

Summary
Membranous nephropathy (MN), the leading cause of nephrotic syndrome in adults, is characterized by the deposition of subepithelial immune deposits that consist mainly of immunoglobulin (Ig)G and complement. Most of the cases are primary or idiopathic (iMN), while only approximately 25% of the cases are secondary to some known disease such as systemic lupus erythematosus, hepatitis B, drugs and malignancies. Most of our knowledge on the pathogenesis of iMN has relied upon old experimental models (i.e. Heymann nephritis) that have shown that immune deposits are formed in situ by the reaction of autoantibodies against the respective podocyte antigen. Recent findings indicate that podocyte proteins also act as an autoantigen in human iMN. The M‐type phospholipase A2 receptor (PLA2R) has been identified as the main target antigen, as it can be found in approximately 70% of iMN patients but only rarely in other glomerulonephritides. Podocytes damage in the experimental model of Heymann nephritis is complement‐mediated. In humans, the presence of complement within the subepithelial deposits is well established, but IgG4, which does not activate complement by classical or alternative pathways, represents the predominant subclass of IgG anti‐PLA2R. Some evidence suggests that IgG4 anti‐PLA2R autoantibodies can bind mannan‐binding lectin (MBL) and activate the lectin complement pathway. A genetic background for iMN has been demonstrated by genome‐wide association studies that have shown highly significant associations of the PLA2R1 and the human leucocyte antigen (HLA)‐DQA1 loci with iMN. In addition to their diagnostic value, anti‐PLA2R antibodies may be useful to monitor disease activity and predict response to treatment.
Keywords: anti‐PLA2R antibody, membranous nephropathy, podocyte, subepithelial deposits IgG4
Introduction
Definition and epidemiology
Membranous nephropathy (MN) is a glomerular disease defined histopathologically by the presence of diffuse thickening of the glomerular capillary wall on light microscopy as a result of an immune complex deposition on the extracapillary side of glomerular basement membrane (GBM). The immune deposits contain the complement fraction C3 and immunoglobulin (Ig)G (mainly IgG4 in idiopathic form) in a peripheral capillary loop pattern, as revealed by immunofluorescence (IF), and appear as electron‐dense subepithelial deposits on electron microscopy (EM).
iMN is considered an organ‐specific autoimmune disease mediated by autoantibodies, which co‐localize with the target antigens to form subepithelial immune complexes (IC), leading to complement activation and the development of proteinuria as a result of podocyte damage.
MN is characterized clinically by non‐selective proteinuria, usually in the nephrotic range (>3·5 g/die), and by a possible progression to end‐stage renal disease (ESRD).
Idiopathic MN (iMN) accounts for approximately 30% of cases of nephrotic syndrome in the Caucasian adult population (European annual incidence, 1·7 per 100 000), and presents a 2 : 1 male‐to‐female predominance and a median age of onset from the fourth to the sixth decades 1, 2, but the disease can affect patients of all ages and ethnic groups. It is one of the main causes of ESRD among primary glomerulonephritis, and 2% of kidney transplant recipients are affected by iMN as primary renal disease 3.
In developed countries, approximately 75% of all MN are idiopathic (or primary); the remaining 20–25% are secondary to different conditions, such as infections (hepatitis B virus, hepatitis C virus, human immunodeficiency virus, malaria, etc.), malignancies (lung, breast, stomach and ovarian cancer, lymphoproliferative disorder, etc.), systemic autoimmune disease [lupus, rheumatoid arthritis (RA), etc.] and drugs (Table 1).
Table 1.
Conditions and drugs associated with membranous nephropathy (secondary forms).
| Common | Uncommon | |
|---|---|---|
| Immune diseases | Systemic lupus erythematosus | Rheumatoid arthritis, Hashimoto thyroiditis, Sjögren's syndrome, psoriasis, sarcoidosis, mixed connective tissue disease, IgG4‐related disease |
| Infections | Hepatitis B virus | Hepatitis C virus, streptococcal infection, malaria, Schistosomiasis, syphilis, leprosy |
| Drugs and toxins | Non‐steroidal anti‐inflammatory drugs (NSAIDs), gold, penicillamine | Captopril, clopidogrel, mercury |
| Tumours | Cancers (bladder, breast, pancreas, prostate, stomach, lung) | Lymphomas, chronic lymphocytic leukaemia |
| Miscellaneous | Diabetes mellitus, renal transplantation | Sickle cell disease, haematopoietic stem cells transplantation |
Pathological and clinical features
Diagnosis of both primary and secondary membranous nephropathy relies on renal biopsy, in particular on the finding of deposits of IgG in a granular pattern along the glomerular capillary loop by IF (Fig. 1), particularly in the earliest stage (I) in which the glomeruli may appear normal on light microscopy 4.
Figure 1.
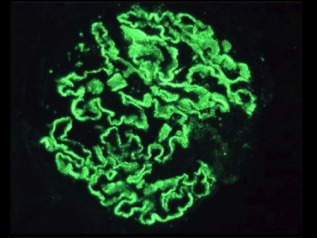
Granular subepithelial deposits of immunoglobulin (Ig)G in a case of idiopathic membranous nephropathy (original magnification ×400).
In the next stages (II–IV) there is a progressive homogeneous thickening of the capillary wall, with the formation of projections of the GBM between deposits called ‘spikes’, until the last stage (V) in which the deposits are not recognizable as they are completely incorporated into the GBM 5.
Some histological features may help in discrimination between the two forms of MN (Table 2).
Table 2.
Distinctive histopathological features of secondary forms of membranous nephropathy.
| Features | |
|---|---|
| Light microscopy | Significant mesangial proliferation |
| Immunofluorescence |
• Positive staining for IgG1 or IgG3, IgA, IgM • Positive staining for C1q and/or C4 • Mesangial immunofluorescence deposition |
| Electron microscopy | Electron‐dense deposits in mesangial and/or subendothelial sites |
Ig: immunoglobulin.
In primary MN, no endocapillary proliferation is usually seen, and the presence of a pronounced mesangial proliferation implies a secondary form of MN.
IgG4 is predominant for unknown reasons in idiopathic MN. A positive staining for non‐IgG4 subclasses (IgG1, IgG3), IgA, IgM or significant staining in the glomerular mesangium suggests a secondary MN, often lupus nephritis class V.
Complement component C3 is also present in approximately 50% of iMN patients, and its presence probably reflects active, ongoing immune deposit formation and complement activation 6.
C4 and C1q, expression of a complement activation through the ‘classical pathway’, are often absent in iMN and their presence is suggestive of secondary membranous nephropathy.
On electron microscopy, the hallmark lesions are subepithelial electron‐dense deposits, effacement of the foot processes and expansion of the glomerular basement membrane due to deposition of new extracellular matrix between the deposits. The electron‐dense deposits reflect the IgG and C3 staining on IF, so in idiopathic form they are not usually seen at subendothelial or mesangial level.
Despite the characteristics described, discrimination between the two forms of MN can sometimes be extremely challenging.
With disease progression, especially in severe proteinuric cases, focal glomerular segmental sclerosis and interstitial tubular damage is observed 7.
iMN is a chronic disease with a variable and unpredictable clinical outcome, which can include spontaneous remissions and relapses. Spontaneous remission occurs in up to 30–40% of cases 8; the remaining two‐thirds of the patients present with persistent proteinuria, and approximately 40% of those will progress within 10 years to ESRD 9. Consequently, treatment with potentially toxic drugs remains controversial. However, an increase of cardiovascular risk, hypercoagulability, major susceptibility to infections and progressive renal failure afflict a severe and persistent nephrotic syndrome.
The identification of reliable prognostic factors (clinical, laboratoristic and/or histological) is extremely important to guide any therapeutic decision. Classical predictors of long‐term progression are baseline proteinuria more than 8 g/day, male sex, important tubular‐interstitial damage and glomerular sclerosis at renal biopsy, age older than 50 years and altered renal function at onset 10.
Achieving complete remission predicts an excellent long‐term renal prognosis and those patients have an almost 100% renal survival at 10 years, whereas the number falls to 90% with partial remission and 45% with no remission 11.
Pathogenesis
From experimental models to identification of human antigenic targets
iMN is an excellent example of disease in which most of the knowledge on the pathophysiological mechanisms is derived from animal experimental studies; in particular, the model of Heymann nephritis 12, which allowed identification of the podocyte target antigen of the autoantibody response in rats and showed that the formation of the IC occurs in situ.
In theory, subepithelial IC deposits can form in three different ways. Antibodies can bind to exogenous antigens that localize on the subepithelial surface because of their cationic charge and small size (Fig. 2); secondly, antigens and antibodies can be trapped as immune complexes on the glomerular filtration wall (Fig. 3); and finally, the antigens could be endogenous constituents of a fixed subepithelial structure, such as a podocyte membrane protein (Fig. 4) 13.
Figure 2.
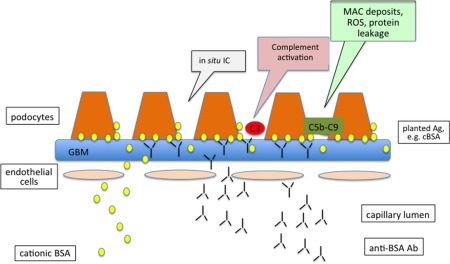
Mechanism of in‐situ subepithelial immune complex formation in early childhood idiopathic membranous nephropathy. Because of its size and charge, modified, the cationic form of bovine serum albumin (BSA) reaches anionic glomerular subepithelial structures and serves as a planted antigen with subsequent formation of immune complexes in situ. The functional impairment represented by proteinuria is the result of formation of the membrane attack complex (C5b‐C9, MAC), which leads to sublethal podocyte injury resulting in the activation of transcription factors encoding for mediators of fibrosis and cytoskeletal podocyte rearrangement. It also increases production of potentially nephritogenic molecules such as reactive oxygen species (ROS), proinflammatory cytokines, proteases and vasoactive molecules.
Figure 3.
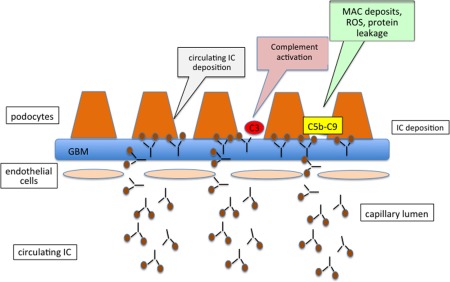
Mechanism of circulating immune complex deposition in membranous nephropathy. Preformed small‐sized circulating immune complexes may traverse the glomerular basement membrane (GBM) and deposit beneath the podocyte. The functional impairment represented by proteinuria is the result of formation of the membrane attack complex (C5b‐C9, MAC), which leads to sublethal podocyte injury resulting in the activation of transcription factors encoding for mediators of fibrosis and cytoskeletal podocyte rearrangement. It also increases production of potentially nephritogenic molecules such as reactive oxygen species (ROS), proinflammatory cytokines, proteases and vasoactive molecules.
Figure 4.
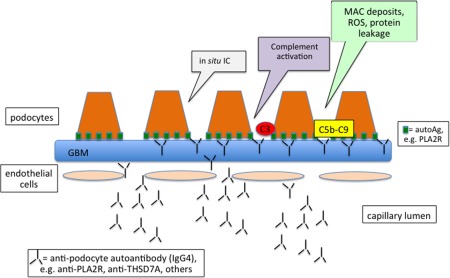
Mechanism of anti‐podocyte autoantibody‐mediated disease in membranous nephropathy. Circulating autoantibodies can target surface‐exposed intrinsic podocyte proteins to form in‐situ immune deposits. The functional impairment represented by proteinuria is the result of formation of the membrane attack complex (C5b‐C9, MAC), which leads to sublethal podocyte injury resulting in the activation of transcription factors encoding for mediators of fibrosis and cytoskeletal podocyte rearrangement. It also increases production of potentially nephritogenic molecules such as reactive oxygen species (ROS), proinflammatory cytokines, proteases and vasoactive molecules.
In the experimental rat model of Heymann nephritis, the subepithelial deposits are formed in situ when circulating antibodies (resulting from either active or passive immunization of the animal) bind an intrinsic antigen in the glomerular capillary wall. This antigen was identified as megalin, a glycoprotein of 516 kDa members of the low‐density lipoprotein (LDL) receptor family present, expressed with clathrin, on the foot process of podocytes 14, 15, 16, 17.
The binding of circulating anti‐megalin antibodies to surface megalin induces complement activation and local generation of the membrane attack complex (MAC, C5b‐C9). The immune complexes are subsequently degraded and form discontinuous subepithelial deposits.
The functional impairment represented by proteinuria is the result of the formation of MAC, which leads to sublethal podocyte injury resulting in the activation of transcription factors encoding for mediators of fibrosis and cytoskeletal podocyte rearrangement. It also increases the production of potentially nephritogenic molecules such as reactive oxygen species (ROS), proinflammatory cytokines, proteases and vasoactive molecules (Fig. 4) 13, 18.
However, megalin is absent in human glomeruli and its human counterpart, the LDL receptor, is not detected in the subepithelial immune deposits of iMN patients 19.
A fundamental step in the identification of antigens involved in human iMN was the discovery by Ronco et al., in 2002, of a human counterpart to passive Heymann nephritis in a neonate born with membranous nephropathy. Maternal antibodies to neutral endopeptidase (NEP), which crossed the placenta and bound to fetal glomerular podocytes, induced the disease antenatally 20, 21. The mother was a carrier of a homozygous deletion in the gene encoding the protein NEP and became alloimmunized to the paternally inherited neutral endopeptidase antigen expressed in the placenta.
This rare form of MN was also the first demonstration of the cardinal role of circulating antibodies against intrinsic podocyte antigens in the pathogenesis of human disease, and the detection of MAC C5b‐C9 in the subepithelial deposits confirmed that the role of complement was also central in human MN.
In 2011, Debiec et al. described another rare form of early childhood MN in which a modified, cationic form of bovine serum albumin (BSA), derived probably from dietary sources and absorbed intact by the immature intestinal tract of infants, served as a planted antigen. The size and charge of this exogenous antigen are responsible for its localization at the glomerular filtration barrier, binding to anionic glomerular structures and the subsequent formation of immune complexes in situ (Fig. 2) 22.
Both NEP and megalin were ruled out as potential antigens in adult idiopathic MN, but recent evidence indicates that the majority of patients with primary MN have circulating autoantibodies to the M‐type phospholipase A2 receptor 1 (PLA2R) 23.
The major human target in iMN: M‐type PLA2R
In 2009, Beck et al. demonstrated the presence of circulating anti‐PLA2R antibodies in 26 of 37 iMN patients, performing Western blotting under non‐reducing conditions with extracts from normal human glomeruli as a source of antigens. They observed a high‐molecular‐weight band which, after partial purification using lectin chromatography, resulted in the mass spectrometric identification of PLA2R, a protein expressed in the podocytes.
Such anti‐PLA2R antibodies were highly specific for idiopathic forms as they were not detected in sera of controls, patients affected by other primary glomerulonephritis or by secondary forms of MN. Circulating anti‐PLA2R antibodies of patients with primary iMN were predominantly, but not exclusively, IgG4. The receptor of PLA2 and IgG4 co‐localized in the immune deposits at subepithelial level, and only the IgG eluted from the biopsy samples of iMN patients reacted with the PLA2R. These results led to the confirmation that the pathogenic mechanisms underlining human iMN were similar to the one responsible of the Heymann nephritis 23.
Circulating autoantibodies to PLA2R were detected in approximately 70% of patient with iMN in all the different studies performed.
PLA2R is a transmembrane glycoprotein of highly glycosylated 185 kDa, and is a member of the mannose receptor (MR) family. All MR family members have a conserved extracellular structure, with an N‐terminal cysteine‐rich domain (Cys‐R), a fibronectin II‐like domain (FNII) and eight to 10 C‐type lectin‐like domains (CTLD) 24. The cytoplasmic domain is short and contains motifs important for the constitutive recycling of these receptors, as all members are internalized with their ligands and recirculate continuously between the membrane and the endosomal compartment. MR family members undergo conformational shifts between a more extended conformation and a compact, folded configuration that may be regulated by pH, oligomerization and/or ligand binding 25. This conformational shift is particularly important, because the target epitopes of the circulating autoantibodies appear to be accessible only in one of the two conformations.
Studies of the different reactivity of iMN patients’ sera to the native and deglycosylated forms of PLA2R show that the autoantibodies recognize a conformational epitope, and studies of high‐throughput capture immunoassay allowed the identification of putative linear epitopes 26.
In 2015, a major immunodominant epitope region was characterized in the three most N‐terminal domains of PLA2R by US 27 and British groups 28, who each used different technical approaches. Two major overlapping epitopes have been identified. The first, characterized by Western blotting of truncated extracellular domains of PLA2R under non‐reducing conditions, is a protein complex consisting of the Cys‐R domain, the FNII domain and the CTLD domain 1 of PLA2R 27. Absence of either the Cys‐R or the CTLD1 domain prevents autoantibody recognition of the remaining domains. This three‐domain complex contains at least one disulphide bond (required for conformational configuration) and completely blocks the reactivity of autoantibodies with the full length PLA2R.
The second overlapping epitope was identified in the Cys‐R domain. Two peptides from the Cys‐R domain showed strong inhibition of autoantibody binding to PLA2R by surface plasmon resonance, with a sequence covering both peptides (31‐mer) producing 85% inhibition of autoantibody binding. Anti‐PLA2R antibody directly binds this 31‐mer peptide under non‐denaturing conditions, and binding is sensitive to reduction because of an internal disulphide bond 28. The identification of these major PLA2R epitopes could enable future therapeutic strategies for iMN patients, including antibody inhibition therapy and immunoadsorption.
The physiological role of PLA2 receptor is unclear. Binding of circulating PLA2 to its receptor participates in both positive and negative regulation of PLA2 functions as well as in its clearance by internalization (endocytosis) of ligand–receptor complex. PLA2R also regulates cellular senescence through the production of ROS and the activation of the DNA‐damage pathways 29.
PLA2R is expressed highly in kidney tissue and is confined almost exclusively to the glomerular podocytes 30.
Predisposing gene variation for idiopathic MN
Apart from rare cases, MN is not a typical hereditary disease in Mendelian terms 31. However, the influence of genetic factors is well established in both rats and humans.
Genome‐wide association studies have shown highly significant associations of the 6p21 HLA‐DQA1 and 2q24 PLA2R1 loci with idiopathic membranous nephropathy in Caucasian European patients. These risk alleles of the two genes had an additive effect for development of idiopathic membranous nephropathy. Patients carrying all four risk alleles had an odds ratio close to 80, compared with individuals who had only the protective alleles 32.
The observation that anti‐PLA2R antibodies were found in 73% of patients with high‐risk genotypical variations, while they were absent in non‐carriers, supports the role of PLA2R as a principal antigenic target in iMN 33. Additional studies are needed to explain how alleles in the PLA2R1 and HLA‐DQA1 loci interact with each other to increase susceptibility to membranous nephropathy.
A possible explanation is that a particular HLA molecule may facilitate the autoimmune response against PLA2R presenting it to T helper type 2 in an aberrant or exuberant way 34.
Additional candidate antigens
Using a proteomic approach involving the use of in‐vitro human podocytes exposed to iMN patient sera, in 2012 Ghiggeri et al. identified various intracellular enzymes [superoxide dismutase 2 (SOD2) aldose reductase AR and α‐enolase] as different targets of circulating autoantibodies in MN 35. These antigens co‐localize in iMN patient biopsies with MAC C5b‐9 and IgG4, but they are not normally expressed highly in normal glomeruli and are not present on the cell surface of podocytes. It is possible that podocyte over‐expression and delocalization of SOD2, and AR may represent an anti‐oxidant response preceding the humoral immune response 35. These characteristics suggest that they are more likely to be neo‐antigens exposed aberrantly on cellular surface after the initial podocyte damage, with the consequent production of autoantibodies that could worsen and/or maintain the podocytes complement‐mediated damage 36. In Heymann nephritis, podocyte‐produced oxygen radicals in the presence of C5b‐9 and mediate glomerular damage. In this light, anti‐SOD2 and anti‐AR antibodies should follow a first autoimmune phase 35.
The prevalence of anti‐SOD2, anti‐AR and anti α‐enolase antibodies in iMN is lower than that of anti‐PLA2R, reaching values of 28, 34 and 43%, respectively, and their specificity appears not comparable to anti‐PLA2R antibodies as they are also present in other autoimmune diseases [anti‐SOD2 in systemic lupus erythematosus (SLE), anti α‐enolase in SLE and RA] 35. Although no strong association with clinical outcome was found for any single autoantibody, follow‐up proteinuria was lower in patients who were negative for all antibodies 35.
In 2014 Tomas and Beck discovered the presence of circulating autoantibodies against THSD7A (thrombospondin type‐1 domain containing 7A), a protein of 250 kD glomerular, in 8–14% of iMN patients’ sera negative for anti‐PLA2R. This protein shares different biochemical characteristics with PLA2R, such as N‐glycosylation, transmembrane localization in glomerular podocytes and serum reactivity only in non‐reducing conditions 37.
Anti‐THSD7A antibodies also appear specific, as they are not detected in healthy individual serum, in patients with secondary MN or different glomerulopathy. Interestingly, these antibodies are mutually exclusive with anti‐PLA2R, suggesting that PLA2R‐associated and THSD7A‐associated MN are two separate molecular entities 37.
Further additional non‐identified autoantigens might exist, as 10–20% of serum samples from iMN patients are negative for both anti‐PLA2R and THSD7A antibodies.
Disease effectors: differences between PLA2R‐associated iMN and Heymann nephritis
Podocyte damage in the Heymann nephritis experimental model is complement‐mediated, as established by the presence of C5b‐9 in the subepithelial deposits and the lack of proteinuria development, with the final depletion of complement‐cascade components or the use of anti‐megalin antibodies inhibiting complement activation 38.
In human disease, the presence of complement factors within the subepithelial deposits is well established, but IgG4, which does not activate complement by classical or alternative pathways, represents the predominant subclass of IgG anti‐PLA2R.
The discrepancy could have two different explanations. The first is based on the observation that both IgG1 and C1q are detected especially in early immune deposits 39, and there are low levels of circulating IgG1 or IgG3 anti‐PLA2R 23 which could, possibly, be responsible for complement activation through the classical pathway.
The second more probable explanation could be that IgG4 anti‐PLA2R autoantibodies can bind mannan‐binding lectin (MBL) and activate the lectin complement pathway. Purified anti‐PLA2R IgG4 autoantibodies from iMN patients have a high ratio of GalNAC (N‐acetylglucosamine) to Gal in a terminal position, an amino acidic sequence not usually present in mammalian carbohydrates, and thereby can bind to C4 through MBL in vitro 40.
In addition to activating complement, anti‐podocyte antibodies might directly change podocyte biology. Soluble PLA2 (sPLa2R) is a potent proinflammatory enzyme. Binding of sPLA2 to its receptor regulates its function positively and negatively depending on the cell type. PLA2R could act as a clearance receptor with potentially anti‐inflammatory activity, which might be affected by an anti‐PLA2R antibody. PLA2R also regulates cellular senescence, and because markers of senescence are over‐expressed in podocytes from patients with iMN, a potential agonistic activity of anti‐PLA2R antibodies should be considered 41.
Anti‐PLA2R antibodies
A few years after the discovery of the role of anti‐PLA2R antibodies in iMN, many studies on their prevalence and clinical significance have been published. Anti‐PLA2R autoantibodies have emerged as a specific and sensitive biomarker of idiopathic membranous nephropathy.
There are at least two commercially available tests for the detection and quantification of circulating anti‐PLA2R antibodies (Euroimmun AG, Lübeck, Germany). The immunofluorescence test, which provides a qualitative and semiquantitative antibody analysis, uses biochips coated with human embryonic kidney cells (HEK293) transfected (or not, to serve as negative control) with the full‐length human PLA2R1 cDNA and incubated with a serum sample from the patient 42. The enzyme‐linked immunosorbent assay (ELISA), based on purified recombinant PLA2R, enables a more quantitative and faster identification of anti‐PLA2R than the immunofluorescence test, although the latter is slightly more sensitive 43. As the specificity of these tests for iMN is close to 100%, some authors suggest not performing a renal biopsy in selected cases (elderly, single kidney or coagulation disorders) if the PLA2R test is positive 44. The sensitivity is, instead, approximately 70–80% in all the population studies carried out to date.
A low prevalence of anti‐PLA2R antibodies is reported in a secondary form of MN related to infection, drug intoxication, graft‐versus‐host disease and tumours, in which it can be difficult to exclude a correlation of iMN with the associated disease 45, 46, 47, 48. In addition, patients with MN related to sarcoidosis or active hepatitis B may show an increased prevalence of anti‐PLA2R antibodies, which suggests that immunological disorders associated with these two diseases might induce or enhance immune response against PLA2R 48, 49.
Detection of the PLA2R antigen in immune deposits in biopsy speciments is also possible with the use of a commercial antibody after a retrieval step to unmask PLA2R epitopes 50. This method appears to be more sensitive than the detection of circulating antibodies, as the PLA2R antigen can still also be found in deposits in some anti‐PLA2R seronegative patients. Several explanations could be proposed, including the possible rapid clearance of circulating antibodies, immunological remission or kidney biopsy performed long after disease onset. In addition, this method allows the retrospective diagnosis of PLA2R‐related MN in archival kidney biopsy samples, especially in patients awaiting a kidney graft, because of the possibility of a recurrence of the primitive disease on the transplanted kidney. Although the detection of PLA2R antigen in biopsy specimens is promising, the measurements of circulating antibodies appear more accessible and simpler to perform.
Anti‐PLA2R antibodies: clinical applications
In addition to the diagnostic significance of the PLA2R antibodies, many studies have shown a correlation between the concentration of circulating antibodies and disease activity expressed in terms of urinary protein output. Anti‐PLA2R antibodies usually become undetectable at spontaneous or treatment‐induced remission and re‐emerge or increase at relapse 23, 51, 52, 53, 54.
Anti‐PLA2R antibody concentration also seems to predict outcome, as high serum levels correlate with a lower probability of spontaneous or treatment‐induced remission and with an increased risk of deterioration in renal function. The time intervals from the start of immunosuppressive therapy to remission are increased in patients with the highest antibody titres 53, 55, 56.
Several studies have shown that partial or complete depletion of anti‐PLA2R antibodies precedes renal remission by several weeks or months 51. The time lag from immunological remission to renal remission probably corresponds to the time required for the restoration of the glomerular filtration barrier. Monitoring of anti‐PLA2R titres could also be relevant in cases of partial remission in which the persistent proteinuria might be due to immunological activity or irreversible podocyte damage, in which a continuation of immunosuppressive therapy would have no benefit.
Furthermore, antibody titres at the end of immunosuppressive treatment seem to predict recurrences, as relapses are observed more frequently in patients with high antibody titres after treatment 57 (Table 3).
Table 3.
Clinical value of anti‐phospholipase A2 receptor (PLA2r) antibodies.
| Features | |
|---|---|
| Diagnostic biomarker |
• High specificity • Good sensitivity |
| Prognostic biomarker | High serum titres correlate with:
|
| Monitoring tool |
• Serum titres correlate with immunological disease activity • Adjustment of immunosuppressive treatment based on immunological activity to limit side effects |
Quantification of anti‐PLA2R antibodies is becoming a monitoring tool of immunological activity of idiopathic membranous nephropathy; however, further studies on larger cohorts of patients are needed to establish and confirm their real utility value in clinical practice.
Disclosure
Authors have no competing interests to disclose.
References
- 1. Tiebosch AT, Wolters J, Frederik PF, van der Wiel TW, Zeppenfeldt E, van Breda Vriesman PJ. Epidemiology of idiopathic glomerular disease: a prospective study. Kidney Int 1987; 32:112–6. [DOI] [PubMed] [Google Scholar]
- 2. Maisonneuve P, Agodoa L, Gellert R et al Distribution of primary renal diseases leading to end‐stage renal failure in the United States, Europe, and Australia/New Zealand: results from an international comparative study. Am J Kidney Dis 2000; 35:157–65. [DOI] [PubMed] [Google Scholar]
- 3. Mallick NP, Short CD, Manos J. Clinical membranous nephropathy. Nephron 1983; 34:209–19. [DOI] [PubMed] [Google Scholar]
- 4. Jennette JC, Heptinstall RH. Heptinstall's pathology of the kidney. Philadelphia, PA: Lippincott Williams & Wilkins, 2007. [Google Scholar]
- 5. Ehrenreich T, Churg J. Pathology of membranous nephropathy. In Pathology Annual 1968, New York, NY, Appleton‐Century‐Crofts, 1968; 3:145–86. [Google Scholar]
- 6. Floege J, Johnson RJ, Feehally J. Comprehensive clinical nephrology, 4th edn Philadelphia, PA: Elsevier, 2010. [Google Scholar]
- 7. Magil AB. Tubulointerstitial lesions in human membranous glomerulonephritis: relationship to proteinuria. Am J Kidney Dis 1995; 25:375–9. [DOI] [PubMed] [Google Scholar]
- 8. Polanco N, Gutiérrez E, Covarsí A et al, Grupo de Estudio de las Enfermedades Glomerulares de la Sociedad Española de Nefrología. Spontaneous remission of nephrotic syndrome in idiopathic membranous nephropathy. J Am Soc Nephrol 2010; 21:697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Glassock RJ. Diagnosis and natural course of membranous nephropathy. Semin Nephrol 2003; 23:324–32. [DOI] [PubMed] [Google Scholar]
- 10. Cattran D. Management of membranous nephropathy: when and what for treatment. J Am Soc Nephrol 2005; 16:1188–94. [DOI] [PubMed] [Google Scholar]
- 11. Elsanjak A, Prabhakar SS. Membranous nephropathy, an update on glomerulopathies – clinical and treatment aspects. Prof. Sharma Prabhakar (Ed.), ISBN: 978‐953‐307‐673‐7, InTech, Available from: http://www.intechopen.com/books/an-update-on-glomerulopathies-clinical-and-treatment-aspects/membranous-nephropathy.
- 12. Van Damme BJ, Fleuren GJ, Bakker WW, Vernier RL, Hoedemaeker PJ. Experimental glomerulonephritis in the rat induced by antibodies directed against tubular antigens. V. Fixed glomerular antigens in the pathogenesis of heterologous immune complex glomerulonephritis. Lab Invest 1978; 38:502–10. [PubMed] [Google Scholar]
- 13. Nangaku M, Couser WG. Mechanisms of immune‐deposit formation and the mediation of immune renal injury. Clin Exp Nephrol 2005; 9:183–91. [DOI] [PubMed] [Google Scholar]
- 14. Heymann W, Hackel DB, Harwood S, Wilson SG, Hunter JL. Production of nephrotic syndrome in rats by Freund's adjuvants and rat kidney suspensions. Proc Soc Exp Biol Med 1959; 100:660–4. [DOI] [PubMed] [Google Scholar]
- 15. Kerjaschki D, Farquhar MG. The pathogenic antigen of Heymann nephritis is a membrane glycoprotein of the renal proximal tubule brush border. Proc Natl Acad Sci USA 1982; 79:5557–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kerjaschki D, Farquhar MG. Immunocytochemical localization of the Heymann nephritis antigen (GP330) in glomerular epithelial cells of normal Lewis rats. J Exp Med 1983; 157:667–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kerjaschki D, Ullrich R, Diem K, Pietromonaco S, Orlando RA, Farquhar MG. Identification of a pathogenic epitope involved in initiation of Heymann nephritis. Proc Natl Acad Sci USA 1992; 89:11179–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nangaku M, Shankland SJ, Couser WG. Cellular response to injury in membranous nephropathy. J Am Soc Nephrol 2005; 16:1195–204. [DOI] [PubMed] [Google Scholar]
- 19. Bruschi M, Candiano G, Murtas C et al Patients with primary membranous nephropathy lack auto‐antibodies against LDL receptor, the homologue of megalin in human glomeruli. Clin Kidney J, 2012; 5:178–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Debiec H, Guigonis V, Mougenot B et al Antenatal membranous glomerulonephritis due to anti‐neutral endopeptidase antibodies. N Engl J Med 2002; 346:2053–60. [DOI] [PubMed] [Google Scholar]
- 21. Debiec H, Nauta J, Coulet F et al Role of truncating mutations in MME gene in fetomaternal alloimmunisation and antenatal glomerulopathies. Lancet 2004; 364:1252–9. [DOI] [PubMed] [Google Scholar]
- 22. Debiec H, Lefeu F, Kemper MJ et al Early‐childhood membranous nephropathy due to cationic bovine serum albumin. N Engl J Med 2011; 364:2101–10. [DOI] [PubMed] [Google Scholar]
- 23. Beck LH Jr, Bonegio RG, Lambeau G et al M‐type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 2009; 361:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Llorca O. Extended and bent conformations of the mannose receptor family. Cell Mol Life Sci 2008; 65:1302–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. East L, Isacke CM. The mannose receptor family. Biochim Biophys Acta 2002; 1572:364–86. [DOI] [PubMed] [Google Scholar]
- 26. Behnert A, Fritzler MJ, Teng B et al An anti‐phospholipase A2 receptor quantitative immunoassay and epitope analysis in membranous nephropathy reveals different antigenic domains of the receptor. PLOS ONE 2013; 8:e61669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kao L, Lam V, Waldman M, Glassock RJ, Zhu Q. Identification of the immunodominant epitope region in phospholipase A2 receptor‐mediating autoantibody binding in idiopathic membranous nephropathy. J Am Soc Nephrol 2015; 26:291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fresquet M, Jowitt TA, Gummadova J et al Identification of a major epitope recognized by PLA2R autoantibodies in primary membranous nephropathy. J Am Soc Nephrol 2015; 26:302–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Augert A, Payré C, de Launoit Y, Gil J, Lambeau G, Bernard D. The M‐type receptor PLA2R regulates senescence through the p53 pathway. EMBO Rep 2009; 10:271–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weis WI, Taylor ME, Drickamer K. The C‐type lectin superfamily in the immune system. Immunol Rev 1998; 163:19–34. [DOI] [PubMed] [Google Scholar]
- 31. Vangelista A, Tazzari R, Bonomini V. Idiopathic membranous nephropathy in 2 twin brothers. Nephron 1988; 50:79–80. [DOI] [PubMed] [Google Scholar]
- 32. Stanescu HC, Arcos‐Burgos M, Medlar A et al Risk HLA‐DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. N Engl J Med 2011; 364:616–26. [DOI] [PubMed] [Google Scholar]
- 33. Lv J, Hou W, Zhou X et al Interaction between PLA2R1 and HLA‐DQA1 variants associates with anti‐PLA2R antibodies and membranous nephropathy. J Am Soc Nephrol 2013; 24:1323–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Salant DJ. Genetic variants in membranous nephropathy: perhaps a perfect storm rather than a straightforward conformeropathy? J Am Soc Nephrol 2013; 24:525–8. [DOI] [PubMed] [Google Scholar]
- 35. Murtas C, Bruschi M, Candiano G et al Coexistence of different circulating anti‐podocyte antibodies in membranous nephropathy. Clin J Am Soc Nephrol 2012; 7:1394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ronco P, Debiec H, Imai H. Circulating antipodocyte antibodies in membranous nephropathy: pathophysiologic and clinical relevance. Am J Kidney Dis 2013; 62:16–9. [DOI] [PubMed] [Google Scholar]
- 37. Tomas NM, Beck LH Jr, Meyer‐Schwesinger C et al Thrombospondin type‐1 domain‐containing 7A in idiopathic membranous nephropathy. N Engl J Med 2014; 371:2277–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cybulsky AV, Rennke HG, Feintzeig ID, Salant DJ. Complement‐induced glomerular epithelial cell injury. Role of the membrane attack complex in rat membranous nephropathy. J Clin Invest 1986; 77:1096–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huang CC, Lehman A, Albawardi A et al IgG subclass staining in renal biopsies with membranous glomerulonephritis indicates subclass switch during disease progression. Mod Pathol 2013; 26:799–805. [DOI] [PubMed] [Google Scholar]
- 40. Ma H, Beck LH, Salant DJ. Membranous nephropathy‐associated anti‐phospholipase A2 receptor IgG4 autoantibodies activate the lectin complement pathway. J Am Soc Nephrol 2011; 22:62A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sis B, Tasanarong A, Khoshjou F, Dadras F, Solez K, Halloran PF. Accelerated expression of senescence associated cell cycle inhibitor p16INK4A in kidneys with glomerular disease. Kidney Int 2007; 71:218–26. [DOI] [PubMed] [Google Scholar]
- 42. Hoxha E, Harendza S, Zahner G et al An immunofluorescence test for phospholipase‐A2‐receptor antibodies and its clinical usefulness in patients with membranous glomerulonephritis. Nephrol Dial Transplant 2011; 26:2526–32. [DOI] [PubMed] [Google Scholar]
- 43. Dahnrich C, Komorowski L, Probst C et al Development of a standardized ELISA for the determination of autoantibodies against human M‐type phospholipase A2 receptor in primary membranous nephropathy. Clin Chim Acta 2013; 421:213–8. [DOI] [PubMed] [Google Scholar]
- 44. Debiec H, Ronco P. Immunopathogenesis of membranous nephropathy: an update. Semin Immunopathol 2014; 36:381–97. [DOI] [PubMed] [Google Scholar]
- 45. Huang X, Qin W, Zhang M, Zheng C, Zeng C, Liu Z. Detection of anti‐PLA2R autoantibodies and IgG subclasses in post‐allogeneic hematopoietic stem cell transplantation membranous nephropathy. Am J Med Sci 2013; 346:32–7. [DOI] [PubMed] [Google Scholar]
- 46. Qin W, Beck LH Jr, Zeng C et al Anti‐phospholipase A2 receptor antibody in membranous nephropathy. J Am Soc Nephrol 2011; 22:1137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nawaz FA, Larsen CP, Troxell ML. Membranous nephropathy and nonsteroidal anti‐inflammatory agents. Am J Kidney Dis 2013; 62:1012–7. [DOI] [PubMed] [Google Scholar]
- 48. Knehtl M, Debiec H, Kamgang P et al A case of phospholipase A(2) receptor‐positive membranous nephropathy preceding sarcoid‐associated granulomatous tubulointerstitial nephritis. Am J Kidney Dis 2011; 57:140–3. [DOI] [PubMed] [Google Scholar]
- 49. Larsen CP, Messias NC, Silva FG, Messias E, Walker PD. Determination of primary versus secondary membranous glomerulopathy utilizing phospholipase A2 receptor staining in renal biopsies. Mod Pathol 2013; 26:709–15. [DOI] [PubMed] [Google Scholar]
- 50. Svobodova B, Honsova E, Ronco P, Tesar V, Debiec H. Kidney biopsy is a sensitive tool for retrospective diagnosis of PLA2R‐related membranous nephropathy. Nephrol Dial Transplant 2013; 28:1839–44. [DOI] [PubMed] [Google Scholar]
- 51. Beck LH Jr, Fervenza FC, Beck DM et al Rituximab‐induced depletion of anti‐PLA2R autoantibodies predicts response in membranous nephropathy. J Am Soc Nephrol 2011; 22:1543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hofstra JM, Beck LH Jr, Beck DM, Wetzels JF, Salant DJ. Anti‐phospholipase A(2) receptor antibodies correlate with clinical status in idiopathic membranous nephropathy. Clin J Am Soc Nephrol 2011; 6:1286–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hofstra JM, Debiec H, Short CD et al Antiphospholipase A2 receptor antibody titer and subclass in idiopathic membranous nephropathy. J Am Soc Nephrol 2012; 23:1735–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Oh YJ, Yang SH, Kim DK, Kang SW, Kim YS. Autoantibodies against phospholipase A2 receptor in Korean patients with membranous nephropathy. PLOS ONE 2013; 8:e62151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kanigicherla D, Gummadova J, McKenzie EA et al Anti‐PLA2R antibodies measured by ELISA predict long‐term outcome in a prevalent population of patients with idiopathic membranous nephropathy. Kidney Int 2013; 83:940–8. [DOI] [PubMed] [Google Scholar]
- 56. Hoxha E, Harendza S, Pinnschmidt H, Panzer U, Stahl RA. Mtype phospholipase A2 receptor autoantibodies and renal function in patients with primary membranous nephropathy. Clin J Am Soc Nephrol 2014; 9:1883–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ruggenenti P, Debiec H, Ruggiero B et al Anti‐phospholipase A2 receptor antibody titer predicts post‐rituximab outcome of membranous nephropathy. J Am Soc Nephrol 2015; 26:2545–58. [DOI] [PMC free article] [PubMed] [Google Scholar]