

Summary
Minimal change nephrosis (MCN) is an important cause of morbidity in children. In spite of successful therapies having been developed in the last three decades, most aspects related to pathogenesis still remain poorly defined. Evolution in basic immunology and results deriving from animal models of the disease suggest a complex interaction of factors and cells starting from activation of innate immunity and continuing with antigen presentation. Oxidants, CD80 and CD40/CD40L have probably a relevant role at the start. Studies in animal models and in human beings also suggest the possibility that the same molecules (i.e. CD80, CD40) are expressed by podocytes under inflammatory stimuli, representing a direct potential mechanism for proteinuria. B and T cells could play a relevant role this contest. Implication of B cells is suggested indirectly by studies utilizing anti‐CD20 monoclonal antibodies as the main therapy. The role of regulatory T cells (Tregs) is supported mainly by results in animal models of nephrotic syndrome (i.e. adriamycin, puromycin, lipopolysaccharide), showing a protective effect of direct Treg infusion or stimulation by interleukin 2 (IL‐2). Limited studies have also shown reduced amounts of circulating Tregs in patients with active MCN cells. The route from bench to bedside would be reduced if results from animal models were confirmed in human pathology. The expansion of Tregs with recombinant IL‐2 and new anti‐CD20 monoclonal antibodies is the beginning. Blocking antigen‐presenting cells with cytotoxic T lymphocyte antigen (CTLA‐4)–Ig fusion molecules inhibiting CD80 and/or with blockers of CD40–CD40 ligand interaction represent potential new approaches. The hope is that evolution in therapies of MCN could fill a gap lasting 30 years.
Keywords: IL‐2, LPS nephropathy, minimal change nephropathy, regulatory T cells
Introduction
Minimal change nephrosis (MCN) in children is characterized by episodes of severe proteinuria and hypoalbuminaemia (serum albumin < 2.5 g/dl) that are often associated with dyslipidaemia and hypercoagulability. It affects two to 10 children per 100 000 per year in western countries, with a prevalence of 16 cases per 100 000 1. Renal pathology justifies the name, because glomeruli appear morphologically normal and without deposition of antibodies. Although most patients respond favourably to steroids, relapse rate is as high as 80% and a long‐term combination of steroids and calcineurin inhibitors is often required to maintain remission 2, 3. Disease remission and prevention of kidney disease progression are the main long‐term care objectives 2, 4, 5.
Characterizing mechanisms of MCN is a challenge. Historically, MCN has been considered a T cell pathology; however, evolution in basic immunology and in therapies of MCN propose a more articulated pathogenesis with participation of innate immunity, B cells and regulatory T cells (Tregs), every single element having a prominent role in different phases of the disease. Probably, a two‐hit mechanism with more regulatory check‐points fit the dynamics of proteinuria more accurately in this setting.
T cell involvement is the historical view
Shaloub et al. 6 first hypothesized that MCN is the renal manifestation of a T cell dysregulation in which cytokines act as modifiers of podocyte ultrastructure. This hypothesis was based on indirect evidence, such as the absence of antibodies in the kidney, sensitivity to steroids and cyclophosphamide, which modify cell‐mediated responses and, finally, the association with Hodgkin's disease. Studies on T cell and cytokine prevalence performed until 2000 substantiated the theory 7, 8, 9, showing an imbalance between CD4+/CD8+/natural killer (NK) cells with the prevalence of the latter two elements 10, 11. However, cytokines have been variably reported based on different methodologies (e.g. peripheral blood mononuclear cells or T cells stimulation versus standing cells, mRNA expression, etc.), and the following main results contributed to reach fragmentary conclusions: (1) tumour necrosis factor (TNF‐α) is increased in serum, T and peripheral blood mononuclear cells and in polymorphonuclear cells 12, 13, 14; (2) interferon (IFN‐γ), IL‐1β, IL‐2, IL‐4 and IL‐6 are low in serum 12, 14, 15, while their synthesis is increased in stimulated peripheral blood mononuclear cells and polymorphonuclear cells 12, 13, 14, 15, 16; and (3) IL‐8 and IL‐12 synthesis have been reported as high and low by different authors 14, 15, 17, 18 (Table 1). Finally, experimental studies in Wistar rats transfected with the IL‐13 gene showed significant proteinuria and pathology changes of human MCN, suggesting that this cytokine is toxic for podocytes 18. Overall, the general idea is that a single cytokine per se cannot be considered pathogenic of MCN and that a complex set of factors and cells may explain the pathology more clearly. It is also evident that immunology in the last few years has obtained impressive advances, and a simplified view on the T cell compartment has been deeply modified by research. Numerous phenotype T cell variants with different function and plasticity of effectors and regulatory cells are now considered a basic element that would be implicated in renal pathology (scheme in Fig. 1).
Table 1.
An overview on data of the literature focusing levels of major cytokines in patients with MCN.
| TNF‐α | IL‐2 | IL‐1β | IL‐4 | IL‐6 | IL‐8 | IL‐10 | IL‐12 | IL‐13 | IFN‐γ | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Surany et al. 1993 | serum + | serum – | serum – | – | |||||||
| Garin et al. 1994 | Cell mRNA + serum + | ||||||||||
| Bustos et al. 1994 | cells + | – | – | ||||||||
| Neuhaus et al. 1995 | serum –PBMC + | PBMC ± | PBMC – | PBMC – | |||||||
| Matsumoto et al. 1999 | PBMC +MCN, IgA | ||||||||||
| Cho et al. 1999 | PBMC + | ||||||||||
| Araya et al. 2009 | PBMC + | T cell – PBMC + | PBMC + | T cell – | mRNA – | PBMC – | PBMC – |
IL = interleukin; IFN = interferon; TNF = tumour necrosis factor; PBMC = peripheral blood mononuclear cells; MCN = minimal change nephrosis; IgA = immunoglobulin A.
Figure 1.
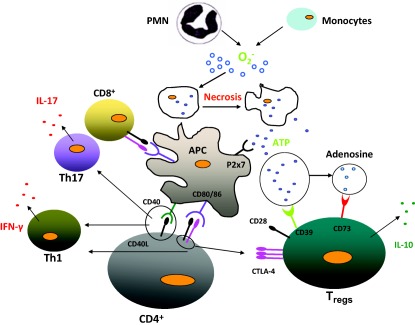
An overview of cells involved in an inflammatory event. Monocytes and polymorphonuclear cells produce oxidants (O2–) in response to an external inflammatory trigger (bacteria, virus, etc.) as a first defence. Adenosine‐5′‐triphosphate (ATP) deriving from necrosis of these cells activates professional antigen‐presenting cells that interact with both CD4+ effectors and CD4+CD25+forkhead box protein 3 (FoxP3) regulatory cells. Both cell lineages (i.e. CD4+ effector and regulatory cells) derive from activation of CD4+ thymocytes upon T cell receptor interaction with major histocompatibility complex (MHC) class II that process proteins deriving from the inflammatory trigger to produce specific cells counterbalancing pathogen effects. The balance between CD4+ and CD4+CD25+FoxP3 regulatory cells drive evolution of the inflammatory event. If regulatory T cells (Tregs) prevail over CD4+ effectors, then ATP is transformed in adenosine that exerts an anti‐inflammatory effect. Interleukin 10 (IL‐10) also plays an anti‐inflammatory role. If CD4+ effectors prevail over Tregs, other inflammatory cells and cytokines expand the inflammatory process. Mechanisms driving the equilibrium between CD4+ and CD4+CD25+FoxP3 regulatory cells are shown in Fig. 2.
Innate and adaptive immunity/B cells
Oxidants
Experimental models and observations in human beings suggest the participation of reactive oxygen species (ROS) in the pathogenesis of MCN. ROS are typical first‐phase reactants produced by polymorphonuclear cells in response to infectious triggers. Bertelli et al. 19 first showed that polymorphonuclear cells produce high quantities of oxidants in children affected by MCN during relapse of proteinuria, due probably to an alteration of a negative regulatory circuit that involves soluble factors deriving from regulatory T cells. In fact, oxidant production was correlated inversely with the amount of Treg expression of apyrase (CD39), the key enzyme that transforms adenosine‐5′‐triphosphate (ATP) in adenosine, the latter metabolite playing an inhibitory role on oxidant production 20. The addition of apyrase to polymorphonuclear cells in vitro reduced oxidants by 40%, while adenosine analogues (2′‐chloroadenosine and 5′‐N‐ethylcarboxamidoadenosine) produced minor effects. Finally, antagonizing ATP efflux with carbenoxolone, or blocking ATP effects with Brilliant Blue G, KN62 and A437089, reduced ROS generation in a comparable manner to apyrase 20.
Indirect evidence for oxidant activity in vivo comes from the observation that in MCN patients a significant part of serum albumin is oxidized, concomitant with proteinuria. Albumin has only recently been recognized as the major anti‐oxidant in serum, a physiological function that evolved over years and that confers a crucial role to this protein during infectious episodes in humans 21. Musante et al. 22 characterized serum albumin in children with MCN by mean of mass spectrometry, and showed chemical modifications of the unique free 34Cys of the protein sequence to a sulphonic group (SO3H) that is the end product of its oxidation, and therefore represents a surrogate biomarker of an oxidative stress 22, 23.
Animal models of nephrosis also support a role of oxidants. Minimal renal lesions evolving to glomerulosclerosis are, in fact, produced in rats by infusion of compounds such as puromycin aminonucleoside (PAN) and adriamycin (ADR) that can be classified as oxidants 24, 25. Metabolic studies and protection by anti‐oxidants support an entirely oxidative stress in these models.
B cells and adaptive immunity
The implication of adaptive immunity and of B cells is suggested by results in animal models and in humans. Although B cells have never been characterized in MCN, the suggestion of their implication derives from studies on the successful use of anti‐CD20 monoclonal antibodies, i.e. rituximab, in patients with MCN 26, 27, 28. However, the therapeutic effect of rituximab has not been explained univocal because, as well as with the CD20 receptor, rituxumab interacts with sphingomyelin phosphodiesterase acid‐like 3b protein (SMPDL) 29 that represents a potential second receptor for the drug. It is interesting that as well as B cells, SMPDL is expressed by T helper type 17 (Th17) 30 and by podocytes. Fornoni et al. 31. demonstrated the binding of rituximab with SMPDL in the raft of podocytes, where it co‐localizes partially with synaptopodin and prevents actin remodelling induced by serum of patients with focal and segmental glomerular sclerosis; this effect furnishes an alternative explanation for the activity of rituximab in nephrotic patients. Conversely, the interaction of rituximab with Th17 cells may also have direct pathogenic implications, as their activation is considered a key element in the pathogenesis of nephrotic syndrome. Other studies demonstrated that rituximab reduces the Th17 cell response in rheumatoid arthritis 32, 33, 34, 35, 36, providing the theoretical basis for its clinical use extension to small vessel vasculitis 37. Overall, the findings above furnish some evidence for a logical connection between rituximab, Th17, SMPDL and nephrotic syndrome that contributes to understanding of the disease mechanisms.
In general, the role of adaptive immunity is supported by findings in animal models of MCN (LPS nephropathy) relative to CD80, a co‐stimulatory molecule expressed by antigen‐presenting cells and by B cells, that is part of the so‐called ‘immunologic synapse’ regulating a balance between immunology activation and its block 38, 39. In fact, CD80 interacts with CD28 on CD4+ cells, mediating their activation into effector cells, and with cytotoxic T lymphocyte antigen 4 (CTLA‐4) on Tregs, mediating the block of maturation towards a T effector (Teff) phenotype with concomitant direct inhibition of the loop ending with antigen presentation 40, 41 (see scheme in Fig. 2). Lipopolysaccharide (LPS) is the mediator of bacterial infection; its infusion in mice is followed by transient proteinuria 42 and induces progressive changes in glomerular architecture that evolve over weeks to glomerulosclerosis, and in some way mimic the natural history of patients with MCN. The pathogenic cascade activated by LPS directly involves CD80 (B7‐1), as shown clearly by Reiser et al. 42 in engineered mice lacking CD80 that are protected from LPS proteinuria 42, thus suggesting that this molecule is the mediator of LPS renal toxicity.
Figure 2.
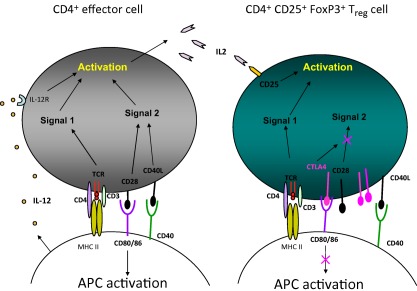
Mechanisms regulating CD4+ and CD4+CD25+forkhead box protein 3 (FoxP3) balance. An antigen‐presenting cell (APC) cell interacts with both CD4+ and CD4+CD25+FoxP3 by means of the same ligands, i.e. the T cell receptor (TCR)/CD4/CD3 that interacts with the major histocompatibility complex (MHC) II complex and CD80/86 and CD40. The former molecules, i.e. TCR/CD4/CD3, are present on both CD4+ and CD4+CD25+FoxP3 and does not represent a regulatory mechanism; this route activates the Signal 1 pathway. At variance, CD80/86 interact with CD4+CD25+FoxP3 cells: in the former case, i.e. CD4+, CD80/86 interacts with CD28 and constitutes a positive stimulus for CD4+ expansion (in black), whereas it interacts preferentially with cytotoxic T lymphocyte antigen 4 (CTLA‐4) in CD4+CD25+FoxP3 and represents an inhibitory mechanism (in purple). Actually, regulatory CD4+CD25+FoxP3 cells contain both CD28 and CTLA‐4 in a ratio of 1 : 2 that implies that negative regulatory mechanisms prevail in these cells and block the Signal 2 pathway that is necessary for activating CD4+ effector cells. CD40–CD40L has the same expression in both cell lineages and does not represent a regulatory mechanism.
CD40 is another co‐stimulatory molecule involved in adaptive immunity. High serum levels of anti‐CD40 antibodies have been reported recently in association with the recurrence of proteinuria in more severe forms of MCN that have developed focal segmental glomerulosclerosis 43. In fact, focal segmental glomerulosclerosis is considered the evolutionary step of MCN, as clinical and pathological observations suggest 44. CD40 belongs to the TNF gene superfamily, expressed preferentially on B lymphocytes, monocytes/macrophages and dendritic cells, and is involved in the adaptive immune response 45. Binding of CD40 to CD40 ligand expressed on T lymphocytes and platelets triggers an inflammatory process with relevant consequences 46. Epitope mapping has suggested altered immunogenicity of CD40 in patients with glomerulosclerosis, and anti‐CD40 antibodies may help to identify patients at high risk of post‐transplant outcome 43. Understanding of the significance of anti‐CD40 antibodies is crucial to understanding the mechanism(s) of proteinuria; one possibility is that saturation of CD40 on antigen‐presenting cells would produce an excess of circulating levels of the shed form of CD40 ligand (sCD40L) free to interact with CD40 in podocytes (Doublier, manuscript in preparation). Altogether, the findings on CD80 and CD40 reflect an overall inflammatory state that would involve both professional antigen‐presenting cells and, potentially, podocytes determining what mimics MCN in early and late phases. There is strong interest in considering both molecules as potential therapeutic targets 47, 48.
The podocyte is an antigen‐presenting cell
During stress conditions, podocytes, the key functional cells in glomeruli, express Toll‐like receptor (TLR‐4), CD80 and CD40 that are the functional markers of a cell devoted to present antigens. Thus, podocytes may be the target of those stimuli that are described in the section above relative to the adaptive processes that characterize the starting phase of immunity. The role of CD80 is crucial, as results from CD80 knock‐out mice have highlighted 42. The basic finding supporting a direct implication of podocytes in LPS nephrosis derives from severe combined immunodeficient (SCID) mice missing both T and B cells that develop proteinuria, implying that in the absence of professional antigen‐presenting cells, TLR‐4 and CD80 in podocytes are possible effectors of proteinuria 42. Findings in humans support podocyte CD80 implication in MCN: (1) urinary levels of CD80 deriving from podocytes are increased in MCN patients during the active phase of the disease 49; and (2) abatacept, a fusion CTLA‐4–Ig molecule that inhibits CD80, is utilized in human settings to reduce proteinuria in some patients with post‐transplant recurrence of focal segmental glomerulosclerosis that is considered an advance state of MCN 48, 50. The expression of podocyte CD80 in renal biopsies of patients is the biomarker for abatacept sensitivity. Overall, these observations suggest that podocytes, probably other antigen‐presenting cells, are activated by immunological stimuli and may produce functional consequences linked directly to proteinuria (see scheme in Fig. 3).
Figure 3.
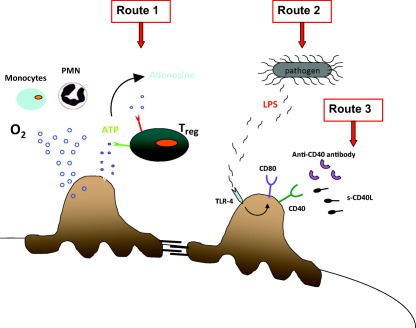
Podocytes may be directly involved in the early immunity response. Oxidants produced by circulating cells in the early immune response may be toxic for podocytes. regulatory T cells (Tregs) reduce the oxidative impact by metabolizing adenosine‐5′‐triphosphate (ATP) to adenosine. Under inflammatory stimuli, podocytes express both Toll‐like receptor (TLR)‐4 and CD80 and become a part of the initial immune response. Podocytes also express CD40 constitutively, whose stimulation by soluble (s)CD40L induces the redistribution and loss of nephrin and increases albumin permeability in isolated rat glomeruli.
Tregs and MCN
Tregs could be involved in MCN as second step in a cascade where the first hit is an acute event involving innate immunity. Data deriving from studies in animal models and in humans support a mechanism in which the modification of Tregs plays a successive regulatory effect.
Human MCN
Studies by Bertelli et al. 20 showed that Tregs (CD39+CD4+CD25+) are reduced in number in patients with active MCN. As already reported, the decrease in Tregs and concomitant low apyrase (CD39) expression has been associated with oxidants. With the exception of this unique observation, no other study has attempted the analysis of Treg expression in nephrotic syndrome. There are, however, a few clinical reports in patients with the immunodeficiency, polyendocrinopathy, enteropathy syndrome (IPEX) who presented an associated MCN 19, 51. Because IPEX is an X‐linked disease due to the mutation of FOXP3, this association supports a direct cause–effect correlation between Tregs and MCN. Of particular interest is the description of a child with missense mutations of forkhead box protein 3 (FoxP3) who developed a mild form of IPEX syndrome associated with MCN 19. Oxidant production in this child was 100 times higher during exacerbation of clinical symptoms and restored to a near‐normal level in remission. Nephrotic syndrome resolved in this patient after bone marrow transplantation that normalized Treg composition.
Animal models
Treg involvement has been studied in experimental models of nephrosis. Le Berre et al. 52 utilized Buffalo/Mna rats that develop glomerulosclerosis spontaneously, showing that pre‐ and post‐transplant proteinuria was reduced by infusion of Tregs; regression of the nephropathy was also obtained. Other authors reported the same protective role of Tregs in adriamycin nephrosis, in which case Tregs were modulated by adenosine or by direct infusion of FoxP3‐transduced T cells 53. More recently, some unexpected results have been obtained by utilizing IL‐2 in LPS nephropathy 54. In this model, IL‐2 has been utilized to enhance Treg proliferation and life span. To escape from the bystander effects of this cytokine due to its wide‐ranging action, the association of IL‐2 and anti‐IL‐2 antibodies was utilized and compared to IL‐2 alone as a stratagem to improve in‐vivo selectivity towards the Treg population 55, 56. In fact, when utilized alone, IL‐2 binds the three α, β and γ chain receptors that are present on several cell lineages (i.e. CD8, NK, Tregs), whereas the γ chain receptor is expressed uniquely by activated CD4+ (Teff) and Tregs. Anti‐IL‐2 bind selectively α and β chain receptors that are saturated in their presence, allowing more IL‐2 to be available for γ chain and free to up‐regulate Tregs. The administration of low doses of IL‐2 coupled to this specific antibody indeed proved to induce high levels of Tregs, being ineffective on CD8 and NK. Polhill et al. 57 induced Treg expansion by IL‐2/IL‐2 antibodies in rats with adriamycin nephrosis and documented improvement of renal function, reduced inflammation and less pathological injury. More articulated are the results of the study by Bertelli et al. 54, who utilized the same approach with IL‐2/IL‐2 antibodies in mice with LPS nephropathy. In fact, IL‐2/IL‐2 antibody administration in mice exposed to LPS had no effect on the progression of the resulting renal damage, while enhancing significantly peripheral and tissutal Treg levels, whereas IL‐2 infused alone elicited some protection despite fewer Tregs. Therefore, there is a dichotomy between Treg levels (higher after IL‐2/anti‐IL‐2 administration) and renal protection (most evident in mice treated with IL‐2 alone), these results partially contradicting a direct role of Tregs and supporting hitherto undefined mechanisms. Evolution in the biology of Tregs are now showing a complex arrow of functions that directly target immunity control and that include phenotypically distinct Treg lines 58, 59, 60. The precise characterization of different Treg phenotypes in patients with MCN is mandatory to gain information on their role in MCN; clinical research into this is needed in the near future.
Tregs in the middle of innate and adaptive immunity
Tregs may play a role in MCN based on their plasticity and multi‐functional effects 61. Basically, these cells are connected to innate immunity by means of CD80 and may, in turn, modulate the levels of pro‐ and anti‐inflammatory compounds. They are, in fact, active in the midst of innate and adaptive immunity and act as a bi‐functional modifier during the entire process. Tregs are a dynamic cell population whose levels can be modified rapidly and made active, with alternative functions, in any inflammatory context 62. At equilibrium, Treg concentration in the periphery is low, but when necessary they are produced from non‐activated CD4+ cells and, in general, follow the same course of all T cells deriving from CD4+CD8+ thymocytes upon reaction with professional presenting cells; in this case CD4+ are expanded in response to a peptide presented by the class II major histocompatibility complex. Starting from CD4+, as well as co‐stimulatory molecules, Treg generation requires transforming growth factor (TGF)‐β and IL‐2; as an alternative evolution the same CD4+ may also differentiate into activated Teff CD4+ CD25– cytotoxic cells in the presence of IL‐7 and induction of IL‐7R (CD127). Active Tregs are able to counteract the inflammatory burst by means of anti‐inflammatory cytokines (IL‐10) and/or by adenosine deriving from the ATP metabolism (see below); nevertheless, as FoxP3 expression is not stable in induced Tregs (iTregs), they can easily revert to an effector lineage in the presence of an inflammatory contest 63. There is evidence that upon continuous exposure to IL‐2 iTregs convert into Th1‐like cells, expressing IFN‐γ, whereas IL‐6 in association with TGF‐β promotes their differentiation in Th17 cells that exert a powerful inflammatory response by the production of IL‐17A and recruitment of polymorphonuclear cells (PMN) (scheme in Fig. 1) Therefore, there are at least three cell subsets (i.e. Treg/Teff/Th17), all deriving from the same progenitor, that play antagonist effects and determine the narrow limit between inflammation and normal status: when the ratio between Teff/Tregs or Th17/Tregs is high the signal is to maintain inflammation and, vice versa, when Tregs are higher than Teff and Th17, inflammation is switched off 64. Co‐stimulatory molecules, CD80 and CD86 (B7‐2) play a key role in this balance by interacting with CD28 that is expressed on both Teff and Tregs and with CTLA‐4, a second ligand up‐regulated in Tregs 65. By interacting with CD28, CD80 and CD86 ligands activate the Signal 2 pathway and induce proliferation of Teffs and alternatively, by interacting with CTLA‐4 they reduce Teff and block Signal 2 while allowing Treg induction via CD25 and IL‐2. The presence of two receptors with opposite functions on Tregs, i.e. CD28 and CTLA‐4, is suggestive of a feedback regulation in which these cells are first switched on by CD28 ligation and in a subsequent step switched off due to the over‐expression of CTLA‐4; this latter is in fact able to compete with CD28, owing to this 100‐fold higher affinity for ligands, and may also suppress their expression by transendocytosis 66.
ATP is another regulatory molecule connected with Tregs, and represents a crucial element between innate immunity and its regulation. It is considered a ‘danger sensor’, depending on its extracellular concentration, that varies from a nanomolar to a micromolar range 67, 68. Two main mechanisms modify ATP levels and drive the cell response. The first mechanism is based on the simultaneous presence of CD39 and CD73 on the surface of Tregs that, together, produce the potent immunosuppressor adenosine from ATP. As CD39 and CD73 are simultaneously highly expressed on Tregs it is clear that these cells have the power to blunt inflammation. Purinergic receptors P2X and P2Y located on the surface of immune cells represent the second mechanism for the ATP regulatory functions. At low ATP concentrations P2X7R promotes dendritic cell maturation and IL‐10 secretion, thus enhancing immune suppression 67, 68; at micromolar concentrations, P2X7R promotes the inflammatory cascade driven by the NLPR3, thus leading to IL‐1‐β secretion.
Conclusions
The pathogenesis of MCN is now considered multi‐factorial. Several experimental results and data from human MCN suggest a start, based on innate immunity with activation of CD80 by professional cells (B cells/TLRs). CD40 and anti‐CD40 molecules could also be involved. Oxidants are up‐regulated in the first phase, and may represent a trigger of pathology if not blunted adequately. The second step is based on alternative evolutions. One possibility is the prevalence of an inflammatory pathway based on the activation, respectively, of Teff by IL‐7 and IL‐2 and of Th17 by IL‐6 and TGF‐β, that produce podocyte lesions. The second possibility is Treg expansion that blunts oxidation by reducing ATP levels and increasing adenosine. While CD80 is pre‐eminent as the regulatory system in the first phase, Tregs and ATP drive the anti‐inflammatory phase. Multi‐point regulation of a crucial event such as inflammation represents a system for ensuring that only motivated triggers turn on the cascade. The failure or reduction of regulatory steps may justify pathology and constitute a basic motif for a transient condition such as proteinuria. Characterizing single cells in each phase of MCN and in relation to therapies is a challenge, and we now have adequate tools for clarifying all single aspects. The route from bench to bedside would be shorter if results from animal models were confirmed in human pathology. Expansion of Tregs with IL‐2 and new anti‐CD20 monoclonal antibodies (ofatumumab) are the beginning. Blockage of antigen‐presenting cells with CTLA‐4–Ig fusion molecules inhibiting CD80 and/or with blockers of CD40–CD40 ligand interaction represents a potential evolution. Considering new molecules and antibodies on the basis of results from studies of the mechanism is an option to be evaluated.
Disclosure
All the authors declare that they have not received support from any company for the submitted work, have no relationships with any company that might have an interest in the submitted work and have no financial or non‐financial interests that may be relevant to the submitted work. They also declare that their spouses, partners or children have not had any financial relationship that may be relevant to the submitted work.
Acknowledgements
This study was supported by funds deriving from ‘Cinque per mille of IRPEF‐Finanziamento della ricerca sanitaria’ and from the Italian Ministry of Health ‘Ricerca Corrente contributo per la ricerca intramuraria’ to Istituto Giannina Gaslini. This work was also supported by the Renal Child Foundation and Fondazione La Nuova Speranza (‘Progetto integrato per la definizione dei meccanismi implicati nella glomerulo sclerosi focale’). The corresponding author certifies that all individuals who contributed significantly to the work have been acknowledged here.
References
- 1. Kidney Disease Improving Global Outcomes (KDIGO) . Clinical practice guideline for glomerulonephritis. Kidney Int 2012; 2:181–5. [Google Scholar]
- 2. Trompeter RS, Lloyd BW, Hicks J, White RH, Cameron JS. Long‐term outcome for children with minimal‐change nephrotic syndrome. Lancet 1985; 1:368–70. [DOI] [PubMed] [Google Scholar]
- 3. Ponticelli C, Rizzoni G, Edefonti A et al A randomized trial of cyclosporine in steroid‐resistant idiopathic nephrotic syndrome. Kidney Int 1993; 43:1377–84. [DOI] [PubMed] [Google Scholar]
- 4. Ghiggeri GM, Catarsi P, Scolari F et al Cyclosporine in patients with steroid‐resistant nephrotic syndrome: an open‐label, nonrandomized, retrospective study. Clin Ther 2004; 26:1411–8. [DOI] [PubMed] [Google Scholar]
- 5. Kyrieleis HA, Lowik MM, Pronk I et al Long‐term outcome of biopsy‐proven, frequently relapsing minimal‐change nephrotic syndrome in children. Clin J Am Soc Nephrol 2009; 4:1593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shalhoub RJ. Pathogenesis of lipoid nephrosis: a disorder of T‐cell function. Lancet 1974; 2:556–60. [DOI] [PubMed] [Google Scholar]
- 7. Karras A, de Montpreville V, Fakhouri F, Grunfeld JP, Lesavre P. Renal and thymic pathology in thymoma‐associated nephropathy: report of 21 cases and review of the literature. Nephrol Dial Transplant 2005; 20:1075–82. [DOI] [PubMed] [Google Scholar]
- 8. Koenecke C, Ukena SN, Ganser A, Franzke A. Regulatory T cells as therapeutic target in Hodgkin's lymphoma. Expert Opin Ther Targets 2008; 12:769–82. [DOI] [PubMed] [Google Scholar]
- 9. Koyama A, Fujisaki M, Kobayashi M, Igarashi M, Narita M. A glomerular permeability factor produced by human T cell hybridomas. Kidney Int 1991; 40:453–60. [DOI] [PubMed] [Google Scholar]
- 10. Fiser RT, Arnold WC, Charlton RK, Steele RW, Childress SH, Shirkey B. T‐lymphocyte subsets in nephrotic syndrome. Kidney Int 1991; 40:913–6. [DOI] [PubMed] [Google Scholar]
- 11. Lama G, Luongo I, Tirino G, Borriello A, Carangio C, Salsano ME. T‐lymphocyte populations and cytokines in childhood nephrotic syndrome. Am J Kidney Dis 2002; 39:958–65. [DOI] [PubMed] [Google Scholar]
- 12. Suranyi MG, Guasch A, Hall BM, Myers BD. Elevated levels of tumor necrosis factor‐alpha in the nephrotic syndrome in humans. Am J Kidney Dis 1993; 21:251–9. [DOI] [PubMed] [Google Scholar]
- 13. Bustos C, Gonzalez E, Muley R, Alonso JL, Egido J. Increase of tumour necrosis factor alpha synthesis and gene expression in peripheral blood mononuclear cells of children with idiopathic nephrotic syndrome. Eur J Clin Invest 1994; 24:799–805. [DOI] [PubMed] [Google Scholar]
- 14. Araya C, Diaz L, Wasserfall C et al T regulatory cell function in idiopathic minimal lesion nephrotic syndrome. Pediatr Nephrol 2009; 24:1691–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Neuhaus TJ, Wadhwa M, Callard R, Barratt TM. Increased IL‐2, IL‐4 and interferon‐gamma (IFN‐gamma) in steroid‐sensitive nephrotic syndrome. Clin Exp Immunol 1995; 100:475–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cho BS, Yoon SR, Jang JY, Pyun KH, Lee CE. Up‐regulation of interleukin‐4 and CD23/FcepsilonRII in minimal change nephrotic syndrome. Pediatr Nephrol 1999; 13:199–204. [DOI] [PubMed] [Google Scholar]
- 17. Garin EH, Blanchard DK, Matsushima K, Djeu JY. IL‐8 production by peripheral blood mononuclear cells in nephrotic patients. Kidney Int 1994; 45:1311–7. [DOI] [PubMed] [Google Scholar]
- 18. Lai KW, Wei CL, Tan LK et al Overexpression of interleukin‐13 induces minimal‐change‐like nephropathy in rats. J Am Soc Nephrol 2007; 18:1476–85. [DOI] [PubMed] [Google Scholar]
- 19. Bertelli R, Trivelli A, Magnasco A et al Failure of regulation results in an amplified oxidation burst by neutrophils in children with primary nephrotic syndrome. Clin Exp Immunol 2010; 161:151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bertelli R, Bodria M, Nobile M et al Regulation of innate immunity by the nucleotide pathway in children with idiopathic nephrotic syndrome. Clin Exp Immunol 2011; 166:55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bruschi M, Candiano G, Santucci L, Ghiggeri GM. Oxidized albumin. The long way of a protein of uncertain function. Biochim Biophys Acta 2013; 1830:5473–9. [DOI] [PubMed] [Google Scholar]
- 22. Musante L, Candiano G, Petretto A et al Active focal segmental glomerulosclerosis is associated with massive oxidation of plasma albumin. J Am Soc Nephrol 2007; 18:799–810. [DOI] [PubMed] [Google Scholar]
- 23. Bruschi M, Grilli S, Candiano G et al New iodo‐acetamido cyanines for labeling cysteine thiol residues. A strategy for evaluating plasma proteins and their oxido‐redox status. Proteomics 2009; 9:460–9. [DOI] [PubMed] [Google Scholar]
- 24. Ghiggeri GM, Cercignani G, Ginevri F et al Puromycin aminonucleoside metabolism by glomeruli and glomerular epithelial cells in vitro . Kidney Int 1991; 40:35–42. [DOI] [PubMed] [Google Scholar]
- 25. Ginevri F, Gusmano R, Oleggini R et al Renal purine efflux and xanthine oxidase activity during experimental nephrosis in rats: difference between puromycin aminonucleoside and adriamycin nephrosis. Clin Sci (Lond) 1990; 78:283–93. [DOI] [PubMed] [Google Scholar]
- 26. Ravani P, Magnasco A, Edefonti A et al Short‐term effects of rituximab in children with steroid‐ and calcineurin‐dependent nephrotic syndrome: a randomized controlled trial. Clin J Am Soc Nephrol 2011; 6:1308–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ravani P, Ponticelli A, Siciliano C et al Rituximab is a safe and effective long‐term treatment for children with steroid and calcineurin inhibitor‐dependent idiopathic nephrotic syndrome. Kidney Int 2013; 84:1025–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ravani P, Rossi R, Bonanni A et al Rituximab in children with steroid‐dependent nephrotic syndrome: a multicenter, open‐label, noninferiority, randomized controlled trial. J Am Soc Nephrol 2015. pii: ASN.2014080799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perosa F, Favoino E, Caragnano MA, Dammacco F. Generation of biologically active linear and cyclic peptides has revealed a unique fine specificity of rituximab and its possible cross‐reactivity with acid sphingomyelinase‐like phosphodiesterase 3b precursor. Blood 2006; 107:1070–7. [DOI] [PubMed] [Google Scholar]
- 30. Bai A, Robson S. Beyond ecto‐nucleotidase: CD39 defines human Th17 cells with CD161. Purinergic Signal 2015. doi: 10.1007/s11302-015-9457-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fornoni A, Sageshima J, Wei C et al Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Transl Med 2011; 3:85ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Park SJ, Shin JI. Another beneficial effect of rituximab on refractory ANCA‐associated vasculitis: the role of interleukin‐17 suppression? Scand J Immunol 2013; 77:221. [DOI] [PubMed] [Google Scholar]
- 33. Pierson ER, Stromnes IM, Goverman JM. B cells promote induction of experimental autoimmune encephalomyelitis by facilitating reactivation of T cells in the central nervous system. J Immunol 2013; 192:929–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vaglio A, Buzio C, Zwerina J. Eosinophilic granulomatosis with polyangiitis (Churg–Strauss): state of the art. Allergy 2913; 68:261–73. [DOI] [PubMed] [Google Scholar]
- 35. van de Veerdonk FL, Lauwerys B, Marijnissen RJ et al The anti‐CD20 antibody rituximab reduces the Th17 cell response. Arthritis Rheum 2011; 63:1507–16. [DOI] [PubMed] [Google Scholar]
- 36. Yamamoto A, Sato K, Miyoshi F et al Analysis of cytokine production patterns of peripheral blood mononuclear cells from a rheumatoid arthritis patient successfully treated with rituximab. Mod Rheumatol 2010; 20:183–7. [DOI] [PubMed] [Google Scholar]
- 37. Stone JH, Merkel PA, Spiera R et al Rituximab versus cyclophosphamide for ANCA‐associated vasculitis. N Engl J Med 2010; 363:221–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bromley SK, Burack WR, Johnson KG et al The immunological synapse. Annu Rev Immunol 2001; 19:375–96. [DOI] [PubMed] [Google Scholar]
- 39. Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28‐mediated signalling co‐stimulates murine T cells and prevents induction of anergy in T‐cell clones. Nature 1992; 356:607–9. [DOI] [PubMed] [Google Scholar]
- 40. Abbas AK, Sharpe AH. T‐cell stimulation: an abundance of B7s. Nat Med 1999; 5:1345–6. [DOI] [PubMed] [Google Scholar]
- 41. Henry J, Miller MM, Pontarotti P. Structure and evolution of the extended B7 family. Immunol Today 1999; 20:285–8. [DOI] [PubMed] [Google Scholar]
- 42. Reiser J, von Gersdorff G, Loos M et al Induction of B7‐1 in podocytes is associated with nephrotic syndrome. J Clin Invest 2004; 113:1390–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Delville M, Sigdel TK, Wei CL et al A circulating antibody panel for pretransplant prediction of FSGS recurrence after kidney transplantation. Sci Transl Med 2014; doi: 10.1126/scitranslmed.3008538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vincenti F, Ghiggeri GM. New insights into the pathogenesis and the therapy of recurrent focal glomerulosclerosis. Am J Transplant 2005; 5:1179–85. [DOI] [PubMed] [Google Scholar]
- 45. Quezada SA, Jarvinen LZ, Lind EF, Noelle RJ. CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol 2004; 22:307–28. [DOI] [PubMed] [Google Scholar]
- 46. Hassan GS, Merhi Y, Mourad W. CD40 ligand: a neo‐inflammatory molecule in vascular diseases. Immunobiology 2012; 217:521–32. [DOI] [PubMed] [Google Scholar]
- 47. Zhang B, Wu T, Chen M, Zhou Y, Yi D, Guo R. The CD40/CD40L system: a new therapeutic target for disease. Immunol Lett 2013; 153:58–61. [DOI] [PubMed] [Google Scholar]
- 48. Yu CC, Fornoni A, Weins A et al Abatacept in B7‐1‐positive proteinuric kidney disease. N Engl J Med 2013; 369:2416–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Garin EH, Diaz LN, Mu W, Wasserfall C, Araya C, Segal M, Johnson RJ. Urinary CD80 excretion increases in idiopathic minimal‐change disease. J Am Soc Nephrol 2009; 20:260–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Alachkar N, Carter‐Monroe N, Reiser J. Abatacept in B7‐1‐positive proteinuric kidney disease. N Engl J Med 2014; 370:1263–4. [DOI] [PubMed] [Google Scholar]
- 51. Baris S, Schulze I, Ozen A et al Clinical heterogeneity of immunodysregulation, polyendocrinopathy, enteropathy, X‐linked: pulmonary involvement as a non‐classical disease manifestation. J Clin Immunol 2014; 34:601–6. [DOI] [PubMed] [Google Scholar]
- 52. Le Berre L, Bruneau S, Naulet J et al Induction of T regulatory cells attenuates idiopathic nephrotic syndrome. J Am Soc Nephrol 2009; 20:57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang YM, Zhang GY, Hu M et al CD8+ regulatory T cells induced by T cell vaccination protect against autoimmune nephritis. J Am Soc Nephrol 2012; 23:1058–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bertelli R, Di Donato A, Cioni M et al LPS nephropathy in mice is ameliorated by IL‐2 independently of regulatory T cells activity. PLOS ONE 2014; 9:e111285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody‐cytokine immune complexes. Science 2006; 311:1924–7. [DOI] [PubMed] [Google Scholar]
- 56. Kim MG, Koo TY, Yan JJ et al IL‐2/anti‐IL‐2 complex attenuates renal ischemia–reperfusion injury through expansion of regulatory T cells. J Am Soc Nephrol 2013; 24:1529–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Polhill T, Zhang GY, Hu M et al IL‐2/IL‐2Ab complexes induce regulatory T cell expansion and protect against proteinuric CKD. J Am Soc Nephrol 2012; 23:1303–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gavin MA, Rasmussen JP, Fontenot JD et al Foxp3‐dependent programme of regulatory T‐cell differentiation. Nature 2007; 445:771–5. [DOI] [PubMed] [Google Scholar]
- 59. Rubtsov YP, Niec RE, Josefowicz S et al Stability of the regulatory T cell lineage in vivo . Science 2010; 329:1667–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shevach EM, Thornton AM. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev 2014; 259:88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hall BM. T cells: soldiers and spies‐the surveillance and control of effector T cells by regulatory T cells. Clin J Am Soc Nephrol 2015: doi: 10.2215/CJN.06620714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 2012; 30:531–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I. Human CD25highFoxp3pos regulatory T cells differentiate into IL‐17‐producing cells. Blood 2008; 112:2340–52. [DOI] [PubMed] [Google Scholar]
- 64. Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev 2014; 13:668–77. [DOI] [PubMed] [Google Scholar]
- 65. Gardner D, Jeffery LE, Sansom DM. Understanding the CD28/CTLA‐4 (CD152) pathway and its implications for costimulatory blockade. Am J Transplant 2014; 14:1985–91. [DOI] [PubMed] [Google Scholar]
- 66. Chen L, Flies DB. Molecular mechanisms of T cell co‐stimulation and co‐inhibition. Nat Rev Immunol 2013; 13:227–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Di Virgilio F. Purinergic signalling in the immune system. A brief update. Purinergic Signal 2007; 3:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Di Virgilio F, Borea PA, Illes P. P2 receptors meet the immune system. Trends Pharmacol Sci 2001; 22:5–7. [DOI] [PubMed] [Google Scholar]