

Abstract
We report herein a facile and efficient method of the construction of the cis-1,2-oxazadecaline system, distinctive of (pre)trichodermamides, aspergillazine A, gliovirin and FA-2097. The formation of the 1,2-oxazadecaline core was accomplished by a 1,2-addition of an αC-lithiated O-silyl ethyl pyruvate oxime to benzoquinone, that is followed by an oxa-Michael ring-closure. The method was successfully applied to the concise total synthesis of trichodermamide A (in gram quantities), trichodermamide B, as well as the first synthesis of trichodermamide C.
The 1,2-oxazadecaline framework is a recurring structural motif of a number of secondary metabolites produced by terrestrial and marine fungi. Examples include trichodermamides A, B, and C (1–3) from Trichoderma virens (A1,2 and B1a) and Eupenicillium sp. (C),3 aspergillazine A (4) from Aspergillus unilateralis,4 as well as the unusual seven-membered epidithiodiketopiperazines pretrichodermamide A (5) from Trichoderma5 and Aspergillus6 spp., N-methylpretrichodermamide B (6) and pretrichodermamide C (7) from Penicillium sp.,7 gliovirin (8) from Trichoderma virens,8 and FA-2097 (N-methylgliovirin, 9) from Eupenicillium abidjanum.9 In addition, the structurally related aspergillazines BE (10–13) from A. unilateralis are presumed to arise from the reductive N–O bond cleavage of aspergillazine A (for B and C) and trichodermamide A (for D and E).4 The structural similarity and co-isolation of aspergillazines, trichodermamides, pretrichodermamides and gliovirin, as well as the facile conversion5 of 5 to 1 suggest a common biogenetic origin of the naturally-occurring 1,2-oxazadecalines.10 The bioactivity of these fungal metabolites remains largely unexplored, primarily due to their scarcity. However, preliminary data attest to their potential as lead compounds for antibiotic and anticancer drug discovery that would be enabled by an efficient synthetic access. For example, gliovirin is a potent inhibitor of the expression of pro-inflammatory enzymes (COX-2, iNOS) and cytokines (TNF-α, IL-2) in T-cells and monocytes/macrophages,11 and was linked to the efficacy of T. virens as a commercial biocontrol agent of several pathogenic fungi.12 FA-2097 (9) is highly active against several drug-resistant anaerobic bacteria, especially Fusobacterium and Bacteroides spp. Both trichodermamides B and C were shown to display significant cytotoxicity towards the human colorectal carcinoma HCT116 cells (IC50 = 0.32 and 0.68 μg/ml, respectively). Significant cytotoxicity was also reported for N-methylpretrichodermamide B. Curiously, trichodermamide A was shown to be completely inactive, indicating that C6-chloro and N-methyl groups may be important for the activity of these compounds.
A notable structural feature common to trichodermamides, pretrichodermamides and gliovirins is the unique and highly functionalized 1,2-oxazadecaline core containing four contiguous stereogenic centers. The synthetically challenging structure and the promising biological activity have attracted significant attention to these secondary metabolites,13 albeit only trichodermamides A and B have been synthesized to date, by Zakarian and Lu (B),14 employing the oxaza-Cope rearrangement,15 and by Joulié and Wan (A and B), stereospecifically from (–)-quinic acid.16
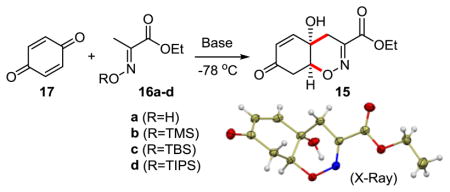
Herein, we report a novel, scalable approach to the construction of the 1,2-oxazadecaline ring system and its application to the concise total synthesis of trichodermamides A, B and C. We envisioned that the synthesis of trichodermamides and the related natural products can be greatly simplified by developing an early-stage 1,2-oxazadecaline core synthesis comprising a 1,2-addition of the C-terminus of the dianionic synthon 14 to benzoquinone, followed by an intramolecular oxa-Michael ring-closure en route to cis-fused bicyclic enone 15 (Scheme 1). Although such a synthesis of the cis-fused 1,2-oxazadecaline system has not, to our knowledge, been reported in the literature, the precedents of 1,2-addition of αC-mono- and αC,O-bislithiated acetophenone oximes to ketones,17 the efficiency of this approach, and the ready availability of benzoquinone and ethyl pyruvate, made it an attractive direction for investigation.
Scheme 1.

Retrosynthetic Analysis of Trichodermamides
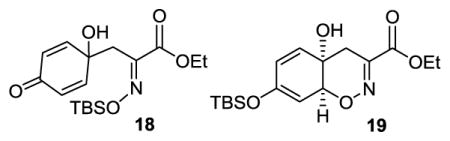
Our initial experiments were met with limited success, as ethyl pyruvate oxime (16a) and benzoquinone (17) did not produce enone 15 under a variety of reaction conditions (Table 1). We then turned our attention to O-silyl oximes 16b–d. While O-TMS and O-TIPS oximes 16b and 16d were ineffective, O-TBS oxime 16c afforded enone 15 in 92% yield with 2 equiv. LiTMP, and in 34% yield with 1 equiv. LiTMP (entries 5 and 6), indicating that 2 equiv. base was required to overcome the coordination of the lithium base to the oxime.17d LiTMP proved to be the base of choice, as no or very little product was observed with other bases. Analysis of the crude reaction mixture by 1H NMR spectroscopy prior to quenching with acetic acid revealed presence of quinol 18 and silyl enol ether 19, along with enone 15, suggesting that 18 and 19 may be intermediates en route to 15. The structure of enone 15 was confirmed by a single crystal X-ray crystallographic analysis. Further, the reaction was successfully scaled up to 10 g of oxime 16c, setting the stage for the synthesis of trichodermamides.
Table 1.
Construction of the 1,2-Oxazadecaline Core of Trichodermamides from Benzoquinone and Ethyl Pyruvate Oximes 16a–d
| |||
|---|---|---|---|
| Entry | Oxime | Base | Yield, %a |
| 1 | 16a | LDA | – |
| 2 | 16a | LHMDS | – |
| 3 | 16a | LiTMP | – |
| 4 | 16b | LiTMP | – |
| 5 | 16c | LiTMP | 92 |
| 6b | 16c | LiTMP | 34 |
| 7c | 16c | LiTMP | 88 |
| 8 | 16c | KHMDS | 11 |
| 9 | 16c | LiHMDS | 6 |
| 10 | 16d | LiTMP | <5 |
Reaction conditions: oxime 16 (1 mmol), 17 (1 mmol), base (2 equiv.) THF (c = 0.16M), −78 °C.
1 equiv of LiTMP was used.
Reaction was carried out on a 40.8 mmol (10 g of oxime 16c) scale.
Our synthesis of trichodermamide A commenced from enone 15, which was subjected to a modified Luche reduction that, under the optimized conditions, was carried out with potassium borohydride to improve the stereoselectivity, and in the presence of acetic acid to suppress polymerization of the allylic alcohol. Methyl and ethyl carbonates 20a and 20b were then prepared in 96% and 85% yields. The trans-configuration of the 1,4-dioxyalkene unit was confirmed by single crystal X-ray crystallographic analysis of 20a. Treatment of carbonate 20b with 5 mol % Pd(PPh3)4 in the presence of N,O-bis(trimethylsilyl)acetamide (BTSA)18 delivered dienol 21 in 90% yield. Installation of the critical C9–OH stereocenter necessitated a regio- and stereoselective epoxidation of the distal double bond of the dienol. While 10 mol % Ti(OiPr)4/tBuOOH afforded a 10 : 1 ratio of the proximal and distal epoxides 22a and 22b, a 1 : 1 ratio was observed with 10 mol % VO(acac)2/PhCMe2OOH. After additional optimization a 1 : 3 ratio of the proximal and distal epoxides 22a and 22b was obtained with 10 mol % N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-ethylenediaminomanganese(III) chloride (23) and iodosobenzene. The epoxidation occurred with complete stereoselectivity at the less hindered convex face of the dienol. The distal epoxide 22b was converted to selenide 24 in a high yield (95%) on treatment with phenylselenol and NaHCO3. The sensitivity of selenide 24 to acid and base necessitated saponification under mild conditions. Ultimately, we found that exposure of 24 to sodium trime-thylsilanolate19 followed by careful neutralization with MsOH resulted in a clean cleavage of the ethyl ester. The subsequent amide coupling with aminocoumarin 25a was effected by HATU in the presence of sym-collidine in a high yield. Surprisingly, the related N-methylaminocoumarin 25b proved resistant to amide coupling under a variety of conditions, indicating that an alternative strategy would be required for the synthesis of trichodermamide C. Oxidation of amide 26 to intermediate selenoxide triggered the [2,3]-sigmatropic rearrangement20 that delivered trichodermamide A (1) in a high yield. The brevity of the synthetic route enabled preparation of 1.1 g of trichodermamide A in 8 steps from enone 15 without the use of protecting groups and with only two chromatographic purifications. The selenoxide [2,3]-sigmatropic rearrangement was previously used by Zakarian and Lu in their synthesis of trichodermamide B.14
We next turned our attention to trichodermamides B and C (Scheme 3). Amide 27 that was envisioned as a common intermediate for both synthetic targets was accessed by a sequence of the ethyl ester cleavage with TMSONa and a HATU-mediated amide coupling with aminocoumarine 25a. A completely regio- and diastereoselective epoxidation of the proximal C6–C7 double bond with peracetic acid delivered epoxide 28 in a nearly quantitative yield. Curiously, Pd(PPh3)4-catalyzed reaction of epoxide 28 with phenylselenol proceeded with overall inversion of configuration and afforded the undesired trans-selenohydrin 29 in a 96% yield, contrary to the observed retention of configuration in the Pd-catalyzed reaction of phenylselenol with an unhindered allylic carbonate.21 Epoxide 28 was, therefore, first treated with Li2CuBr4 to give the corresponding trans-bromohydrin that, on treatment with phenylselenol and 1,8-bis(dimethylamino)naphthalene (30), and subsequent tosylation, afforded the desired cis-selenotosylate 31 in a 68% yield over three steps. The selenoxide [2,3]-sigmatropic rearrangement was induced by the oxidation of selenide 31. Finally, a nucleophilic displacement of the tosylate with CaCl2 in DMSO afforded trichodermamide B (2) in 11 steps from ketone 15.
Scheme 3. Synthesis of Trichodermamides B and Ca.

aReagents and conditions: a. TMSONa, CH2Cl2, 3 Å MS, then MsOH, MeOH; b. 25a, HATU, sym-collidine, DMF; c. MeI, 18-crown-6, K2CO3, acetone, 40 °C; d. N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-ethylenediaminomanganese(III) chloride (23) (10 mol %), PhIO, CH2Cl2; e. PhSeH, K2CO3, THF, 0 °C; f. H2O2, pyridine, THF, 0 °C; g. AcOOH, NaHCO3, CH2Cl2; h. Pd(PPh3)4 (10 mol %), PhSeH, PhMe; i. Li2CuBr4, THF; j. PhSeH, 1,8-bis(dimethylamino)naphthalene (Proton Sponge®, 30), THF, 0 °C; k. Ts2O, pyridine, 0 °C; l. H2O2, pyridine, THF, 0 °C; m. CaCl2, DMSO.
For the synthesis of trichodermamide C amide 27 was first subjected to the N-methylation that under the optimized conditions proceeded in 95% yield with iodomethane in the presence of 18-crown-6 and K2CO3. The Mn(salen)-catalyzed epoxidation of the distal C8–C9 double bond was followed by a trans-selective ring-opening of the intermediate distal allylic epoxide with phenylselenol. The oxidatively-induced selenoxide rearrangement completed the first synthesis of trichodermamide C (3) in 9 steps from enone 15.
In conclusion, we have developed a new gram-scale synthesis of the cis-fused 1,2-oxazadecaline enone 15 from readily available starting materials, ethyl pyruvate and benzoquinone, and successfully applied it to the short syntheses of trichodermamides A, B and C. The strategies described herein should prove useful for the future synthesis of related 1,2-oxazadecaline natural products.
Supplementary Material
Figure 1.

1,2-Oxazadecaline Fungal Metabolites.
Scheme 2. Synthesis of Trichodermamide Aa.

aReagents and conditions: a. KBH4, CeCl3, AcOH/THF (1:1), 0 °C; b. MeOC(O)Cl or EtOC(O)Cl, pyridine, PhMe/CH2Cl2 (2 : 1); c. Pd(PPh3)4 (10 mol %), N,O-bis(trimethylsilyl)acetamide (BTSA), PhMe; d. N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-ethylenediaminomanganese(III) chloride (23) (10 mol %), PhIO, cyclohexane/PhCF3 (1:1); e. PhSeH, NaHCO3, THF, 0 °C; f. TMSONa, CH2Cl2, 3 Å MS, then MsOH; g. 25a or 25b, HATU, sym-collidine, DMF; h. H2O2, pyridine, THF, 0 °C.
Acknowledgments
Financial support by the NIGMS (SC3GM105579), the Welch Foundation (AX-1788), the Max and Minnie Tomerlin Voelcker Fund, and UTSA is gratefully acknowledged. Mass spectroscopic analysis was supported by a grant from the NIMHD (G12MD007591).
Footnotes
Notes
The authors declare no competing financial interests.
Experimental and spectral details for all new compounds and all reactions reported. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Garo E, Starks CM, Jensen PR, Fenical W, Lobkovsky E, Clardy J. J Nat Prod. 2003;66:423–426. doi: 10.1021/np0204390. [DOI] [PubMed] [Google Scholar]; (b) Li DL, Chen YC, Tao MH, Li HH, Zhang WM. Helv Chim Acta. 2012;95:805–809. [Google Scholar]
- 2.Pencillazine, isolated by Lin et al. in 2000, was later confirmed to be identical to trichodermamide A: Lin Y, Shao Z, Jiang G, Zhou S, Cai J, Vrijmoed LLP, Jones EBG. Tetrahedron. 2000;56:9607–9609.Shao CL, Wang CY, Gu YC, Cai JW, Deng DS, She ZG, Lin YC. Lett Org Chem. 2009;6:387–391.
- 3.Davis RA, Longden J, Avery VM, Healy PC. Bioorg Med Chem Lett. 2008;18:2836–2839. doi: 10.1016/j.bmcl.2008.03.090. [DOI] [PubMed] [Google Scholar]
- 4.Capon RJ, Ratnayake R, Stewart M, Lacey E, Tennant S, Gill HJ. Org Biomol Chem. 2005;3:123–129. doi: 10.1039/b413440k. [DOI] [PubMed] [Google Scholar]
- 5.Seephonkai P, Kongsaeree P, Prabpai S, Isaka M, Thebtaranonth Y. Org Lett. 2006;8:3073–3075. doi: 10.1021/ol061046l. [DOI] [PubMed] [Google Scholar]
- 6.Zhou Y, Debbab A, Mandi A, Wray V, Schulz B, Mueller WEG, Kassack MU, Lin WH, Kurtan T, Aly AH, Proksch P. Eur J Org Chem. 2013:894–906. [Google Scholar]
- 7.Orfali RS, Aly AH, Ebrahim W, Abdel-Aziz MS, Müller WEG, Lin WH, Daletos G, Proksch P. Phytochem Lett. 2015;11:168–172. [Google Scholar]
- 8.Stipanovic RD, Howell CR. J Antibiot. 1982;35:1326–1330. doi: 10.7164/antibiotics.35.1326. [DOI] [PubMed] [Google Scholar]
- 9.(a) Miyamoto C, Yokose K, Furumai T, Maruyama HB. J Antibiot. 1982;35:374–377. doi: 10.7164/antibiotics.35.374. [DOI] [PubMed] [Google Scholar]; (b) Yokose K, Nakayama N, Miyamoto C, Furumai T, Maruyama HB, Stipanovic RD, Howell CR. J Antibiot. 1984;37:667–669. doi: 10.7164/antibiotics.37.667. [DOI] [PubMed] [Google Scholar]
- 10.Stipanovic RD, Howell CR, Hedin PA. J Antibiot. 1994;47:942–944. doi: 10.7164/antibiotics.47.942. [DOI] [PubMed] [Google Scholar]
- 11.Rether J, Serwe A, Anke T, Erkel G. Biol Chem. 2007;388:627–637. doi: 10.1515/BC.2007.066. [DOI] [PubMed] [Google Scholar]
- 12.Howell CR, Stipanovic RD. Can J Microbiol. 1983;29:321–324. [Google Scholar]
- 13.Magnus P, Mitchell IS. Tetrahedron Lett. 1998;39:9131–9134.Wan X, Doridot G, Joullié MM. Org Lett. 2007;9:977–980. doi: 10.1021/ol062993x.Donald JR, Edwards MG, Taylor RJK. Tetrahedron Lett. 2007;48:5201–5204.For a review, see: Gu Z, Saito T, Zakarian A. Chem Heterocycl Compd. 2012;48:11–16.
- 14.Lu C-D, Zakarian A. Angew Chem, Int Ed. 2008;47:6829–6831. doi: 10.1002/anie.200801652. [DOI] [PubMed] [Google Scholar]
- 15.Zakarian A, Lu C-D. J Am Chem Soc. 2006;128:5356–5357. doi: 10.1021/ja0609710. [DOI] [PubMed] [Google Scholar]
- 16.Wan X, Joullié MM. J Am Chem Soc. 2008;130:17236–17237. doi: 10.1021/ja807011b. [DOI] [PubMed] [Google Scholar]
- 17.(a) Henoch FE, Hampton KG, Hauser CR. J Am Chem Soc. 1969;91:676–681. [Google Scholar]; (b) Park CA, Beam CF, Kaiser EM, Kaufman RJ, Henoch FE, Hauser CR. J Heterocycl Chem. 1976;13:449–453. [Google Scholar]; (c) Hussein AQ, Jarrar AA, Madi AS. Heterocycles. 1989;29:1681–1687. [Google Scholar]; (d) Jarrar AA, Hussein AQ, Madi AS. J Heterocycl Chem. 1990;27:275–278. [Google Scholar]
- 18.(a) Tsuji J, Yamakawa T, Kaito M, Mandai T. Tetrahedron Lett. 1978:2075–2078. [Google Scholar]; (b) Trost BM, Verhoeven TR, Fortunak JM. Tetrahedron Lett. 1979:2301–2304. [Google Scholar]; (c) Trost BM, Lautens M, Peterson B. Tetrahedron Lett. 1983;24:4525–4528. [Google Scholar]
- 19.(a) Laganis ED, Chenard BL. Tetrahedron Lett. 1984;25:5831–5834. [Google Scholar]; (b) Lovrić M, Cepanec I, Litvić M, Bartolinčić A, Vinković V. Croat Chem Acta. 2007;80:109–115. [Google Scholar]
- 20.(a) Sharpless KB, Lauer RF. J Am Chem Soc. 1972;94:7154–7155. [Google Scholar]; (b) Sharpless KB, Young MW, Lauer RF. Tetrahedron Lett. 1973;22:1979–1982. [Google Scholar]; (c) Reich HJ. J Org Chem. 1975;40:2570–2572. [Google Scholar]
- 21.Waetzig SR, Tunge JA. Chem Commun. 2008:3311–3313. doi: 10.1039/b806949b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.