

Summary
The gene PIK3CD codes for the catalytic subunit of phosphoinositide 3‐kinase δ (PI3Kδ), and is expressed solely in leucocytes. Activating mutations of PIK3CD have been described to cause an autosomal dominant immunodeficiency that shares clinical features with common variable immunodeficiency (CVID). We screened a cohort of 669 molecularly undefined primary immunodeficiency patients for five reported mutations (four gain‐of‐function mutations in PIK3CD and a loss of function mutation in PIK3R1) using pyrosequencing. PIK3CD mutations were identified in three siblings diagnosed with CVID and two sporadic cases with a combined immunodeficiency (CID). The PIK3R1 mutation was not identified in the cohort. Our patients with activated PI3Kδ syndrome (APDS) showed a range of clinical and immunological findings, even within a single family, but shared a reduction in naive T cells. PIK3CD gain of function mutations are more likely to occur in patients with defective B and T cell responses and should be screened for in CVID and CID, but are less likely in patients with a pure B cell/hypogammaglobulinaemia phenotype.
Keywords: B cells, common variable immunodeficiency, hypogammaglobulinaemia, PI3Kδ, primary immunodeficiency
Introduction
Genetic defects of the immune system impair host defence and lead to an increased susceptibility to recurrent, severe or unusual infections. There are more than 200 Mendelian traits described to cause immunodeficiency 1. Defects affecting the humoral branch of the adaptive immune system are either caused by intrinsic dysfunction of B cells and their capacity to produce specific neutralizing antibodies or by insufficient T cell help in the germinal centres of lymph nodes and tonsils. In humans, this dysfunction may manifest as a syndrome known as common variable immunodeficiency (CVID), characterized by hypogammaglobulinaemia and recurrent infections, predominantly of the respiratory tract and the gut 2.
Activating mutations in p110δ, the catalytic subunit of phosphoinositide 3‐kinase δ (PI3Kδ) have been demonstrated to cause activated PI3Kδ syndrome (APDS, MIM615513), with an autosomal dominant pattern of inheritance. PI3Kδ belongs to the class IA phospho‐inositide‐3‐kinases (PI3Ks) and is expressed exclusively in leucocytes 3, 4. The protein is composed of a regulatory subunit, p85, and the catalytic subunit p110δ which phosphorylates PI(4,5)P2 to PI(3,4,5)P3 (PIP3) at the plasma membrane of lymphocytes upon receptor activation leading to downstream Akt/mTOR signalling with a consequent impact on T (and B) cell activation and fate 5.
In APDS, increased Akt/mTOR signalling leads to an immune dysregulation and an immunodeficiency, characterized by respiratory infections, bronchiectasis and autoimmune cytopenias 6, 7. Loss of function mutations in phosphatidylinositol 3‐kinase regulatory (PIK3R1) encoding the p85α regulatory subunit also result in hyperactivation of PI3K signalling with the same phenotype resulting from PIK3CD gain of function mutations, namely immunodeficiency, lymphoproliferation, poor antibody responses and expansion of senescent CD8+ T cells 8, 9. Immunological findings described previously in APDS include B cell lymphopenia with relatively increased transitional B cell numbers and reduced immunoglobulin (Ig)G, but elevated IgM levels in serum 6, 7, features that are shared partially with CVID 10. The differential diagnosis of APDS also extends to combined immunodeficiency (CID) or ‘atypical’ severe combined immunodeficiency (SCID) (defined as immunodeficiency due to mutations in SCID‐causing genes in patients with a presentation different from typical SCID and Omenn syndrome and T cell levels above 500 cells/μl 11, 12). CID patients present above the age of 1 year with clinical features that can include bronchiectasis, autoimmune cytopenia, recurrent and prolonged viral infection, lymphopenia, restricted antibody response and Epstein–Barr virus (EBV)‐associated lymphoproliferation 13. Clinical overlap with other primary antibody deficiencies, including X‐linked agammoglobulinaemia and hyper‐IgM syndrome, has been noted 14.
PI3K has been implicated previously in haematological malignancies, including B cell lymphomas 15, 16, 17. Correspondingly, patients with activating mutations of PI3Kδ have been described to exhibit benign and malignant lymphoproliferative disease, often in association with EBV viraemia 7. Among eight APDS patients reported by Kracker and co‐authors, two developed B cell lymphoma 18. Crank and co‐workers identified another pathogenic activating mutation in the p110δ subunit in patients with hyper‐IgM syndrome who also developed lymphoproliferative syndromes, although not in association with EBV in their cohort 19.
Among the genetically defined immunodeficiencies which are not lethal in infancy, APDS is of particular interest because commercially available inhibitors of PI3K may represent a specific therapeutic option 15, 20. Previous reports did not explore the incidence of PIK3CD and PIK3R1 mutations in patients with undefined hypogammaglobulinaemia. We therefore probed for the four published PIK3CD gain‐of‐function mutations and one splice site mutation in PIK3R1 in a large European cohort of predominantly CVID patients and patients with other primary antibody deficiencies.
Methods
Patients
A total of 669 immunodeficiency patients, mainly from continental Europe, were included in this screen: 610 patients diagnosed with CVID, 10 patients with an autoimmune lymphoproliferative syndrome (ALPS) phenotype but no identifiable defect in the Fas‐apoptotic pathway, 10 patients with a diagnosis of hyper‐IgM syndrome, 10 patients with a specific antibody deficiency, six patients with a combined immunodeficiency (CID) phenotype 11, five patients with selective IgA deficiency, two patients with agammaglobulinaemia and 16 patients with other minor antibody deficiencies.
The gender distribution of the cohort was almost equal, 49% male and 51% female. Almost 14% were diagnosed at age ≤ 10 years, 20% at age > 10–≤ 20 years, 41% at age > 20–≤ 40 years, 22% at age > 40–≤ 60 years, and 3% at age > 60 years. Patients in this multi‐centre cohort were diagnosed according to the criteria of the European Society for Immunodeficiency (ESID) and the Pan‐American Group for Immunodeficiency (PAGID) (available at www.esid.org). A total of 416 patients were recruited at the University Medical Centre Freiburg in Germany, 112 at Oslo University Hospital in Norway and 141 at the Royal Free Hospital in London, UK.
All individuals donated samples following written informed consent. This study was approved by the ethics review board of the Albert Ludwig University Freiburg, Germany (protocols 239/99_BG, 251/13_KW and 282/11_SE version 140023), the research ethics committee of the Royal Free Hospital and Medical School, London, UK (protocols #04/Q0501/119_AM03 for affected individuals, #07/H0720/182 for family members and #08/H0720/46 for healthy controls) and the regional committees for medical and health research ethics, Norway (reference number REK 2.2007.201).
Target mutations
Four gain‐of‐function mutations in PIK3CD encoding the catalytic p110δ subunit were targeted for screening, namely: C→A at cDNA position c.1002 resulting in the amino acid substitution N334K in the C2 domain; G→A at position c.1573 with consequent E525K substitution in the helical domain; G→A at position c.3061 resulting in an E1021K substitution in the C‐lobe of the kinase domain [6,7]; and T→C at position c.1246, resulting in the amino acid substitution C416R in the C2 domain 19. The study population was screened further for a splice site mutation in PIK3R1 corresponding to c.1425 + 1 that results in skipping of exon 11 of the gene with in‐frame deletion of the nucleotides encoding the amino acid residues 434–475 of p85α, which constitute part of the inter‐SH2 domain common to PIK3R1 isoforms 8, 9. No search for new possible mutations was undertaken in this study.
Pyrosequencing
Pyrosequencing technology was employed to detect the five mutations under investigation. Assays were designed using the PyroMark Assay software version 2·0·1·15 (Qiagen, Hilden, Germany). All primers were obtained from Apara Biosciences GmbH (Denzlingen, Germany). Target genomic DNA fragments were amplified by polymerase chain reaction (PCR) using the primers detailed in Supporting information, Table S1; 5 pmol each of the forward and reverse primers, one being biotinylated, were employed to amplify the fragments of interest in a final reaction volume of 12 μl. PCR products were rendered single‐stranded according to an established protocol 21. Three pmol of the sequencing primers provided in Supporting information, Table S1 were used in each case to carry out pyrosequencing of the amplified fragments on the PyroMark Q96 MD apparatus (Qiagen). Samples were amplified and sequenced in 96‐well plates.
Immunophenotyping
Measurement of immunoglobulin levels and lymphocyte immunophenotyping was performed at the Centre of Chronic Immunodeficiency, University Medical Centre Freiburg, Germany, and the Institute of Immunity and Transplantation at the University College London. Paediatric immunological investigations on the UK patients were performed at Great Ormond Street Hospital, London.
Results
A total of 669 immunodeficiency patients were screened for the reported gain‐of‐function mutations in PIK3CD, N334K, E524K, E1021K and C416R, and a splice site mutation in PIK3R1. We identified five patients with the heterozygous E1021K mutation in PIK3CD: a family of three siblings (family A) and two sporadic cases. None of the other mutations were observed. Results of the mutation screen are presented in Table 1. Despite being first‐degree relatives, with an identical mutation, the three siblings in family A, referred to further as siblings 1, 2 and 3, displayed diverse clinical and immunological features reminiscent of CVID, as detailed below and in Table 2. The two sporadic cases with the E1021K mutation were diagnosed with CID, distinguished from the three siblings by their elevated IgM levels, and are referred to below as sporadic patients (SP) 1 and 2.
Table 1.
Overview of screen for reported PI3Kδ and p85α mutations in 669 immunodeficiency patients from continental Europe
| Screened mutations | ||||||
|---|---|---|---|---|---|---|
| PIK3CD | PIK3R1 | |||||
| Diagnosis* | Number of patients | E1021K | E525K | N334K | C416R† | c.1425 + 1 |
| CVID | 610 | 3 | 0 | 0 | 0 | 0 |
| ALPS‐phenotype | 10 | 0 | 0 | 0 | 0 | 0 |
| HIGM | 10 | 0 | 0 | 0 | 0 | 0 |
| SPAD | 10 | 0 | 0 | 0 | 0 | 0 |
| sIgAD | 5 | 0 | 0 | 0 | 0 | 0 |
| Agammaglobulinaemia | 2 | 0 | 0 | 0 | 0 | 0 |
| CID‐phenotype | 6 | 2 | 0 | 0 | 0 | 0 |
| Others | 16 | 0 | 0 | 0 | 0 | 0 |
| Total | 669 | 5 | 0 | 0 | 0 | 0 |
ALPS = autoimmune lymphoproliferative syndrome. †Mutation screen was performed in 627 samples. CVID = common variable immunodeficiency; CID = combined immunodeficiency; HIGM = hyper‐immunoglobulin (Ig)M syndrome; SPAD = specific antibody deficiency; sIgAD = selective IgA deficiency; others = other minor forms of hypogammaglobulinaemia.
Table 2.
Clinical and immunological features of activated PI3Kδ syndrome (APDS) patients
| Sibling 1 | Sibling 2 | Sibling 3 | Sporadic patient 1 | Sporadic patient 2 | |
|---|---|---|---|---|---|
| Current Age (years) | 26 | 21 | 20 | 15 | 9 |
| Sex | Male | Female | Female | Male | Male |
| Age at diagnosis of antibody deficiency (years) | 5 | 1 | 1 | 9 | 3 |
| Chest infections | Yes | Yes | Yes | Yes | Yes |
| Bronchiectasis | Yes (minimal) | Yes | No | Yes | Yes |
| CVID enteropathy | No | Yes | No | No | Yes |
| Chronic rhinosinusitis | No | Yes | Yes | No | No |
| Asthma | No | Yes | No | No | No |
| Lymphadenopathy | Yes | Yes | Yes | Yes | Yes |
| Splenomegaly | Yes | No | Yes | Yes | Yes |
| EBV viraemia | Yes | No | No | No | No |
| Other clinical features | Nil | Eczema | Exocrine pancreatic dysfunction |
Thyroiditis, AIHA, ITP |
Mucocutaneous candidiasis,systemic CMV infection,adenovirus viraemia |
| On Ig replacement | No | Yes | Yes | Yes | Yes |
| IgG at initial diagnosis prior to antibody replacement (g/l) (NR) |
4·4 (4·9–16·1) |
0·54 (3·1–13·8) |
0·95 (3·1–13·8) |
1·78 (4·9–16·1) |
1·42 (4·2–16·1) |
| IgM at initial diagnosis prior to antibody replacement (g/l) (NR) |
0·96 (0·5–2·0) |
0·95 (0·5–2·0) |
1·26 (0·5–2·0) |
4·86 (0·5–2·0) |
0·52 (0·5–2·0) |
|
Total lymphocyte count (×106/l) (NR) |
1345 (1000–2800) |
1005 (1000–2800) |
946 (1000–2800) |
584 (1100–5900) |
1220 (1100–5900) |
|
CD3+ T cells (×106/l) (NR) |
1035 (700–2100) |
800 (700–2100) |
718 (700–2100) |
314 (700–4200) |
1076 (700–4200) |
|
CD4+ T cells (×106/l) (NR) |
490 (300–1400) |
403 (300–1400) |
322 (300–1400) |
180 (300–2000) |
226 (300–2000) |
|
CD8+ T cells (×106/l) (NR) |
522 (200–900) |
370 (200–900) |
368 (200–900) |
100 (300–1800) |
839 (300–1800) |
| CD4/CD8 ratio | 0·94 | 1·09 | 0·88 | 1·72 | 0·27 |
|
Naïve T cell (CD4+ CD45RA+) % (NR) |
19 (33–66) |
24 (33–66) |
14 (33–66) |
14·3 (46–77) |
7·4 (46–77) |
|
Memory T cell (CD4+CD45RO+) % (NR) |
84 (30–58) |
79 (30–58) |
88 (30–58) |
44 (13–30) |
86·5 (13–30) |
|
B cell count (×106/l) (NR) |
225 (100–500) |
41 (100–500) |
138 (100–500) |
28 (100–500) |
62 (100–500) |
|
Switched memory B cell % (IgD‐ CD27+) |
36·1 | 1·7 | 3·5 | 27·4 | NA |
|
Transitional B cell % (CD24+ CD38+) |
0·1 | 90·9 | 67·3 | 0·07 | NA |
NR = normal range; Ig = immunoglobulin; CVID = common variable immunodeficiency; AIHA = autoimmune haemolytic anaemia; IT = idiopathic thrombocytopenic purpura; EBV = Epstein–Barr virus; CMV = cytomegalovirus.
Case histories
CVID siblings (family A)
The eldest sibling, a male now aged 26 (sibling 1), was unwell from the first year of life with frequent respiratory tract infections. He suffered epiglottitis at 18 months of age and was ventilated for almost 2 weeks; at that time he was found to be IgA‐deficient. He continued to experience recurrent sinus, ear and respiratory infections and was diagnosed with mild bronchiectasis at the age of 2 years. His immune system was not fully re‐evaluated until his sister (sibling 2 – see below) was diagnosed with antibody deficiency; at that time sibling 1 was aged 5 years. He was found to be borderline IgG‐deficient (4·4 g/l) with normal IgM (see Table 2). He was commenced on intravenous immunoglobulin (IVIG) replacement, which he discontinued at age 16 years. He has been prescribed co‐trimoxazole prophylaxis, but currently does not take it. He still suffers from occasional respiratory infections and has minimal bronchiectasis (Fig. 1). He also has splenomegaly (15 cm) and a persistent low‐level EBV viraemia.
Figure 1.
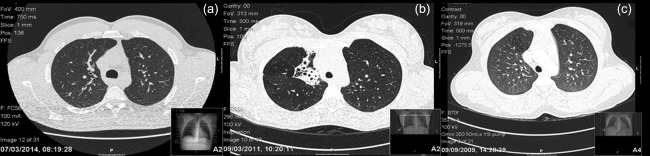
Spectrum of bronchial disease in family A siblings. Chest computed tomography (CT) scans of siblings 1, 2 and 3 (a,b,c) showing minimal bronchiectasis in sibling 1 (a), moderate bronchiectasis in sibling 2 (b) and unaffected lung tissue in sibling 3 (c).
The second sibling (sibling 2), a 21‐year‐old female, has suffered since infancy from recurrent lower respiratory infections, chronic rhinosinusitis and colitis. Identified initially to be antibody‐deficient with very low levels of IgG (0·54 g/l), but normal IgM (Table 2), she was diagnosed subsequently with CVID. She remains on IVIG infusions and prophylactic antibiotics and so far has moderate bronchiectasis (Fig. 1). Recent respiratory pathogens identified include Streptococcus pneumoniae and Haemophilus influenzae; she has previously required eradication therapy for Pseudomonas aeruginosa. She also has mild eczema. A particular issue for sibling 2 has been bowel inflammation with diarrhoea. Biopsies of the stomach and colon have demonstrated a polyclonal lymphocytic infiltrate within the lamina propria, associated with meshworks of CD21+ follicular dendritic cells. A lymphoid nodule has been discovered within the stomach, while in the colon, among a background inflammatory change and ulceration, there was possible epithelial infiltration by IgM+ lymphocytes. Although these infiltrating cells were not monoclonal, on one occasion a VH4 clone was discovered in peripheral blood on immunoglobulin heavy chain analysis. There is no other evidence of lymphoproliferative disease and PCR for EBV is negative; the spleen is at the upper limit of normal (13 cm). In 2014, sibling 2 had a norovirus gastrointestinal infection, which persisted for several weeks but eventually cleared in stool spontaneously (defined as consistently negative viral PCRs for several months); this correlated with a reduction in clinical symptoms, but some diarrhoea still persisted. After clearance of the norovirus infection she was commenced on a tapering dose of prednisolone and maintenance mesalazine, with good clinical response.
The youngest female sibling (sibling 3), now aged 20 years, was diagnosed with hypogammaglobulinaemia at the age of 1 year (IgG 0·95 g/l), again with normal IgM (Table 2). She was commenced on immunoglobulin replacement at that time. She had a significant varicella zoster infection at the age of 2·5 years with a large number of lesions, but did not require hospitalization. She had an episode of Henoch–Schonlein purpura at the age of 11 and was also diagnosed in the same year with molluscum contagiosum. This particularly affected the buttocks and hips and lasted for approximately 1 year. However, she has suffered only occasional respiratory tract infections and has a normal computed tomography (CT) scan of the chest (Fig. 1). She continues to suffer from pharyngitis intermittently and is symptomatic from a perennial rhinitis. She also has splenomegaly (15·3 cm) and mild thrombocytopenia (current platelet count 124 × 109/l), but EBV viraemia has not been demonstrated. Sibling 3's clinical picture is dominated currently by acquired exocrine pancreatic dysfunction with malabsorption, which was diagnosed at approximately 6 years of age. She passes multiple greasy stools per day despite supplementation with pancrelipase, and was found recently to be deficient in vitamin E [4·6 µmol/l (NR 11.6–46.4 µmol/l)]. She does not have diabetes mellitus and has no evidence of inflammation of the gastrointestinal tract on multiple endoscopies.
This clinical heterogeneity correlates with differences in immunological findings. In particular, the proportion of CD27+ IgD class‐switched memory B cells differs significantly between the siblings, with the lowest levels seen in sibling 2, who has the highest burden of sinopulmonary infection, bronchiectasis and inflammatory bowel disease (Fig. 1 and Table 2). Transitional B cell percentages demonstrate a reciprocal pattern, with the highest percentage reported in sibling 2. The male sibling (sibling 1) has a relatively normal B cell profile. All three siblings demonstrate reduction in the proportion of CD4+ cells expressing CD45RA+ (sibling 1, 19%, sibling 2, 24%, sibling 3, 14% versus a normal reference range of 33–66%).
Importantly, both parents of the three siblings are healthy and tested negative in blood for the E1021K mutation in p110δ.
Sporadic CID patients
SP1 is the only child of non‐consanguineous German parents, now aged 15 years. At 6 years of age he developed his first bacterial pneumonia. A second pneumonia, in combination with purulent sinusitis and otitis media, manifested at 6 years of life. One month later, a first episode of severe autoimmune haemolytic anaemia (AIHA) occurred (nadir Hb 4·9 g/dl), which at this time responded to short‐term steroid treatment. Aside from some mild upper respiratory infections his further clinical course was stable until 8 years of life, when he was admitted with an abscess of a cervical lymph node, which required surgical intervention (cultures positive for S. pneumoniae) and bilateral bacterial pneumonia. Splenomegaly was noted for the first time and progressed up to his umbilicus within a few months (Fig. 3). Shortly after, he suffered another episode of AIHA which has relapsed multiple times (often in response to respiratory viral infection) ever since.
Figure 3.
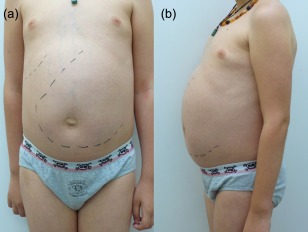
Massive splenomegaly in sporadic patient 1 (SP1). Ventral (a) and lateral (b) images of abdomen in SP1 showing massive splenomegaly.
Severely reduced IgG levels (1·78 g/l) and elevated polyclonal IgM (4·86 g/l) were recognized at 9 years of age and IgG replacement therapy was initiated. A CT scan at this time identified significant bronchiectasis and mediastinal lymphadenopathy, but no interstitial lung disease (Fig. 2). At age 10 years, idiopathic thrombocytopenic purpura (ITP) and neutropenia occurred and mycophenolate mofetil (MMF) was introduced as a steroid sparing agent. Chronic lymphopenia (below 1000 × 106/l) was documented in all available blood counts since age 9 and was present before the introduction of any immunosuppressive treatment.
Figure 2.
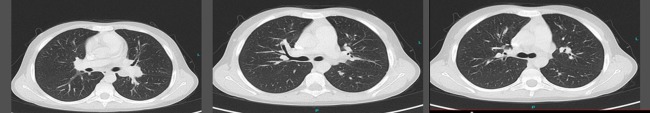
Lung disease in sporadic patient 1 (SP1). Axial chest computed tomography (CT) scans of SP1 demonstrating pronounced bronchiectasis, mediastinal lymphadenopathy, peribronchial inflammation but no interstitial disease.
Lymphocyte subset analysis over time demonstrated progressive B and T cell lymphopenia including loss of naive T cells but normal T cell proliferation to mitogens. Total lymphocyte, T and B cell counts dropped from 1650, 1472 and 93 × 106/l in 2008 to 584, 314 and 28 × 106/l in 2013, respectively (Table 2). Among his reduced total B cells the fractions of plasmablasts and CD21‐low B cells were relatively increased. At 11 years, attacks of AIHA and ITP worsened and were controlled only transiently by various immunosuppressive regimens, including steroid pulses, rituximab and mTOR pathway inhibition with sirolimus. Both ITP and AIHA improved eventually after splenectomy at 12 years of age. Prior to this splenectomy he also developed maturation arrest of granulopoiesis, which was treated with granulocyte–colony stimulating factor (G‐CSF). However, this condition also improved post‐splenectomy. The previous use of sirolimus for 6 months did not result in a reduction of his spleen size or improvement of cellular phenotype and was discontinued when the patient developed hypertension and neutropenia. Histological sections of resected lymph nodes, spleen and liver are provided in Supporting information, Fig. S1.
SP2 is the first child of non‐consanguineous Polish parents. He is now aged 9 years, with a younger healthy sibling. He had severe enteropathy with follicular hyperplasia presenting as chronic diarrhoea from the age of 2 months. He suffered from severe recurrent lower respiratory tract infections from the age of 7 months (Fig. 4). Onset of lymphoproliferation was from age 2 years onwards, when he first presented with generalized lymphadenopathy and marked splenomegaly. He was placed on subcutaneous immunoglobulin (SCIG) replacement therapy after hypogammaglobulinaemia which was recognized at age 3 and the frequency of his previous recurrent bacterial pneumonias improved. Other infectious complications included systemic cytomegalovirus (CMV) infection at the age of 4 years, mucocutaneous candidiasis at 5 years and chronic adenovirus viraemia detected at 8 years of age.
Figure 4.
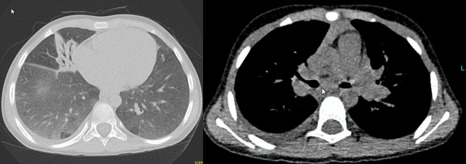
Lung disease in sporadic patient 2 (SP2). Axial chest computed tomography (CT) scans of SP2 demonstrating mucus impaction of the left middle lobe, moderate bronchiectasis, interstitial disease and enlargement of mediastinal lymph nodes.
Immunologically, he had profound hypogammaglobuinaemia (IgG 1·42 g/l and IgA undetectable) at age 3 years and an initially normal IgM (0·52 g/l) level that increased to 3·34 g/l by 8 years of age. His blood counts showed mild thrombocytopenia 86 × 109/l, a reduction of total B cell, CD4+ and naive T cell counts, but normal T cell proliferation to mitogens.
Both SP1 and SP2 were classified clinically as cases of CID based on the presence of severe immune dysregulation, chronic systemic infections despite immunoglobulin substitution, T cell lymphopenia and progressive loss of naive T cells.
Discussion
We identified five patients with the heterozygous gain of function mutation E1021K in the gene PIK3CD encoding PI3Kδ in a large multi‐centre European cohort of 669 patients diagnosed with antibody deficiencies. Three siblings from the same family had been diagnosed previously with CVID and two sporadic cases were diagnosed previously as CID.
Our findings contrast with the higher mutation frequency established by Lucas et al. [7] and Angulo et al. [6]. The latter group had excluded a founder effect in their UK/Ireland cohort 6. We therefore believe that the low frequency we have observed is explained by the specific cohort we have studied, consisting mainly of antibody‐deficient patients.
Indeed, our report is the first to document APDS among patients with CVID and highlights the need to consider APDS in the differential diagnosis of this condition.
Our findings underline the variability of the APDS phenotype, even within one family. Importantly, APDS may present without elevation of IgM and be diagnosed as CVID, emphasizing the importance of screening patient cohorts with this diagnosis. The phenotype in this family corresponded to features associated with CVID such as persistent hypogammaglobulinaemia, respiratory infection, bronchiectasis, splenomegaly and low‐level EBV viraemia. Importantly, even the sibling with relatively few infections (sibling 1) has potentially premalignant risk factors (EBV viraemia and splenomegaly). Similarly, his more clinically affected sister (sibling 2) shows epithelial lymphocytic infiltration in the colonic mucosa.
The two sporadic patients carrying a mutant PIK3CD allele were diagnosed previously with CID, involving both the B and T cell compartment. Bronchiectasis was pronounced in both cases and lymphoproliferation was a common feature. Both had low numbers of mature and naive T cells together with low B cell counts, low IgG levels and elevated IgM. Their clinical and immunological phenotype greatly resembled previously reported APDS patients 6, 7 with respiratory infections, bronchiectasis humoral immunodeficiency and progressive B cell lymphopenia with normal or reduced T cells, reduction in class‐switched memory B cells, increased IgM and reduced IgG2 levels. This profile of combined immunodeficiency, lymphoproliferation and inflammation is shared with patients carrying the splice site mutation in PIK3R1 with subsequent hyperactivation of PI3K due to impaired association with p110δ 8, 9, although we did not identify any patients with this mutation in our cohort.
APDS shares clinical parallels with other antibody deficiencies that exhibit a combined immunodeficiency phenotype such as hyper‐IgM syndrome 22, as demonstrated by the identification of PIK3CD mutations in patients diagnosed with this condition 19. The cohort investigated here included only a limited number of hyper‐IgM syndrome patients, and none carried any of the five reported APDS mutations. Autoimmune phenomena, enteropathies and blood dyscrasias feature strongly in lipopolysaccharide‐responsive and beige‐like anchor protein (LRBA) deficiency, another condition with a profile of combined immunodeficiency and autoimmunity 23, 24, while massive EBV‐driven B cell lymphoproliferation and possibly EBV‐associated lymphoma is characteristic of deficiencies of interleukin 2‐inducible T cell kinase (ITK) 25, 26, 27, CD27 28, 29 and X‐linked lymphoproliferative disease caused by mutations in SH2D1A 30.
The triad of severe immunodeficiency, microthrombocytopenia and eczema with often high IgE levels sets Wiskott–Aldrich syndrome apart from the above, but a clinical overlap with APDS is worth noting 31, 32. The recently described autosomal dominant syndrome of cytotoxic T lymphocyte‐associated protein 4 (CTLA4) deficiency shares a clinical picture of hypogammaglobulinaemia, lymphoproliferation, progressive loss of B cells, recurrent infections and prominent autoimmune features 33, 34 with APDS, although patients with CTLA4 deficiency tend to have a later onset of disease 33.
Taking note of the phenotypical heterogeneity of APDS, the following features are observed most frequently and should alert the clinician to a possible diagnosis: sporadic or autosomal dominant transmission, bacterial and viral infections, lymphadenopathies, normal or often increased serum IgM kevels, increased frequency of transitional B cells, decreased numbers of naive CD4 and CD8 T cells and increased numbers of CD8 effector/memory T cells.
In summary, we have demonstrated that APDS, although rare within our cohort of primary immunodeficiency patients, has diverse clinical and immunological characteristics (even within a single family), making it hard to differentiate from other primary immunodeficiencies. Of particular concern is the risk of malignancy in this syndrome, and thus screening for PIK3CD mutations and consideration of targeted treatment is warranted in patients with a current diagnosis of CVID or CID.
We conclude that activating mutations in PIK3CD may cause immunodeficiency in patients with a complex phenotype combining defective B and T cell responses, but are less likely to be discovered in patients where only the humoral arm of the immune response is affected and naive T cell numbers are normal. APDS should be considered in the differential diagnosis of CVID and CID.
Disclosure
The authors declare that they have no disclosures.
Author contributions
M. E., D. S., J. H., Z. E. and A. D. carried out the experiments in the laboratories of A. N. and B. G.; D. M. L. and C. S. provided clinical data on patients. A. S. G. performed histological examinations. D. M. L., C. S., B. F., U. S., S. B. and B. G. contributed patient samples. J. W., S. F. J. and D. S. prepared DNA aliquots. M. E., D. M. L. and C. S. drafted the manuscript with contributions from D. S; B. G. conceived the study. S. F. J., B. F., U. S., A. N., S. B. and B. G. reviewed the manuscript and M. E. prepared the manuscript for submission. We thank Sarita Workman for her contribution.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Table S1. Primer sets for pyrosequencing of PIK3CD and PIK3R1 mutations.
Fig. S1. Representative tissue sections of lymph node (a), spleen (b–f) and liver (g,h) from sporadic patient 1 (SP1). (a) Low magnification of a lymph node showing an expansion of the paracortical area [haematoxylin and eosin (H&E)]. Congested high endothelial venules and pale‐stained dentritic cells are prominent, while lymph follicles with germinal centres are absent. (b) Hyperplasia of the red pulp in the removed enlarged spleen. The white pulp comprises periarteriolar lymphoid sheets (PALS) but is depleted of follicles (H&E). (c–f) Immuohistochemistry of splenic tissue sections. (c) CD3‐positive T cells of PALS but (d) no CD20‐positive B cells are detected in the splenic white pulp. (e) Disproportional increase in IgA‐ versus (f) IgG‐positive plasma cells in the red pulp. (g,h) Wedge biopsy of the liver showing a nodular pattern resulting from an expansion of hyperplastic hepatocytes adjacent to atrophic liver cell plates consistent with diffuse nodular regenerative hyperplasia. (g) Discrete condensation of reticulin fibres surround small nodules without formation of true fibrous septa (Gomori's reticulin stain). (h) Focal steatosis of hepatocyte nodules with features of non‐alcoholic fatty liver disease (H&E).
Acknowledgements
This study was supported by the Excellence Initiative of the German Research Foundation (GSC‐4, Spemann Graduate School) and by the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung) grants 01EO0803 and 01GM1111B.
References
- 1. Al‐Herz W, Bousfiha A, Casanova JL et al Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Front Immunol 2014; 5:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grimbacher B, Hutloff A, Schlesier M et al Homozygous loss of ICOS is associated with adult‐onset common variable immunodeficiency. Nat Immunol 2003; 4:261–8. [DOI] [PubMed] [Google Scholar]
- 3. Vanhaesebroeck B, Welham MJ, Kotani K et al P110delta, a novel phosphoinositide 3‐kinase in leukocytes. Proc Natl Acad Sci USA 1997; 94:4330–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kok K, Geering B, Vanhaesebroeck B. Regulation of phosphoinositide 3‐kinase expression in health and disease. Trends Biochem Sci 2009; 34:115–27. [DOI] [PubMed] [Google Scholar]
- 5. Okkenhaug K. Signaling by the phosphoinositide 3‐kinase family in immune cells. Annu Rev Immunol 2013; 31:675–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Angulo I, Vadas O, Garcon F et al Phosphoinositide 3‐kinase delta gene mutation predisposes to respiratory infection and airway damage. Science 2013; 342:866–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lucas CL, Kuehn HS, Zhao F et al Dominant‐activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat Immunol 2014; 15:88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lucas CL, Zhang Y, Venida A et al Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med 2014; 211:2537–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deau MC, Heurtier L, Frange P et al A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest 2014; 124:3923–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yong PF, Thaventhiran JE, Grimbacher B. ‘A rose is a rose is a rose,’ but CVID is not CVID common variable immune deficiency (CVID), what do we know in 2011? Adv Immunol 2011; 111:47–107. [DOI] [PubMed] [Google Scholar]
- 11. Felgentreff K, Perez‐Becker R, Speckmann C et al Clinical and immunological manifestations of patients with atypical severe combined immunodeficiency. Clin Immunol 2011; 141:73–82. [DOI] [PubMed] [Google Scholar]
- 12. Roifman CM, Somech R, Kavadas F et al Defining combined immunodeficiency. J Allergy Clin Immunol 2012; 130:177–83. [DOI] [PubMed] [Google Scholar]
- 13. van der Burg M, Gennery AR. Educational paper. The expanding clinical and immunological spectrum of severe combined immunodeficiency. Eur J Pediatr 2011; 170:561–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Conley ME, Dobbs AK, Farmer DM et al Primary B cell immunodeficiencies: comparisons and contrasts. Annu Rev Immunol 2009; 27:199–227. [DOI] [PubMed] [Google Scholar]
- 15. Bauer TM, Patel MR, Infante JR. Targeting PI3 kinase in cancer. Pharmacol Ther 2015; 146:53–60. [DOI] [PubMed] [Google Scholar]
- 16. Chonmaitree T, Revai K, Grady JJ et al Viral upper respiratory tract infection and otitis media complication in young children. Clin Infect Dis 2008; 46:815–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kusel MM, de Klerk NH, Holt PG, Kebadze T, Johnston SL, Sly PD. Role of respiratory viruses in acute upper and lower respiratory tract illness in the first year of life: a birth cohort study. Pediatr Infect Dis J 2006; 25:680–6. [DOI] [PubMed] [Google Scholar]
- 18. Kracker S, Curtis J, Ibrahim MA et al Occurrence of B‐cell lymphomas in patients with activated phosphoinositide 3‐kinase delta syndrome. J Allergy Clin Immunol 2014; 134:233–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crank MC, Grossman JK, Moir S et al Mutations in PIK3CD can cause hyper‐IgM syndrome (HIGM) associated with increased cancer susceptibility. J Clin Immunol 2014; 34:272–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Conley ME, Fruman DA. Genetics. Can cancer drugs treat immunodeficiency? Science 2013; 342:814–5. [DOI] [PubMed] [Google Scholar]
- 21. Royo JL, Hidalgo M, Ruiz A. Pyrosequencing protocol using a universal biotinylated primer for mutation detection and SNP genotyping. Nat Protoc 2007; 2:1734–9. [DOI] [PubMed] [Google Scholar]
- 22. Qamar N, Fuleihan RL. The hyper‐IgM syndromes. Clin Rev Allergy Immunol 2014; 46:120–30. [DOI] [PubMed] [Google Scholar]
- 23. Lopez‐Herrera G, Tampella G, Pan‐Hammarstrom Q et al Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet 2012; 90:986–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alangari A, Alsultan A, Adly N et al LPS‐responsive beige‐like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. J Allergy Clin Immunol 2012; 130:481–8.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huck K, Feyen O, Niehues T et al Girls homozygous for an IL‐2‐inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV‐associated lymphoproliferation. J Clin Invest 2009; 119:1350–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Serwas NK, Cagdas D, Ban SA et al Identification of ITK deficiency as a novel genetic cause of idiopathic CD4+ T‐cell lymphopenia. Blood 2014; 124:655–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bienemann K, Borkhardt A, Klapper W, Oschlies I. High incidence of Epstein–Barr virus (EBV)‐positive Hodgkin lymphoma and Hodgkin lymphoma‐like B‐cell lymphoproliferations with EBV latency profile 2 in children with interleukin‐2‐inducible T‐cell kinase deficiency. Histopathology 2015; 67:607–16. [DOI] [PubMed] [Google Scholar]
- 28. van Montfrans JM, Hoepelman AI, Otto S et al CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. J Allergy Clin Immunol 2012; 129:787–93.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Salzer E, Daschkey S, Choo S et al Combined immunodeficiency with life‐threatening EBV‐associated lymphoproliferative disorder in patients lacking functional CD27. Haematologica 2013; 98:473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Booth C, Gilmour KC, Veys P et al X‐linked lymphoproliferative disease due to SAP/SH2D1A deficiency: a multicenter study on the manifestations, management and outcome of the disease. Blood 2011; 117:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cleland SY, Siegel RM. Wiskott–Aldrich Syndrome at the nexus of autoimmune and primary immunodeficiency diseases. FEBS Lett 2011; 585:3710–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Imai K, Morio T, Zhu Y et al Clinical course of patients with WASP gene mutations. Blood 2004; 103:456–64. [DOI] [PubMed] [Google Scholar]
- 33. Schubert D, Bode C, Kenefeck R et al Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med 2014; 20:1410–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kuehn HS, Ouyang W, Lo B et al Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 2014; 345:1623–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Table S1. Primer sets for pyrosequencing of PIK3CD and PIK3R1 mutations.
Fig. S1. Representative tissue sections of lymph node (a), spleen (b–f) and liver (g,h) from sporadic patient 1 (SP1). (a) Low magnification of a lymph node showing an expansion of the paracortical area [haematoxylin and eosin (H&E)]. Congested high endothelial venules and pale‐stained dentritic cells are prominent, while lymph follicles with germinal centres are absent. (b) Hyperplasia of the red pulp in the removed enlarged spleen. The white pulp comprises periarteriolar lymphoid sheets (PALS) but is depleted of follicles (H&E). (c–f) Immuohistochemistry of splenic tissue sections. (c) CD3‐positive T cells of PALS but (d) no CD20‐positive B cells are detected in the splenic white pulp. (e) Disproportional increase in IgA‐ versus (f) IgG‐positive plasma cells in the red pulp. (g,h) Wedge biopsy of the liver showing a nodular pattern resulting from an expansion of hyperplastic hepatocytes adjacent to atrophic liver cell plates consistent with diffuse nodular regenerative hyperplasia. (g) Discrete condensation of reticulin fibres surround small nodules without formation of true fibrous septa (Gomori's reticulin stain). (h) Focal steatosis of hepatocyte nodules with features of non‐alcoholic fatty liver disease (H&E).