

Summary
In 40% of cases of classical Hodgkin lymphoma (cHL), Epstein–Barr virus (EBV) latency‐II antigens [EBV nuclear antigen 1 (EBNA1)/latent membrane protein (LMP)1/LMP2A] are present (EBV+cHL) in the malignant cells and antigen presentation is intact. Previous studies have shown consistently that HLA‐A*02 is protective in EBV+cHL, yet its role in disease pathogenesis is unknown. To explore the basis for this observation, gene expression was assessed in 33 cHL nodes. Interestingly, CD8 and LMP2A expression were correlated strongly and, for a given LMP2A level, CD8 was elevated markedly in HLA‐A*02– versus HLA‐A*02+ EBV+cHL patients, suggesting that LMP2A‐specific CD8+ T cell anti‐tumoral immunity may be relatively ineffective in HLA‐A*02– EBV+cHL. To ascertain the impact of HLA class I on EBV latency antigen‐specific immunodominance, we used a stepwise functional T cell approach. In newly diagnosed EBV+cHL, the magnitude of ex‐vivo LMP1/2A‐specific CD8+ T cell responses was elevated in HLA‐A*02+ patients. Furthermore, in a controlled in‐vitro assay, LMP2A‐specific CD8+ T cells from healthy HLA‐A*02 heterozygotes expanded to a greater extent with HLA‐A*02‐restricted compared to non‐HLA‐A*02‐restricted cell lines. In an extensive analysis of HLA class I‐restricted immunity, immunodominant EBNA3A/3B/3C‐specific CD8+ T cell responses were stimulated by numerous HLA class I molecules, whereas the subdominant LMP1/2A‐specific responses were confined largely to HLA‐A*02. Our results demonstrate that HLA‐A*02 mediates a modest, but none the less stronger, EBV‐specific CD8+ T cell response than non‐HLA‐A*02 alleles, an effect confined to EBV latency‐II antigens. Thus, the protective effect of HLA‐A*02 against EBV+cHL is not a surrogate association, but reflects the impact of HLA class I on EBV latency‐II antigen‐specific CD8+ T cell hierarchies.
Keywords: classical Hodgkin lymphoma, Epstein–Barr virus, genetic associations, HLA class I, T cell immunity
Introduction
Epstein–Barr virus (EBV) is a ubiquitous, persistent B cell‐trophic virus that typically establishes a benign infection of minimal clinical consequence. However, in a minority of hosts, EBV is associated with particular malignancies. In these conditions, EBV resides within the malignant cell in a restricted state of latent antigen expression. The frequency of association and expression of latent proteins is distinctive to the type of cancer. In classical Hodgkin lymphoma (cHL), approximately 40% of cases are associated with EBV (EBV+cHL), and virus expression in the Hodgkin–Reed–Sternberg (HRS) cells is limited to the EBV nuclear antigen 1 (EBNA1) and latent membrane proteins (LMP1 and LMP2)1, 2, 3. These are collectively termed EBV latency‐II antigens, a pattern of expression also observed in undifferentiated nasopharyngeal carcinoma. EBV+cHL is characterized by intact human leucocyte antigen (HLA) class I antigen processing and presentation 4, 5, 6, 7, enabling successful treatment of relapsed EBV+cHL by adoptive immunotherapy targeting EBV latency‐II antigens 8, 9, 10, 11.
A well‐established hierarchy exists among CD8+ T cell responses that target EBV latency antigens. In particular, EBV latency‐III antigens EBNA3A/3B/3C are immunodominant, whereas the EBV latency‐II antigens (EBNA1/LMP1/LMP2A) are subdominant and more challenging to detect without in‐vitro expansion 12, 13. However, the impact of HLA class I on EBV latency antigen‐specific CD8+ T cell immunity has not been determined systematically. Interestingly, large epidemiological and genomewide association studies have consistently reported differential HLA class I susceptibility to EBV+cHL (Supporting information, Table S1) 14, 15, 16, 17. In western European populations, HLA‐A*01 and HLA‐B*37 are associated with increased susceptibility to EBV+cHL, while HLA‐A*02 is associated with protection 15, 16, 17. By contrast, the HLA‐A*02 subtype HLA‐A*0207, which presents HLA‐A*0201‐restricted LMP2A‐derived peptides poorly 18, is over‐represented in northern Chinese EBV+cHL patients 19. Non‐HLA‐linked genetic susceptibility loci have also been identified for cHL, as has a single nucleotide polymorphism (SNP) found in association with an HLA class II locus. However, these associations were not specific for EBV+cHL 14, 20, 21.
The aim of this study was to understand the role of HLA class I in the pathogenesis of EBV+cHL. The presentation of viral peptide determinants by HLA‐A*02 and non‐HLA‐A*02 molecules provides a potential mechanistic link between EBV latency‐II‐specific CD8+ T cell immunity and the described genetic associations with EBV+cHL. However, there are many genes with diverse functions in close proximity to HLA class I. Therefore, such associations may simply reflect linkage disequilibrium between HLA class I and the ‘true’ predisposition locus. To distinguish these possibilities, we analysed the impact of HLA‐A*02 and non‐HLA‐A*02 molecules on EBV latency‐II antigen‐specific effector CD8+ T cell immunity in EBV+cHL.
Materials and methods
Sample cohorts
Blood samples and diseased tissue from newly diagnosed cHL patients and blood samples from healthy participants were acquired as part of an Australasian Leukaemia and Lymphoma Group prospective observational study. EBV association was confirmed via EBV‐encoded‐RNA in‐situ hybridization (EBER‐ISH), as described previously 22. Peripheral blood mononuclear cells (PBMCs) were isolated and cryopreserved in 90% fetal bovine serum (FBS) with 10% dimethylsulphoxide (DMSO). This study conformed to the Declaration of Helsinki and was approved by the Human Research Ethics Committees at all participating institutions. Written informed consent was obtained in all cases.
Digital multiplex gene expression by NanoString nCounter
Nucleic acid was extracted from 33 cHL formalin‐fixed paraffin‐embedded (FFPE) diseased node tissues (17 EBV‐vecHL, 16 EBV+cHL) using a RecoverAll Total Nucleic Acid Extraction Kit (Life Technologies, Paisley, UK). Gene expression profiling was conducted using the nCounter platform (NanoString Technologies). All analyses were performed using NCounter software. For normalization, gene expression data were controlled internally to the mean of the positive control probes to account for interassay variability. Gene normalization was performed using the geometric mean of four housekeeper genes [phosphoglycerate kinase 1 (PGK1), glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH), phosphoglycerate mutase 1 (PGAM1), ornithine decarboxylase anti‐zyme 1(OAZ1)], selected as per the manufacturer's recommendation.
Ex‐vivo EBV‐specific CD8+ T cell responses
Peptide pools (17‐mers overlapping by 10 amino acids) were synthesized to span the entire lengths of LMP1, LMP2A and BamHI Z fragment leftward open reading frame 1 (BZLF1) (Synbiosci, San Francisco, CA, USA and Mimotopes, Notting Hill, VIC, Australia). Each individual peptide in each pool was used at a final concentration of 2 μg/ml to stimulate EBV‐specific CD8+ T cells. Initially, PBMCs from 19 pretreatment EBV‐seropositive cHL patients (eight EBV+cHL, 11 EBV–cHL) were assayed. Subsequently, PBMCs from 14 EBV‐seropositive healthy volunteers were analysed at a separate site. PBMCs were resuspended in culture medium containing the co‐stimulatory antibodies αCD28 and αCD49d (1 μg/ml each; BD Pharmingen, Franklin Lakes, NJ, USA), brefeldin A (Golgi Plug, 10 μg/ml; BD Biosciences), monensin (Golgi Stop, 0.7 μg/ml; BD Biosciences) and fluorochrome‐labelled αCD107a. Each sample was divided into five stimulations: LMP1, LMP2A, BZLF1, an unstimulated control (co‐stimulation only) and a positive control comprising either Staphyloccoccus enterotoxin B (0·1 μg/ml; Sigma‐Aldrich, St Louis, MO, USA) or phorbol myristate acetate (10 ng/μl) with ionomycin (2 μg/ml). Cells were cultured overnight at 1–2 × 106 cells/ml in a 37°C, 5% CO2 incubator. The following day, PBMCs were washed, labelled with a viability dye to enable dead cell exclusion and surface‐stained for CD3, CD4 and CD8. Cells were then fixed/permeabilized and stained intracellularly for interferon (IFN)‐γ and tumour necrosis factor (TNF)‐α. All staining procedures were conducted as described previously 23. Samples were acquired on an LSRII flow cytometer (BD Biosciences) and data analysis was performed using FlowJo version 9·2 (TreeStar Inc., Ashland, OR, USA). The gating strategy followed previously published standard practice and is shown in Supporting information, Fig. S1 24.
LMP2A‐specific CD8+ T cell expansion using monogenic HLA class I cell lines
Nine healthy EBV‐seropositive HLA‐A*02 heterozygotes (three HLA‐A*01/A*02, three HLA‐A*02/A*03 and three HLA‐A*01/A*02/B*08) were tested. PBMCs were stimulated with HLA class I‐deficient, EBV‐infected 721·221 lymphoblastoid cell lines transfected separately with HLA‐A*01, A*02, A*03 or B*08. Procedures were adapted from a previously published protocol 25. Each transfected 721·221 cell line was pulsed with overlapping LMP2A peptide pools spanning the entire protein (Synbiosci). Peptides were divided into six pools (Supporting information, Table S2), each comprising 10–11 peptides at a final individual concentration of 10 μg/ml. These smaller pools at higher concentrations were used to maximize the antigen‐specific stimulus (a single pool of all 65 peptides at this high concentration would have been toxic). The corresponding unpulsed cells served as baseline controls in all experiments. Cultures were expanded for 10 days under standard conditions. The relative strength of the HLA class I‐restricted EBV‐specific CD8+ T cell response was assessed using intracellular cytokine staining for IFN‐γ after a 5‐h restimulation. Samples were acquired on a fluorescence activated cell sorter (FACS)Canto flow cytometer (BD Biosciences) and data analysis was performed using FlowJo version 9·2 (TreeStar Inc.).
In‐vitro LMP2A‐specific CD8+ T cell cytotoxicity
EBV‐specific T cells were expanded by repeated stimulation with autologous EBV‐transformed B cells for 6–8 weeks using a published expansion protocol 26, 27 in four EBV‐seropositive healthy control donors (two HLA‐A*02+ and two HLA‐A*02–). Direct lysis of autologous EBV‐transformed B cells was confirmed in all cases, demonstrating successful expansion of cytotoxic EBV‐specific T cells. LMP2A‐specific cytotoxicity was quantified using autologous carboxyfluorescein succinimidyl ester (CFSE)‐labelled phytohaemagglutinin (PHA) blasts (target cells) incubated with 2 μg/ml of peptides from each of six LMP2A overlapping peptide pools (Supporting information, Table S2) in duplicate. The ratio of effector to target cells was 20 : 1. After 6 h incubation, the number of CFSE‐labelled targets remaining was determined by comparison to a constant number of CountBright absolute counting beads (ThermoFisher, Scoresby, Australia). LMP2A‐specific lysis was calculated for each well relative to the unpulsed control sample.
HLA class I‐restricted EBV latency antigen‐specific CD8+ T cell responses
Ex‐vivo CD107ab+CD8+ T cell responses to HLA class I‐restricted peptides were assayed in 30 healthy EBV‐seropositive donors. PBMCs were resuspended in culture medium containing monensin (GolgiStop, 0.7 μg/ml; BD Biosciences), αCD107a‐fluorescein isothiocyanate (FITC) and αCD107b‐FITC (BD Pharmingen), and incubated for 5 h with peptide (2 μg/ml). Samples were acquired on a FACSCanto flow cytometer (BD Biosciences) and data analysis was performed using FlowJo version 9·2 (TreeStar Inc.). To minimize bias, an equivalent number of ‘predicted’ and ‘defined’ (31 and 30, respectively) peptides were included. The peptides were derived from LMP1/LMP2A or EBNA3A/3B/3C and restricted by one of the following HLA class I allotypes: HLA‐A*01, A*02, A*03, A*11, A*24, B*07, B*08, B*35, B*37, B*44 or B*60 (B*40:01). Equivalent numbers of defined epitopes covering comparable numbers of HLA class I alleles were used for the immunodominant EBNA3A/3B/3C (15 peptides, six alleles) and the subdominant LMP1/LMP2A (15 peptides, seven alleles) proteins. Combined, these alleles cover 75% of HLA‐A and 57% of HLA‐B alleles in the Australian population 28. Peptides (and references) are listed in Supporting information, Table S3. All defined epitopes were validated previously in functional assays. For HLA class I alleles with few or no known epitopes, predicted peptides were identified in silico (http://www.syfpeithi.de) using a previously described algorithm [29]. The ‘SYFPEITHI’ scores of defined epitopes were used to delineate a threshold score for the predicted peptides.
HLA class I peptide binding of algorithm‐predicted peptides
Binding of predicted HLA‐A*03‐restricted peptides was confirmed using HLA‐A*03‐expressing T2 cell lines, as described previously 30. Expression of stabilized HLA‐A*03 on the cell surface after peptide pulsing was assessed by flow cytometry. Results are reported as the mean fluorescence intensity above the negative (no peptide) control.
Binding of predicted HLA‐A*01‐restricted peptides was confirmed by in‐vitro HLA‐A*0101‐peptide complex refolding and thermal stability analysis by circular dichroism (CD) spectroscopy. Competent Rosetta DE3 Escherichia coli cells were used to produce the HLA‐A*0101 and beta 2‐microglobulin (β2M) chains, as described previously 31. For a 1‐l refold, 30 mg of HLA‐A*0101 was mixed with 30 mg of β2M and 4 mg of peptide at 37°C for 15 min. The mixture was then added to cold refold buffer [50 mM Tris pH 8, 2 mM ethylenediamine tetraacetic acid (EDTA), 400 mM L‐arginine, 6 mM cysteamine hydrochloride and 4 mM cystamine]. Refolds were stirred at 4°C for > 1 h. Dialysis was carried out against 10 mM Tris pH 8.1 until the conductivity of the refolds was <2 mS/cm. The refolds were then filtered, purified by ion exchange using a Poros50HQTM column and gel filtered into phosphate‐buffered saline (PBS) using a Superdex200HRTM column. Protein quality was analysed by Coomassie‐stained sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE). After ion exchange, the central five fractions around the maximum HLA class I peak (by SDS gel) from each refold were selected for gel filtration. After gel filtration, the central four fractions around the maximum HLA class I peak from each refold were selected for analysis by spectrometry (A280 nm) to determine the final protein yield. Two positive control peptides from cytomegalovirus, VTEHDTLLY 32 and YSEHPTFTSQY 25, and a negative control peptide with a binding sequence optimal for HLA‐A*0201 (ALAAAAAAV), were used in addition to the 13 predicted HLA‐A*01‐restricted EBV‐derived peptides.
Thermal stability of HLA‐A*0101/β2M/peptide complexes was assessed by CD spectroscopy. Data were collected on an Aviv Model 215 spectropolarimeter (Aviv Biomedical Inc., Lakewood, NJ, USA) equipped with a Peltier thermostatted cell holder using a 0·1‐cm quartz cell. Proteins were dissolved in 75 mM NaCl, 20 mM , pH 7·5. Melting curves were recorded in 0·5°C intervals from 4°C up to a maximum temperature when protein aggregation was observed with settings resulting in an average heating rate of ∼30°C/h. Values were corrected to a calibration curve recorded with the temperature measured in the cell. Melting curves were analysed assuming a two‐state trimer‐to‐monomer transition from the native (N) to unfolded (U) conformation N3 ↔ 3U with an equilibrium constant K = [U]3/[N3] = F/[3c2 (1‐F)3], where F and c are the degree of folding and protein concentration, respectively. Data were fitted as described 33 using the non‐linear least‐squares routine of Origin version 7·5 (OriginLab, Northampton, MA, USA). Fitted parameters were the melting temperature TM, at which 50% of proteins are in the folded and unfolded state, van't Hoff's enthalpy ΔHvH at the transition midpoint and the slope and Θ‐intercept of the native baseline assumed as a linear function of the temperature. As all protein complexes aggregated at various degrees of unfolding, the ellipticity of the unfolded state was set as a constant of −4400 degrees cm2 dmol−1; this value resulted from fitting melting curves of LMP2ALTE and positive control peptide VTEHDTLLY‐containing complexes, which showed the least aggregation tendency, and is in good agreement with values reported for other thermally denatured proteins 34. For all peptides, the coefficient of determination for fitted curves versus measurements was r 2 > 0·99.
Statistical analysis
Combined (but not summed) IFN‐γ, TNF‐α and CD107a responses to EBNA1/LMP1/LMP2A/BZLF1 in patients with cHL and healthy volunteers were compared using a linear mixed‐effects model (with a random effect for subject). This enabled comparison of groups while accounting for the correlation induced by measuring multiple (but not necessarily comparable) responses in the same individual 35. Using spice (software version 5·3, downloaded from http://exon.niaid.nih.gov), individual and polyfunctional IFN‐γ, TNF‐α and CD107a responses in patients with cHL were compared by Wilcoxon's signed‐rank tests and a partial permutation test, as described previously 36. Healthy donor ex‐vivo CD107ab+CD8+ T cell responses against defined and predicted HLA class I‐restricted peptides from LMP1/LMP2A and EBNA3A/3B/3C were compared using the Mann–Whitney U‐test. In cases where responses against multiple peptides were measured in the same individual, individual responses were considered independent. Significance above zero was determined using a one‐sample t‐test with one‐tailed P‐values. Cytotoxicity was compared using Fisher's exact test with a two‐tailed P‐value. Statistical analysis was performed using GraphPad Prism version 5·0 (Graphpad Software Inc., San Diego, CA, USA) and spss statistics version 19 (IBM, Armonk, NY, USA).
Results
Intratumoral CD8 : LMP2A ratios are enriched in HLA‐A*02– EBV+cHL
Gene expression was quantified in 33 cHL diseased node FFPE tissues (17 EBV–cHL, 16 EBV+cHL) using NanoString nCounter 37. The expression levels of CD8 and β2M as markers of immune effector and antigen presentation, respectively, and the EBV latency II genes (EBNA1, LMP1 and LMP2A with the EBV‐lytic gene BZLF1 as a comparator) were measured. CD8 expression correlated with β2M in the entire cHL cohort (r = 0·5906, P = 0·0003), consistent with intratumoral CD8 being proportional to antigen presentation. In the EBV+cHL subgroup, the β2M–CD8 correlation was more marked (r = 0·7176, P = 0·0024, Fig. 1a). In this subgroup, β2M also correlated with LMP2A levels (r = 0·5685, P = 0·0216, Fig. 1b). Accordingly, CD8 expression correlated with LMP2A (r = 0·7938, P = 0·0004, Fig. 1c). No correlations were found for the other EBV genes with either CD8 or β2M, and there was no difference in expression of CD8 or β2M between EBV+ and EBV–cHL.
Figure 1.
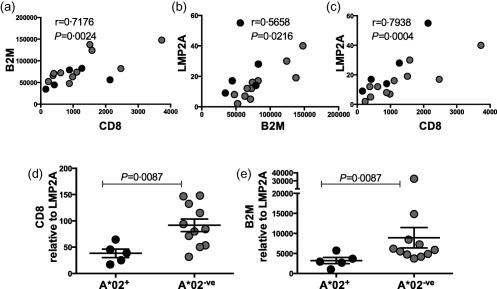
Gene expression in Epstein–Barr virus (EBV)+ classical Hodgkin lymphoma (cHL) diseased tissue. (a) Correlation of beta 2‐microglobulin (β2M) and CD8 expression levels. (b) Correlation of latent membrane protein (LMP)2A and β2M expression levels. (c) Correlation of LMP2A and CD8 expression levels. Spearman's correlation = r. (d) CD8 expression relative to LMP2A in human leucocyte antigen (HLA)‐A*02+ compared to HLA‐A*02– patients. (e) β2M expression relative to LMP2A in HLA‐A*02+ compared to HLA‐A*02– patients. Error bar represents standard error of the mean. Solid black circles represent HLA‐A*02+ patients. Grey circles represent HLA‐A*02– patients.
Both EBNA1 and LMP1 are known to encode for antigenic peptides that expand IL‐10‐secreting regulatory CD4+ T cells 38, 39. However, no correlations were found between the EBV latency II genes and expression of either CD4 or the regulatory T cell markers IL‐10, lymphocyte‐activation gene 3 (LAG3) and forkead box protein 3 (FoxP3) [P = not significant (n.s.)].
Comparisons of HLA‐A*02+ (n = 5) and HLA‐A*02– (n = 11) patients with EBV+cHL showed no difference in expression levels of β2M, CD8, LMP2A or any of the other EBV genes. However, CD8 and β2M (normalized to the expression of LMP2A) were elevated markedly in HLA‐A*02– versus HLA‐A*02+ patients (P = 0·0087, P = 0·0087 Fig. 1d,e). This was not observed when expression was normalized to the other EBV latency II genes. These results demonstrate that, for a given LMP2A expression level, there is more CD8 and β2M expression within the malignant lymph node in HLA‐A*02– compared to HLA‐A*02+ patients with EBV+cHL.
HLA‐A*02+ patients with EBV+cHL exhibit increased responses to EBV latency‐II antigens relative to HLA‐A*02– patients
The observation that CD8 is relatively enriched within HLA‐A*02– EBV+cHL nodes suggests that EBV latency‐II‐specific effector CD8+ T cell immunity is relatively ineffective in these patients. To investigate the impact of HLA‐A*02 on EBV latency‐II‐specific effector CD8+ T cell immunity, a series of functional assays were performed. Initially, we tested CD8+ T cell immunity against relevant EBV latency antigens in pretherapy cHL patients, with EBV tissue status and HLA class I as covariates. Previous studies in cHL have not evaluated the contribution of non‐defined subdominant epitopes to the total CD8+ T cell response or assessed whether HLA class I alleles impact the magnitude of the response 40, 41. Effector molecule testing in these studies was also limited to IFN‐γ production.
First, we tested 19 cHL patients (Table 1). Blood samples were collected at diagnosis prior to therapy. Age and gender were not significantly different between subgroups, which were also comparable for Ann Arbor stage and prognostic score 42. To ensure that multi‐faceted CD8+ effector T cell immunity was assayed, we used polychromatic flow cytometry to measure ex‐vivo IFN‐γ and TNF‐α production together with CD107a mobilization. Overlapping peptide pools spanning EBNA1 and the latent proteins LMP1 and LMP2A were used to ensure comprehensive antigenic coverage. A peptide pool spanning the EBV lytic protein BZLF1 (not expressed by EBV+ HRS cells) was included as a comparator. Peptide stimulations were conducted overnight to enhance sensitivity, and highly stringent gating strategies were employed to maximize specificity (Supporting information, Fig. S1). A linear mixed‐effects model was used to incorporate and compare all three effector markers and any compounding significance while accounting for multiple measurements within a given individual (represented in Fig. 2a as summed percentages of IFN‐γ, TNF‐α and CD107a responses). spice software, designed specifically for post‐cytometric complex multivariate data sets, was used to conduct Wilcoxon signed‐rank tests and permutation comparisons on individual (Fig. 2b,c) and polyfunctional (Fig. 2d,e) responses.
Table 1.
Characteristics of classical Hodgkin lymphoma (cHL) patients tested ex vivo for CD8+ T cell responses
| Patient no. | EBER‐ISH | Age | Gender | ‘B’ symptoms | Stage | Prognostic score* | Classical subtype | HLA‐A*02 | HLA class I |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Negative | 79 | F | Yes | III | 3 | NS | Negative | A*01/*30, B*51/*62 |
| 2 | Negative | 25 | M | Yes | IIIb | 3 | NS | Negative | A*11/*11, B*60/*60 |
| 3 | Negative | 79 | F | No | III | 1 | NS | Negative | A*01/*31, B*57/*61 |
| 4 | Negative | 33 | F | Yes | IIb | 2 | LD | Negative | A*02/*25, B*62/*63 |
| 5 | Negative | 55 | M | No | II | 3 | MC | Positive | A*02/*03, B*13/*51 |
| 6 | Negative | 54 | M | Yes | IVb | 5 | NS | Positive | A*02/*31, B*44/*60 |
| 7 | Negative | 22 | M | Yes | IVb | 5 | Classical unspecified | Positive | A*01/*02, B*35/*57 |
| 8 | Negative | 21 | M | No | IV | 3 | NS | Positive | A*02/*03, B*07/*51 |
| 9 | Negative | 27 | M | No | III | 3 | NS | Positive | A*02/*03, B*07/*51 |
| 10 | Negative | 20 | F | No | IVa | 1 | Classical unspecified | Positive | A*02/*02, B*07/*35 |
| 11 | Negative | 20 | F | Yes | IIIb | 3 | NS | Positive | A*01/*02, B*08/*49 |
| 12 | Negative | 18 | M | No | III | 2 | NS | Negative | A*11/*24, B*18/*60 |
| 13 | Positive | 48 | M | No | II | 2 | LR | Negative | A*24/*24, B*18/*51 |
| 14 | Positive | 40 | M | No | II | 1 | MC | Negative | A*01/*24, B*08/*51 |
| 15 | Positive | 40 | M | Yes | IVb | 5 | NS | Negative | A*01/*03, B*35/*75 |
| 16 | Positive | 33 | M | Yes | II | 2 | NS | Positive | A*02/*29, B*07/B*44 |
| 17 | Positive | 25 | F | No | IIa | 0 | NS | Positive | A*02/*02, B*44/*44 |
| 18 | Positive | 49 | M | No | IV | 4 | NS | Positive | A*01/*02, B*37/*57 |
| 19 | Positive | 21 | F | No | IIa | 1 | NS | Positive | A*02/*66, B*41/*44 |
*Hasenclever 42. LD = lymphocyte‐depleted; MC = mixed‐cellularity; NS = nodular sclerosing; LR = lymphocyte‐rich; M = male; F = female; HLA = human leucocyte antigen; EBER‐ISH = Epstein–Barr virus‐encoded‐RNA in‐situ hybridization.
Figure 2.
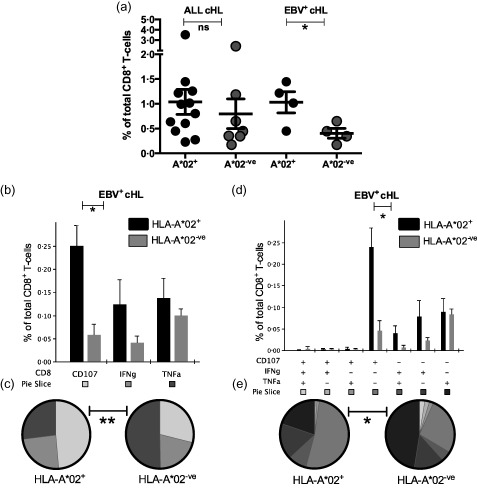
Ex‐vivo Epstein–Barr virus (EBV)‐specific CD8+ T‐cell responses in patients with classical Hodgkin lymphoma (cHL) . (a) Summed percentages of ex‐vivo latent membrane protein (LMP)1/2A‐specific CD8+ T cell responses defined by interferon (IFN)‐γ, tumour necrosis factor (TNF)‐α and CD107a. Error bars represent standard error of the mean. P‐values were calculated using a linear mixed effects model. (b–c) Comparison of individual effector molecules in human leucocyte antigen (HLA)‐A*02+ versus HLA‐A*02‐ve patients with EBV+cHL. (b) Percentage of total CD8+ T cells specific for LMP1/2A. (c) Percentage of the total LMP1/2A‐specific CD8+ T cell subset. (d–e) Comparison of polyfunctional LMP1/2A‐specific CD8+ T cell responses in HLA‐A*02+ versus HLA‐A*02‐ve patients with EBV+cHL. (d) Percentage of total CD8+ T cells specific for LMP1/2A. (e) Percentage of the total LMP1/2A‐specific CD8+ T cell subset. **P < 0·01; *P < 0·05; P > 0·05 = not significant.
Global effector CD8+ T cell responses (defined as combined IFN‐γ, TNF‐α and CD107a) were compared for the EBV latency II proteins (EBNA1, LMP1, LMP2A). Responses were analysed as total EBV latency II (summed EBNA1, LMP1/2A‐specific), LMP1/2A (summed LMP1/2A‐specific) and individual protein responses. No significant differences were found between patients with EBV+cHL and EBV–cHL. Furthermore, response magnitudes across all patients with cHL independent of EBV tissue status were equivalent irrespective of HLA‐A*02 expression (Fig. 2a). In addition, the relevant EBV‐specific CD8+ T cell responses in patients with EBV–cHL were not influenced by HLA‐A*02 status.
Interestingly, the combined IFN‐γ, TNF‐α and CD107a CD8+ T cell responses were significantly greater in HLA‐A*02+ compared to HLA‐A*02‐ve patients with EBV+cHL for the LMP1/2A proteins (P = 0·0371, Fig. 2a). Although elevated response levels were seen in HLA‐A*02+ EBV+cHL patients for total EBV latency II (P = 0·0753) and individual EBNA1, LMP1 and LMP2A proteins (P = 0·3329, P = 0·0525 and P = 0·0983, respectively), these did not reach significance.
We then compared individual immune effector molecules stratified by HLA‐A*02 status using spice. Although all three functional markers were elevated in HLA‐A*02+ compared to HLA‐A*02– patients with EBV+cHL for LMP1/2A proteins (CD107a, 4·3‐fold; IFN‐γ, 3·0‐fold; TNF‐α, 1·4‐fold), only CD107a reached significance (P = 0·007, Fig. 2b). Across all LMP1/2A‐specific effector CD8+ T cells, the proportion of individual effector molecules was significantly different between HLA‐A*02+ and HLA‐A*02– patients (P = 0·0041, Fig. 2c). By polyfunctional analysis, this significant increase was attributable to CD107a single‐positive CD8+ T cells (CD107a+IFN‐γ‐TNF‐α‐; P = 0·007, Fig. 2d) and the proportion of polyfunctional immune effectors within all LMP1/2A‐specific CD8+ T cells was also significantly different (P = 0·043, Fig. 2e). In HLA‐A*02+ EBV+cHL patients, the majority of immune effector cells were CD107a single‐positive CD8+ T cells. No differences (P = n.s.) were observed for BZLF1‐specific CD8+ T cell responses between any patient categories.
In line with our findings that differences between HLA‐A*02+ and HLA‐A*02– patients were detectable only in EBV+cHL, analysis of 14 healthy age‐/sex‐matched controls (using the linear mixed‐effects model to evaluate comprehensive CD8+ T cell responses to the EBV latency‐II proteins) revealed no significant differences between HLA‐A*02+ (n = 7) and HLA‐A*02– (n = 7) individuals (Supporting information, Fig. S2a). In patients with cHL, global effector CD8+ T cell responses were greater for LMP2A compared with LMP1 (P = 0·016) and EBNA1 (P < 0·0001) (Supporting information, Fig. S2b). Similarly, for healthy controls global effector CD8+ T cell responses were greater for LMP2A compared with LMP1 (P = 0·011) and EBNA1, although the latter did not reach statistical significance (Supporting information, Fig. S2c).
Thus, by ex‐vivo analysis, we observed modest differences in CD8+ T cell responses against relevant EBV latency‐II proteins between HLA‐A*02+ and to HLA‐A*02– patients with EBV+cHL. These results were not observed in EBV–cHL patients or healthy participants. In line with our tissue expression data, these results suggest that EBV latency‐II‐specific effector CD8+ T cell immunity may be less effective in HLA‐A*02– EBV+cHL.
LMP2A‐specific responses restricted by HLA‐A*02 achieve greater magnitudes than those restricted by other HLA class I molecules
Ex‐vivo assays are less sensitive than in‐vitro expansion for the detection of low‐frequency EBV‐specific CD8+ T cells 43. Indeed, low‐frequency responses can be detected via in‐vitro expansion in patients receiving immunosuppressive therapies 13. Furthermore, up to four different HLA‐A/B molecules can potentially present relevant EBV‐derived epitopes in each individual, adding a confounding layer of complexity to single allele‐based effects.
To overcome these limitations, we generated mutant HLA class I‐negative 721·221 cell lines transfected individually with HLA‐A*01, HLA‐A*02, HLA‐A*03 and HLA‐B*08. Previous work with these cell lines demonstrated an absence of HLA‐A*01‐restricted responses to endogenously processed EBV latency antigens. However, this system was biased preferentially towards the immunodominant EBNA3A/3B/3C antigens that are not expressed by HRS cells 25. In order to use this single allele system to evaluate responses relevant to the EBV+cHL setting, transfected 721·221 cells were pulsed with LMP2A peptides. LMP2A was chosen for this purpose, as it is the most immunogenic of the three EBV genes expressed in EBV+cHL and because it was the only gene we found to associate significantly with HLA‐A*02 in diagnostic tissues. Overlapping peptides divided into six pools spanning the entire LMP2A protein were used to ensure inclusion of the total CD8+ T cell response restricted by each HLA class I molecule, regardless of the specific target epitopes (Supporting information, Table S2). PBMCs from healthy EBV‐seropositive participants were stimulated separately with each peptide pool to expand LMP2A‐specific CD8+ T cells. The results were summed for each allele. Nine EBV‐seropositive donors were tested, all heterozygous for HLA‐A*02. Other HLA class I donor alleles were selected based on the literature. Notably, HLA‐A*01 has been associated consistently with an increased incidence of EBV+cHL 15, 16, 17. HLA‐B*08 is known to be in linkage disequilibrium with HLA‐A*01 44, and a functionally defined HLA‐B*08‐restricted LMP2A‐derived epitope was identified recently 45. Finally, HLA‐A*03 was chosen as a relatively common HLA class I allele in Australia. Together, these four alleles represent > 75% of the Australian population 46. Accordingly, donors 1–3 were HLA‐A*01/A*02, donors 4–6 were HLA‐A*02/A*03 and donors 7–9 were HLA‐A*01/A*02/B*08.
IFN‐γ‐specific CD8+ T cells inhibit in‐vitro transformation of EBV‐infected B cells 47, 48. The cytokine may also play a critical role in the pathogenesis of cHL via the canonical Janus kinase–signal transducer and activator of transcription (JAK–STAT) pathway known to be over‐expressed in HRS cells 49. Therefore, identifying any differences in IFN‐γ‐specific CD8+ T cell responses has important implications. In‐vitro expansion permitted more sensitive discrimination of the impact of HLA class I status on IFN‐γ+ responses. As shown in Fig. 3, HLA‐A*02 uniformly expanded more LMP2A‐specific IFN‐γ+ CD8+ T cells. The total response for each HLA class I allele (i.e. all HLA‐A*01, all HLA‐A*02, all HLA‐A*03 and all HLA‐B*08) was significantly greater than zero only for HLA‐A*02 (P = 0·0034). Thus, in an in‐vitro system with few confounding influences, HLA‐A*02 generated more LMP2A‐specific IFN‐γ+ CD8+ T cells compared to HLA‐A*01, HLA‐A*03 and HLA‐B*08.
Figure 3.
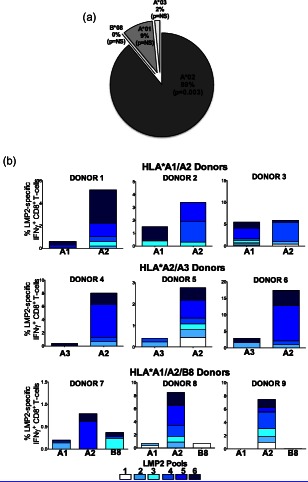
Human leucocyte antigen (HLA) class I‐restricted expansion of latent membrane protein (LMP)2A‐specific interferon (IFN) γ+CD8+ T cells. (a) Percentages of total HLA class I‐restricted LMP2A‐specific CD8+ T cell responses generated in vitro from healthy Epstein–Barr virus (EBV) seropositive HLA‐A*02 heterozygotes (n = 9). Only HLA‐A*02‐restricted responses were significantly above zero. (b) The combined expansion of LMP2A‐specific IFN‐γ+CD8+ T‐cells is shown for all donors from the three HLA‐A*02 heterozygote combinations. Each bar depicts the percentage of LMP2A‐specific IFN‐γ+CD8+ T cells expanded from one of the six peptide pools. The peptide pools are described in Supporting information, Table S2.
Of note, peptide pool 5 generated the greatest response (37% of the total HLA‐A*02‐restricted responses) and contained the sequences of two known HLA‐A*02 epitopes (FLYALALLL, LLWTLVVL) 12. Peptide pool 6 contained the sequences of three known HLA‐A*02 epitopes (CLGGLLTMV, LTAGFLIFL and LLSAWILTA) and generated 22% of the total HLA‐A*02‐restricted responses. Peptide pool 4 also generated 22% and contained the sequence of an identified HLA‐A*02 epitope (GLGTLGAAL). Pools 1–3 did not contain known HLA‐A*02 epitopes and generated 4, 8 and 7% of the total HLA‐A*02‐restricted responses, respectively.
To confirm that functional LMP2A‐specific responses restricted by HLA‐A*02 achieve greater magnitudes than those restricted by other HLA class I molecules in conventional cytotoxicity assays, we took advantage of a published expansion protocol that generates EBV‐specific CD8+ T cells for therapeutic use 26, 27. This method uses autologous EBV‐transformed B cells as antigen‐presenting cells, thus not only is it physiologically relevant but it expands a broad range of EBV latent antigen‐specific CD8+ T cells across multiple HLA class I alleles. The latter is an important point, because it permits a comparison of HLA‐A*02 versus non‐HLA‐A*02 LMP2A‐specific CD8+ T cells in a setting in which other EBV latent antigens, including those presented by other HLA class I alleles, are also presented. Consistent with earlier results, minimal killing was observed in non‐HLA‐A*02 participants (<5% mean LMP2A‐specific killing), whereas strong killing was observed in HLA‐A*02 individuals (>54% mean LMP2A‐specific killing; P < 0·0001; Supporting information, Fig. S3).
Prediction and verification of HLA class I‐binding EBV latency antigen‐derived peptides
Next, we assessed the impact of multiple HLA class I alleles on the hierarchy of CD8+ T cell responses against the spectrum of EBV latency proteins. For immunodominant proteins, we tested EBNA3A/3B/3C (very few EBNA2 and EBNA‐LP epitopes have been identified) 50. Based on our earlier findings, subdominant proteins were restricted to LMP1/LMP2A and not EBNA1. Initially, we selected ‘defined’ epitopes shown previously to elicit CD8+ effector T cell immunity. For EBNA3A/3B/3C proteins, several such epitopes presented by a variety of HLA class I molecules are known. By contrast, for LMP1/LMP2A, there are relatively few defined HLA class I epitopes, with the exception of peptides presented by HLA‐A*02 12, 51, 52. As HLA‐A*01, HLA‐A*03 and HLA‐B*37 have each been associated with differential susceptibility to EBV+ lymphomas 15, 16, 17, 53, we aimed to increase the number of defined EBV latency antigen‐derived epitopes restricted by these HLA class I molecules. To achieve this, we screened for potential HLA class I binding peptides using the bioinformatic algorithm ‘SYFPEITHI’ 29. Thirty functionally defined epitopes (Supporting information, Table S3) were scored by the algorithm in order to define the parameters by which predicted peptides were to be selected. Defined epitopes had a ‘SYFPEITHI’ score ranging from 7 to 36 (mean: 23·4). To reduce the likelihood of predicted peptides being unable to bind to the relevant HLA class I molecule, we selected a cut‐off value that excluded the bottom 20th centile of the defined epitopes (‘SYFPEITHI’ score: ≥ 21). A total of 31 predicted peptides were used with HLA class I peptide binding scores ≥ 21 (range = 21–31; mean score = 26·1).
Binding of predicted HLA‐A*03‐restricted peptides was confirmed using the HLA class I stabilization assay on live T2 cells, which lack the transporter associated with antigen processing, transfected with HLA‐A*03 (Fig. 4a) 30. Using this approach, we confirmed that all 10 predicted peptides bound HLA‐A*03. As no stable HLA‐A*01‐transfected T2 cell line was available, binding was confirmed for all predicted HLA‐A*01‐restricted peptides using in‐vitro HLA‐A*0101‐peptide refolding and thermal stability CD analysis. The maximum mAU at 280 nm for each 1l HLA‐A*0101 refold is shown in Fig. 4b. For all peptides, the purity of the peak containing properly refolded HLA‐A*0101‐peptide was confirmed by SDS gel electrophoresis. All the peptides, with the exception of the negative control, generated detectable amounts of conformationally intact HLA‐A*0101. Melting temperatures (TM) ranged between 40°C and 72°C (Fig. 4c), indicating that all the HLA‐A*0101‐peptide variants were stable at or above body temperature. Values for van't Hoff's enthalpy of unfolding ranged from −110 to −700 KJ mol−1, with more negative values indicating a higher ratio of folded to misfolded peptide. Consistent with the indications from the refolding data, the positive control peptides and LMP2AESE were the most stable complexes overall (i.e. highest overall TM range and most negative van't Hoff's enthalpy of unfolding values). Peptides LMP2ALTE and LMP1LLA were also towards the high end of the stability range. Although the other peptides tested demonstrated a range of different stabilities, all the HLA‐A*0101‐peptide variants generated a CD signal consistent with the presence of properly refolded HLA class I. The predicted HLA‐B*37‐restricted peptides are identified in Supporting information, Table S3 and Fig. 5, but HLA class I binding was not tested directly, as neither the in‐vitro HLA‐B*37‐peptide refolding system nor a stable HLA‐B*37‐transfected T2 cell line was available.
Figure 4.
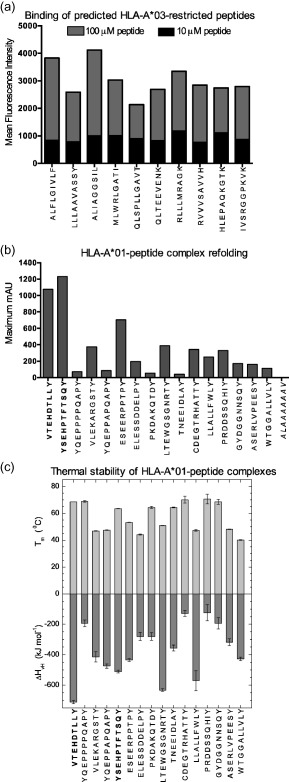
Human leucocyte antigen (HLA) class I peptide binding assays for algorithm‐predicted peptides. (a) HLA‐A*03 binding of predicted peptides was confirmed using a T2 cell line transfected with HLA‐A*03. Peptides were added at concentrations of 10 μM or 100 μM, and HLA class I expression was measured by flow cytometry analysis using an αHLA‐A*03 antibody. Data are shown as the mean fluorescence intensity above the negative control. (b) HLA‐A*01 binding of predicted peptides was confirmed by in‐vitro refolding. Maximum mAU values are shown. Positive control peptides are in shown in bold type. The negative control peptide is shown in italics. (c) Thermal stability with respect to melting temperature (upper panel) and van't Hoff's enthalpy of unfolding (lower panel). Error bars represent standard error of the respective parameter based on fitting each set of measured data. Positive control peptides are shown in bold type.
Figure 5.
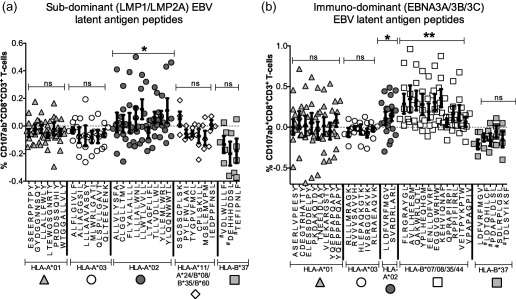
Hierarchy of ex‐vivo human leucocyte antigen (HLA) class I‐restricted CD8+ T cell responses to known and predicted peptides derived from latent membrane protein (LMP)1/LMP2A and EBNA3A/3B/3C. The ex‐vivo CD107ab+CD8+ T cell responses to individual peptides are shown. Complete epitope sequences are listed together with the restricting allotypes; further details are shown in Supporting information, Table S3. Error bars represent standard error of the mean. #Predicted HLA‐B*37‐restricted peptides with unconfirmed binding. (a) Subdominant LMP1/LMP2A‐derived epitope‐specific responses. (b) Immunodominant EBNA3A/3B/3C‐derived epitope‐specific responses. The P‐values indicate significance of combined peptide responses above zero: **P < 0·01; *P < 0·05; P > 0·05 = not significant.
LMP1/LMP2A‐specific CD8+ T cell responses are confined to HLA‐A*02
The defined and predicted peptide epitopes listed in Supporting information, Table S3 were used to investigate the HLA class I hierarchy of ex‐vivo effector CD8+ T cell responses to immunodominant and subdominant EBV latency proteins. Blood samples were obtained for this purpose from 30 healthy participants (mean age = 44 years; female/male ratio: 15 : 15). CD107 was selected as the single functional readout, as it was the only functional marker to reach significance in the previous ex‐vivo assays reported above. As we were testing for individual and not pooled (total) peptide responses, CD107b was combined with CD107a to enhance sensitivity.
Initially, we compared CD107ab+CD8+ T cell responses against 30 subdominant LMP1/LMP2A peptides (15 predicted and 15 defined, presented by nine different HLA class I allotypes; Fig. 5a). The defined epitopes were presented by HLA‐A*02, A*11, A*24, B*08, B*35 or B*60. Each of these alleles is present at a carrier frequency of > 10% in the Australian population. The predicted peptides were presented by HLA‐A*01, A*03 or B*37. The rationale for focusing on these three HLA class I alleles has already been outlined. Within the Australian population, the total frequencies of HLA‐A and HLA‐B allotypes that restrict these peptides (defined and predicted) are ∼75 and ∼33%, respectively.
We were unable to detect ex‐vivo responses significantly above zero for any of the individual peptides, regardless of HLA class I restriction (Fig. 5a). However, by grouping the individual peptide responses into the total response for each HLA class I allele, we found that the HLA‐A*02‐restricted responses were significantly greater than zero (P = 0·016). This was not observed for HLA‐A*01, HLA‐A*03, HLA‐B*37 or the group of defined peptides for HLA‐A*11, A*24, B*08, B*35 and B*60. These results indicate that, despite their modesty in the ex‐vivo setting, HLA‐A*02‐restricted responses are dominant for LMP1/LMP2A.
Next, we compared ex‐vivo CD8+ T cell responses to the immunodominant EBV latency‐III proteins EBNA3A/3B/3C, which are not expressed in EBV+cHL. To keep the analysis comparable with EBV latency‐II antigens, we used 31 peptides (16 predicted and 15 defined, presented by eight different allotypes; Fig. 5b). As with LMP1/LMP2A, we included peptides presented by HLA‐A*01, A*02, A*03 and B*37, including a defined EBNA3A‐derived HLA‐A*03‐restricted epitope and a defined EBNA3C‐derived HLA‐B*37‐restricted epitope. Other defined EBNA3A/3B/3C‐derived epitopes were presented by HLA‐B*07, B*08, B*35 or B*44, which account for ∼50% of HLA‐B alleles in Australian populations.
As shown in Fig. 5b, we were able to detect responses to EBNA3A/3B/3C that were significantly greater than zero for one of two HLA‐A*02‐restricted peptides (EBNA3ASVR) and five of 11 HLA‐B*07/B*08/B*35/B*44‐restricted peptides (EBNA3AFLR, QAK, YPL, EBNA3CEGG, KEH). None of the other alleles generated responses significantly greater than zero. The total responses to peptides presented by HLA‐A*02 and HLA‐B*07/B*08/B*35/B*44 were both significantly greater than zero (P = 0·0126 and P < 0·0001, respectively). The magnitudes of the HLA‐B*07/B*08/B*35/B*44‐restricted EBNA3A/3B/3C‐specific responses were significantly greater than the HLA‐A*02‐restricted EBV latency‐II responses (P = 0·0008).
In summary, we were able to detect ex‐vivo responses to EBNA3A/3B/3C‐derived peptides restricted by several HLA class I allotypes. By contrast, responses to EBV latency‐II peptides were detected only in the context of HLA‐A*02. Thus, the immunodominant EBV latency‐III proteins generate strong CD8+ T cell responses through a broad range of HLA class I allotypes, whereas the subdominant proteins expressed in EBV+cHL generate modest responses through a single common allotype, namely HLA‐A*02.
Discussion
In this study, we investigated the immunopathology underpinning the decreased frequency of HLA‐A*02 carriers observed in EBV+cHL. The results indicate that this is not a surrogate association, but rather reflects the impact of HLA class I on the established hierarchy of CD8+ T cell responses against EBV latency‐II antigens. To reach these conclusions, we used a series of distinct but complementary approaches.
Our initial investigation assessed relevant gene expression in the diseased tissues of cHL patients. Intriguingly, we observed a strong correlation between LMP2A and CD8 in diagnostic EBV+cHL lymph nodes. Importantly, for a given level of LMP2A, intratumoral CD8 expression was higher in HLA‐A*02‐ve compared to HLA‐A*02+ EBV+cHL patients. These data imply that CD8+ T cell anti‐tumoral immunity is relatively ineffective in HLA‐A*02– EBV+cHL. This is consistent with a study that found intratumoral LMP2A‐specific CD8+ T cells difficult to detect in EBV+cHL 40.
To test if the presence of HLA‐A*02 impacts anti‐tumoral immunity in EBV+cHL, we assessed ex‐vivo responses to relevant EBV latency antigens in pretherapy cHL patients. In order to obtain an unbiased and more comprehensive assessment of CD8+ T cell immunity with high sensitivity and specificity, we used overlapping EBNA1/LMP1/LMP2A peptide pools and measured both cytokine production (IFN‐γ and TNF‐α) and degranulation (CD107a) 35, 36. Even a modest cohort size was sufficient to observe significantly reduced LMP1/LMP2A antigen‐specific CD8+ T cell immunity in newly diagnosed HLA‐A*02– versus HLA‐A*02+ EBV+cHL patients. No differences between HLA‐A*02+ and HLA‐A*02– immune responses were observed in either EBV–cHL patients or healthy participants. These findings suggest that diminished immune‐surveillance as a result of HLA class I hierarchies may be pertinent only to the pathogenesis of EBV+cHL. Although this approach enabled an unbiased evaluation of relevant anti‐tumoral immunity in EBV+cHL, ex‐vivo assays are less sensitive than in‐vitro expansion for the detection of low‐frequency EBV‐specific CD8+ T cells 43. Furthermore, these results were confounded by the presence of additional HLA‐A/B molecules potentially masking single allele‐based effects.
To overcome these limitations and specifically test the impact of individual HLA class I alleles on relevant EBV‐specific CD8+ T cell immunodominance hierarchies, peptide‐pulsed single HLA class I‐restricted cell lines were used to expand LMP2A‐specific CD8+ T cells in vitro from EBV‐seropositive healthy HLA‐A*02 heterozygotes. In‐vitro expansion would permit more sensitive discrimination of the impact of HLA class I status on CD8+ T cell immunodominance hierarchies than ex‐vivo analysis. IFN‐γ‐specific CD8+ T cells inhibit in‐vitro transformation of EBV‐infected B cells 47, 48. The cytokine exerts its effects via the canonical JAK–STAT pathway that is known to be over‐expressed in HRS cells 49, and therefore the IFN‐γ‐specific CD8+ T cell response is likely to be important in the pathogenesis of EBV+cHL. Thus IFN‐γ production was chosen as a highly relevant in‐vitro measure of effector function. Strikingly, approximately 90% of all HLA class I‐restricted LMP2A‐specific IFN‐γ‐producing CD8+ T cell responses generated were elicited through HLA‐A*02 compared to the A*01, A*03 and B*08 allotypes. Previous work using these cell lines assessed the response to endogenously processed EBV latency‐III antigens. In contrast, our novel approach enabled us to narrow our assessment to the EBV+cHL‐relevant protein LMP2A 25. From our ex‐vivo analysis (and consistent with previous work in healthy EBV‐seropositive individuals) 12, LMP2A‐specific IFN‐γ+ effector CD8+ T cell responses were modest, but none the less higher than for LMP1 or EBNA1. These data implicate LMP2A as a critical target for effector CD8+ T cells in the immunopathogenesis of EBV+cHL. This is consistent with the correlation between LMP2A, but not LMP1 or EBNA1, and intratumoral CD8 expression, with data showing that LMP1 and EBNA1 encode for peptides that stimulate IL‐10‐secreting regulatory T cells 38, 39, and with the observation that EBNA1 predominantly (but not exclusively) generates a CD4+ effector T cell response 12.
To substantiate the emerging hypothesis, we tested the influence of HLA class I on the hierarchy of CD8+ T cell responses across the spectrum of EBV latency antigens in healthy seropositive donors. The immunodominant (EBNA3A/3B/3C) and subdominant (LMP1/LMP2A) viral antigens were compared. To enable a comprehensive evaluation, in addition to functionally defined epitopes, we adopted computational analysis to predict novel HLA class I‐restricted epitopes. Predicted epitopes were selected using stringent criteria. The prerequisite for a predicted peptide to be a potentially immunogenic CD8+ T cell epitope is an ability to bind to an HLA class I molecule (although the magnitude of binding is not in itself an accurate forecast of immunogenicity) 54. Extensive assays were used to confirm HLA class I binding in all cases, with the exception of HLA‐B*37‐restricted LMP1/LMP2A peptides for which validated reagents were lacking. Equal numbers of defined and predicted peptides with equivalent HLA class I coverage for the immunodominant and subdominant EBV latency proteins were tested in a large number of healthy EBV‐seropositive donors. Strikingly, EBNA3A/3B/3C‐specific CD8+ T cell responses were stimulated by peptides presented by numerous HLA class I allotypes. By contrast, the relatively modest EBV latency‐II‐specific CD8+ T cell responses were confined largely to HLA‐A*02.
These findings indicate that HLA class I impacts EBV latency‐II antigen‐specific CD8+ T cell response hierarchies and provide a mechanistic basis for the immunopathogenesis of EBV+cHL. In EBV+ HRS cells, antigen presentation is intact and restoration of relevant EBV‐specific T cell immunity can induce EBV+cHL regression 8, 9, 10, 11. Indeed, HLA class I expression is more evident in EBV+cHL than EBV–cHL 7, 16. The lack of selection pressure to down‐regulate HLA class I in virus‐associated cHL indicates that this strategy is not required to evade tumour‐associated CD8+ T cell immunity. These results provide a mechanism by which HRS cells can avoid CD8+ T cell‐mediated immune attack despite intact antigen presentation.
The factors that determine CD8+ T cell immunodominance have largely been investigated in animal models and include antigen abundance, temporal antigen presentation, CD8+ T cell precursor frequency, the priming environment and T cell receptor (TCR)/peptide‐HLA class I binding affinity 55. Here, we demonstrate that HLA class I restrictions influence established EBV latency antigen‐specific CD8+ T cell hierarchies. One explanation for this observation might be preferential handling of EBV latency‐II antigens by the HLA‐A*02 processing pathway. Notably, for human cytomegalovirus, strong viral epitope‐specific responses restricted by HLA‐A*02 and HLA‐A*01 have been identified 56, suggesting that the influence of HLA class I on CD8+ T cell immunodominance may be virus‐dependent. Further studies are required to address the issue.
A limitation of this study is that the findings do not discriminate between reduced immune reactivity being due to diminished antigen recognition of non‐HLA‐A*02 latency‐II antigens, or alternatively an anergic phenotype or a reduced frequency of memory T cell precursors. After acute EBV infection, a large CD8+ T cell response is elicited which contracts subsequently into a smaller memory pool 57, 58, 59. Interestingly, it is known that at least one HLA‐A*02‐restricted lytic epitope reactivity present in the acute phase consistently disappears once infection resolves 57. Although this phenomenon has not been observed for latent epitope reactivity, we cannot rule out definitively that non‐HLA‐A*02 latency‐II antigens induced a T cell response in the acute phase, which was subsequently extinguished in the memory phase. Alternatively, non‐HLA‐A*02 latency‐II antigen‐specific CD8+ T cells may be relatively enriched in inhibitory receptors such as programmed cell death protein 1 (PD‐1), relative to HLA‐A*02 latency‐II antigen‐specific CD8+ T cells. Although there are major histocompatibility complex (MHC) class I multimers for HLA‐A*02 latency‐II antigen‐specific CD8+ T cells, the lack of defined epitopes means there are no such reagents to directly visualize and phenotype non‐HLA‐A*02 latency‐II antigen‐specific CD8+ T cells.
Population‐based studies have demonstrated a positive association between infectious mononucleosis and cHL 60. However, this does not necessarily implicate EBV in the aetiology of cHL. It may, instead, reflect a predisposition to a particular clinical response to primary EBV infection, perhaps as a consequence of a predating immune impairment. In addition, transformation of EBV latency‐II‐expressing benign B cells may represent an initiating event in the pathogenesis of EBV+cHL. EBV‐infected germinal centre B cells have been shown to express the EBV latency‐II pattern 61, 62, identical to that seen in the HRS cells of EBV+cHL, which are known to have an atypical germinal centre derivation 63. In this situation, differential EBV latency‐II‐specific effector CD8+ T cell immune surveillance might contribute to the pathogenesis of EBV+cHL by attenuating immune‐mediated destruction of premalignant B cells. Using functional assays that removed the confounding influence of co‐expressed HLA alleles, we confirmed that HLA class I status indeed conferred differential levels of EBV latency‐II‐specific effector CD8+ T cell immunity in healthy seropositive participants. Here, those peptide pools (4–6) that contained HLA‐A*02 restricted defined LMP2A epitopes contributed > 80% of the HLA‐A*02 CD8+ T cell response, whereas the remaining pools (1–3) provided < 20%. However, reduced ex‐vivo HLA‐A*02– global (i.e. mediated through all co‐expressed HLA‐alleles) LMP1/LMP2A‐specific effector CD8+ T cell responses were observed only in EBV+cHL patients. These results suggest that EBV latency‐II‐specific effector CD8+ T cell immune surveillance is particularly relevant to the pathogenesis of EBV+cHL. Further investigations into associations with disease outcome may be informative; however, given the high response rate in cHL very large numbers of uniformly treated patients would be required to be sufficiently powered.
Collectively, our data provide new insights into the immunopathogenesis of EBV+cHL and suggest that even modest CD8+ T cell responses directed against tumour‐associated viral proteins may reduce the incidence of malignant disease. Further studies are required to determine if similar mechanisms are applicable to other malignancies with EBV latency‐II expression patterns, such as extranodal natural killer (NK)/T cell lymphoma and undifferentiated nasopharyngeal carcinoma 64, 65.
Disclosure
The authors declare no disclosures.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Flow cytometry analysis gating strategy.
Fig. S2. Ex‐vivo Epstein–Barr virus (EBV)‐specific CD8+ T cell responses in healthy controls and comparison between responses against EBV latency‐II proteins.
Fig. S3. In‐vitro latent membrane protein (LMP) 2A‐specific CD8+ T cell cytotoxicity compared between HLA‐A*02 and non‐human leucocyte antigen (HLA)‐A*02 healthy control participants.
Table S1. Previously published human leucocyte antigen (HLA) class I associations with Epstein–Barr virus (EBV)+ classical Hodgkin lymphoma (cHL).
Table S2. Latent membrane protein (LMP)2A overlapping peptide pools.
Table S3. Predicted and defined peptide epitopes from Epstein–Barr virus (EBV) latent proteins.
Acknowledgements
This study was conducted under the auspices of the Australasian Leukaemia and Lymphoma Group. K. J. and C. K. were supported by the Leukaemia Foundation (Australia). M. K. G. is supported by the Leukaemia Foundation of Queensland. D. A. P. is a Wellcome Trust Senior Investigator. S. R. B is an NHMRC Principal Research Fellow (APP1021452).
References
- 1. Young L, Alfieri C, Hennessy K et al Expression of Epstein–Barr virus transformation‐associated genes in tissues of patients with EBV lymphoproliferative disease. N Engl J Med 1989; 321:1080–5. [DOI] [PubMed] [Google Scholar]
- 2. Deacon EM, Pallesen G, Niedobitek G et al Epstein–Barr virus and Hodgkin's disease: transcriptional analysis of virus latency in the malignant cells. J Exp Med 1993; 177:339–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gandhi MK, Tellam JT, Khanna R. Epstein–Barr virus‐associated Hodgkin's lymphoma. Br J Haematol 2004; 125:267–81. [DOI] [PubMed] [Google Scholar]
- 4. Young LS, Rickinson AB. Epstein–Barr virus: 40 years on. Nat Rev Cancer 2004; 4:757–68. [DOI] [PubMed] [Google Scholar]
- 5. Murray PG, Constandinou CM, Crocker J, Young LS, Ambinder RF. Analysis of major histocompatibility complex class I, TAP expression, and LMP2 epitope sequence in Epstein–Barr virus‐positive Hodgkin's disease. Blood 1998; 92:2477–83. [PubMed] [Google Scholar]
- 6. Lee SP, Constandinou CM, Thomas WA et al Antigen presenting phenotype of Hodgkin Reed–Sternberg cells: analysis of the HLA class I processing pathway and the effects of interleukin‐10 on Epstein–Barr virus‐specific cytotoxic T‐cell recognition. Blood 1998; 92:1020–30. [PubMed] [Google Scholar]
- 7. Huang X, van den Berg A, Gao Z et al Expression of HLA class I and HLA class II by tumor cells in Chinese classical Hodgkin lymphoma patients. PLOS ONE 2010; 5:e10865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bollard CM, Aguilar L, Straathof KC et al Cytotoxic T lymphocyte therapy for Epstein–Barr virus+ Hodgkin's disease. J Exp Med 2004; 200:1623–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Khanna R, Moss D, Gandhi M. Technology insight: applications of emerging immunotherapeutic strategies for Epstein–Barr virus‐associated malignancies. Nat Clin Pract Oncol 2005; 2:138–49. [DOI] [PubMed] [Google Scholar]
- 10. Bollard CM, Gottschalk S, Leen AM et al Complete responses of relapsed lymphoma following genetic modification of tumor‐antigen presenting cells and T‐lymphocyte transfer. Blood 2007; 110:2838–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bollard CM, Gottschalk S, Torrano V et al Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein–Barr virus latent membrane proteins. J Clin Oncol 2014; 32:798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hislop AD, Taylor GS, Sauce D, Rickinson AB. Cellular responses to viral infection in humans: lessons from Epstein–Barr virus. Annu Rev Immunol 2007; 25:587–617. [DOI] [PubMed] [Google Scholar]
- 13. Jones K, Nourse JP, Morrison L et al Expansion of EBNA1‐specific effector T cells in posttransplantation lymphoproliferative disorders. Blood 2010; 116:2245–52. [DOI] [PubMed] [Google Scholar]
- 14. Urayama KY, Jarrett RF, Hjalgrim H et al Genome‐wide association study of classical Hodgkin lymphoma and Epstein–Barr virus status‐defined subgroups. J Natl Cancer Inst 2012; 104:240–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hjalgrim H, Rostgaard K, Johnson PC et al HLA‐A alleles and infectious mononucleosis suggest a critical role for cytotoxic T‐cell response in EBV‐related Hodgkin lymphoma. Proc Natl Acad Sci USA 2010; 107:6400–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Niens M, Jarrett RF, Hepkema B et al HLA‐A*02 is associated with a reduced risk and HLA‐A*01 with an increased risk of developing EBV+ Hodgkin lymphoma. Blood 2007; 110:3310–5. [DOI] [PubMed] [Google Scholar]
- 17. Huang X, Kushekhar K, Nolte I et al HLA associations in classical Hodgkin lymphoma: EBV status matters. PLOS ONE 2012; 7:e39986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee SP, Tierney RJ, Thomas WA, Brooks JM, Rickinson AB. Conserved CTL epitopes within EBV latent membrane protein 2: a potential target for CTL‐based tumor therapy. J Immunol 1997; 158:3325–34. [PubMed] [Google Scholar]
- 19. Huang X, Hepkema B, Nolte I et al HLA‐A*02:07 is a protective allele for EBV negative and a susceptibility allele for EBV positive classical Hodgkin lymphoma in China. PLOS ONE 2012; 7:e31865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Enciso‐Mora V, Broderick P, Ma Y et al A genome‐wide association study of Hodgkin's lymphoma identifies new susceptibility loci at 2p16.1 (REL), 8q24.21 and 10p14 (GATA3). Nat Genet 2010; 42:1126–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cozen W, Timofeeva MN, Li D et al A meta‐analysis of Hodgkin lymphoma reveals 19p13.3 TCF3 as a novel susceptibility locus. Nat Commun 2014; 5:3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gandhi MK, Lambley E, Burrows J et al Plasma Epstein–Barr virus (EBV) DNA is a biomarker for EBV‐positive Hodgkin's lymphoma. Clin Cancer Res 2006; 12:460–4. [DOI] [PubMed] [Google Scholar]
- 23. Freel SA, Lamoreaux L, Chattopadhyay PK et al Phenotypic and functional profile of HIV‐inhibitory CD8 T cells elicited by natural infection and heterologous prime/boost vaccination. J Virol 2010; 84:4998–5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mahnke YD, Roederer M. OMIP‐001: quality and phenotype of Ag‐responsive human T‐cells. Cytometry A 2010; 77:819–820. [DOI] [PubMed] [Google Scholar]
- 25. Brennan RM, Burrows SR. A mechanism for the HLA‐A*01‐associated risk for EBV+ Hodgkin lymphoma and infectious mononucleosis. Blood 2008; 112:2589–90. [DOI] [PubMed] [Google Scholar]
- 26. Gandhi MK, Wilkie GM, Dua U et al Immunity, homing and efficacy of allogeneic adoptive immunotherapy for posttransplant lymphoproliferative disorders. Am J Transplant 2007; 7:1293–9. [DOI] [PubMed] [Google Scholar]
- 27. Vickers MA, Wilkie GM, Robinson N et al Establishment and operation of a Good Manufacturing practice‐compliant allogeneic Epstein–Barr virus (EBV)‐specific cytotoxic cell bank for the treatment of EBV‐associated lymphoproliferative disease. Br J Haematol 2014; 167:402–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Witt C, Sayer D, Christiansen F. HLA‐A, ‐B and ‐DRB1 allele frequencies in a population from Western Australia. Hum Immunol 2004; 861–2. [Google Scholar]
- 29. Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 1999; 50:213–9. [DOI] [PubMed] [Google Scholar]
- 30. Bell MJ, Burrows JM, Brennan R et al The peptide length specificity of some HLA class I alleles is very broad and includes peptides of up to 25 amino acids in length. Mol Immunol 2009; 46:1911–7. [DOI] [PubMed] [Google Scholar]
- 31. Garboczi DN, Hung DT, Wiley DC. HLA‐A2‐peptide complexes: refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc Natl Acad Sci USA 1992; 89:3429–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brooimans RA, Boyce CS, Popma J et al Analytical performance of a standardized single‐platform MHC tetramer assay for the identification and enumeration of CMV‐specific CD8+ T lymphocytes. Cytometry A 2008; 73:992–1000. [DOI] [PubMed] [Google Scholar]
- 33. Greenfield NJ. Analysis of circular dichroism data. Methods Enzymol 2004; 383:282–317. [DOI] [PubMed] [Google Scholar]
- 34. Venyaminov S, Baikalov IA, Shen ZM, Wu CS, Yang JT. Circular dichroic analysis of denatured proteins: inclusion of denatured proteins in the reference set. Anal Biochem 1993; 214:17–24. [DOI] [PubMed] [Google Scholar]
- 35. McCulloch CE, Searle SR. Generalized, linear, and mixed models. New York: Wiley, 2001. [Google Scholar]
- 36. Roederer M, Nozzi JL, Nason MC. SPICE: exploration and analysis of post‐cytometric complex multivariate datasets. Cytometry A 2011; 79:167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Geiss GK, Bumgarner RE, Birditt B et al Direct multiplexed measurement of gene expression with color‐coded probe pairs. Nat Biotechnol 2008; 26:317–25. [DOI] [PubMed] [Google Scholar]
- 38. Voo KS, Peng G, Guo Z et al Functional characterization of EBV‐encoded nuclear antigen 1‐specific CD4+ helper and regulatory T cells elicited by in vitro peptide stimulation. Cancer Res 2005; 65:1577–86. [DOI] [PubMed] [Google Scholar]
- 39. Marshall NA, Vickers MA, Barker RN. Regulatory T cells secreting IL‐10 dominate the immune response to EBV latent membrane protein 1. J Immunol 2003; 170:6183–9. [DOI] [PubMed] [Google Scholar]
- 40. Chapman AL, Rickinson AB, Thomas WA, Jarrett RF, Crocker J, Lee SP. Epstein–Barr virus‐specific cytotoxic T lymphocyte responses in the blood and tumor site of Hodgkin's disease patients: implications for a T‐cell‐based therapy. Cancer Res 2001; 61:6219–26. [PubMed] [Google Scholar]
- 41. Gandhi MK, Lambley E, Duraiswamy J et al Expression of LAG‐3 by tumor‐infiltrating lymphocytes is coincident with the suppression of latent membrane antigen‐specific CD8+ T‐cell function in Hodgkin lymphoma patients. Blood 2006; 108:2280–9. [DOI] [PubMed] [Google Scholar]
- 42. Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin's disease. International prognostic factors project on advanced Hodgkin's disease. N Engl J Med 1998; 339:1506–14. [DOI] [PubMed] [Google Scholar]
- 43. Moor RJ, Morrison LE, Moss DJ, Tscharke DC. Use of CD107‐based cell sorting ex vivo to enrich subdominant CD8+ T cells in culture. Immunol Cell Biol 2007; 85:546–50. [DOI] [PubMed] [Google Scholar]
- 44. Evseeva I, Nicodemus KK, Bonilla C, Tonks S, Bodmer WF. Linkage disequilibrium and age of HLA region SNPs in relation to classic HLA gene alleles within Europe. Eur J Hum Genet 2010; 18:924–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tsang ML, Munz C. Cytolytic T lymphocytes from HLA‐B8+ donors frequently recognize the Hodgkin's lymphoma associated latent membrane protein 2 of Epstein Barr virus. Herpesviridae 2011; 2:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Middleton D, Menchaca L, Rood H, Komerofsky R. New allele frequency database: http://www.allelefrequencies.net. Tissue Antigens 2003; 61:403–7. [DOI] [PubMed] [Google Scholar]
- 47. Lotz M, Tsoukas CD, Fong S, Carson DA, Vaughan JH. Regulation of Epstein‐Barr virus infection by recombinant interferons. Selected sensitivity to interferon‐gamma. Eur J Immunol 1985; 15:520–5. [DOI] [PubMed] [Google Scholar]
- 48. Shi Y, Lutz CT. Interferon–gamma control of EBV‐transformed B cells: a role for CD8+ T cells that poorly kill EBV‐infected cells. Viral Immunol 2002; 15:213–25. [DOI] [PubMed] [Google Scholar]
- 49. Ikeda H, Old LJ, Schreiber RD. The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev 2002; 13:95–109. [DOI] [PubMed] [Google Scholar]
- 50. Rickinson AB, Moss DJ. Human cytotoxic T lymphocyte responses to Epstein–Barr virus infection. Annu Rev Immunol 1997; 15:405–31. [DOI] [PubMed] [Google Scholar]
- 51. Meij P, Leen A, Rickinson AB et al Identification and prevalence of CD8(+) T‐cell responses directed against Epstein–Barr virus‐encoded latent membrane protein 1 and latent membrane protein 2. Int J Cancer 2002; 99:93–9. [DOI] [PubMed] [Google Scholar]
- 52. Edwards RH, Sitki‐Green D, Moore DT, Raab‐Traub N. Potential selection of LMP1 variants in nasopharyngeal carcinoma. J Virol 2004; 78:868–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Subklewe M, Marquis R, Choquet S et al Association of human leukocyte antigen haplotypes with posttransplant lymphoproliferative disease after solid organ transplantation. Transplantation 2006; 82:1093–100. [DOI] [PubMed] [Google Scholar]
- 54. Bihl F, Frahm N, Di Giammarino L et al Impact of HLA‐B alleles, epitope binding affinity, functional avidity, and viral coinfection on the immunodominance of virus‐specific CTL responses. J Immunol 2006; 176:4094–101. [DOI] [PubMed] [Google Scholar]
- 55. Yewdell JW, Bennink JR. Immunodominance in major histocompatibility complex class I‐restricted T lymphocyte responses. Annu Rev Immunol 1999; 17:51–88. [DOI] [PubMed] [Google Scholar]
- 56. Gandhi MK, Khanna R. Human cytomegalovirus: clinical aspects, immune regulation, and emerging treatments. Lancet Infect Dis 2004; 4:725–38. [DOI] [PubMed] [Google Scholar]
- 57. Hislop AD, Annels NE, Gudgeon NH, Leese AM, Rickinson AB. Epitope‐specific evolution of human CD8(+) T cell responses from primary to persistent phases of Epstein–Barr virus infection. J Exp Med 2002; 195:893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Woodberry T, Suscovich TJ, Henry LM et al Differential targeting and shifts in the immunodominance of Epstein–Barr virus‐specific CD8 and CD4 T cell responses during acute and persistent infection. J Infect Dis 2005; 192:1513–24. [DOI] [PubMed] [Google Scholar]
- 59. Callan MF, Fazou C, Yang H et al CD8(+) T‐cell selection, function, and death in the primary immune response in vivo . J Clin Invest 2000; 106:1251–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hjalgrim H, Askling J, Rostgaard K et al Characteristics of Hodgkin's lymphoma after infectious mononucleosis. N Engl J Med 2003; 349:1324–32. [DOI] [PubMed] [Google Scholar]
- 61. Thorley‐Lawson DA, Gross A. Persistence of the Epstein–Barr virus and the origins of associated lymphomas. N Engl J Med 2004; 350:1328–37. [DOI] [PubMed] [Google Scholar]
- 62. Babcock GJ, Hochberg D, Thorley‐Lawson AD. The expression pattern of Epstein–Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 2000; 13:497–506. [DOI] [PubMed] [Google Scholar]
- 63. Kuppers R. B cells under influence: transformation of B cells by Epstein–Barr virus. Nat Rev Immunol 2003; 3:801–12. [DOI] [PubMed] [Google Scholar]
- 64. Tang M, Lautenberger JA, Gao X et al The principal genetic determinants for nasopharyngeal carcinoma in China involve the HLA class I antigen recognition groove. PLOS Genet 2012; 8:e1003103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li X, Fasano R, Wang E, Yao KT, Marincola FM. HLA associations with nasopharyngeal carcinoma. Curr Mol Med 2009; 9:751–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Flow cytometry analysis gating strategy.
Fig. S2. Ex‐vivo Epstein–Barr virus (EBV)‐specific CD8+ T cell responses in healthy controls and comparison between responses against EBV latency‐II proteins.
Fig. S3. In‐vitro latent membrane protein (LMP) 2A‐specific CD8+ T cell cytotoxicity compared between HLA‐A*02 and non‐human leucocyte antigen (HLA)‐A*02 healthy control participants.
Table S1. Previously published human leucocyte antigen (HLA) class I associations with Epstein–Barr virus (EBV)+ classical Hodgkin lymphoma (cHL).
Table S2. Latent membrane protein (LMP)2A overlapping peptide pools.
Table S3. Predicted and defined peptide epitopes from Epstein–Barr virus (EBV) latent proteins.