

Abstract
The purpose of this pilot study was to determine whether macronutrient content (low-fat v. high-fat diet) influences an indicator of advanced glycation end products (AGE), Nε carboxymethyl-lysine (CML), in the context of a 1-d, high-AGE diet. The effect of the diets on inflammatory markers was also assessed. A total of nineteen overweight and obese adults (nine men and ten women) without known disease were recruited to participate in a crossover challenge of a high-fat, high-AGE (HFHA) and low-fat, high-AGE (LFHA) diet. In each phase patients had fasting blood drawn, followed by consumption of a high-fat or low-fat breakfast test meal, then three postprandial blood draws at 1, 2 and 3 h after consuming the test meal. After consuming high-AGE meals for the remainder of the day, participants returned the next day for a follow-up analysis. A different pattern in the 3-h post-meal CML and soluble receptor for AGE response to the two diets was observed (P = 0·01 and 0·05, respectively). No change in serum CML was observed following consumption of a LFHA breakfast (535 (25th–75th percentile 451–790) to 495 (25th–75th percentile 391–682) ng/ml; P = 0·36), whereas a rise in CML occurred after the HFHA breakfast (463 (25th–75th percentile 428–664) to 578 (25th–75th percentile 474–865) ng/ml; P = 0·05). High sensitivity C-reactive protein and high molecular weight adiponectin were not affected by either diet. These findings suggest that dietary CML may not be as important in influencing serum CML as other dietary factors. In addition, acute exposure to dietary CML may not influence inflammation in adults without diabetes or kidney disease. This is contrary to previous findings.
Keywords: Advanced glycation end products, High-fat diet, Low-fat diet, Obesity, Inflammation, Glycotoxins, Maillard reaction products
The Western diet today favours inflammation and the development of chronic diseases. Theories for the mechanism behind this observational finding include dietary imbalance of n-3 and n-6 fatty acids, consumption of high-fat diet, inadequate intake of micronutrients and plant foods, excessive intake of refined or processed foods and an imbalance in consumption of dietary acid and base(1). A less well-known theory purports that cooking of food produces advanced glycation end products (AGE), compounds formed when sugars react with amino acids or other substances forming glycosylated molecules, which are thought to favour oxidative stress when absorbed(2–5). AGE include pentosidine, Nε-carboxymethyl-lysine (CML), hydroimidazolone, furosine, glucosepane and many others. CML is the most widely studied AGE and is regarded as a good proxy measurement of AGE load in the body on the basis of early studies(6–8).
AGE are produced very slowly within biological systems as blood glucose interacts with body proteins and other molecules, but AGE may also enter the body via dietary consumption, although only about 10 % of dietary AGE are absorbed(9,10). AGE may increase oxidative stress via affinity for the receptor for AGE, which initiates a cascade of events, leading to increased arterial endothelial dysfunction and possible diabetic complications(3). The soluble receptor for AGE (sRAGE) is thought to act as a decoy receptor, aiding the body in removal of AGE(11,12).
Several studies on food have demonstrated that dietary AGE are present in high concentrations in foods exposed to high-temperature cooking methods, such as deep-frying, broiling, roasting, baking and grilling(13–18). Additionally, foods that are high in protein and high in fat that are cooked using these methods, particularly fried foods, tend to be particularly high in AGE(16,19,20). However, there is little consensus about appropriate measurement techniques for quantifying AGE in foods(17,21–23) and whether endogenous or dietary AGE found in a heat-processed western diet may negatively impact inflammation(3,21). Despite low levels of absorption, several studies have found decreased serum CML in participants (both human and animal) following consumption of reduced AGE diets(6,10,20,24–33). In addition, a study by Tan et al.(34) found a correlation in patients with diabetes between dietary AGE and C-reactive protein (CRP), an indicator of inflammation. Stirban et al.(35) found that a high-AGE test meal significantly increased 6-h postprandial CRP in patients with diabetes, whereas Negrean et al.(28) did not note this effect in patients with diabetes, nor did Poulsen et al.(36) in healthy, overweight participants. However, there have been no studies to compare the effect of consumption of a high-fat diet with a low-fat diet when both are elevated in AGE on serum CML and sRAGE. Also, the effect of dietary AGE on serum AGE is not clear, and two recent studies by Semba et al.(37,38) found no relationship between dietary CML intake and serum CML.
Current research suggests that dietary AGE increase oxidative and carbonyl stress, which may cause endothelial dysfunction and contribute to cellular damage related to diabetes, CVD and chronic kidney disease(3,19,20,25,30,31,39–41). Increased body fat and body weight are associated with both higher and lower levels of AGE (measured by serum CML)(38,42–44), but increased body fat is generally associated with decreased levels of sRAGE(45–50). As low-fat diet is often recommended for weight control in obese and overweight persons, it is important to determine whether a low-fat diet prepared using cooking methods such as baking, broiling, roasting and toasting would contribute to AGE load and negatively affect inflammatory cytokines thought to be influenced by AGE.
The purpose of this study was to determine whether a low-fat diet cooked using high-heat methods has equivalent effects on serum CML and pro- and anti-inflammatory cytokines when compared with a high-fat diet cooked using high-heat cooking methods. In addition, we hoped to further determine how serum CML responds to a dietary AGE load and whether sRAGE would be up-regulated in response to an AGE load.
Methods
Study design and participants
This pilot study was a crossover, 1-d dietary trial involving nineteen community-dwelling, overweight or obese adults (BMI of 27–35 kg/m2; overweight or class 1 obesity) aged 20–45 years. This study was conducted according to the guidelines laid down in the Declaration of Helsinki, and all procedures involving human subjects/patients were approved by the Texas Woman’s University Institutional Review Board’s Human Subjects Committee. Written informed consent was obtained from all patients/subjects. Participants consumed either a low-fat, high-AGE (LFHA) or high-fat, high-AGE (HFHA) diet first, followed by a 2–4-week wash-out phase, and then consumed the opposite diet.
Participants were instructed to adhere to their usual level of activity during the study period, and information on any supplements or medications taken by participants was obtained. Exclusion criteria included self-reported hypertension, dyslipidaemia, kidney disease, diabetes, heart disease and metabolic disease. Smokers were also excluded. Participants were advised to take no supplements for at least 1 week before the study and to avoid anti-inflammatory drugs. Participants taking medications that could affect blood glucose, lipids or inflammation were excluded.
Anthropometric, biochemical measurements and study protocol
On day 1 of the study, participants reported to the lab for anthropometric measurements. A portable stadiometer was used to measure height to the nearest one-quarter inch, and a beam scale was used to measure weight to the nearest 0·5 pound. Height and weight measurements were converted to centimetres and kilograms for reporting, and BMI was calculated (kg/m2). Waist circumference was measured with a non-stretch tape measure to the nearest 0·5 cm at the narrowest part between the lower costal (tenth) rib border and iliac crest, using the navel as a marker. Percentage of body fat was measured using bioelectrical impedance analysis (Tanita electronic scale; Tanita).
Five blood draws were conducted during each phase of the study. The first blood draw was carried out at baseline during the first trip to the lab with follow-up blood draws to include 1, 2 and 3 h after consumption of the first test breakfast meal. After completion of the 3-h postprandial blood draw, participants consumed lunch. They received an afternoon snack, dinner and a final bedtime snack over the course of the remainder of the day. Participants returned to the lab the next morning for another fasting blood draw.
After the 2–4-week wash-out period, participants returned for the same procedure using the other study diet not yet consumed. Anthropometric measurements were taken immediately before the second dietary intervention period by the same personnel. The same protocol was used for both dietary interventions. The order of meals was switched at random for test groups beginning the trial so that an almost equal number of subjects completed the low-fat diet first (n 10) compared with those consuming the high-fat diet first (n 9).
Diet intervention
Participants received two approximately iso-energetic, study diets (LFHA: 9121 kJ; HFHA 8955 kJ): one containing 23 % of energies from fat and the other with 42 % of energies from fat.
The fatty acid distribution of both diets was 2:1:1 (MUFA: PUFA:SFA). High-AGE cooking methods (broiling, roasting, baking and frying) were used for the preparation of both diets. (see Table 1).
Table 1.
Macronutrient and advanced glycation end products (AGE) content of study diets low-fat, high-AGE and high-fat, high-AGE diet*,†
| Meals | Low-fat diet | AGE (kU) | AGE (mg) | High-fat diet | AGE (kU) | AGE (mg) |
|---|---|---|---|---|---|---|
| Breakfast | 30 g Rice Krispies cereal | 600 | 0·41 | 30 g Life cereal | 394 | 0·74 |
| 250 ml fat-free milk | 4 | 0·05 | 250 ml whole milk | 12 | 0·13 | |
| 25 g (1 slice) whole-wheat toast | 21 | 0·78 | 25 g (1 slice) whole-wheat toast | 21 | 0·78 | |
| 2·5 g tub margarine: Smart Balance | 156 | 0 | 5 g tub margarine: Smart Balance | 311 | 0 | |
| 90 g smoked deli ham | 2114 | 0·19 | 20 g bacon, microwaved | 1805 | 0·09 | |
| 85 g banana | 8 | 0·05 | 85 g banana | 8 | 0·05 | |
| 120 ml 100 % apple juice | 3 | 0·04 | 120 ml 100 % apple juice | 3 | 0·04 | |
| Total AGE meal | 2906 | 1·52 | Total AGE meal | 2554 | 1·83 | |
| Lunch | Toasted cheese sandwich: 25 g × 2 whole-wheat bread | 42 | 1·56 | Toasted cheese sandwich: 25 g × 2 whole-wheat bread | 42 | 1·56 |
| 30 g reduced-fat (2 %) Cheddar cheese | 737 | 0·17 | 30 g regular Cheddar cheese | 1657 | 0·35 | |
| 2·5 g tub margarine: Smart Balance | 156 | 0 | 5 g tub margarine: Smart Balance | 311 | 0 | |
| 50 g carrot sticks | 5 | 0 | 50 g Carrot sticks | 5 | 0 | |
| 240 ml chicken noodle soup | 4 | 2·06 | 240 ml chicken noodle soup | 4 | 2·06 | |
| 100 g baked apples with 15 ml honey | 46 | 0 | 100 g apples – no skin | 13 | 0 | |
| 250 ml Coke | 7 | – | 250 ml Diet Coke | 3 | – | |
| Total AGE meal | 997 | 3·79 | Total AGE meal | 2035 | 3·97 | |
| Dinner | 90 g broiled chicken breast | 5245 | 0·36 | 90 g pan-fried chicken breast | 4444 | 0·46 |
| 100 g grilled California veggies (broccoli, carrots, cauliflower) | 226 | 0 | 100 g grilled California veggies (broccoli, carrots, cauliflower) | 226 | 0 | |
| (1·25 tsp maize oil) | 150 | 0·01 | (1·25 tsp maize oil) | 150 | 0·01 | |
| 100 g roasted potatoes | 218 | 0·08 | 50 g fried potatoes | 347 | 0·03 | |
| 35 g whole grain roll | 29 | 0·49 | 35 g whole grain roll | 29 | 0·49 | |
| 2·5 g tub margarine: Smart Balance | 156 | 0 | 5 g tub margarine: Smart Balance | 311 | 0 | |
| 60 g frozen fruit bar | 11 | – | 60 g frozen fruit bar | 11 | – | |
| 120 ml orange juice | 7 | 0·2 | ||||
| 360 ml Diet Coke (12 oz can) | 4 | – | 360 ml Diet Coke (12 oz can) | 4 | – | |
| Total AGE meal | 6046 | 1·14 | Total AGE meal | 5522 | 0·99 | |
| Snack 1 | 30 g roasted cashews (1 oz) | 2942 | 1·02 | 30 g roasted almonds (1 oz) | 1995 | 1·02 |
| 250 ml fat-free milk | 4 | 0·05 | 250 ml whole milk | 12 | 0·13 | |
| With 1 tbsp strawberry syrup | – | |||||
| Total AGE snack | 2946 | 1·07 | Total AGE snack | 2007 | 1·15 | |
| Snack 2 | 20 g cocktail peanuts | 1667 | 0·68 | 28 g cocktail peanuts | 2333 | 0·94 |
| 30 g Snyder’s Pretzel Minis | 537 | 0·06 | 30 g goldfish-shaped cheese crackers | 653 | 0·03 | |
| 120 ml apple juice | 3 | 0·04 | ||||
| Total AGE snack | 2207 | 0·78 | Total AGE snack | 2986 | 0·97 | |
| Total AGE | 2906 + 997 + 6046 + 2946 + 2207 = 15 102 kU | 15 102 | 8·3 | 2554 + 2035 + 5522 + 2007 + 2985 = 15 098 kU | 15 098 | 8·9 |
| Total energy (kJ) | 9121 kJ (2180 kcal) | 8955 kJ (2140 kcal) | ||||
| % Fat | 23 % (58 g) (2:1:1, MUFA:PUFA:SFA) | 42 % (103 g) (2:1:1, MUFA:PUFA:SFA) | ||||
| Protein | 109 g (20 %) | 98 g (18 %) | ||||
| Carbohydrate | 319 g (57 %) | 218 g (40 %) | ||||
| Added sugars | 62 g | 12 g | ||||
| Vitamin E (AT) | 9·3 mg | 16·4 mg | ||||
| Vitamin C | 137·6 mg | 74·6 mg |
AT, α tocopherol.
Numbers in AGE column (kU) represent the calculated AGE value of the food item from tables in the article by Uribarri et al.(14), which employs ELISA.
Numbers in AGE column (mg) represent the estimated calculated AGE value of the food item from tables in the article by Hull et al.(13), which employs HPLC.
Breakfast and lunch were consumed in the laboratory. Dinner, afternoon and bedtime snacks were consumed in the laboratory or boxed and given to the participant to be consumed at home, according to participants’ need.
Dietary analysis
Dietary macronutrient content was calculated initially using Nutritionist Pro (Axxya) and then reanalysed using Nutrition Data System for Research 2014. Then, 100-g frozen samples of the diets were sent for proximate analysis by an independent laboratory (Pope Labs). Analysis of the diet sample is shown in Table 2.
Table 2.
Comparison of test diets: calculated macronutrient values v. experimentally determined values
| Diet A (LFHA) | Diet B (HFHA) | |||||||
|---|---|---|---|---|---|---|---|---|
| Pope Labs | Calculated | Pope Labs | Calculated | |||||
| g | % | g | % | g | % | g | % | |
| Fat | 54 | 24 | 58 | 23 | 78 | 36 | 103 | 42 |
| Protein | 98 (0·52 % N) | 19 | 109 | 20 | 102 (0·48 % N) | 21 | 98 | 18 |
| Carbohydrate | 302 | 59 | 318 | 57 | 213 | 43 | 218 | 40 |
| Energy (kJ) | 8435 | 9121 | 8176 | 8955 | ||||
| Energy (kcal) | 2018 | 2180 | 1956 | 2140 | ||||
LFHA, low-fat, high-advanced glycation end products (AGE) diet; HFHA, high-fat, high-AGE diet.
Participants were allowed to eat only the study foods during each 1-d intervention period.
Biochemical analysis
Blood samples were immediately put on ice, allowed to sit for 10–15 min to facilitate clotting and centrifuged at 1200 rpm for 15 min. Sera were separated, aliquoted and stored at −80°C. Sera were analysed for glucose, TAG, total cholesterol (TC) and HDL using commercially available kits (Stanbio Laboratory). High molecular weight (HMW) adiponectin was measured using ELISA (ALPCO) (within- and between-assay CV: 5 and 7 %, respectively). CML (Microcoat) (within- and between-assay CV: 5 and 13 %, respectively), CRP (ALPCO) (within- and between-assay CV: 10 and 8 %, respectively) and sRAGE (BioVendor) (within- and between-assay CV: 6 and 15 %, respectively) were also measured using ELISA.
Statistical analyses
Male and female baseline characteristics were compared with two-sample t tests or the Wilcoxon’s rank sum test. Linear mixed model repeated measures analysis was utilised to analyse this crossover design, which assessed the effect of the diet intervention (low-fat diet v. high-fat diet), the effect of diet sequence and the interaction between diet effect and sequence. To compare the response pattern between diets for the 0–3-h time-course, a repeated factor for diet and time and the time by diet interaction were included in the model. A significant time by diet interaction indicates differing time-courses between diets. Pre-diet v. the next day post-diet responses were analysed similarly. Because of the high variability between subjects in serum CML, sRAGE, TAG, CRP and adiponectin, these variables were log transformed before parametric analysis. One participant had very high levels of CRP, but because an intent-to-treat model was employed, this data was kept in the analysis and log-transformed. Data were analysed with and without this data point, and the results were not changed. AUC for hours 0–3 was calculated using the trapezoidal rule. AUC low-fat diet v. high-fat diet comparisons were made with the Wilcoxon’s signed rank test. Results are presented as median and 25th–75th percentile or mean values and standard deviations, unless specified otherwise. A two-sided P value < 0·05 was considered statistically significant. Analyses were performed with SAS version 9.4 (SAS Institute).
Results
Anthropometric measurements and biochemical analyses of the study participants at baseline are listed in Table 3. Some individual participants showed fluctuations in body weight over the course of the study, but there were no overall significant changes in weight, BMI or percentage of body fat over the 2–4-week study period. Anthropometric and biochemical variables were compared between males and females enrolled in the study. Males and females were different with regard to body fat composition (males: 27 (sd 4) %; females: 41 (sd 4) %; P < 0·0001) but were otherwise similar. Data from males and females were grouped together for the remaining analyses.
Table 3.
Anthropometric measurements and selected biochemical analyses of study participants at baseline: all participants, males only, females only (Mean values and standard deviations; medians and 25th–75th percentiles)
| All | Males (n 9) | Females (n 10) | |||||
|---|---|---|---|---|---|---|---|
| Mean | sd | Mean | sd | Mean | sd | P | |
| Age (years) | 31·4 | 8·0 | 34·2 | 9·8 | 28·9 | 5·3 | 0·28 |
| Weight (kg) | 88·7 | 8·3 | 88·9 | 6·7 | 88·4 | 9·9 | 0·89 |
| BMI (kg/m2) | 30·5 | 2·7 | 29·7 | 2·8 | 31·3 | 2·5 | 0·92 |
| Waist circumference (cm) | 101·0 | 9·0 | 101·0 | 11·0 | 100·0 | 8·0 | 0·92 |
| Body fat (%) | 34·0 | 8·0 | 27·0 | 4·0 | 41·0 | 4·0 | <0·01 |
| Glucose (mmol/l) | 5·16 | 0·56 | 5·16 | 0·72 | 5·11 | 0·33 | 0·92 |
| TC (mmol/l) | 5·52 | 1·01 | 5·57 | 1·06 | 5·46 | 1·01 | 0·92 |
| HDL (mmol/l) | 1·24 | 0·36 | 1·27 | 0·47 | 1·24 | 0·23 | 0·65 |
| Median | 25th–75th percentile | Median | 25th–75th percentile | Median | 25th–75th percentile | ||
| TAG (mmol/l) | 0·94 | 0·53–1·29 | 0·86 | 0·59–1·54 | 0·95 | 0·53–0·99 | 0·59 |
| CML (ng/ml) | 518 | 425–803 | 478 | 459–612 | 719 | 425–808 | 0·43 |
| sRAGE (pg/ml) | 538 | 388–676 | 430 | 354–578 | 589 | 497–676 | 0·11 |
| hsCRP (µg/ml) | 1·4 | 0·2–3·6 | 0·3 | 0·2–1·6 | 2·0 | 0·7–4·0 | 0·10 |
| HMW adiponectin (µg/ml) | 3·2 | 1·4–4·0 | 3·2 | 1·5–3·3 | 3·8 | 1·2–4·4 | 0·59 |
TC, total cholesterol; CML, Nε-carboxymethyl-lysine; sRAGE, soluble receptor for advanced glycation end products; hsCRP, high sensitivity C-reactive protein; HMW, high molecular weight.
The effect of diet sequence and the interaction between diet effect and sequence were tested, and no carryover effect was observed.
Participants tolerated both the HFHA and LFHA diets well and consumed all of the study foods in most cases. One participant reported indigestion, which she attributed to consumption of diet cola, and another participant reported vomiting a small amount following consumption of cashew nuts. Otherwise, both study diets were well tolerated.
CML levels following consumption of the LFHA breakfast meal rose from a fasting median of 518 (25th–75th percentile 425–803) ng/ml to a peak of 553 (25th–75th percentile 425–799) ng/ml at 2 h postprandial, then decreased to 495 (25th–75th percentile 391–682) ng/ml by 3 h postprandial and rose to 538 (25th–75th percentile 403–740) ng/ml the next day (see Table 4). By contrast, CML levels following consumption of the HFHA breakfast meal rose from a fasting median of 463 (25th–75th percentile 428–664) ng/ml to a presumed peak of 578 (25th–75th percentile 474–865) ng/ml at 3 h, before declining slightly to 565 (25th–75th percentile 426–769) ng/ml the next day. The pattern of change in CML following the LFHA meal was generally more complex with greater variability from person to person. At 3 h postprandial, only the HFHA diet showed a pattern of significant change in CML compared with baseline (interaction P = 0·05). By contrast, the change in CML following the LFHA diet was not significant at 3 h (P = 0·36) or even at the numerical peak at 2 h (interaction P = 0·32). However, the response to the two diets over time was significantly different (interaction P = 0·01) (see Table 4 and Fig. 1). sRAGE levels following consumption of LFHA breakfast meals decreased from a fasting median of 556 (25th–75th percentile 402–676) pg/ml to 515 (25th–75th percentile 379–642) pg/ml by 1 h, to 492 (25th–75th percentile 330–623) pg/ml by 2 h and to 484 (25th–75th percentile 313–580) pg/ml by 3 h (time effect P = 0·02). The changes at 2 and 3 h were statistically significant (P < 0·01). Following the HFHA breakfast meal also sRAGE decreased significantly from baseline to 1 to 2 to 3 h postprandially (time effect P = 0·008) and then rose the next day, though not significantly. The response pattern in sRAGE to the diets differed significantly (interaction P = 0·05) (see Table 4 and Fig. 2). AUC for sRAGE at 0, 1, 2 and 3 h did not differ significantly between LFHA and HFHA diets.
Table 4.
Nε-carboxymethyl-lysine (CML) and soluble receptor for advanced glycation end products (sRAGE) response to low-fat, high-advanced glycation end products (AGE) diet (LFHA) v. high-fat, high-AGE diet (HFHA) diets at baseline, 1, 2, 3 and 24 h: CML, sRAGE (Medians and 25th–75th percentiles)
| 0 h | 1 h | 2 h | 3 h | 24 h | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Variables | Median | 25th–75th percentile |
Median | 25th–75th percentile |
Median | 25th–75th percentile |
Median | 25th–75th percentile |
Median | 25th–75th percentile |
P interaction between diet and hour (0–3) |
| LFHA diet | |||||||||||
| CML (ng/ml) | 535 | 451–790 | 541 | 362–813 | 553 | 425–799 | 495 | 391–682 | 538 | 403–740 | 0·01*† |
| sRAGE (pg/ml) | 556 | 402–676 | 515 | 379–642 | 492 | 330–623 | 484 | 313–580 | 541 | 382–672 | 0·05*‡ |
| HFHA diet | |||||||||||
| CML (ng/ml) | 463 | 428–664 | 607 | 450–682 | 552 | 448–731 | 578 | 474–865 | 565 | 426–769 | 0·01*† |
| sRAGE (pg/ml) | 546 | 388–756 | 467 | 360–710 | 460 | 325–664 | 443 | 322–604 | 548 | 392–788 | 0·05*‡ |
Statistically significant (P ≤ 0·05).
Represents the interaction between diet and time (i.e. a difference in diet response over the time-course) in CML from 0 to 3 h postprandial.
Represents the interaction between diet and time (i.e. a different in diet response over the time course) in sRAGE from 0 to 3 h postprandial.
Fig 1.
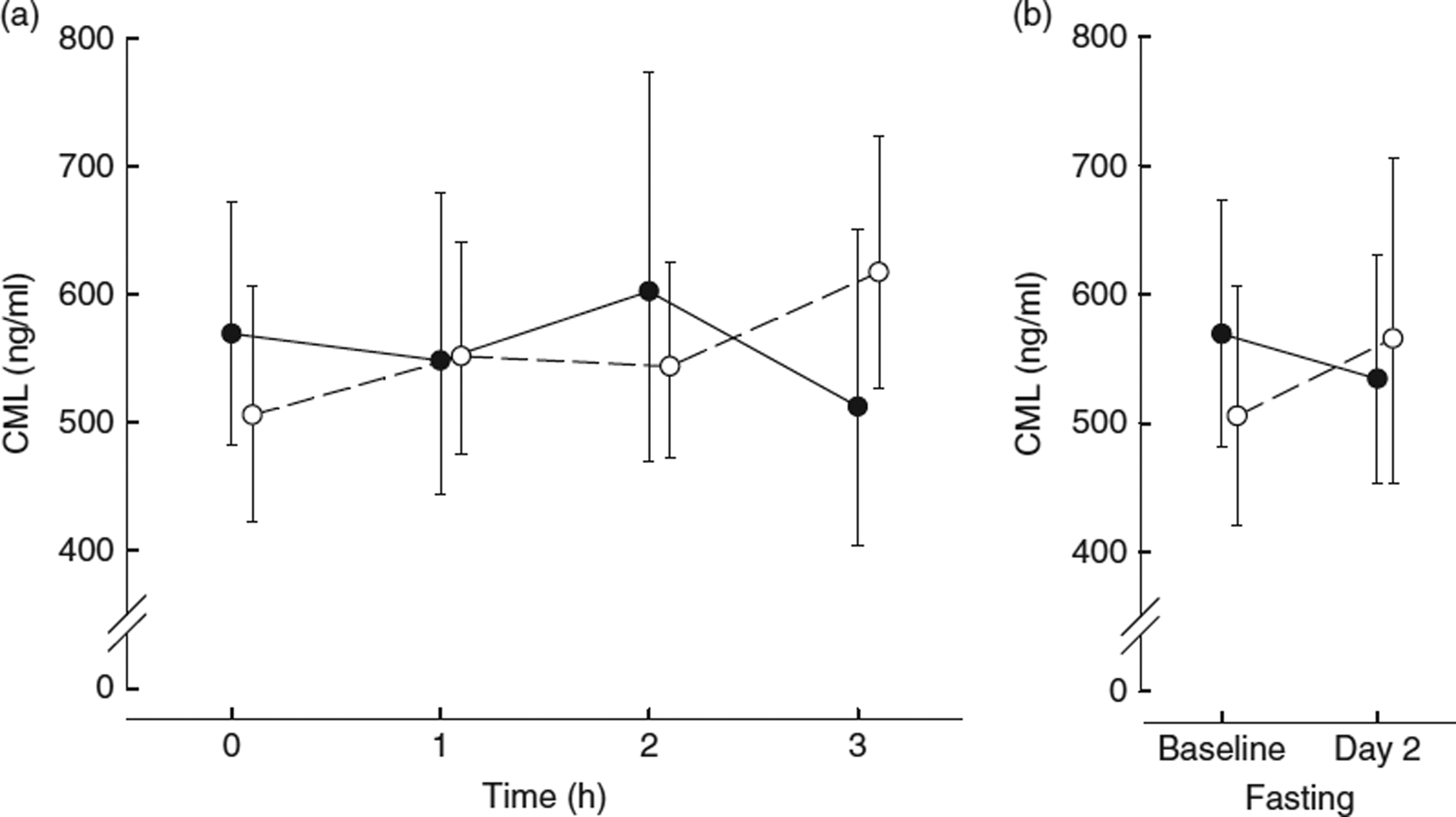
(a) Nε-carboxymethyl-lysine (CML) 3-h time-course:  , low-fat, high-AGE diet v.
, low-fat, high-AGE diet v.  , high-fat, high-AGE diet; (b) fasting CML levels before diet (baseline day 1) and after diet (day 2). Values are geometric means and 95% CI.
, high-fat, high-AGE diet; (b) fasting CML levels before diet (baseline day 1) and after diet (day 2). Values are geometric means and 95% CI.
Fig 2.
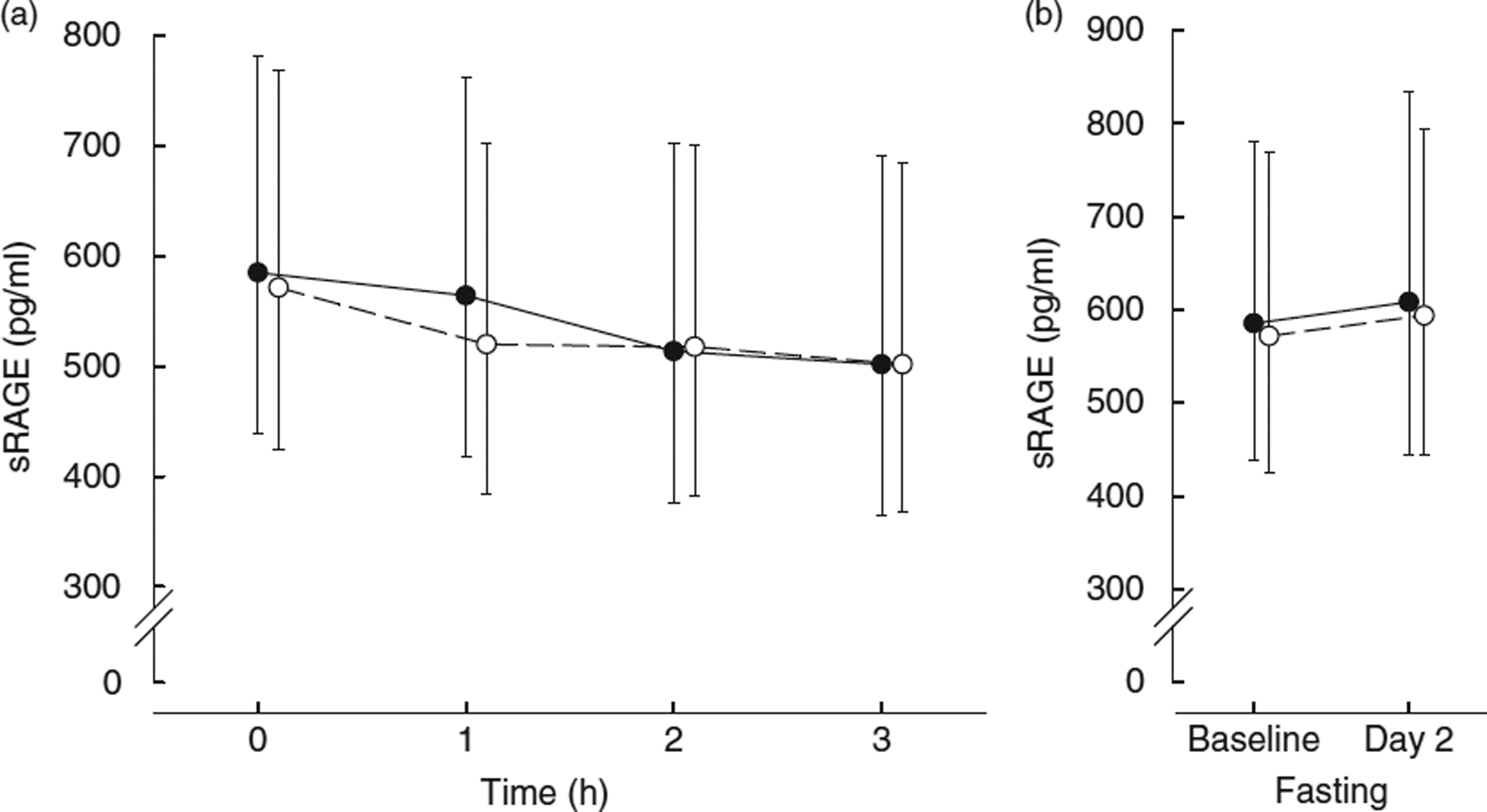
(a) Soluble receptor for AGE (sRAGE) 3-h time-course: , low-fat, high-AGE diet v. , high-AGE diet; (b) fasting sRAGE levels before diet (baseline day 1) and after diet (day 2). Values are geometric mean and 95% CI.
Other biochemical variables changed little from baseline to fasting the next day; however, TC decreased significantly for participants consuming the HFHA diet (HFHA: baseline: 5·83 (sd 1·24) mmol/l, post: 5·52 (sd 1·13) mmol/l, P = 0·02; LFHA: baseline: 5·28(sd 0·75) mmol/l, post: 5·34 (sd 1·04) mmol/l). The response in cholesterol to the diets was different (P = 0·03). High sensitivity CRP (hsCRP, a pro-inflammatory indicator) and HMW adiponectin (an anti-inflammatory indicator) did not differ following consumption of either diet (P = 0·61 and 0·92, respectively) (see Table 5). The 0 v. 24 h changes were not statistically different within either diet.
Table 5.
Response to low-fat, high advanced glycation end products (AGE) diet (LFHA) v. high fat, high AGE diet (HFHA) diets at baseline and 24 h: glucose, cholesterol, HDL, TAG, high molecular weight (HMW) adiponectin and C-reactive protein (CRP) (Medians and 25th–75th percentiles)
| LFHA diet | HFHA diet | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 0 h | 24 h | 0 h | 24 h | ||||||
| Variables | Median | 25th–75th percentile |
Median | 25th–75th percentile |
Median | 25th–75th percentile |
Median | 25th–75th percentile |
P† |
| Glucose (mmol/l) | 5·16 | 4·71–5·49 | 5·05 | 4·77–5·33 | 4·99 | 4·94–5·55 | 4·93 | 4·77–5·55 | 0·50 |
| TC (mmol/l) | 5·28 | 4·74–5·88 | 5·36 | 4·61–6·03 | 5·28 | 4·71–7·07 | 5·18 | 4·74–6·03 | 0·03* |
| HDL (mmol/l) | 1·14 | 0·98–1·50 | 1·24 | 1·06–1·40 | 1·17 | 1·04–1·30 | 1·14 | 1·01–1·48 | 0·63 |
| TAG (mmol/l) | 0·94 | 0·50–1·13 | 0·88 | 0·52–1·18 | 1·05 | 0·45–1·54 | 0·94 | 0·63–1·32 | 0·22 |
| HMW adiponectin (µg/ml) | 2·9 | 1·5–4·0 | 3·0 | 1·4–3·7 | 2·9 | 1·4–3·9 | 2·6 | 1·3–3·9 | 0·92 |
| hsCRP (µg/ml) | 1·1 | 0·3–3·6 | 1·0 | 0·5–2·9 | 1·0 | 0·2–2·9 | 1·1 | 0·2–2·3 | 0·61 |
TC, total cholesterol; hsCRP, high sensitivity CRP
Statistically significant (P < 0·05).
Represents the interaction between diet and time (i.e. a difference in diet response over time) from 0 to 24 h postprandial.
Discussion
This study is the first to compare consumption of a low-fat and high-fat diet, each high in AGE, in terms of serum AGE, sRAGE and indicators of inflammation. Our study had some limitations, including small sample size, the presence of many participants with dyslipidaemia despite the screening to identify healthy participants and the short duration of the study.
Another potential limitation of this study is the use of circulating CML as a proxy measure of AGE and a primary end point. There remains controversy within the realm of AGE research regarding the following: whether CML is the most significant AGE physiologically; whether it negatively impacts health; the extent to which circulating CML is affected by dietary CML; and the validity of ELISA as a method to measure it(19,20,21,42,51–55). Because multiple human studies suggest an association between consumption of heat-treated foods rich in CML and risk factors for chronic illness such as endothelial dysfunction and inflammation(25,28–30,32,33,35,41,56), we felt it important to assess how a low-fat v. a high-fat meal impacts the postprandial response to CML. In addition, because several studies by the same group, which use ELISA to measure CML, have found a rise in circulating CML following consumption of a high-AGE diet(10,28,30,35), we wanted to attempt to replicate this finding using the same methodology. The AGE content of the diets was calculated using the tables created by Uribarri et al.(14). However, we separately estimated the AGE content of the diet using tables created by Hull et al.(13), which use HPLC, choosing similar foods from that database when exact matches were not available, and found little difference in AGE content between the two diets using this method (8·3 mg for the LFHA diet v. 8·9 mg for HFHA diet (a 6 % difference) (see Table 1). In spite of these limitations, the findings are important in several ways.
Even though both diets were calculated to be the same in AGE and used almost the same foods with fruit juices added to the low-fat diet in order to compensate for decreased energy due to less fat, the measured AGE content of the total LFHA diet, which was quantified using ELISA, was about 15 % higher in AGE compared with the AGE content in the high-fat diet (data not shown). It is possible that this measurement was inaccurate due to the limitations of ELISA in quantifying CML in food substrates(51). The calculated AGE content of the test meals using the tables of Hull et al.(13) showed the HFHA test meal to be about 17 % higher in CML compared with the LFHA test meal; however, the food lists of Hull were significantly limited in scope and often did not contain exact matches to the foods used in this study; thus, these calculations may not be accurate. The foods served in the breakfast test meals were designed to be almost identical, only differing in fat content. The meat offering in the LFHA test meal was smoked deli ham, and the HFHA meal contained bacon. Whole milk was used in place of skimmed milk in the HFHA meal. Slightly more margarine was offered in the HFHA meal. Finally, the breakfast cereals were made from different grains, with the HFHA meal containing an oat and maize-based cereal, whereas the LFHA cereal was rice-based; however, both cold cereals were prepared from moistened grains, which were subsequently cooked at high temperatures. Because of the strong similarity between the breakfast meals and the methods of preparing them, it is probable that the meals were very similar in AGE content. However, whereas consumption of the LFHA diet was associated with a non-significant postprandial decrease in serum CML with no rise in CRP or reduction in adiponectin at 24 h, the HFHA diet was associated with a significant rise in postprandial serum CML with no rise in CRP or reduction in adiponectin at 24 h.
Because we used fruit juices to balance the energy intake between the diets while attempting to hold protein steady and change only the quantity – not type – of fat, we may have introduced more inhibitors of AGE formation into the total diet. Inhibitors of AGE formation in cooked food include plant compounds such as flavonoids, phenolic acids, terpenes and vitamins(19,57) as well as acid content, moisture and cooking times and temperatures(19). However, because we did not cook the foods together with the fruit juices, it is unlikely that these compounds inhibited formation of AGE in the food products used for the test diet. In addition, the fruit juice type and amount was identical at the breakfast test meal. Thus, the reason for the postprandial rise in CML with the HFHA diet is not explained by differences in fruit juice consumption.
It is possible that consumption of the high-fat meal resulted in greater peroxidation of unsaturated fatty acids, which led to higher serum CML levels. However, the HFHA meal contained more vitamin E; thus, one might expect that lipid peroxidation of the fats ingested would have been minimised, and postprandial CML levels would have been similar between the two diets.
Antioxidant content of the diets might also have affected serum CML. The antioxidant content was necessarily different between the two diets. Specifically, the α-tocopherol content of the HFHA diet was 43 % higher than that of the LFHA diet (16 v. 9 mg). The opposite relationship was true of the vitamin C content of the two diets: the LFHA diet had a 46 % higher content compared with the HFHA diet (138 v. 75 mg). Therefore, although the antioxidant content of the diets differed, whereas the HFHA diet was lower in vitamin C, it was higher than the LFHA diet in vitamin E. Higher α-tocopherol was present in the HFHA breakfast test meal compared with LFHA, but vitamin C content was identical between the two meals. It seems unlikely that these antioxidant differences resulted in higher circulating CML following consumption of the HFHA diet.
It is possible that the study results were confounded by previously ingested AGE compounds, which were gradually degraded by intestinal microbiota in the large intestine and later contributed to a rise in serum CML. However, the participants were fasting and did not report previous digestive problems; thus, they might be supposed to have typical gut transit times, which should have allowed minimal presence of these compounds present from previous meals. In addition, one might expect the effect to be similar between diet phases.
Finally, it is possible that the participants in this study were aware of their group assignment and altered their dietary pattern accordingly. Participants were not told of their group assignment; however, in a feeding study of this nature, true blinding is not possible. Because the foods served in both arms of the trial seem very similar to the lay person (2 % Cheddar v. whole milk Cheddar on a grilled cheese sandwich with the same bread; one slice of toast with 2·5 or 5 g of margarine; whole milk v. skimmed milk), several participants wondered aloud to the primary researcher regarding their study diet, seemingly unaware of their group assignment. Thus, although it is possible that participants altered their home diets based on group assignment, we do not think this occurred to any appreciable or consistent extent. Because all study foods were provided during the day of study and energy intake was substantial, we do not think there was likely to be much eating of foods outside of the study diet. Most participants reported feeling that it was ‘a lot of food’. Although it is possible that recent AGE intake could influence CML levels during the study, we attempted to limit this effect by requiring fasting and asking participants to keep a 3-d food record before beginning the study.
Thus, the mechanism for the rise in CML following the HFHA meal remains unclear. However, a possible explanation is that the higher fat content of the breakfast test meal caused delayed gastric emptying, allowing longer time for hydrolysis of the AGE-containing proteins, making CML more available for absorption. With documented low absorption of dietary AGE for reasons that are not entirely clear but may relate to the resistance of AGE to break down by enzymes or acid(10), absorption in two diets and meals that are similar in AGE content may relate more to other characteristics of the meal affecting digestion and absorption, rather than solely the AGE content of the diet.
These findings suggest that the contribution of dietary AGE to circulating AGE may be modulated by dietary factors such as phytochemicals, dietary fat or other unidentified factors. These various dietary factors may affect circulating AGE by modulating absorption and/or metabolism. Our breakfast test meal had a lower calculated AGE content than that of the low-fat, high-AGE dinner test meal evaluated by Poulsen(36). However, like Poulsen, we also found no significant change in serum CML following consumption of the LFHA diet. We did find an increase in CML following the HFHA meal, but we found no difference in CML by 24 h. Uribarri et al.(30) demonstrated increases in serum AGE at 90 min following a single AGE-rich beverage challenge, whereas we found no such effect at 1 or 2 h for either high-AGE dietary challenge. Stirban et al.(35) found significant decreases in adiponectin following a single high-AGE meal, but we found no such effect. In contrast, we found that CRP and adiponectin were very stable in the presence of both diets. Because CRP has a half-life of about 19 h and has stable concentrations over time, which are not thought to be influenced by fasting v. eating(58), one might not expect changes in CRP within such a short time period. However, synthesis rates of CRP can increase rapidly, thus producing strong, rapid shifts in CRP in response to inflammatory stimuli(58). Finally, Negrean et al.(28) reported an increase in CRP with the administration of a single very high-AGE test meal. AGE are theorised to increase inflammation via up-regulation of NFκB and TNFα(7,59); thus, one might theorise that these molecules would up-regulate low-grade inflammation rapidly in the presence of high AGE intake. However, we did not find any effect of either diet on hsCRP.
In addition, sRAGE decreased in response to both study diets. sRAGE is thought to act as a decoy receptor and bind to excess AGE. However, the decrease in sRAGE does not suggest an up-regulation of sRAGE in response to a higher AGE dietary load.
It is not clear why with healthful fats included in both test diets, very similar micronutrient profiles and almost identical foods being purchased whenever possible, there would be a trend towards decrease in CML in the presence of the LFHA diet and a rise in CML after consumption of the HFHA diet. We suggest that this result could relate to delayed gastric emptying with the higher fat content of the HFHA diet. This finding suggests that serum CML may not be substantially influenced by dietary AGE content in healthy adults at all given such low absorption. Our results are supported by the findings of Semba et al.(37) who reported that estimation of dietary AGE using dietary records did not correlate with serum AGE levels. In this study, intake of foods thought to be high in AGE also did not correlate with serum CML. Finally, Semba et al.(42) recently published a longer trial of a well-controlled high-AGE diet in healthy adults and found no effect of dietary CML on inflammatory mediators or endothelial dysfunction.
This study is limited in its size, scope and methods. To uncover whether dietary AGE influences short-term circulating levels of AGE, feeding of radioactively labelled CML and other dietary AGE could be performed followed by several hours of postprandial monitoring. In addition, such a feeding protocol would need to be carried out in the context of various dietary modifications: high-fat, low-AGE; HFHA; LFHA; and low-fat, low-AGE. Beyond the effect of macronutrient content on AGE absorption, the presence of dietary inhibitors of AGE formation in the diet would need to be evaluated for its impact on serum CML. How dietary AGE influences circulating AGE and markers of oxidative stress and inflammation in the context of diabetes as well as renal impairment also needs to be more closely assessed. Ideally, measurement of CML or other AGE in study diets should be performed via HPLC. The work of food scientists assessing how CML and other Maillard reaction products are influenced in food by cooking temperature, time, macronutrients and inhibitors indicates that the AGE content of foods is highly complex and is affected by the amount of oil and carbohydrate in a cooked food, heating temperature, heating time, acidity and the presence or absence of polyphenols(15,16,19). As complex as this reaction may be in foods, we should not suppose that the digestion, absorption and physiology of AGE in the body is any less complex.
Our results suggest that at least in the very short term, dietary CML alone is not very important to serum CML, sRAGE, CRP and HMW adiponectin. Thus, there is insufficient evidence at this time to recommend the public to modify their intake of AGE to improve health status. Before such recommendations could be made, we need reliable methods of estimating AGE content of foods, knowledge of which AGE have the greatest physiological impact on health outcomes (if any) and how cooking and food preparation methods affect formation of AGE.
Acknowledgments
This study is partially supported by NIH NCATS Clinical and Translational Science Award UL1TR001105.
K. E. D. designed the study including the menu, prepared the foods, ran the biochemical analyses, initially ran and reviewed the statistics and prepared the manuscript. C. P. consulted on the study design, reviewed the statistics and manuscript and provided editorial assistance. P. V. consulted on the study design, providing technical assistance on the laboratory analyses, reviewed the statistics and manuscript and provided editorial assistance. S. J. consulted on the study design, reviewed the statistics and manuscript and provided editorial assistance. B. A.-H. ran the statistics and provided editorial assistance in preparing the manuscript. V. I. consulted on the study design, particularly the dietary arm, reviewed the statistics and manuscript, provided editorial support and assisted with obtaining IRB approval.
Footnotes
The authors declare that there are no conflicts of interest.
References
- 1.Cordain L, Eaton SB, Sebastian A, et al. Origins and evolution of the Western diet: health implications for the 21st century. Am J Clin Nutr. 2005;81:341–354. doi: 10.1093/ajcn.81.2.341. [DOI] [PubMed] [Google Scholar]
- 2.Kalousova M, Zima T, Tesar V, et al. Advanced glycooxidation end products in chronic disease – clinical chemistry and genetic background. Mutat Res. 2005;579:37–46. doi: 10.1016/j.mrfmmm.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 3.Vlassara H, Striker GE. AGE restriction in diabetes mellitus: a paradigm shift. Nat Rev Endocrinol. 2011;7:426–539. doi: 10.1038/nrendo.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Semba RD, Nicklett EJ, Ferrucci L. Does accumulation of advanced glycation end products contribute to the aging phenotype? J Gerontol A Biol Sci Med. 2010;65:963–975. doi: 10.1093/gerona/glq074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barlovic DP, Soro-Paavonen A, Jandeleit-Dahm KA. RAGE biology, atherosclerosis and diabetes. Clin Sci. 2011;121:43–55. doi: 10.1042/CS20100501. [DOI] [PubMed] [Google Scholar]
- 6.Cai W, He J-C, Zhu L, et al. Oral glycotoxins determine the effects of calorie restriction on oxidant stress, age-related diseases, and lifespan. Am J Pathology. 2008;173:327–336. doi: 10.2353/ajpath.2008.080152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 8.Requena JR, Ahmed MU, Fountain CW, et al. Carboxyethylethanolamine, a biomarker of phospholipids modification during the Maillard reaction in vivo. J Biol Chem. 1997;272:17453–17479. doi: 10.1074/jbc.272.28.17473. [DOI] [PubMed] [Google Scholar]
- 9.Sgarbieri VC, Amaya J, Tanaka M, et al. Nutritional consequences of the Maillard reaction: amino acid availability from fructose-lysine and fructose-tryptophan in the rat. J Nutr. 1973;103:657–663. doi: 10.1093/jn/103.5.657. [DOI] [PubMed] [Google Scholar]
- 10.Koschinsky T, He C-J, Mitsuhashi T, et al. Orally absorbed reactive glycation products (glycotoxins): an environmental risk factor in diabetic nephropathy. Proc Natl Acad Sci. 1997;94:6474–6479. doi: 10.1073/pnas.94.12.6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raucci A, Cugusi S, Antonelli A, et al. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10) FASEB J. 2008;22:3716–3727. doi: 10.1096/fj.08-109033. [DOI] [PubMed] [Google Scholar]
- 12.Kalea AZ, Schmidt AM, Hudson BI. Alternative splicing of RAGE: roles in biology and disease. Front Biosci. 2011;16:2756–2770. doi: 10.2741/3884. [DOI] [PubMed] [Google Scholar]
- 13.Hull GL, Woodside JV, Ames J, et al. N-carboxymethyl lysine content of foods commonly consumed in a Western-style diet. Food Chem. 2012;131:170–174. [Google Scholar]
- 14.Uribarri J, Woodruff S, Goodman S, et al. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J Am Diet Assoc. 2010;110:911–916. doi: 10.1016/j.jada.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delgado-Andrade C, Seiquer I, Haro A, et al. Development of the Maillard reaction in foods cooked by different techniques: intake of Maillard-derived compounds. Food Chem. 2010;122:145–153. [Google Scholar]
- 16.Srey C, Hull GLJ, Connolly L, et al. Effect of inhibitor compounds on n-epsilon-carboxymethyllysine and n-epsilon carboxyethyllysine formation in model foods. J Agric Food Chem. 2010;58:12036–12041. doi: 10.1021/jf103353e. [DOI] [PubMed] [Google Scholar]
- 17.Delgado-Andrade C, Seiquer I, Navarro MP, et al. Maillard reaction indicators in diets usually consumed by the adolescent population. Mol Nutr Food Res. 2007;51:341–351. doi: 10.1002/mnfr.200600070. [DOI] [PubMed] [Google Scholar]
- 18.Goldberg T, Weijing C, Peppa M, et al. Advanced glycoxidation end products in commonly consumed foods. J Am Diet Assoc. 2004;104:1287–1291. doi: 10.1016/j.jada.2004.05.214. [DOI] [PubMed] [Google Scholar]
- 19.Poulsen MW, Hedegaard RV, Andersen JM, et al. Advanced glycation endproducts in food and their effects on health. Food and Chem Toxicol. 2013;60:10–37. doi: 10.1016/j.fct.2013.06.052. [DOI] [PubMed] [Google Scholar]
- 20.Uribarri J, Cai W, Sandu O, et al. Diet-derived advanced glycation end products are major contributors to the body’s AGE pool and induce inflammation in healthy subjects. Ann N Y Acad Sci. 2005;103:461–466. doi: 10.1196/annals.1333.052. [DOI] [PubMed] [Google Scholar]
- 21.Ames JM. Evidence against dietary advanced glycation endproducts being a risk to human health. Mol Nutr Food Res. 2007;51:1085–1090. doi: 10.1002/mnfr.200600304. [DOI] [PubMed] [Google Scholar]
- 22.Drusch S, Faist V, Erbersdobler HF, et al. Determination of N-carboxymethyllysine in milk products by a modified reversed phase HPLC method. Food Chem. 1999;65:547–553. [Google Scholar]
- 23.Assar SH, Moloney C, Lim M, et al. Determination of N epsilon (carboxymethyl) lysine in food systems by ultra performance liquid chromatography-mass spectrometry. Amino Acids. 2009;36:317–326. doi: 10.1007/s00726-008-0071-4. [DOI] [PubMed] [Google Scholar]
- 24.Cai W, Gao Q-D, Zhu L, et al. Oxidative stress-inducing carbonyl compounds from common foods: novel mediators of cellular dysfunction. Mol Med. 2002;8:337–346. [PMC free article] [PubMed] [Google Scholar]
- 25.Vlassara H, Cai W, Crandall J, et al. Inflammatory mediators are induced by dietary glycotoxins, a major risk factor for diabetic angiopathy. Proc Natl Acad Sci. 2002;99:15596–15602. doi: 10.1073/pnas.242407999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandu O, Song K, Cai W, et al. Insulin resistance and type 2 diabetes in high-fat fed mice are linked to high glycotoxin intake. Diabetes. 2005;54:2314–2319. doi: 10.2337/diabetes.54.8.2314. [DOI] [PubMed] [Google Scholar]
- 27.Diamanti-Kandarakis E, Piperi C, Kalafoutis A, et al. Increased levels of serum advanced glycation end-products in women with polycystic ovarian syndrome. Clin Endocrinol. 2005;62:37–43. doi: 10.1111/j.1365-2265.2004.02170.x. [DOI] [PubMed] [Google Scholar]
- 28.Negrean M, Stirban A, Stratmann B, et al. Effects of low and high advanced glycation endproduct meals on macro and microvascular endothelial function and oxidative stress in patients with type 2 diabetes mellitus. Am J Clin Nutr. 2007;85:1236–1243. doi: 10.1093/ajcn/85.5.1236. [DOI] [PubMed] [Google Scholar]
- 29.Stirban A, Negrean M, Götting C, et al. Dietary advanced glycation endproducts and oxidative stress. Ann N Y Acad Sci. 2008;1126:276–279. doi: 10.1196/annals.1433.042. [DOI] [PubMed] [Google Scholar]
- 30.Uribarri J, Negrean M, Sander D, et al. Single oral challenge by advanced glycation end products acutely impairs endothelial function in diabetic and non diabetic subjects. Diabetes Care. 2007;30:2579–2582. doi: 10.2337/dc07-0320. [DOI] [PubMed] [Google Scholar]
- 31.Vlassara H, Cai W, Goodman S, et al. Protection against loss of innate defenses in adulthood by low advanced glycation products (AGE) intake: role of the anti-inflammatory AGE receptor-1. J Clin Endocrinol Metab. 2009;94:4483–4491. doi: 10.1210/jc.2009-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harcourt B, Sourris KC, Coughlan MT, et al. Targeted reduction of advanced glycation improves renal function in obesity. Kidney Int. 2011;80:190–198. doi: 10.1038/ki.2011.57. [DOI] [PubMed] [Google Scholar]
- 33.Uribarri J, Cai W, Ramdas M, et al. Restriction of advanced glycation end products improves insulin resistance in type 2 diabetes: potential role of AGER1 and SIRT1. Diabetes Care. 2011;34:1610–1616. doi: 10.2337/dc11-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan KC, Chow WS, Tam S, et al. Association between acute-phase reactants and advanced glycation end products in type 2 diabetes. Diabetes Care. 2004;27:223–228. doi: 10.2337/diacare.27.1.223. [DOI] [PubMed] [Google Scholar]
- 35.Stirban A, Negrean M, Stratmann B, et al. Benfotiamine prevents macro and microvascular endothelial dysfunction and oxidative stress following a meal rich in advanced glycation end products in individuals with type 2 diabetes. Diabetes Care. 2006;29:2064–2071. doi: 10.2337/dc06-0531. [DOI] [PubMed] [Google Scholar]
- 36.Poulsen MW, Bak MJ, Andersen JM, et al. Effect of dietary advanced glycation end products on postprandial appetite, inflammation, and endotehthelial activation in healthy, overweight adults. Eur J Nutr. 2014;53:661–672. doi: 10.1007/s00394-013-0574-y. [DOI] [PubMed] [Google Scholar]
- 37.Semba RD, Ang A, Talegawkar S, et al. Urinary carboxymethyl-lysine, a major advanced glycation end product in adults: the Energetics Study. Eur J Clin Nutr. 2012;66:3–9. doi: 10.1038/ejcn.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Semba RD, Arab L, Sun K, et al. Fat mass is inversely associated with serum carboxymethyl-lysine, an advanced glycation end product, in adults. J Nutr. 2011;141:1726–1730. doi: 10.3945/jn.111.143172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ueda S, Yamagishi S, Matsui T, et al. Serum levels of advanced glycation end products (AGEs) are inversely associated with the number and migratory activity of circulating endothelial progenitor cells in apparently healthy subjects. Cardiovasc Ther. 2011;30:249–254. doi: 10.1111/j.1755-5922.2011.00264.x. [DOI] [PubMed] [Google Scholar]
- 40.Tahara N, Yamagishi S, Takeuchi M, et al. Positive association between serum level of glyceraldehyde-derived advanced glycation end products and vascular inflammation evaluated by [18F] Fluorodeoxyglucose positron emission tomography. Diabetes Care. 2012;35:2618–2625. doi: 10.2337/dc12-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luevano-Contreras C, Garay-Sevilla ME, Wrobel K, et al. Dietary advanced glycation end products diminishes inflammation markers and oxidative stress in patients with type 2 diabetes mellitus. J Clin Biochem Nutr. 2013;52:22–26. doi: 10.3164/jcbn.12-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Semba RD, Gebauer SK, Baer DJ, et al. Dietary intake of advanced glycation end products did not affect endothelial function and inflammation in healthy adults in a randomized controlled trial. J Nutr. 2014;144:1037–1042. doi: 10.3945/jn.113.189480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Diamanti-Kandarakis E, Piouka A, Livadas S, et al. Antimullerian hormone is associated with advanced glycosylated end products in lean women with polycystic ovary syndrome. Eur J Endocrinol. 2009;160:847–853. doi: 10.1530/EJE-08-0510. [DOI] [PubMed] [Google Scholar]
- 44.Yamagishi S, Adachi H, Nakamura K, et al. Positive association between serum levels of advanced glycation end products and the soluble form of receptor for advanced glycation end products in nondiabetic subjects. Metabolism. 2006;55:1227–1231. doi: 10.1016/j.metabol.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 45.Davis KE, Prasad C, Vijayagopal P, et al. Serum soluble receptor for advanced glycation end products correlates inversely with measures of adiposity in young adults. Nutr Res. 2014;34:478–485. doi: 10.1016/j.nutres.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 46.Selvin E, Halushka M, Rawlings AM, et al. sRAGE and risk of diabetes, cardiovascular disease and death. Diabetes. 2013;62:2116–2121. doi: 10.2337/db12-1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giorgis T, D’Adamo E, Gianni C, et al. Could receptors for advanced glycation end products be considered cardiovascular risk markers in obese children? Antioxid Redox Signal. 2012;17:187–191. doi: 10.1089/ars.2012.4525. [DOI] [PubMed] [Google Scholar]
- 48.Vazzana N, Guagnana MT, Cuccurullo C, et al. Endogenous secretory RAGE in obese women: association with platelet activation and oxidative stress. J Clin Endocrinol Metab. 2012;97:E1726–E1730. doi: 10.1210/jc.2012-1473. [DOI] [PubMed] [Google Scholar]
- 49.Norata GD, Garlaschelli K, Grigori L, et al. Circulating soluble receptor for advanced glycation end products is inversely associated with body mass index and waist/hip ratio in the general population. Nutr, Metab, Cardiovasc Dis. 2009;19:129–134. doi: 10.1016/j.numecd.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 50.Koyama H, Shoji T, Yokoyama H, et al. Plasma level of endogenous secretory RAGE is associated with components of the metabolic syndrome and atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:2587–2593. doi: 10.1161/01.ATV.0000190660.32863.cd. [DOI] [PubMed] [Google Scholar]
- 51.Ames J. Determination of N-(carboxymethyl) lysine in foods and related systems. Ann N Y Acad Sci. 2008;1126:20–24. doi: 10.1196/annals.1433.030. [DOI] [PubMed] [Google Scholar]
- 52.Mittelmaier S, Pischetsrieder M. Multistep ultrahigh performance liquid chromatography/tandem mass spectrometry analysis for untargeted quantification of glycating activity and identification of most relevant glycation products. Anal Chem. 2011;83:9660–9668. doi: 10.1021/ac2025706. [DOI] [PubMed] [Google Scholar]
- 53.Tessier FJ, Birlouez-Aragon I. Health effects of dietary Maillard reaction products: the results of ICARE and other studies. Amino Acids. 2012;42:1119–1131. doi: 10.1007/s00726-010-0776-z. [DOI] [PubMed] [Google Scholar]
- 54.Hanssen NMJ, Engelen L, Ferreira I, et al. Plasma levels of advanced glycation endproducts Nε-(carboxymethyl)lysine, Nε(carboxyethyl)lysine, and pentosidine are not independently associated with cardiovascular disease in individuals with or without type 2 diabetes: the Hoorn and CODAM studies. J Clin Endocrinol Metab. 2013;98:E1369–E1373. doi: 10.1210/jc.2013-1068. [DOI] [PubMed] [Google Scholar]
- 55.Piroddi M, Palazzetti I, Quintaliani G, et al. Circulating levels and dietary intake of the advanced glycation end product marker CML in chronic kidney disease patients on a conservative predialysis therapy: a pilot study. J Ren Nutr. 2011;21:329–339. doi: 10.1053/j.jrn.2010.06.024. [DOI] [PubMed] [Google Scholar]
- 56.Birlouez-Aragon I, Saavedra G, Tessier FJ, et al. A diet based on high-heat-treated foods promotes risk factors for diabetes mellitus and cardiovascular diseases. Am J Clin Nutr. 2010;91:1220–1226. doi: 10.3945/ajcn.2009.28737. [DOI] [PubMed] [Google Scholar]
- 57.Wu CH, Huang SM, Lin J-A, et al. Inhibition of advanced glycation endproduct formation by foodstuffs. Food Funct. 2011;2:224–234. doi: 10.1039/c1fo10026b. [DOI] [PubMed] [Google Scholar]
- 58.Pepys MB, Hirschfield GM. C-reactive protein: a critical update. J Clin Invest. 2003;111:1805–1812. doi: 10.1172/JCI18921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramasamy R, Vannucci SJ, Yan SSD, et al. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration and inflammation. Glycobiology. 2005;15:16R–28R. doi: 10.1093/glycob/cwi053. [DOI] [PubMed] [Google Scholar]