Chaillou S, Merigout P, Talbotec J, Daniel-Vedele F. Quantitative trait loci analysis of nitrogen use efficiency in Arabidopsis. Plant Physiol. 2003;131:345–358. doi: 10.1104/pp.102.010785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng Y, Cao LY, Wu WM, Shen XH, Zhan XD, Zhai RR, et al. Mapping QTLs for nitrogen-deficiency tolerance at seedling stage in rice (Oryza sativa L.) Plant Breed. 2010;129:652–656. doi: 10.1111/j.1439-0523.2009.01728.x. [DOI] [Google Scholar]
- 17.Wei D, Cui K, Ye G, Pan J, Xiang J, Huang J, et al. QTL mapping for nitrogen-use efficiency and nitrogen-deficiency tolerance traits in rice. Plant Soil. 2012;359:281–295. doi: 10.1007/s11104-012-1142-6. [DOI] [Google Scholar]
- 18.Zhao C, Zhou LH, Zhang Y, Zhu Z, Chen T, Zhao Q, et al. QTL mapping for seedling traits associated with low-nitrogen tolerance using a set of advanced backcross introgression lines of rice. Plant Breed. 2014;133(2):189–195. doi: 10.1111/pbr.12123. [DOI] [Google Scholar]
- 19.Hoffmann A, Maurer A, Pillen K. Detection of nitrogen deficiency QTL in juvenile wild barley introgression lines growing in a hydroponic system. BMC Genet. 2012;13:88. doi: 10.1186/1471-2156-13-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mickelson S, See D, Meyer FD, Garner JP, Foster CR, Blake TK, et al. Mapping of QTL associated with nitrogen storage and remobilization in barley (Hordeum vulgare L.) leaves. J Exp Bot. 2003;54:801–812. doi: 10.1093/jxb/erg084. [DOI] [PubMed] [Google Scholar]
- 21.Joppa LR, Du C, Hart GE, Hareland GA. Mapping gene(s) for grain protein in tetraploid wheat (Triticum turgidum L.) using a population of recombinant inbred chromosome lines. Crop Sci. 1997;37:1586–1589. doi: 10.2135/cropsci1997.0011183X003700050030x. [DOI] [Google Scholar]
- 22.Laperche A, Brancourt-Hulmel M, Heumez E, Gardet O, Hanocq E, Devienne-Barret F, et al. Using genotype x nitrogen interaction variables to evaluate the QTL involved in wheat tolerance to nitrogen constraints. Theor Appl Genet. 2007;115:399–415. doi: 10.1007/s00122-007-0575-4. [DOI] [PubMed] [Google Scholar]
- 23.Habash DZ, Bernard S, Schondelmaier J, Weyen J, Quarrie SA. The Genetics of nitrogen use in hexaploid wheat: N utilization, development and yield. Theor Appl Genet. 2007;114:403–419. doi: 10.1007/s00122-006-0429-5. [DOI] [PubMed] [Google Scholar]
- 24.Fontaine JX, Ravel C, Pageau K, Heumez E, Dubois F, Hirel B, et al. A quantitative genetic study for elucidating the contribution of glutamine synthetase, glutamate dehydrogenase and other nitrogen-related physiological traits to the agronomic performance of common wheat. Theo Appl Genet. 2009;119:645–662. doi: 10.1007/s00122-009-1076-4. [DOI] [PubMed] [Google Scholar]
- 25.Quraishi UM, Abrouk M, Murat F, Pont C, FouFoucrier S, Desmaizieres G, et al. Cross-genome map based dissection of a nitrogen use efficiency ortho-metaQTL in bread wheat unravels concerted cereal genome evolution. Plant J. 2011;65(5):745–756. doi: 10.1111/j.1365-313X.2010.04461.x. [DOI] [PubMed] [Google Scholar]
- 26.Maranville JW, Clark RB, Ross WM. Nitrogen efficiency in grain sorghum. J Plant Nutr. 1980;2:577–589. doi: 10.1080/01904168009362800. [DOI] [Google Scholar]
- 27.Youngquist JB, Bramel-Cox P, Maranville JW. Evaluation of alternative screening criteria for selecting nitrogen-use efficient genotypes in sorghum. Crop Sci. 1992;32(6):1310–1313. doi: 10.2135/cropsci1992.0011183X003200060002x. [DOI] [Google Scholar]
- 28.Senthilvel S, Vinod KK, Malarvizhi P, Maheswaran M. QTL and QTL× environment effects on agronomic and nitrogen acquisition traits in rice. J Integr Plant Biol. 2008;50(9):1108–1117. doi: 10.1111/j.1744-7909.2008.00713.x. [DOI] [PubMed] [Google Scholar]
- 29.Menz MA, Klein RR, Mullet J, Obert JA, Unruh NC, Klein PE. A high-density genetic map of Sorghum bicolor (L.) Moench based on 2926 AFLP(R), RFLP and SSR markers. Plant Mol Biol. 2002;48:483–499. doi: 10.1023/A:1014831302392. [DOI] [PubMed] [Google Scholar]
- 30.Jaccoud D, Peng K, Feinstein D, Kilian A. Diversity arrays: a solid state technology for sequence information independent genotyping. Nucleic Acids Res. 2001;29(4):E25. doi: 10.1093/nar/29.4.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mace ES, Xia L, Jordan DR, Halloran K, Parh DK, Huttner E, et al. DArT markers: diversity analyses and mapping in Sorghum bicolor. BMC Genomics. 2008;9:26. doi: 10.1186/1471-2164-9-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paterson AH, Bowers JE, Bruggmann R. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457:551–556. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- 33.Mace E, Jordan D. Location of major effect genes in sorghum (Sorghum bicolor (l.) Moench). Theor Appl Genet. 2010; doi:10.1007/s00122-010-1392-8. [DOI] [PubMed]
- 34.Mace E, Jordan D. Integrating sorghum whole genome sequence information with a compendium of sorghum QTL studies reveals uneven distribution of QTL and of gene-rich regions with significant implications for crop improvement. Theor Appl Genet. 2011;123:169–191. doi: 10.1007/s00122-011-1575-y. [DOI] [PubMed] [Google Scholar]
- 35.Varshney RK, Nayak SN, May GD, Jackson SA. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 2009;27:522–530. doi: 10.1016/j.tibtech.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 36.Zou G, Zhai G, Feng Q, Yan S, Wang A, Zhao Q, et al. Identification of QTLs for eight agronomically important traits using an ultra-high-density map based on SNPs generated from high-throughput sequencing in sorghum under contrasting photoperiods. J Exp Bot. 2012;63(15):5451–5462. doi: 10.1093/jxb/ers205. [DOI] [PubMed] [Google Scholar]
- 37.Huang X, Feng Q, Qian Q, Zhao Q, Wang L, Wang A, et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009;19:1068–1076. doi: 10.1101/gr.089516.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Bucker ES, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6(5):e19379. doi: 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poland JA, Brown PJ, Sorrells ME, Jannink JL. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. Plos One. 2012;7:e32253. doi: 10.1371/journal.pone.0032253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dugas DV, Monaco MK, Olsen A, Klein RR, Kumari S, Ware D. Functional annotation of the transcriptome of Sorghum bicolor in response to osmotic stress and abscisic acid. BMC Genomics. 2011;12:514. doi: 10.1186/1471-2164-12-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davidson RM, Gowda M, Moghe G, Lin H, Vaillancourt B, Shiu SH. Comparative transcriptomics of three Poaceae species reveals patterns of gene expression evolution. Plant J. 2012;71:492–502. doi: 10.1111/j.1365-313X.2012.05005.x. [DOI] [PubMed] [Google Scholar]
- 42.Yazawa T, Kawahigashi H, Matsumoto T, Mizuno H. Simultaneous transcriptome analysis of Sorghum and Bipolaris sorghicola by using RNA-seq in combination with de novo transcriptome assembly. PLoS One. 2013;8:e62460. doi: 10.1371/journal.pone.0062460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gelli M, Duo Y, Konda AR, Zhang C, Holding DR, Dweikat IM. Identification of differentially expressed genes between sorghum genotypes with contrasting nitrogen stress tolerance by genome-wide transcriptional profiling. BMC Genomics. 2014;15:179. doi: 10.1186/1471-2164-15-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Makita Y, Shimada S, Kawashima M, Kondou-Kuriyama T, Toyoda T, Matsui M. MOROKOSHI: Transcriptome Database in Sorghum bicolor. Plant Cell Physiol. 2015;56:e6. doi: 10.1093/pcp/pcu187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maranville JW, Madhavan S. Physiological adaptations for nitrogen use efficiency in sorghum. Plant Soil. 2002;245:25–34. doi: 10.1023/A:1020660504596. [DOI] [Google Scholar]
- 46.Haussmann B, Mahalakshmi V, Reddy BVS, Seetharama N, Hash CT, Geiger HH. QTL mapping of stay-green in two sorghum recombinant inbred populations. Theor Appl Genet. 2002;106:133–142. doi: 10.1007/s00122-002-1012-3. [DOI] [PubMed] [Google Scholar]
- 47.Joshi AK, Pandya JN, Buhecha KV, Dave HR, Pethani KV, Dangaria CJ. Grain yield in pearl millet in relation to source size and proximity to sink. Photosynthetica. 2003;41:157–159. doi: 10.1023/A:1025841320024. [DOI] [Google Scholar]
- 48.Littell RC, Milliken GA, Stroup WW, Wolfinger RD. SAS system for mixed models. Cary, NC, USA: SAS Institute Inc; 1996. [Google Scholar]
- 49.Holland JB, Nyquist WE, Cervantes-Martinez CT. Estimating and interpreting heritability for plant breeding: an update. Plant Breed Rev. 2003;22(22):9–112. [Google Scholar]
- 50.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J. Accurate whole genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang J. Inclusive composite interval mapping of quantitative trait genes. Acta Agron Sin. 2009;35:239–245. doi: 10.3724/SP.J.1006.2009.00239. [DOI] [Google Scholar]
- 52.Wang S, Basten CJ, Zeng ZB. Windows QTL Cartographer 2.5. Raleigh, NC: Department of Statistics, North Carolina State University; 2007. [Google Scholar]
- 53.Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138:963–971. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McCouch SR, Cho YG, Yano M, Paul E, Blinstrub M, Morishima H, et al. Report on QTL nomenclature. Rice Genet Newsl. 1997;14:11–13. [Google Scholar]
- 55.Rieseberg LH, Widmer A, Arntz AM, Burke JM. The genetic architecture necessary for transgressive segregation is common in both natural and domesticated populations. Philos trans royal soc London B. 2003;358:1141–1147. doi: 10.1098/rstb.2003.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Landbeck MV. Untersuchungen zur genetischen verbesserung der anbaueigung von körnermais unter produktionsbedingungen mit verringerter sticksoffversorgung. PhD thesis. Universität Hohenheim. 1995. [Google Scholar]
- 57.Below FE, Cazetta JO, Seebauer JR. Carbon/nitrogen interactions during ear and kernel development of maize. In: Physiology and modelling kernel set in maize. CSSA special publication 2000; no. 29. p15–24.
- 58.McCullough DE, Aguilera A, Tollenaar M. N uptake, N partitioning, and photosynthetic N-use efficiency of an old and a new maize hybrid. Can J Plant Sci. 1994;74:479–484. doi: 10.4141/cjps94-088. [DOI] [Google Scholar]
- 59.Ritter KB, Jordan DR, Chapman SC, Godwin ID, Mace ES, McIntyre CL. Identification of QTL for sugar-related traits in a sweet x grain sorghum (Sorghum bicolor L. Moench) recombinant inbred population. Mol Breed. 2008;22:367–384. doi: 10.1007/s11032-008-9182-6. [DOI] [Google Scholar]
- 60.Srinivas G, Satish K, Madhusudhana R, Seetharama N. Exploration and mapping of microsatellite markers from subtracted drought stress ESTs in Sorghum bicolor (L.) Moench. Theor Appl Genet. 2009;118:703–717. doi: 10.1007/s00122-008-0931-z. [DOI] [PubMed] [Google Scholar]
- 61.Feltus FA, Hart GE, Schertz KF, Casa AM, Kresovich S, Abraham S, et al. Alignment of genetic maps and QTLs between inter- and intraspecific sorghum populations. Theor Appl Genet. 2006;112:1295–1305. doi: 10.1007/s00122-006-0232-3. [DOI] [PubMed] [Google Scholar]
- 62.Shiringani AL, Frisch M, Friedt W. Genetic mapping of QTLs for sugar-related traits in a RIL population of Sorghum bicolor L. Moench. Theor Appl Genet. 2010;121:323–336. doi: 10.1007/s00122-010-1312-y. [DOI] [PubMed] [Google Scholar]
- 63.Brown PJ, Klein PE, Bortiri E, Acharya CB, Rooney WL, Kresovich S. Inheritance of inflorescence architecture in sorghum. Theor Appl Genet. 2006;113:931–942. doi: 10.1007/s00122-006-0352-9. [DOI] [PubMed] [Google Scholar]
- 64.Childs KL, Miller FR, Cordonnier-Pratt MM, Pratt LH, Morgan PW, Mullet JE. The sorghum photoperiod sensitivity gene, Ma3, encodes a phytochrome B. Plant Physiol. 1997;113:611–619. doi: 10.1104/pp.113.2.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Duan K, Yi K, Dang L, Huang H, Wu W, Wu P. Characterization of a sub-family of Arabidopsis genes with the SPX domain reveals their diverse functions in plant tolerance to phosphorus starvation. Plant J. 2008;54:965–975. doi: 10.1111/j.1365-313X.2008.03460.x. [DOI] [PubMed] [Google Scholar]
- 66.Xu D, Duan X, Wang B, Hong B, Ho T, Wu R. Expression of a late embryogenesis abundant protein gene, HVA7, from Barley confers tolerance to water deficit and salt stress in transgenic rice. Plant Physiol. 1996;110:249–257. doi: 10.1104/pp.110.1.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hirner A, Ladwig F, Stransky H, Okumoto S, Keinath M, Harms A, et al. Arabidopsis LHT1 is a high-affinity transporter for cellular amino acid uptake in both root epidermis and leaf mesophyll. Plant Cell. 2006;18:1931–1946. doi: 10.1105/tpc.106.041012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen L, Bush DR. LHT1, a lysine and histidine specific amino acid transporter in Arabidopsis. Plant Physiol. 1997;115(3):1127–1134. doi: 10.1104/pp.115.3.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pereira MG, Lee M. Identification of genomic regions affecting plant height in sorghum and maize. Theor Appl Genet. 1995;90:380–388. doi: 10.1007/BF00221980. [DOI] [PubMed] [Google Scholar]
- 70.Rami JF, Dufour P, Trouche G, Fliedel G, Mestres C, Davrieux F, et al. Quantitative trait loci for grain quality, productivity, morphological and agronomical traits in sorghum (Sorghum bicolor L. Moench) Theor Appl Genet. 1998;97:605–616. doi: 10.1007/s001220050936. [DOI] [Google Scholar]
- 71.Hart GE, Schertz KF, Peng Y, Syed NH. Genetic mapping of Sorghum bicolor (L.) Moench QTLs that control variation in tillering and other morphological characters. Theor Appl Genet. 2001;103:1232–1242. doi: 10.1007/s001220100582. [DOI] [Google Scholar]
- 72.Torrigiani P, Bressanin D, Ruiz KB, Tadiello A, Trainotti L, Bonghi C, et al. Spermidine application to young developing peach fruits leads to a slowing down of ripening by impairing ripening-related ethylene and auxin metabolism and signaling. Physiol Plant. 2012;146:86–98. doi: 10.1111/j.1399-3054.2012.01612.x. [DOI] [PubMed] [Google Scholar]
- 73.Feng HY, Wang ZM, Kong FN, Zhang MJ, Zhou SL. Roles of carbohydrate supply and ethylene, polyamines in maize kernel set. J Integr Plant Biol. 2011;53:388–398. doi: 10.1111/j.1744-7909.2011.01039.x. [DOI] [PubMed] [Google Scholar]
- 74.Wang Z, Xu Y, Wang J, Yang J, Zhang J. Polyamine and ethylene interactions in grain filling of superior and inferior spikelets of rice. Plant Growth Regul. 2012;66:215–228. doi: 10.1007/s10725-011-9644-4. [DOI] [Google Scholar]
- 75.Xu WW, Subudhi PK, Crasta OR, Rosenow DT, Mullet JE, Nguyen HT. Molecular mapping of QTLs conferring stay-green in grain sorghum (Sorghum bicolor L. Moench) Genome. 2000;43:461–469. doi: 10.1139/gen-43-3-461. [DOI] [PubMed] [Google Scholar]
- 76.Crasta OR, Xu WW, Nguyen HT, Rosenow DT, Mullet J. Mapping of post flowering drought resistance traits in grain sorghum: association between QTLs influencing premature senescence and maturity. Mol Genet. 1999;262:579–588. doi: 10.1007/s004380051120. [DOI] [PubMed] [Google Scholar]
- 77.Klein RR, Mullet JE, Jordan DR, Miller FR, Rooney WL, Menz MA, et al. The effect of tropical sorghum conversion and inbred development on genome diversity as revealed by high-resolution genotyping. Crop Sci. 2008;48:S12–S26. doi: 10.2135/cropsci2007.06.0319tpg. [DOI] [Google Scholar]
- 78.Lin YR, Schertz KF, Paterson AH. Comparative analysis of QTLs affecting plant height and maturity across the Poaceae, in reference to an interspecific sorghum population. Genetics. 1995;141:391–411. doi: 10.1093/genetics/141.1.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Graham D, Lessman KJ. Effect of height on yield and yield components of two isogenic lines of Sorghum vulgare. Crop Sci. 1966;6:372–374. doi: 10.2135/cropsci1966.0011183X000600040024x. [DOI] [Google Scholar]
- 80.Dennis DT, Blakeley SD. Carbohydrate Metabolism. In: Buchanan BB, Gruissem W, Jones RL, editors. Biochemistry & molecular biology of plants. Rockville: American Society of Plant Physiologists; 2000. pp. 630–675. [Google Scholar]
- 81.Murray SC, Sharma A, Rooney WL, Klein PE, Mullet JE, Mitchell SE. Genetic improvement of sorghum as a biofuel feedstock: I. QTL for stem sugar and grain nonstructural carbohydrates. Crop Sci. 2008;48:2165–2179. doi: 10.2135/cropsci2008.01.0016. [DOI] [Google Scholar]
- 82.Multani DS, Briggs SP, Chamberlin MA, Blakeslee JJ, Murphy AS, Johal GS. Loss of an MDR transporter in compact stalks of maize br2 and sorghum dw3 mutants. Science. 2003;302:81–84. doi: 10.1126/science.1086072. [DOI] [PubMed] [Google Scholar]
- 83.Bouche N, Fait A, Zik M, Fromm H. The root-specific glutamate decarboxylase (GAD1) is essential for sustaining GABA levels in Arabidopsis. Plant Mol Biol. 2004;55(3):315–325. doi: 10.1007/s11103-004-0650-z. [DOI] [PubMed] [Google Scholar]
- 84.Tao YZ, Henzell RG, Jordan DR, Butler DG, Kelly AM, McIntyre CL. Identification of genomic regions associated with stay green in sorghum by testing RILs in multiple environments. Theor Appl Genet. 2000;100:1225–1232. doi: 10.1007/s001220051428. [DOI] [Google Scholar]
- 85.Brown PJ, Rooney WL, Franks C, Kresovich S. Efficient mapping of plant height quantitative trait loci in a sorghum association population with introgressed dwarfing genes. Genetics. 2008;180:629–637. doi: 10.1534/genetics.108.092239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yamaya T, Obara M, Nakajima H, Sasaki S, Hayakawa T, Sato T. Genetic manipulation and quantitative-trait loci mapping for nitrogen recycling in rice. J Exp Bot. 2002;53:917–925. doi: 10.1093/jexbot/53.370.917. [DOI] [PubMed] [Google Scholar]