

Abstract
Heterocyclic N-oxides have emerged as potent compounds with anticancer, antibacterial, antihypertensive, antiparasitic, anti-HIV, anti-inflammatory, herbicidal, neuroprotective, and procognitive activities. The N-oxide motif has been successfully employed in a number of recent drug development projects. This review surveys the emergence of this scaffold in the mainstream medicinal chemistry with a focus on the discovery of the heterocyclic N-oxide drugs, N-oxide-specific mechanisms of action, drug-receptor interactions and synthetic avenues to these compounds. As the first review on this subject that covers the developments since 1950s to date, it is expected that it will inspire wider implementation of the heterocyclic N-oxide motif in the rational design of new medicinal agents.
Keywords: Heterocyclic N-oxides, nitric oxide mimics, bioisosteres, hypoxia, factor Xa inhibitors, coagulation cascade direct thrombin inhibitors, antichagasic agents, antimalarials
1. INTRODUCTION
1.1. Background
Small organic molecules have been the mainstay of medicine and biology both as therapeutic agents and as probes used to investigate complex biological processes. [1, 2] Groups of structurally similar chemical agents that exert influence on these processes can be described as “privileged scaffolds”. [3] Modification of these frameworks by replacement [4, 5] of functional groups with bioisosteric units, [6] often results in an adjustment of pharmacological and physico-chemical properties, and can ultimately lead to development of new drugs. [7] Heterocyclic N-oxidation of N-heterocyclic bioactive compounds is an excellent example, where introduction of small structural changes (in this case – N-oxidation) can lead to improvement of the desired properties, and in some cases to emergence of unrelated but beneficial therapeutic outcomes. [8] Historically, heterocyclic N-oxides were often perceived as side products of hepatic metabolism of N-heterocyclic therapeutic agents. [9] Emergence of heterocyclic N-oxides in the mainstream drug development is partly credited to the unusual discovery of antihypertensive and anti-alopecia agent minoxidil by Upjohn in the early 1960s. Since then, a growing number of antibacterial, [10] antiviral (anti-HIV), [11] anticancer, [12] anti-protozoal [13] and anti-fungal [14] heterocyclic N-oxides have reached advanced drug discovery stages. In addition to pharmaceutical applications, a number of heterocyclic N-oxides have found use as agrochemicals [15] and also in cosmetics industry. [16]
1.2. Scope of the review
This review focuses on the discovery, biological activities, synthesis and some pharmacological profiles of heterocyclic N-oxide drugs and related emerging bioactive compounds. Topics related to the central theme, such as structure-activity relationships, historical perspectives and drug-receptor interactions will also be discussed. The review is organized according to the various heterocyclic N-oxide drugs and provides insights into the drug development process, as well as the current status of these agents.
2. GENERAL MODES OF ACTION OF BIOACTIVE HETEROCYCLIC N-OXIDES
2.1. Principal modes of action
Although the initial medicinal chemistry of heterocyclic N-oxides dates back to 1950, the full implementation of this scaffold in drug discovery is still at its infancy. There are several emerging areas, where these compounds hold significant promise. In particular, heterocyclic N-oxides have emerged as valuable anticancer, antibacterial, antihypertensive, antiparasitic, anti-HIV, anti-inflammatory, herbicidal, neuroprotective, and procognitive agents. There are four principal modes of action that distinguish the heterocyclic N-oxide motif with respect to each therapeutic area. The details of each mode of action are discussed vide infra in the sections dedicated to the corresponding bioactive agents.
2.2. Nitric oxide (NO) mimics
Nitric oxide (NO) is a small molecule that has been implicated in a number of important processes in living systems. These processes (including neuromodulation as well as neurotransmission) have been the subject of drug discovery research for several decades. Chemical moieties that are isoelectronic to nitric oxide have been shown to elicit NO-like functions. [17] For instance the N–O moiety in a variety of heterocyclic N-oxides can elicit nitric oxide-like effects, acting as a nitric oxide agonist. For instance, just like NO, minoxidil and its derivatives are potent ATP-dependent potassium channel openers. [18] Hence, the vasodilating and hypertrichosis activities of these drugs have been explained by these effects, e.g. in the case of minoxidil known to induce opening of K-ATP channels, leading to vasodilation and hair growth stimulation. [19]
2.3. Bioisosteres of the carbonyl group
The relatively high electron density of the heterocyclic N-oxide oxygen makes these compounds strong hydrogen bond acceptors. As a consequence, a number of heterocyclic N-oxide motifs have been used as potent bioisosteric replacements for the carbonyl function. For instance in the cases of direct thrombin as well as p38 MAP kinase inhibitors, the heterocyclic N-oxide motifs were shown to be superior over their carbonyl counterparts. [20] X-ray crystallographic data suggest that the N–O group forms critical hydrogen bonding networks, which cause conformational changes at the active site that lead to allosteric modulation of the enzyme function. In the case of thrombin inhibitors, replacement of the pyrazinone group with a pyridine N-oxide moiety led to superior inhibitory activities. [21] This review discusses further examples of heterocyclic N-oxides as bioisosteres for the carbonyl group section (3.4, 3.7).
2.4. Hypoxic-selective cytotoxins
Certain heterocyclic N-oxides, e.g. tirapazamines and their derivatives have been shown to undergo selective reduction to highly reactive free radicals under the action of multiple reductases. [22] These free radicals confer inherent cytotoxicity through DNA strand breaks and/or topoisomerase poisoning. [23] Recent research offers evidence of involvement of both hydroxyl and benzotriazinyl radicals in this pathway. [24] This review discusses the involvement of heterocyclic N-oxides as free radical sources during hypoxic-selective DNA damage sections (3.6, 3.8, 3.9 and 3.10).
2.5. Nitric oxide donors
A number of heterocyclic N-oxides, e.g. furoxans and diazetine N-oxides can be regarded as masked NO donors. Thus, under thiol mediation, these molecules have been shown to produce nitric oxide. [25] The nitric oxide released by these agents has been linked to the activation of the soluble guanylyl cyclase-cGMP pathway. [26] Consequently, there is a growing number of reports on the potential of these agents in the area of cardiovascular diseases. [27] The NO produced by these agents has also been shown to elicit neuroprotective and procognitive effects. [26] Furthermore, it has been shown that the furoxan moiety could be electronically modulated to produce controlled dosage of nitric oxide, which can be critical for nitric oxide therapeutics, especially since different dosages of nitric oxide can potentially produce different responses. [25] This review discusses some aspects of the N-oxide-mediated NO generation and the SAR (structure–activity relationship) studies of the NO donor N-oxides sections (3.8).
3. CASE STUDIES OF HETEROCYCLIC N-OXIDE DRUGS AND EMERGING BIOACTIVE COMPOUNDS
3.1. Minoxidil and kopexil
3.1.1. Introduction
Minoxidil (1) and kopexil (2) are both pyrimidine N-oxide drugs, whose development is a clear example of serendipity. Minoxidil is currently used for the treatment of any form of alopecia, – a condition characterized by hair loss, usually due to androgenic miniaturization. Kopexil on the other hand, can be viewed as a structural analogue of minoxidil, and is also known to treat alopecia. However, it has not been approved by the FDA and in most European countries, and there are no indications that an approval has been sought. Chemically, minoxidil and kopexil are 2,4-diamino pyrimidine-3-N-oxide with minoxidil containing a piperidinyl unit at C-6 (Figure 1).
Figure 1.

Minoxidil and structural analogues
3.1.2. Discovery of minoxidil
The development of minoxidil (1) as a drug dates back to the early 1960. [28] Researchers at Upjohn searched for an active agent that could lower the blood pressure in animal models (rats and dogs). In this quest, a large number of compounds were screened and N,N-diallylmelamine (3) emerged as the original lead compound. The discovery of N,N-diallylmelamine as the lead compound is very unusual, as it was initially developed as an additive to reduce photodegradation in plastics. Subsequently, it was tested against gastric ulcers.
Although the performance of compound 3 in the gastric ulcer tests was mediocre, compound 3 was found to lower the blood pressure in both dogs and rats. Subsequent structural optimization of 3 led to simplification of the melamine scaffold by omission of one of the nitrogen atoms in the aromatic ring and introduction of piperidinyl moiety. Interestingly, the Upjohn team was intrigued by the time lag between administration of the simplified version and the onset of action of the drug. As a result, it was postulated that the effective ingredient is most probably a product of hepatic metabolism. Detailed biochemical experiments revealed a hepatic oxidative modification of pyrimidine backbone, which indeed, was responsible for the antihypertensive activity. However, the exact nature and location of the oxygenated group in this molecule was unknown at the time. The results led the introduction of various oxygenation patterns into the lead compound. After several years of experimentation, the antihypertensive activity of the molecule was linked to the O-sulfated metabolite 4. [29] However, this analogue was highly unstable and too difficult to synthesize at that time. [30] The closely related N-oxide 2, whose activities were comparable to the O-sulfated analogue 4 made it through clinical trials. The drug was so potent that its administration was limited only to those cases, where other vasodilators failed. In addition to its antihypertensive activity, several cases of unusual side effects were reported, such as severe hair growth at unusual locations. This unexpected effect led to investigation of minoxidil as a possible treatment of hair loss, and the results from clinical trials strongly supported the observation.
3.1.3. Pharmacological characteristics of minoxidil and kopexil
Minoxidil is well absorbed unchanged from the gastrointestinal tract with levels reaching a maximum after 1 h of oral administration and declining rapidly with a serum elimination half-life of 1.1–4.2 h. [31] The rate of clearance of unchanged minoxidil has been estimated at 1584 mL/min. Reports also show that approximately 67% of minoxidil is metabolized in the liver via conjugation to glucuronic acid (that is via the formation of minoxil-o-glucuronide). [32] Also, 12–20% of the drug is excreted renally in unchanged form. Multiple experiments have identified that the magnitude of antihypertensive activity by this drug is related to the hypertension present, which implies, the blood pressure response declines with reduced blood pressures and there is little to no response at pretreatment diastolic blood pressure of 85 mmHg. Hence, minoxidil is known to have negligible hypotensive activity in healthy individuals. Reports also show that the concentration of minoxidil does not correlate well with the magnitude or duration of the elicited action. This effect has been explained to be partly due to the persistence of the drug at the receptor sites. Also, despite peak plasma concentration levels within 1 h, peak blood pressure reduction occurs within 2–8 h after oral administration, with a slow recovery of the action within 11 h to 5 days. Because of this effect, the drug is usually prescribed once daily.
3.1.4. Mechanism of action of minoxidil
The mechanism of action of minoxidil has been debated for a period of more than 30 years. Several theories have been postulated to explain the differences in the duration between peak plasma level of the drug and the onset of the elicited action. This disparity has been correlated partly to the residence time of the drug at receptor sites and also to the formation and involvement of an active metabolite. Several experiments have identified the O-sulfated minoxidil to be the active metabolite. [33] The conversion of minoxidil to its O-sulfated analogue in rats has been ascribed to the action of at least four enzymes known as sulfo-transferases. The activity of these enzymes has been demonstrated in animal models (rats), as well as in human liver, [34] platelets, [35] and the epidermal keratinocytes. [36] There are also reports of concrete biochemical evidence of the sulfation of minoxidil by transferases, which were isolated from human scalp skin. [37] In addition, there are reports showing mRNA expression of the sulfo-transferases in human epidermal keratinocytes. [38] In clinical settings, the levels of these enzymes have been reported to be higher in men, who responded to minoxidil treatment compared to those, who did not respond. [39] Therefore, it is logical to conclude minoxidil sulfate is the active metabolite responsible for antihypertensive activity, as well as hypertrichosis activity observed after administration of minoxidil. In addition, minoxidil and its O-sulfate metabolites have been shown to elicit nitric oxide-like activities. For instance, these drugs are known ATP-sensitive K-channel openers. The function of these channels has been linked to the control and regulation of vasodilation. [40] Thus, the mechanism of action of minoxidil may involve hepatic or epidermal keratinocyte sulfo-transferases-mediated conversion of parent minoxidil to its sulfate metabolite, which in turn causes the opening of the plasma membrane adenosine triphosphate (ATP)-sensitive potassium channels (K-ATP channels). The opening of this channel leads to relaxation of the vascular smooth muscles, and hence propagation of vasodilation. In addition to the smooth muscles, potassium channels are also located in a variety of tissues and cell types including cells of the heart, pancreas, and the central nervous system, where they couple intracellular metabolic variations to the electrical activity of the plasma membrane. The K-ATP channels are also widely recognized to be involved in hypertrichosis (severe hair growth). Thus, upon topical administration, the sulfo-transferases of the epidermal keratinocyctes convert minoxidil to it sulfated analogue. The sulfated analogue then interact with the K-ATP channels leading to relaxation of the vascular smooth muscles or triggering a cascade of events via the central nervous system. [40] This vasodilation leads to the increase of oxygenation and nutrients levels in blood and at hair follicles. This in turn triggers shortening of the telogen phase (resting phase) leading to premature entry of the resting hair follicle into anagen (active phase), which then results in hair thickening and growth. [16] Given the structural similarity of kopexil to minoxidil, it is believed that the mode of action of kopexil is also related to ATP potassium channel activation possibly through the formation of kopexil sulfate, although no detailed mechanism of action of kopexil has been elucidated.
3.1.5. Current uses of minoxidil and kopexil
Minoxidil is direct-acting peripheral arteriolar vasodilator, which is used orally only in the cases of severe hypertension. More than 95% of all minoxidil sold today is used by the cosmetic industry principally for hair restoration. It is sold under the trade name Loniten, when used to treat hypertension and as Rogaine (which is a 2 or 5% solution in propylene glycol), when used to promote hair regrowth, especially in male-pattern baldness. Other trade names include acvogain, regaine, lipogaine, avacor physician’s formulation and vanarx. Kopexil, on the other hand, which is marketed under the trade amnexil is known to elicit similar antihypertensive and hypertrichotic effects as minoxidil.
3.1.6. Synthesis of minoxidil
There are a number of methods of assembly of the pyrimidine N-oxide core that is shared by minoxidil and kopexil. One of the synthetic routes to minoxidil reported by Upjohn involves conversion of cyanomalonate to piperidinyl cyanomalonamide 5 by direct amidation using piperidine. [41] The resulting amide could be treated with trimethyloxonium tetrafluoroborate to afford the corresponding enol ether. The reaction of this enol ether affords enamine 6 which could be converted to minoxidil (1) via treatment with hydroxylamine (Figure 2). Alternatively, minoxidil (1) was synthesized from barbiturate 7, which was first treated with phosphoryl chloride and then ammonia to afford chloropyrimidine 8. Compound 8 was then be oxidized to N-oxide 10 and reacted with piperidine to give minoxidil (1). [42] In a modification of the procedure, chloride 8 was first reacted with 2,4-dichlorophenol, and then with piperidine to afford desoxyminoxidil (9) that was subsequently oxidized to minoxidil. [43]
Figure 2.

3.2. Chlordiazepoxide
3.2.1. Introduction
Chlordiazepoxide is another important and early example of a heterocyclic N-oxide drug. Chlordiazepoxide belongs to the benzodiazepine drug family (Figure 3). Benzodiazepines are known for their remarkable psychoactive properties, which result from their interactions with the neurotransmitter gamma-aminobutyric acid (GABA) receptor. [44]
Figure 3.

General structure of the benzodiazepines and chlordiazepoxide.
Chlorodiazepoixde is the first discovered member of this class of drugs. Some of the other members include: chlorazepate, diazepam (valium), flurazepam, lorazepam, oxazepam adinazolam and midazolam. Chlordiazepoxide is the only heterocyclic N-oxide benzodiazepine. It is marketed under the trade names Elenium, Angirex, Libotryp Lubrium Mesural, Multum, Novapam, Rosolid, Silibrin, Sonimen and Tropium.
3.2.2. Discovery of chlordiazepoxide
The discovery of the benzodiazepines in general and chlordiazepoxide in particular is credited to Leo Henryk Sternbach, who was working at Hoffmann-La Roche’s Research facility in Nutley, New Jersey. [45] As a postgraduate student while in Poland in the early 1930s, Sternbach engaged in the synthesis of several heptoxdiazine compounds, – a research which was geared towards the development of synthetic dyes. Inspired by the tremendous success in the development of chlorpromazine and early reports on meprobamate, Sternbach resumed research on his heptoxdiazines hoping to contribute to the emerging area of psychopharmacology in the mid-1950s.
In this quest, he re-characterized one of his heptoxdiazines and made a library of this class of compounds. However, pharmacological assays of these compounds as tranquilizers and sedative agents were largely unsuccessful. Compounds 13 were then reacted with primary amines in an effort to synthesize their amine derivatives 14 (Figure 4). In the midst of this work, questions arose on the exact structural identity of compounds 13 and 14. Further characterization of compounds 13 and 14 revealed that indeed compound 13 and 14 were misassigned as benzoheptoxdiazines, but were rather analogues of quinazoline N-oxides 15 and 16 respectively. Additional set of experiments involving mass analysis unequivocally confirmed that compounds 13 and 14 were actually quinazoline N-oxides. With this new slew of compounds further biological tests were carried out; however, the results were disappointing in the sedative and tranquilizer screenings, and the project was suspended. In the spring of 1957, during a laboratory cleanup, Sternbach and co-workers found a few hundred milligrams of crystals in the crystallizing dish used in 1955, which have been left during their synthesis of compound 16. It also came to his recollection that this compound had not been tested in 1955. Since this compound was pure, it was submitted for testing, although with the anticipation of a negative result. Surprisingly, the compound showed interesting properties in six tests that were commonly used for tranquillizer and sedative screening. [46] Puzzled by the disparity of the UV and IR spectra data of this compound with the quinazoline N-oxides synthesized earlier, Sternbach and co-workers decided to use chemical degradation studies to determine the correct structure of this compound (Figure 5). The degradation studies of the compound were carried out by first reducing with phosphorous trichloride and then by treatment of the mixture with an acid. Result showed that the lead compound was not a quinazoline N-oxide but benzodiazepine-4-oxide 12.
Figure 4.

General structure of benzoheptodiazines and other intermediates encountered in the discovery of chlordiazepoxide.
Figure 5.

Degradation studies of chlordiazepoxide. [46]
These exciting results led to the synthesis of a new set of benzodiazepine N-oxides. Unfortunately, none of these analogues had a superior activity compared to the initial lead. Sternbach and co-workers also reported that chlordiazepoxide easily hydrolyzed, and the hydrolysis product had a comparable activity. Active research in this field in the past four decades has led to a discovery of several additional benzodiazepines, some of which have been approved for clinical use.
3.2.3. Current uses of chlordiazepoxide
Chlordiazepoxide is used in the treatment of anxiety disorders including obsessive-compulsive disorder (OCD), phobia, and panic attacks. It is also used in the treatment of acute alcohol withdrawal and as a short-term relieve for fear and anxiety before surgery.
3.2.4. Pharmacological characteristics of chlordiazepoxide
Chlordiazepoxide is the first and only heterocyclic N-oxide member of the benzodiazepine drug family in clinical use. When administered orally, chlordiazepoxide is rapidly and completely absorbed, while intramuscular injection results in slow and erratic adsorption. Approximately 96% of the drug binds to plasma in the first 8 hrs and plasma elimination half-life in healthy individuals ranges from 5 to 30 hours. It is metabolized into a complex mixture of biotransformed pharmacologically active, demoxepam and desmethyldiazepam. It is also known that the active metabolite (desmethyldiazepam) has an elimination half-life 36 to 300 hours. This feature makes chlordiazepoxide a long circulating drug. The drug is excreted with urine both in the unchanged (1–2%) and conjugate (3–6%) forms. Multiple dose therapy with chlordiazepoxide leads to accumulation of the parent, as well as the active metabolites. The rate and extent of the accumulation is known to vary considerably and increases with age. [47]
3.2.5. Mechanism of action of chlordiazepoxide and related benzodiazepines
It has been known for several decades that all benzodiazepines elicit their effect via a centralized mechanism, which involves the allosteric modulation of the GABAA receptor function (Figure 6). [48] To fully understand this mechanism it is important to describe the nature of the binding events within this receptor. γ-Amino-butyric acid (GABA) is the main inhibitory neurotransmitter that controls the central nervous systems. Its receptor known as the γ-aminobutytyric acid receptor complex (GABAA complex) consists of five protein subunits that rings a central chlorine ion channel. The majority of these receptor subunits are α, β and γ, which are known to have different isoforms. [49] It has been demonstrated that these isoforms are responsible for the different sensitivities of different benzodiazepines. [50] GABA binds to the β subunits and most benzodiazepines bind at the interface of the γ subunit and any of the α1, α 2, α3, or α5 subunits. [51] It is also known that benzodiazepines with affinity for α1 subunit elicit sedative activity while those with affinity for the α2, or α3, subunits produce anxiolytic effects. [52]
Figure 6.

γ-Aminobutyric acid (GABA) receptor complex
(Reprinted with permission from Ref. 53. Copyright 2008 Nature Publishing Group).
Some studies have also showed that binding to the α5 subunit plays a major role in memory processes. [54] The chloride ion channel is controlled by the GABA binding site on this receptor. When GABA binds to its receptor, a conformational change in this receptor favors the entry of chloride ions into the cell through the channel. This results in hyperpolarization of the cell membrane, consequently increasing the threshold and inhibitory effect on action potential. Thus, binding of chlordiazepoxide to the GABAA receptor complex (at the interface of the γ and any of α2, or α3) induces an allosteric modulation at the receptor sites, which locks the receptor into a conformation for optimal GABA binding. [55] This results in the amplification of the inhibitory effect hence dampened emotions and relaxation of muscles. [52]
3.2.6. Synthesis of chlordiazepoxide
The synthesis of chlordiazepoxide published by Sternbach and co-workers in 1961 is still used today for the synthesis of analogues of this compound (Figure 7). The synthesis starts with the reaction of 2-amino-5-chlorobenzophenone 21 with hydroxylamine to form oxime 22. Treatment of oxime 22 with chloroacetyl chloride in the presence of acetic acid produces quinazoline N-oxide 16, which was first observed during the development stages of this drug. Finally, methylamine treatment of 16 results in chlordiazepoxide through an interesting ring expansion.
Figure 7.

Synthetic route to chlordiazepoxide. [56]
3.3. Otamixaban: direct factor Xa inhibitor
3.3.1. Introduction
Otamixaban 26 is a small molecule experimental antithrombotic agent, discovered by Sanofi. It is used for the management of acute coronary syndrome (ACS). Like other thromboembolic diseases, ACS, is characterized by obstruction of the coronary arteries due to inappropriate formation of a thrombus. Otamixaban is known to elicit its effects through reversible direct inhibition of a critical serine protease coagulant factor Xa (FXa). [57] This coagulant factor is located at the confluence of two important pathways: the contact activation pathway (intrinsic) and the tissue factor pathway (extrinsic) of the blood coagulation cascade. FXa catalyzes activation of prothrombin to thrombin via the prothrombinase complex. The activation of thrombin potentiates a cascade of events leading to the formation of a blood clot. [58] Thus FXa is an important pharmacological target for modulating thrombosis in vertebrates. Chemically, otamixaban belongs to the class of amidine-containing direct FXa inhibitors, which are designed to mimic the arginine residue of the natural substrate prothrombin. This compound 26 contains a 4-carbomoylphenyl 4-pyridine N-oxide that is connected via a peptide bond to a benzamidinyl β-aminoester unit 26.
3.3.2. Discovery of otamixaban
The discovery of otamixaban as a direct inhibitor for factor Xa (FXa) started with the identification of β-lactam compound 23 and evolved through the stages of N-p-nitrobenzamido-β-amino acid 24, then the main lead compound 25, and, finally heterocyclic N-oxide 26 (otamixaban) that had superior properties and activity profiles (Figure 8). [8]
Figure 8.

Discovery of otamixaban (26).
3.3.3. Action on the blood coagulation cascade
Blood coagulation in vertebrates is a complex series of orchestrated processes involving plasma proteins, serine protease enzymes and cellular receptors that play dynamic roles in providing delicate balancing acts leading to thrombosis and hemostasis (Figure 9). [59] The interior of blood vessels are lined by cells of the endothelium. These cells are specially designed to execute two functions: to promote blood fluidity [60] or in the case of a tissue injury, they act to promote blood coagulation at the wound site through the mediation of a cascade of enzymes and cofactors, and also prevent excessive propagation of this coagulation beyond the site by a feedback regulatory mechanism. In the case of injury, vessel disruption or cytokine inflammatory stimulation, bleeding occurs. Hemostatic plug formation is initiated upon exposure of the subendothelium at the injured site. Vasoconstriction of this region occurs due to the release of a protein known as thromboxane A2 (a major enzymatic product from platelet activation, which causes vessel walls to constrict). [61] Another protein known as von Willebrand factor (VWF) which is secreted by the endothelial cells, binds to exposed collagen fibers of the site. Platelets then adhere to the site through their interaction with the bound von Willebrand factor. Additional platelets are activated, and deposit around the site by a process known as platelet aggregation. Fibrinogen molecules form bridges between adjacent platelets, which accrue the area until all bleeding stops. The unstable loosely aggregated platelets and fibrinogen must then be consolidated. This is achieved through generation of thrombin (via the coagulation cascade) at the injured site. A glycosylated protein known as tissue factor (TF), which is released from subendothelial stores and the platelets initiates the coagulation cascade. [62] When the tissue factor (TF) binds to another protein factor VII in a 1:1 ratio, an activated complex known as TF-FVIIa is formed via limited proteolysis. The tissue TF/VIIa complex can then convert factor X or factor IX to their corresponding activated serine proteases via cleavage of an activation peptide (i.e. factor Xa or factor IXa). This marks initiation of the coagulation cascade and once the pathway starts, activation of factor X is immediately stopped by an inhibitor produced by the endothelial cells, known as tissue factor pathway inhibitor (TFPI). [63] The activated factor IXa formed by TF/FVIIa now binds to its cofactor known as factor VIIIa, which is usually located on phospholipid surfaces. The binding of factor IXa to factor VIIIa results in further activation of factor X to factor Xa. The activation of FXa initiates what is known as the common pathway for thrombin activation. This factor Xa binds its cofactor, factor Va and together with calcium ions on phospholipid surfaces forms prothrombinase complex. [64] This enzyme complex catalyzes the conversion of prothrombin to thrombin by cleavage of an activation peptide. The formation of minute amounts of thrombin has been shown to be enough to start the coagulation mechanism. Thrombin can also be formed via an intrinsic mechanism, whereby small amount of thrombin activates factor XI to factor XIa. Then factor XIa and factor VIIIa can activate factor X to factor Xa. This model of the coagulation cascade explains how thrombin activates two important cofactors, – factor V and factor VIII. The activation of factors V and VIII by thrombin result in further amplification of the coagulation activity via increased activity of prothrombinase complex. Fibrinogen, the main structural protein element of blood clot, is produced in the liver as dimeric units composed of three pairs of protein chains Aα, Bβ and γ, known to be linked at their N-termini. [65] Fibrinogen contains three globular domains, a central E domain connected by two identical D domains. Thrombin from the above cascade cleaves small peptides on fibrinogen known as fribinopeptides A and B from the Aα, and Bβ chains respectively. This ultimately leads to the formation of fibrin monomers that assemble into half-staggered, side-to-side fashion (known as protofibrils) stabilized by nonbonding interactions between the fibrin molecules. The protofibrils merge laterally into thicker fibrin and form unstable fibrin clots. The clot is finally stabilized by the action of thrombin that activates factor XIII to factor XIIIa. Factor XIIIa then form a peptide bond between the glutamic acid and lysine side chains in fibrin to create some permanence in the fibrin fibers.
Figure 9.

A model of the coagulation cascade highlighting the otamixaban inhibition of factor Xa.
In addition, factor XIIIa covalently crosslinks plasminogen and antiplasmin (e.g alpha-2-antiplasmin) in forming a fibrin clot. [66] This feature of factor XIIIa is important in finalizing the wound healing and tissue repair process. There are regulatory mechanisms that limit and clear a blood clot at the injury sites after healing. The clot removal process known as fibrinolysis involves a zymogen (plasminogen), a number of activators and several inhibitors. One of the most important of these activators is the tissue plasminogen activator (tPA), a product of the endothelial cell. During coagulation, plasminogen produced in the liver is entrapped within the crosslinked clot. tPA and other enzymes convert plasminogen to plasmin, which interacts with antithrombin III resulting in the breakdown of the blood clot. [67]
The regulation of the coagulation cascade can also be initiated when thrombin binds to the endothelial cell receptor known as thrombomodulin. Once thrombin binds to thrombomodulin receptor, its ability becomes limited to two pathways, one of which is the protein C pathway. The thrombin/thrombomodulin receptor complex activates protein C to its active form. The active form of protein C together with its co-factor protein S and calcium ions deactivates factors Va and VIIIa. Once these factors are deactivated, any further clot growth is prevented, thus concluding the coagulation cascade. Individuals with disorders caused by malfunctioning of the regulatory process described above may develop one of the following thromboembolic diseases: pulmonary embolism (PE), acute coronary syndrome (ACS), and deep vein thrombosis (DVT). These disease conditions result from thrombus formation in the coronary arteries, pulmonary veins, and the deep veins respectively. At present, treatment of any form of thromboembolic disease includes intravenous heparin, low molecular weight heparins (LMWH), oral vitamin K antagonist (coumadin), intravenous glycoprotein IIb/IIIa antagonists, and direct thrombin inhibitors (argatroban and bivalirudin). These current therapies have certain limitations with respect to safety and efficacy. [68] As a result, development of alternative methods to combat these deadly diseases has been pursued. It is important to note that within the coagulation cascade, the only entry point leading to the formation of thrombin involves factor Xa.
Thus, development of direct inhibition of factor Xa represents an important strategy of pharmacological intervention. Heterocyclic N-oxide drug otamixaban was developed as a specific direct inhibitor of factor Xa. The development of otamixaban commenced in 1994 with the first unsuccessful attempt in screening a large compounds library at Rhone-Poulenc Rorer, now known as Sanofi-Aventis. After this failure to identify any lead compound with a Factor Xa inhibitory activity, the company adopted a more rational strategy, which was based on the results from identification of a previously discovered potent inhibitor of serine protease elastase. [69] Since factor Xa is also a serine protease, it was hypothesized that β-lactams 27 would act as suicide inhibitors of FXa (Fugure 10). The important structural elements in this β-lactam design include introduction of a meta-substituted benzamidine unit and an N-p-nitrobenzoyl substitution, as in compound 23. The N-p-nitrobenzoyl group in 23 was expected to activate the β-lactam moiety towards irreversible acylation of Ser195, which is located at the active site of FXa, hence, disabling the enzyme.
Preliminary in silico docking experiments revealed that both the styryl side chain and the p-nitrobenzoyl groups satisfy the expected binding requirements for FXa. The synthesis of racemic compound 23 was thus carried out. It commenced with the construction of the β-lactam moiety in 23. This unit was synthesized by condensation of thiopyridyl ester 28 and imine 29 (Figure 11). This reaction afforded the desired β-lactam intermediate 30 as a 95:5 mixture of trans and cis isomers. [70] The oxidative removal of the p-methoxybenzyl unit afforded β-lactams 31a and 31b, which were easily separated. Compound 31a (trans) was then acylated with p-nitrobenzoyl chloride to give diamide 32a. All attempts to convert 32a to the desired amidine target resulted in the opening of the labile β-lactam unit leading to acid 24a. [8] Interestingly, compound 24a showed inhibitory activity against human FXa at submicromolar levels (Ki = 2.4 μM). In addition, the simplified racemic ester 25 that lacked the nitro group, caused inhibition of FXa with Ki 0.34 μM. The enantiomers of 25 were resolved and tested independently, and the results showed that isomer 25 (trans) inhibited FXa with Ki = 0.16 μM, while 25 (cis) had no activity against FXa. At that point, lactam 25 (trans) began to be considered as the lead compound. The identification of the main lead compound 25 (trans-diastereomer), led to the investigation of its absolute configuration and also an optimized asymmetric method for the synthesis. The absolute configuration of the active enantiomer was established to be 2R,3R, and the methyl ester function was also found to be necessary for the binding to FXa. In addition to the methyl ester as in the lead compound 25, a biphenyl moiety (compound 35) showed further increase in the inhibitory activity. The reduction of the olefinic unit in the styryl moiety to give ester 36 did not improve the activity. Further structure activity relationship studies revealed that the styryl moiety in 35 could be replaced with a smaller methyl group without a compromise in the inhibitory activity (Figure 12). [71] Thus, a program directed towards the synthesis of analogues with variability in the benzoyl unit but with conservation of the ester and benzamidine units was initiated.
Figure 11.

First attempted synthesis of β-lactam 23. [70]
Figure 12.

Synthesis of otamixaban analogues. [71]
The new analogues were tested in the factor IIa and trypsin inhibitory activity assays. The synthesis of these analogues was carried out using an optimized procedure, which resulted in the synthesis of the active enantiomer. The details of this synthetic strategy are discussed in section 5 below). Since most of the analogues showed significant activity against trypsin, the ratio of Ki for trypsin to that of FXa was also established as a yardstick for activity selection. Table 1 summarizes some of the results for selected analogues. It is worth noting that the introduction of electron-deficient or basic groups onto the biphenyl moiety in compound 38 led to the significant improvement in the activity against FXa, as well as selectivity, as seen for compounds 39 and 40. The introduction of imidazole showed improved selectivity, albeit with a reduction in the FXa activity. Other electron-deficient groups of the pyridine class showed some dramatic increase in the potency, as illustrated by compounds 26, 42–45. The best results were obtained for compound 26 (otamixaban) that contains 4-pyridyl N-oxide unit.
Table 1.
Optimization studies leading to development of otamixaban. [72]

| |||||
|---|---|---|---|---|---|
| Compounds | R | Ki(FXa) (nM) | Ki(FIIa) (nM) | Ki(Trypsin) (nM) | Ki(trypsin)/Ki(FXa) ratio |
| 38 |

|
5.3 | 3,250 | 69 | 13 |
| 39 |

|
1.0 | 2,650 | 69 | 69 |
| 40 |

|
3.0 | 3,950 | 170 | 57 |
| 26 |

|
0.5 | 4,000 | 300 | 600 |
| 42 |

|
2.0 | >4,000 | 130 | 65 |
| 43 |

|
0.58 | >4000 | 36 | 62 |
| 44 |

|
0.5 | >4,000 | 76 | 152 |
| 45 |

|
2.7 | >4,000 | 168 | 62 |
This compound was then chosen for the advanced stages of the drug development.
The X-ray structure of compound 26 (otamixaban) in the active site of FXa was obtained (Figure 13). [72]
Figure 13.

X-ray structure of otamixaban in FXa active site highlighting water bound via N-oxide moieties (represented by red spheres)
(Reprinted with permission from Ref. 72. Copyright 2002 Elsevier Science Ltd.).
Otamixaban 26 binds in a fully extended conformation, showing a bridging hydrogen bonding between Asp189 with the benzamidine moiety. This benzamidine moiety also hydrogen bonds to the carbonyl oxygen of Gly219. Other interactions in this pocket include hydrogen bonding between the amide carbonyl oxygen from the inhibitor and the NH of Gly219 and extensive van der Waals association between Try99, Phe174 and Trp215 with pyridine N-oxide ring. The N-oxide moiety is also hydrogen bonded to water (purple circles), and the combination of all these favorable interactions is responsible for the excellent potency and selectivity of otamixaban. [72]
3.3.4. Current status of otamixaban
Otamixaban is an experimental drug, which was intended for treatment of Non-STE acute coronary syndrome (NSTE-ACS). However, the drug did not perform better than current therapy with heparin and Merck’s Integrilin (eptifibatide) in reducing mortality or new heart attacks in patients with non-ST elevation acute coronary syndrome (NSTE-ACS). Hence, otamixaban has been withdrawn from clinical trial. [73]
3.3.5. Pharmacological characteristics of otamixaban
The in vitro pharmacological profile of otamixaban 26 characterized by Chu and coworkers revealed a highly selective inhibitory activity against FXa compared to the other relevant serine protease. [72] The inhibitory activity (Ki) of compound 26 for FXa and the other serine protease are 0.5 nM (FXa), 3956 nM (thrombin, FIIa), 301 nM (trypsin), >18491 nM (activated protein C, APC), 656 nM (plasmin), and 8681 nM (tissue plasminogen activator, tPA). The selectivity measured as a ratio of Ki of enzyme normalized to Ki of FXa are 1, >7900, 600, >36000, 1300 and > 17000 for the respective enzymes. [74] In addition, these studies also showed that the introduction of the N-oxide significantly increased the inhibitory activity and selectivity for FXa compared to the biphenyl and the pyridine derivatives. The inhibitory effect of otamixaban on the physiologically relevant complex of prothrombinase/FXa at phosphilipid interface and platelets in the presence of calcium and FVa was studied. The results revealed a dose dependent inhibitory activity for thrombin generation with IC50 = 1.38 nM for prothrombinase/FXa and 2.55 nM for platelets, respectively. Experiments performed to assess otamixaban anticoagulation response in different species show that the anticoagulation response decreases in the order rabbits > humans > primates > rats and dogs. [72] In particular, humans are 2.5 times more sensitive to compound 26 compared to dogs. Other experiments performed to quantify the effect of compound 26 on platelets aggregation revealed that it has no inhibitory effect on platelet aggregation up to 100 μM concentration. [72] This experiment suggested that otamixaban therapy would not affect normal primary hemostasis and could offer some inherent advantage (e.g. reduced risk of bleeding).
3.3.6. Synthesis of otamixaban
The asymmetric synthesis of otamixaban started with deprotonation of β-amino- ester 46 followed by alkylation with 3-cyanobenzyl bromide (Figure 14). [72] Upon treatment with benzoic acid the reaction mixture furnishes the benzoate salt of amino ester 47. The reaction mixture is then basified and subjected to a peptide coupling reaction with 4-(4′-pyridinyl)benzoic acid. This sequence afforded 48, which was oxidized to N-oxide 49. The nitrile was then converted into benzamidine by way of acid-mediated methanolysis followed by a reaction with ammonia.
Figure 14.

Synthesis of otamixaban. [72]
Reaction conditions: (a) i) LiHMDS, THF, −20 °C; ii) 3-cyanobenzyl bromide; iii) benzoic acid, water/toluene. (b) i) aq. Na2CO3; ii) 4-(4′-pyridinyl)benzoic acid; iii) TBTU, NMM, DMF. (c) MMPP, CH2Cl2/H2O. (d) i) HCl/MeOH; ii) NH3.
3.4. Pyridine N-oxide derivatives: emerging direct thrombin inhibitors
3.4.1. Discovery of pyridine N-oxide direct thrombin inhibitors
Thromboembolism is one of the major causes of morbidity and mortality worldwide. Thus, new strategies to combat this disease have been actively sought in the past several decades. In addition to the growing number of direct factor Xa inhibitors, such as otamixaban, there is also interest in the development of direct thrombin inhibitors. Towards this end, pyrazinone-based small-molecule peptidomimetics have been reported as direct thrombin inhibitors (Figure 15). [21] However, these molecules suffered from extensive metabolic oxidation that adversely affected their pharmacological profiles. [75] To address this issue, pyridine N-oxide was proposed as an alternative functional group that can retain the critical hydrogen-bonding network with Gly216 in the active site of the enzyme. Thus, the original pyrazinone core was replaced by pyridine N-oxide, which was shown to resist metabolic oxidation that plagued the pyrazinones. [76]
Figure 15.

Evolution of pyridine N-oxide class of thrombin inhibitors.
(2× APPT refers to the ability to double the activated partial thromboplastin time in human plasma
Derivatives with pyridine N-oxide ring bearing electron deficient substituents, such as Cl and CN, exhibited higher inhibitory activity. It was also shown that the non-N-oxide derivatives were much less potent than the corresponding N-oxides lending support to the proposed model that entails active participation of the N-oxide unit in hydrogen-bonding.
To further test this hypothesis, the X-ray structure of one of the inhibitors in the active site of the enzyme was obtained. Indeed, the structure clearly demonstrated the proposed hydrogen-bonding network with Gly-216 within the active site (Figure 16).
Figure 16.

X-ray structure of thrombin inhibitor 53 in the thrombin active site
(Reprinted with permission from Ref 76. Copyright 2005 Elsevier Ltd.).
Further SAR studies were carried out with the goal of optimization of the P1 groups on the already optimized P2 template (Table 1). Towards this end, several compounds were synthesized and evaluated for their thrombin inhibitory potency and also for their functional ability to double the activated partial thromboplastin time (2× APPT) in human plasma. [77] The native unsubstituted benzyl amide 54 (Ki = 150 nM) was chosen as the starting point. The introduction of an N-linked triazole function in the ortho-position of the benzyl group afforded inhibitors with both good intrinsic and functional potency. Upon installation of a 2-aminomethyl substituent 55 significant improvement in potency was observed. Furthermore, introduction of meta-chloro substituent also yielded significant improvement in intrinsic potency. However, studies have shown that increase in lipophilicity of this inhibitor leads to erosion of functional activity [78] as a result these analogues were not considered. The combination of the ortho-azole and the meta-chloro substituents yielded very potent thrombin inhibitors 56. For instance compound 61 displayed a 1 pM Ki against thrombin, with a 2× APTT of 70nM. Similarly, this additive effect was also observed with other aminomethyl and chlorine substituents. Compound 61 showed the highest potency amongst all of the analogues synthesized. [76]
3.4.2. Pharmacological properties of P2 pyridine N-oxide direct thrombin inhibitors
The pharmacokinetics of the pyridine N-oxide thrombin inhibitors were evaluated orally using dogs as animal models. [76] Thus, oral administration of compound 57 showed peak plasma levels of 0.82 mM and a half-life of 1.5 hr, while peak plasma levels of 0.7 mM with t1/2 of 1.3 hr were attained for compound 59. All other analogues displayed inferior pharmacokinetics profiles. Hence, further structural optimization of the P1 unit was focused on the aminomethyl analogues 57 and 59. Metabolic studies of these analogues using human and dog microsomes showed that the benzylic sites were susceptible to oxidation and N-dealkylated metabolites were formed as primary products.
Attenuation of this oxidative metabolism was achieved with α-methyl benzylic analogues 62–65 and 66. The more active analogue 63 displayed approximately 8-fold loss in binding potency compared to compound 62, but maintained good anticoagulant activity (Ki = 3.2 nM, 2× APTT = 360 nM). This structural modification indeed translated into desirable improvement in the pharmacokinetic profile (Cmax = 2.6 μM, t1/2 = 4.5 hr). Other structural modifications (Table 3) did not lead to any improvement in the overall pharmacological profile. Hence, compound 63 was the best analogue that showed a perfect balance between potency and pharmacokinetics. The increase in binding potency of compound 65 and 66 compared to compound 54 has been attributed to the hydrogen bonding participation of Ser195 of the active site and the P1 substituent on these inhibitors.
Table 3.
Pharmacological profiles of pyridine N-oxide direct thrombin inhibitors 62–66. [76]
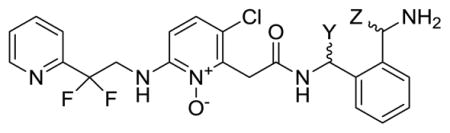
| ||||||
|---|---|---|---|---|---|---|
| Compound | Y | Z | Ki (nM) | 2× APTT | Cmax (μM) | t1/2 (hr) |
| 46 | H | H | 0.4 | 0.13 | 0.82 | 1.5 |
| 62 | Me | H | 260 | __ | __ | __ |
| 63 | Me | H | 3.2 | 0.36 | 2.6 | 4.5 |
| 64 | H | Me | 20 | 5.9 | __ | __ |
| 65 | H | Me | 0.45 | 0.27 | 0.1 | 2.0 |
| 66 | CH2OH | H | 0.48 | 0.23 | 0.34 | 1.5 |
3.4.3. Synthesis of P2 pyridine N-oxide thrombin inhibitors
These inhibitors were synthesized starting with the Boc-protection of 2-amino-6-methylpyridine 67 followed by regioselective chlorination to give chloropyridine 68 (Figure 17). [76] Compound 68 was metallated at the 2-methyl group with LDA and then treated with diethyl carbonate to give pyridyl acetate 69. Alkylation of compound 69 with 2,2-difluoro-2-(2-pyridyl)ethyl-trifluoromethanesulfonate (70) established the P2–P3 subunits of compound 71. Ester hydrolysis and the carbodiimide-mediated attachment of the P1 units were followed by Boc-deprotection and N-oxidation to give the P2 pyridine N-oxide thrombin inhibitors of type 72. The cyano derivatives could be similarly prepared starting with 2-amino-5-cyano-6-methyl pyridine.
Figure 17.

Synthesis of P2 pyridine N-oxide direct thrombin inhibitors. [76]
Reaction conditions: (a) Boc2O; (b) NCS, DCE. (c) LDA, diethyl carbonate, THF. (d) NaH, DMF. (e) 1N LiOH, THF. (f) P1-NH2, EDCl, HOAt, DMF. (g) TFA, CH2Cl2; (h) mCPBA, DCE.
3.5. Ancriviroc: HIV-1 entry inhibitor
3.5.1. Introduction
Ancriviroc 75 is a heterocyclic N-oxide investigated as an HIV-1 entry inhibitor. Current approaches to the management of human immunodeficiency virus type 1 (HIV-1) involve use of combinations of reverse transcriptase, integrase and protease inhibitors. [79] An important area of the antiretroviral drug development includes the point at which the virus enters its host cell. It has been known that HIV-1 enters the targeted macrophages and T-cells through the interaction of its gp120 and gp41 with the CD4 antigen receptors on the surface of the cells. In addition to the interaction with the CD4 receptors, there is also the participation of an important co-receptor known as CXCR4 in T-lymphocytes, and CCR5 in macrophages (Figure 18). [80] These co-receptors belong to the G-protein-coupled-receptor superfamily for which small molecule antagonists have been developed.
Figure 18.

Attachment of HIV to a CD4+ T-helper cell: (1) the gp120 viral protein attaches to CD4. (2) gp120 variable loop attaches to a co-receptor, either CCR5 or CXCR4. (3) HIV enters the cell. [81]
(Source: National Institute of Allergy and Infectious Diseases).
The native ligands to CCR5 include chemokines RANTES, MIP-1α and MIP-1β, which are known to inhibit the HIV-1 infection. [82] It has also been reported that many individuals, who are HIV positive but do not develop AIDS, have a mutation in the CCR5 receptor that makes the receptors defective, thus preventing the viron entry. [83] This finding lends support to the hypothesis that inhibition of CCR5 can provide protection against HIV-1. This discovery spurred efforts towards the development of small molecule antagonists for CCR5. [84] Amongst them, is the heterocyclic N-oxide known as ancriviroc (75), which was first reported by Schering-Plough in 2002. [85]
3.5.2. Discovery of ancriviroc as a CCR5 antagonist
The discovery of ancriviroc started with the screening of a large number of compounds from Schering-Plough’s compound library using the high-throughput 125I-labeled RANTES binding assay. [85] This effort led to the identification of compound 73 which was a CCR5 receptor antagonist with Ki = 1.0 μM (Figure 19). However, this compound was a more powerful antagonist for muscarinic M2 receptor (Ki = 1.3 nM). The initial efforts in this discovery were directed towards suppressing the muscarinic antagonism, while at the same time increasing antagonism for CCR5 receptor. Further structure activity relationship studies led to the refinement of the original structure, and, ultimately to the discovery of compound 74 (Ki(CCR5) = 66 nM; Ki(M2) = 1323 nM). [86]
Figure 19.

Discovery of ancriviroc.
The investigation of the effect of the position and identity of the substituents on the phenyl ring revealed that the para-position was optimal and other oxygenated and halogen substituent could be used in place of bromide. The results showed that p-chloro, p-iodo and some oxygen containing small groups had similar potency as compound 76 (Table 4). [85] However, their oral bioavailability was poor. This was linked to the possibility of oxidative metabolism at the benzylic sites. As a result, SAR studies were focused on introducing moieties at this benzylic site that would either block or reduce its metabolic involvement. Optimization studies revealed that a methoxime linker improved both the CCR5 antagonist potency (by 6-fold) and the blood plasma levels. These studies also show that the alkyl groups on the oxime function were limited to methyl and ethyl, as bigger groups led to decrease in potency. The Z-oxime isomer was more potent than the corresponding E-isomer. After the optimization of the geometry and alkyl substitution on the oxime, efforts were focused on the possible metabolic outcome of the drug. Experiments showed that the dimethylbenzamide moiety as in compound 74 was severely metabolized by the liver into a number of oxidized metabolites. The replacement of the methyl groups at the 2,6-positions led to a slight decrease in CCR5 binding potency. However, upon changing the benzamide to a nicotinamide, while maintaining the 2,6-dimethyl substituents, gave a potent CCR5 antagonist with significantly improved rat plasma levels after oral administration. Other substituents in the nicotinamide moiety did not increase either binding potency or bioavailability of the drug. Further metabolic studies of this analogue showed that the nicotinamide unit was significantly less metabolized compared to the 2,6-dimethylbenzamides. However, a major metabolite emerged with (M+16) mass unit. This was interpreted as N-oxidation of the nicotinamide core. This observation necessitated the expansion of the library by synthesis of new analogues with different N-oxide patterns. Further testing revealed that the new analogue compound 75 exhibited 2- to 3-fold higher oral plasma levels in the rat pharmacokinetic screen and also possessed increased binding potency. Compound 75 was, therefore, selected for advanced clinical studies.
Table 4.
Optimization of the CCR5 antagonists en route to ancriviroc. [85]

| |||||
|---|---|---|---|---|---|
| Compounds | Ar1 | R2 | Ki (nM) | IC50 | Rat PK (10 mg/kg PO) AUC0–6 (hr μg/mL) |
| 76 |

|
Me | 2.0 | 1.2 | 1.4 |
| 77 |

|
Me | 18 | 4 | 3.0 |
| 78 |

|
Me | 1.1 | 0.5 | 1.9 |
| 79 |
|
Et | 1.1 | 0.2 | 2.1 |
| 80 |

|
Et | >30 | NA | NA |
| 75 |

|
Et | 2.1 | 0.6 | 6.5 |
| 81 |

|
Et | 16 | 1.7 | 3.2 |
| 82 |

|
Et | >30 | NA | NA |
3.5.3. Pharmacological characterization of ancriviroc
The pharmacological profile of ancriviroc has been characterized in animal models (rats, monkeys and dogs) by Palani and co-workers. [87] The results showed that compound 75 is well absorbed in both rats and monkeys with oral bioavailability of 63% in rats, 52% in monkeys and 92% in dogs. The peaks plasma concentration of compound 48 at 12 and 24 h after oral administration were 330 and 16 nM in rats and 300 and 40 nM in monkeys respectively. The main route of excretion was found to be through urine for dogs and rats and through the bile in monkeys. The excreted metabolite was shown to the de-ethylated oxime, and no E/Z-isomerization of the oxime was observed in vivo. In addition, compound 75 showed negligible affinity (less than 15% inhibition at concentrations of 2–20 μM) for muscarinic M2 receptors or to the other closely related chemokine receptors, which include CCR1, CCR2, CCR3 and CCR7. [88] Despite the favorable potency and pharmacological characteristics, ancriviroc was withdrawn from clinical trials due to affinity for the hERG ion channel, when administered at high doses. [89] The inhibition of hERG leads to prolongations of the QT interval that predispose to life-threatening cardiac arrhythmias of the torsades de pointes type. [90]
3.5.4. Mechanism of action of ancriviroc
Ancriviroc was designed to be a noncompetitive allosteric antagonist of chemokine receptor CCR5. This receptor mediates viral entry into the host cell by binding to the CD4-Gp120 complex. Formation of CD4-Gp120 complex at the attachment of HIV leads to a conformation change, which exposes the binding site for CCR5. However, when CCR5 inhibitors (such as Ancriviroc) are present, the binding of CCR5 N-terminus and extracellular loops to Gp120 is blocked, which results in inhibition of the viron entry, [91] thus preventing HIV-1 replication.
3.5.5. Synthesis of ancriviroc
All structural congeners en route to the discovery of ancriviroc (75) were synthesized by a unified strategy developed by Palani and coworkers (Figure 20). The synthesis starts with protection of isonipecotic acid 83 as the trifluoroacetamide. The resulting acid was converted to acyl chloride 84 by treatment with thionyl chloride. The Friedel-Crafts reaction of acid chloride 84 with excess bromobenzene in the presence of AlCl3 afforded ketone 85. The ketone was protected as an ethylene acetal, and the trifluoroacetamide group was removed. The free secondary amine was allowed to react with N-Bocpiperidine-4-one in the presence of Ti(OiPr)4 and concomitant treatment of the resulting intermediate with diethylaluminum cyanide gave aminonitrile 87. The cyanide group was displaced by treatment with methylmagnesium bromide to give compound 88. Subsequent acetal cleavage under acidic conditions resulted in the concomitant Boc-deprotection. The Boc group was reinstalled to give ketone 89. Ketone 89 was converted to oxime 90, which was further transformed into ancriviroc by means of Boc-deprotection and a carbodiimide-mediated acylation.
Figure 20.

Synthesis of ancriviroc and its analogues. [87]
Reaction conditions and reagents: (a) TFAA (excess), reflux, 4 hr. (b) SOCl2 (excess), 12 hr. (c) AlCl3, bromobenzene (excess), reflux. (d) ethylene glycol, TsOH, toluene, reflux. (e) MeOH, H2O, K2CO3, r.t. 12 hr. (f) N-Boc-piperidine-4-one, Ti(OiPr)4, DCE, r.t. 12 hr, then Et2AlCN, r.t. 3 hr. (g) MeMgBr, THF, r.t., 2 hr. (h) 6M HCl, EtOAc, r.t. 12 hr. (i) Boc2O, 10% NaOH, Et2O, r.t., 12 h. (j) NH2OR, HCl, NaOAc, MeOH, r.t., 24 hr. (k) TFA, CH2Cl2, r.t., 2 h. (l) EDCl, DIEA, HOBt, CH2Cl2, r.t., 12 hr.
3.6. Tirapazamines: bioreductively-activated antitumor prodrugs
3.6.1. Introduction
Tirapazamine is an experimental anticancer prodrug that was developed as a hypoxic-selective cytotoxin for solid tumors. [92] This prodrug undergoes selective bioactivation catalyzed by multiple reductases at low oxygen tension (characteristic of solid tumor) to oxidizing radicals that react with DNA [22] leading to DNA damage via strand breakage and/or topoisomerase II poisoning. [23] Under oxygen-rich conditions, such as in normal cells, the free radical produced by tirapazamine is oxidized, reverting to the prodrug. Thus, this molecule is ideally suited for anticancer therapy, as it conveys excellent selectivity based on the intrinsic mechanism. Chemically, tirapazamine belongs to the class of benzotriazine di-N-oxides 92 (Figure 21).
Figure 21.

General structure of tirapazamines.
3.6.2. Discovery of tirapazamine
Tirapazamine was originally developed in a screening program directed towards the discovery of new herbicides. The first anticancer clinical use of tirapazamine was reported in 1986 by Zeman and coworkers. [93] However, the first synthesis of tirapazamine was reported by Robbins and Schofield as a derivative of 1, 2, 4-benzotriazines as early as in 1957. [94] The most important characteristic that distinguished tirapazamine was its relatively high hypoxic cytotoxicity ratio (HCR). HCR is defined as the ratio of drug concentration under aerobic and hypoxic conditions to yield approximately the same survival. Before the discovery of tirapazamine as hypoxic-selective anticancer agent is described, it is important to briefly discuss the physiology associated with hypoxia and survival of solid tumors.
Solid tumor cells transcribe a discrete set of genes in response to hypoxic stress under oxygen deprivation. [95] This hypoxic stress stimulates the tumor cell towards activation of hypoxia-inducible factor-1 HIF-1 (the main transcription factor regulating gene expression under hypoxia). The HIF-1 is a heterodimer, composed of α and β subunits. Under usual oxygen rich conditions, the alpha subunits have been shown to be hydroxylated at a conserved proline residue by enzymes known as HIF-prolyl-hydroxylases. This hydroxylation is a critical signal that leads to their ubiquitination [96] and, hence destruction by the proteasomal cascade. However, under oxygen-deprived conditions (hypoxia), such as in solid tumor, the activity of HIF prolyl-hydroxylases is inhibited, since oxygen is known to be a co-substrate of these enzymes. [97] This inhibition causes stabilization of the α subunit in HIF-1, which then translocates into the nuclear compartment and binds DNA as a heterodimer with the β subunit. The binding event occurs at the hypoxia-response element (HRE). This binding event triggers overexpression of genes necessary for tumor survival under such conditions. Some of these include glycolysis enzymes, which allow ATP-synthesis in an oxygen-independent manner, and vascular endothelial growth factor (VEGF) that promotes angiogenesis leading to abnormal tumor vasculature. Therapeutic intervention employing radiation that is aimed at damaging the DNA has severe limitations, due to resistance of tumor especially at such low oxygen tension. This resistance under oxygen deprivation was first described by Gray and co-workers. [98] They showed that upon irradiation, the biological lesion react with oxygen. Oxygen, which has the highest electron affinity in the cell, reacts extremely rapidly with the free radical, thus making the damage permanent. In the absence of oxygen, (e.g. in a solid tumor) most of the free radicals produced on DNA and other biological targets can be reverted by proton transfer due to the participation of non-proteinogenic sulfhydryls in the cells (Figure 22). [99]
Figure 22.

Schematic representation of radiation-induced DNA damage in aerobic conditions and thiol-mediated DNA repair under hypoxic conditions.
The chemical restitution, or DNA repair pathway, which is characteristic of tumor, spurred efforts towards the development of alternative methods to produce free radicals under such hypoxic and reducing conditions. In 1986, Zerman and co-workers embarked on a search for small non-natural molecules, as well as natural products capable of producing free radicals selectively in a solid tumor under hypoxia. In this quest, mitomycin, misonidazole and tirapazamine were tested on a number of cancer cell lines. Their results showed that tirapazamine exhibited superior balance between hypoxic potency and selectivity for tumor cell compared to mitomycin and misonidazole. They investigated the use of tirapazamine in combination with radiotherapy, and their result in vivo experiments showed that non-toxic doses of tirapazamine enhanced radiation-induced tumor cell kill, when the drug was given between 1 hr before and 2 hr after the radiation dose. [93]
Tirapazamine entered clinical trials as a combination therapy with radiotherapy for the treatment of advanced head and neck cancer. [100] Although the drug did not complete phase III clinical trials due to poor extravascular penetration at the tumor site, it is a perfect therapeutical model that is used in the development of new selective-hypoxia agents. [101] Since then, several efforts to improve the activity and the pharmacological properties of tirapazamine have resulted in diverse analogues with some conservation of the benzotriazinyl backbone. For instance, Hay and coworkers studied influence of the ring substitution on the activity of tirapazamine. They established that ring substituents on tirapazamine analogues can be used to predictably modulate the single electron reduction potential of parent tirapazamine, – an important attribute that dictates the hypoxic-cytotoxic efficacies. In addition, they showed that greater selectivity could be achieved with substituents that are weakly electron-donating, such as halogens with single electron reduction potentials of E(1) values between −450 to −510 mV. However, in their studies, tirapazamine outperformed all new analogues in terms of the balance between hypoxic-cytotoxicity and solubility. [102] Continuous efforts in this field have been directed towards the development of more potent and selective analogues with improved penetrability in solid tumors. More recent structure activity relationship (SAR) studies carried out using a spatially resolved pharmacokinetic/pharmacodynamics model, which employs tissue penetrability explicitly in lead optimization, revealed analogues with improved pharmacological properties. This method developed by Hicks and coworkers showed that two new lead compounds SN30000 (93) and SN29751 (94) have superior performance in hypoxic-selective cytotoxicity compared to parent tirapazamine (Figure 23). [103] For instance, metabolic studies of 93 and 94 in HT29 (colon cancer) cultures showed that each compound undergoes a hypoxia-dependent loss with a concomitant formation of their corresponding 1-oxides (95, 96, 97). With the exception of SN30000, a trace amount of the non-oxide 98 was formed. Formation of the 1-oxide metabolite confirms that these drugs operate via a similar mechanism to the parent tirapazamine. Comparatively, SN30000 showed highest rate constant for bioreductive consumption (Kmet 1.53 ± 0.21 min−1) followed by tirapazamine (1.3 ± 0.04 min−1) and the lowest for SN29751 (0.87±0.03 min−1). The reduced analogues of these parent N-oxides showed substantially reduced potencies and lower selectivity as hypoxic cytotoxins compared to the parent prodrugs. Further studies carried out with eight cell lines in order to test the scope of hypoxia-selective cytotoxicity of the compounds showed that SN30000 was more potent across all the cell lines than tirapazamine and SN 29751. The order of tumor penetrability calculated as a diffusion coefficient across HT29 multicellular layers was: SN30000 > SN 299751 > tirapazamine. Thus, the new analogue SN 30000 has superior hypoxic potency and selectivity. [103]
Figure 23.

Structures of tirapazamine analogues and their metabolites. [103]
Beatriz and coworkers [104] reported the synthesis and hypoxia-selective cytotoxicity of a series of 1,4-di-N-oxide derivatives of 99 and 100 (Figure 24). From their studies, a derivative 99-Cl with R1 = Cl, exhibited good cytotoxicity against the NCI 60 cell panel with a mean GI50 = 0.07 μM. Also, in an effort to improve the poor extravascular transport of tirapazamine, Hu and coworkers [105] reported the synthesis and evaluation of 3-aryl amino and 3-(alkoxymethylamino)benzotriazine-1,4-dioxide derivatives 101 and 102 by introducing lipophilic groups at the C-3 amino group of tirapazamine. Most of the analogues of this compound were more potent than tirapazamine in the tumor cell lines assay, and some of them exhibited greater hypoxia selectivity. In addition, SAR studies revealed that the introduction of an aromatic group at the C-3 amino 101 had a favorable effect on hypoxic cytotoxic activity, and a positive correlation between hypoxic activity and lipophilicity was demonstrated as well.
Figure 24.

Tirapazamine-based hypoxic-selective cytotoxins. [103]
Along these lines, Hu and coworkers also reported the synthesis and evaluation of a series of 3-phenyl-2-ethylthio-(or 2-ethylsulfonyl)-quinoxaline-1,4-dioxide derivatives by replacement of the 2-cyano group in 104 with ethylthio or ethylsulfonyl groups. The 2-ethylsulfonyl derivatives 106 displayed moderate to good antiproliferative activities under hypoxic conditions, while the corresponding 2-ethylthio derivatives showed almost no activity in a cell-based assay. These results support the observation that electron-withdrawing moieties are necessary for hypoxic activities, due to the modulation of one-electron reduction potentials of the molecules. Based on this finding, analogues of type 103, e.g. 3-(4-bromophenyl)-2-(ethylsulfonyl)-6-methylquinoxaline-1,4-dioxide, were synthesized that exhibited good antiproliferative activities in extensive cell lines in hypoxia, and also showed good inhibition of SMMC-7721 tumor growth in a dose-dependent manner using a human tumor xenograft mice model. [106] In order to further improve the potency and selectivity in hypoxia, Hu and coworkers reported a series of analogues of type 103 with a variation of the R1 and R2 substituents. Upon evaluation of these analogues across diverse cell lines, the most potent compound was identified with R1 = CH3 and R2 = Cl, i.e. 7-methyl-3-(3-chlorophenyl)-quinoxaline-2-carbonitrile 1,4-dioxide. This analogue displayed significant cytotoxic activity against BEL-7402, HepG2, HL-60, NCI-H460, HCT-116 and CHP126 cell lines in hypoxia with IC50 values ranging from 0.31 to 3.16 μM. [107]
Other strategies to improve the therapeutic efficacy of tirapazamine include ligation to endogeous ligand inhibitors whose receptors are native to tumor environment. [108] One worthy example is the chemical ligation of tirapazamine to indoleamine 2,3-dioxygenase (IDO) inhibitor. [109] IDO is an enzyme that catalyzes the metabolism of L-tryptophan through the kynurenine pathway. [110]
The depletion of L-tryptophan has been linked to halted growth of microbes as well as T-cells. Thus IDO is known as an immunomodulator. It has been demonstrated that tumor cells escape the immune system by engaging in the overexpression of IDO, which depletes L-Trp. Indeed, a wide range of human cancers such as prostatic, colorectal, pancreatic, cervical, gastric, ovarian, head, lung, are known to overexpress human IDO (hIDO). [111] The combination of hypoxic cytotoxin with IDO inhibitors has been postulated to be effective for hypoxic-neoplastic cells. An example of IDO-tirapazamine hybrid inhibitor is shown in figure 25.
Figure 25.

Tirapazamine analogue chemically-ligated to an indoleamine 2, 3-dioxygenase (IDO) inhibitor (IDO) and its mono-N-oxide derivative.
These hybrid inhibitors consist of three important modules: the hypoxia-selective cytotoxin unit, a linker, and an IDO inhibitor unit. It was demonstrated that hybrid inhibitors 107 and 108 were potent non-competitive IDO inhibitors. The tirapazamine-monoxide hybrid 108 was shown to be a stronger inhibitor than the di-N-oxide analogue 107. The hypoxia-selective cytotoxicity of both was compared to unmodified tirapazamine, and the results showed that compound 108 was indeed the most potent of them. However, compound 107 was still more potent than tirapazamine, confirming a cumulative effect originating from the presence of the IDO-inhibitor units.
There are also tirapazamine derivatives that contain attached DNA-targeting agents. These compounds were designed based on the hypothesis that tirapazamine toxicity occurs principally in the nuclear matrix of the cell [112] and that any metabolism or toxicity outside the nuclear compartment contribute to the unwanted side effects of the drug. [113] To test this hypothesis, tirapazamine was chemically linked to acridine and other agents known to target DNA (Figure 26). These compounds have been tested in comparison to tirapazamine. [114]
Figure 26.

Tirapazamine chemically-ligated to a DNA targeting unit.
The results demonstrated that attaching tirapazamine to acridine, e.g. as in compound 109, indeed led to increase in the potency by two orders of magnitude compared to unmodified tirapazamine depending on the cell investigated, while also retaining its selectivity towards hypoxic cells. The report also showed that tirapazamine undergoes bioreduction while bound to acridine chromophore on DNA.
3.6.3. Unified mechanism of action of tirapazamine and related analogues
The unified mechanism of the hypoxic-selective cytotoxicity by tirapazamine and its analogues involves the metabolic action of multiple reductases to form a radical anion species. In the presence of oxygen (normal cells and tissues), this radical anion is back-oxidized to the parent compound. Under hypoxic conditions on the other hand, the radical abstracts a proton from DNA, thus inducing a single and double strand cleavage in a pathway, which is not completely understood. Studies show that under hypoxia, the protonated tirapazamine radical undergoes disproportionation forming a nontoxic two electron species 1,2,4-benzotriazin-3-amine 1-oxide, and also decays by a first order process forming a DNA damaging radical. The exact identity of the DNA damaging radical involved in this process has been a subject of debate. Some studies show evidence supporting decay of the protonated tirapazamine radical to a hydroxyl radical, [24] while others support the loss of water and concomitant formation of benzotriazinyl radical. [24]
Either way, the abstraction of a proton from DNA by the hydroxyl or benzotriazinyl radical produces DNA radicals, which then lead to strand breaks. [115] At the level of the strand cleavage, some studies have supported the second involvement of tirapazamine in the oxidation of DNA radical to form similar oxidized products as in oxygen-induced radiation cell death (Figure 27). [116] This finding is based on the demonstration of oxidized phosphoglycolate residues in DNA incubated under anoxic conditions with tirapazamine radical-generating system (i.e. a mixture of tirapazamine, xanthine and xanthine oxidase). [117] The finding has been supported by isotope-transfer and kinetic experiments, which shows the oxidation of photolytically generated DNA radical by tirapazamine under anaerobic conditions. [118] Thus, the radical-induced cleavage of DNA leads to major perturbations in the genes responsible for tumor survival under hypoxia and hence cell death. [119]
Figure 27.

Proposed bioreductive mechanism of action of tirapazamine resulting in DNA cleavage. [24a]
3.6.4. Synthesis of tirapazamines
Tirapazamine and their analogues were prepared in two steps that involve a condensation of nitroaniline with cyanamide, followed by cyclization of the resulting guanidine with sodium hydroxide (Figure 28). The resulting 1-oxide was further oxidized to afford the di-N-oxide 116. The mono-N-oxide could be diazotized then chlorinated with POCl3. The resulting 2-chloro derivatives could be reacted with primary amines to afford 2-substituted tirapazamine derivatives 118. This method is widely used to build the benzotriazinyl backbone of all tirapazamine analogues. The benzotriazinyl core was also been prepared by reaction of o-nitroaniline with triphosgene (Figure 29). This reaction afforded an isocyanate which was then reacted with ammonia to afford urea derivative 121. The urea derivative was then cyclized using sodium hydroxide, providing a hydroxy group at the C-3 of the benzotriazine moiety of 122.
Figure 28.

Synthetic routes to tirapazamine analogues. [102]
Reaction conditions: (a) NH2CN, HCl, then 30% NaOH. (b) CF3CO3H, CF3CO2H, CH2Cl2. (c) i) NaNO2, CF3CO2H; ii) DMF, POCl3. (d) RNH2.
Figure 29.

Alternative synthetic routes to tirapazamine analogues. [102]
Reaction conditions: (a) triphosgene. (b) i) NH3; ii) NaOH, then AcOH. (c) i) POCl3; ii) R2NH2.
The chlorination of this compound with phosphorus oxychloride followed by reaction with various primary amines afforded a library of 1-oxide compounds with an amino group at C-3. Upon N-oxidation with peracid, this sequence furnished a family of tirapazamine derivatives.
The tricyclic analogues were prepared following the above protocol but starting with bicyclic o-nitroanilines 124 (Figure 30). To introduce non-heteroatom substituents at C-3, the corresponding 3-aminotriazine-1-oxide was diazotized in the presence of trifluoroacetic acid following by chlorination or iodination of the intermediate phenol. Iodination could be achieved by reaction of the corresponding diazonium with diiodomethane, while phosphorus oxychloride could be used to install the 3-chloro substituent.
Figure 30.

Synthetic routes to tricyclic tirapazamine analogues. [103]
Reaction conditions: (a) NH2CN, HCl, then 30% NaOH. (b) i) NaNO2, CF3CO2H; ii) DMF, POCl3 heat; (c) i) tBuNO2, CH2I2, CuI, THF, reflux; ii) allyl alcohol, Pd(OAc)2 (cat.), nBu4NBr, NaHCO3, DMF, heat; ii) morpholine, MeOH, then NaBH3CN, AcOH. (d) CF3CO2H, CF3CO3H, CH2Cl2.
The Heck reaction of these 3-halotriazines with allyl alcohol afforded an aldehyde which was subjected to reductive amination in the presence of morphine to give mono-N-oxide 127. The oxidation of 127 by trifluoroperacetic acid yielded the desired SN 30000 analogue 128. Other tirapazamine derivatives were also made using this protocol.
3.7. Aminopyridines and 1,7-naphthyridine N-oxides: emerging p38α mitogen-activated protein kinase (MAPK) inhibitors
3.7.1. Introduction
Mitogen-activated protein kinases (MAPKs) are constituents of a multifunctional-signaling cascade that controls complex cellular processes such as differentiation, proliferation and apoptosis. [120] Members of the MAP kinase family share some common sequence homology with a conserved structural motif. They are activated upon phosphorylation of threonine or tyrosine residues (located in the activation loop). Different MAPKs respond to different extracellular stimuli and each activates distinct, although often overlapping sets of cellular targets. For instance, extracellular signal-regulated kinases (ERKs) are activated by growth factors, cytokines, and carcinogens, among other stimuli, while the c-Jun N-terminal kinases (JNKs) and p38α are activated by such stimuli as inflammatory signals, heat shock and radiation. [121]
Activated p38α MAP kinase has been linked to the phosphorylation of a range of intracellular protein substrates that regulate such processes as inflammation, cell motility, gene expression, and cell death. One of these important processes is biosynthesis of tumor necrosis factor-α (TNF-α) and the interleukins e.g. IL-1β. [122] The pathophysiology associated with overexpression of genes responsible for the uncontrolled biosynthesis of TNF-α and interleukins IL-1β has been shown to be related to the progression of many inflammatory diseases, e.g. rheumatoid arthritis, psoriasis, and inflammatory bowel disease. [123] Thus, selective modulation of p38α MAP kinase activity has been considered as an important therapeutic strategy to combat a wide range of inflammatory and autoimmune diseases.
The discovery of pyridylimidazole-based p38α inhibitor 129 in 1994 stimulated the development of a number of compounds with p38 MAPK inhibitory activities (Figure 31). [124] The possibility of obtaining crystal structures of the inhibitors in the active site of p38 MAP kinases ushered in a new method for robust and rapid validation of prospective compounds with improved p38 MAP inhibitory activities. One of them was Merck’s inhibitor 130, which has a dihydroquinolinone core with two pendant aromatic rings that are critical for the enzymatic potency. This compound displayed a significant activity with IC50 = 0.74 nM and a selectivity for p38 over ERK1 and JNK1-3 by >100000 fold. [125] Other compounds include VX-745 (Vertex) 131, [126] (Bayer), [127] BIRB 796 (Boehringer Ingelheim), [128] SD006 (Pfizer), [129] AMG-548 (Amgen), [130] and SCIO-469 (Scios). [131]
Figure 31.

Path to the discovery of aminopyridine and naphthyridine N-oxide-based p38 MAP kinases inhibitors.
These compounds have been reported to have comparable or in some cases improved potencies and lower liabilities than the original pyridylimidazole analogues. Among these compounds, VX-745 131 was the first orally bioavailable drug that progressed through phase IIb clinical trials. This compound was shown to have anti-inflammatory properties in rodent models and was used to establish a concrete proof-of-principle in rheumatoid arthritis patients. However, it was discontinued due to adverse neurological effects in animal models. [20] In 2009, aminopyridine N-oxides 132 and 133 [132] were reported, based on structural similarity to Bayer’s pyridinone 131, which had p38 MAP kinase inhibitory activity at submicromolar levels. More recently, information on the crystal structure of compound 130 in the active site of unphosphorylated p38 MAPK was used for the rational design and optimization of 1,7 naphthyridine N-oxides 134. [133] The N-oxide moieties in these compounds were proven to be absolutely crucial for the potency of these analogues. The following section discusses the rational discovery of aminopyridine and 1,7-napthpyridine N-oxides 134 as potent and selective p38 MAP kinase inhibitors.
3.7.2. Discovery of aminopyridine and naphthyridine N-oxides as potent and selective p38 MAPK inhibitors
The process leading to the discovery of heterocyclic N-oxides 132 and 134 as potent and selective p38 MAP kinase inhibitors commenced with the realization of the structural intricacies of the ATP binding pockets of unphosphorylated p38 MAP kinase. Studies of the X-ray structure of Merck’s inhibitor 130 in the unphosphorylated ATP p38 MAPK active site revealed three important binding features: (1) the hydrophobic interactions between the difluoroaryl moiety and hydrophobic pocket I; (2) the bridging hydrogen bonds between the carbonyl group of the lactam unit and the NH groups of both Met106 and Gly110; (3) the second hydrophobic interaction between the dichloroaryl unit and hydrophobic pocket II. It has been shown that the bridging hydrogen bonding contact between the NHs of Gly110 and Met109 and the carbonyl group of the inhibitor 130 induces a conformational change in the hinge region of the enzyme. [121] This conformational flexibility due to the presence of a small Gly110, is unique to p38α and p38β, despite high degree of structural homology of the kinase superfamily. Other related kinases have bulkier residues at this position, which has been shown to prevent this conformational change. Thus, the high degree of selectivity of inhibitor 130 has been attributed to this unique interaction with the active site.
In an effort to further improve the selectivity and to generate new p38 MAP kinase inhibitors, Lumeras and coworkers [133] proposed to introduce the N-oxide moiety as a bioisosteric replacement of the carbonyl function (Figure 32). Based on Bayer’s inhibitor 131 as the original lead compound, they proposed replacing the pyridinone function with a pyridine N-oxide, as in compound 132. From the core structure of 135, it was postulated that the formation of intramolecular hydrogen bonding between the aromatic amino group and the carbonyl oxygen would lead to a pseudobicyclic scaffold, which gives the inhibitor a suitable arrangement for optimal binding within the active site of the enzyme.
Figure 32.

p38 MAPK inhibitor 131 and bioisosteric aminopyridine N-oxide 135.
As shown in figure 33, this arrangement allows for the N-oxide oxygen to form suitable hydrogen bond bridges between the NH of Met109 and the NH of Gly110 in the hinge region and the aromatic units to interact with the two hydrophobic pockets of the enzymes. This binding was expected to induce similar conformational change, which is characterized by the flipping of Gly110 and resulting in good potency and improved selectivity.
Figure 33.

(a) Schematic representation of key interactions observed in the X-ray structure of dihydroquinolinone-based inhibitor 130 within p38 α active site. (b) Schematic prediction of the key interactions of aminopyridine N-oxide inhibitor 135 in the active site of p38α.
(Reprinted with permission from Ref. 133. Copyright 2011 American Chemical Society).
Based on the X-ray studies, a simplified non–N-oxide analogue was designed and its ability to inhibit to inhibit p38 MAPK was tested in vitro. It was shown that the non-N-oxide had some activity with IC50 = 2.2 μM in enzymatic assay. Upon oxidation of the nitrogen atom, an improvement in the activity was seen with IC50 = 0.97 μM in enzymatic assay and IC50 = 1.63 μM in cellular assay (lipopolysaccharide-induced TNFα release in THP-1 cells). [133] These results corroborate the hypothesis that N-oxide participates in the bridging hydrogen bond network within the active site (Figure 34). To explore the full potential of the system, Lumeras and coworkers [133] investigated the optimal requirement for the hydrophobic pockets of the active sites. For hydrophobic pocket I, the 2,4-difluorophenyl ring was considered optimum based on the performance of dihydroquinolinone-based inhibitor 130. For hydrophobic pocket II, an ortho methyl and chloride substituents were found suitable. The above SAR studies led to the identification of compound 132 and 133 with potent p38α inhibitory activity. [133] These compounds were also shown to have the ability to effectively inhibit the production of TNFα in LPS-stimulated THP-1 cells as well as in human whole blood.
Figure 34.

X-ray overlay of compounds 132 (shown in light violet) and 130 (shown in yellow) in within p38 α active site. The conformational change of Gly110 can be visualized in both proteins, showing significant hydrogen bond interactions between the NHs of Met109/Gly110 and N-oxide oxygen (in 132) or the amide carbonyl (in 130). Both compounds occupy in a similar manner the deep hydrophobic pocket I (selectivity pocket) that is not accessed by ATP.
(Reprinted with permission from Ref. 133. Copyright 2011 American Chemical Society).
Comparatively, compound 132 displayed p38α inhibitory activity with IC50 = 21 nM and LPS induced release of TNFα in human whole blood (HWB) with IC50 = 170 nM, while compound 133 with an ortho chloride substituent inhibited p38a with IC50 = 23 nM, but with a slight shift in the cellular assay (TNFα in HWB IC50 = 291 nM). To confirm the formation of a suitable binding network, compound 132 was co-crystallized with unphosphorylated p38α. Indeed, the X-ray structure revealed the proposed binding modes between the N-oxide and the NHs of Met109 and Gly110 resulting in the desired conformational change characterized by a Gly109 flipping. An overlay of this X-ray structure with that of 130 in p38α shows some distinct similarities in the key hydrogen bonding interactions with the hinge region of the enzyme. [133] Additionally, the proposed pseudobicyclic scaffold formed via hydrogen bonding of the arylamino group and the carbonyl group in compound 132 was clearly established in the crystal structure. This pseudobicyclic scaffold showed some similarities to the bicyclic system of inhibitor 130 within the active site of the enzyme. To explore the full potentials of this aminopyridine N-oxide system, Lumeras and coworkers investigated the 1,7-napthpyridine N-oxide system, as in 134, in place of the pseudobicyclic backbone. [133] This replacement was expected to convey similar conformational change in the hinge region of the protein and at the same time to reinforce the hydrophobic requirement of the binding pockets. In this quest, compound 134 was synthesized and its p38 MAPK inhibitory activity was compared to its corresponding non-N-oxide analogues. It was again showed that the presence of N-oxide group in this compound significantly increased the inhibitory potency. As expected, the optimized structure of for the naphthyridine N-oxide 134 was found to superior balance between its potency and pharmacokinetics and also excellent behavior in a chronic model of arthritis.
3.7.3. Pharmacological characteristics of aminopyridine N-oxide and naphthyridine N-oxide
Pharmacological studies showed that compound 132 and 134 attained peak plasma levels of 4.7 μM and 5.3 μM at tmax = 0.75 and 3.5 hr respectively after oral administration to rats. [133] Plasma half-life of 7.4 and 1.6 hr with a clearance rate of 16.7 and 2.7 μL·min−1·kg−1 were achieved respectively. The slightly lower bioavailability of compound 134 (38%) compared to 75% of compound 132 is compensated by its low clearance rate compared to the high clearance rate for 132. The efficacies of both N-oxides (132 and 134) were tested in vivo based on rat lipopolysaccharide (LPS)-induced TNFα and in an adjuvant-induced arthritis (AIA) assay as a chronic disease model. These results were compared with reference standards including BIRB-796 and VX 745. In the rat-induced TNFα model, the compounds were dosed orally 1 hr prior to LPS administration, and the amount of TNFα in plasma was determined after 1.5 h, which coincides with peak TNFα production. [125] Results from these experiments showed that both compound 132 and 134 exhibited dose dependence in the inhibition of TNFα production with ED50 = 1.03 and 0.5 mg/kg, respectively, compared to 7.9 mg/kg for BIRB-796. In addition, upon arthritis induction, naphthyridine N-oxide 134 (administered orally once daily for 7 days) showed a superior dose-dependent inhibition of arthritis progression with ED50 < 1mg/kg compared to ED50 = 4.5 mg/kg for aminopyridine N-oxide 132, and 8.9 and > 100 for BIRB 796 and VX745, respectively. In this evaluation, the naphthyridine N-oxide clearly demonstrated superior pharmacological performance compared to aminopyridine 132 and other closely related inhibitors. With respect to cardiovascular safety, both compounds 132 and 134 were reported to show negligible effect on the inhibition of the human ether-à-go-go-related gene (hERG) channels (4.7 % at 3 μM) that are expressed in human embryonic kidney (HEK) cells. Furthermore, compound 134 exhibited a low oxidative metabolic rate in rat (9%) and human (<5%) microsomes and did not show significant inhibition for any of the relevant P450 isoforms (at 10μM). [133]
3.7.4. Synthesis of aminopyridine N-oxide and naphthyridine N-oxide
The aminopyridine N-oxides were synthesized in 7 steps (Figure 35) starting from N-protected aminopyridine 136. Lithiation of compound 136 under cryogenic conditions followed by addition of the corresponding aldehydes afforded corresponding alcohol 137. Benzylic oxidation of the secondary alcohol with MnO2 followed by hydrolysis of the pivalamide group afforded amino-ketopyridine 138. The amino-ketopyridine was converted to the corresponding N-oxide 139 using mCPBA. Treatment of this N-oxide with phosphoryl tribromide furnished key 2-bromopyridine derivative 140, which could be subjected to standard palladium-catalyzed coupling reactions with boronic acids or ester derivatives. [134] Other C–C bond-forming reactions were reported in the synthesis of sterically demanding analogues. N-Oxidation of the pyridine nitrogen in 141 affords the desired aminopyridine N-oxide derivatives 142.
Figure 35.

Synthesis of aminopyridine N-oxide p38α MAP kinase inhibitors. [133]
Reaction conditions: (a) nBuLi, TMEDA, Et2O, R1CHO, −78 to 0 °C, 5h, 35–65 %. (b) MnO2, CHCl3, r.t., 24–48 h, 85–100 %. (c) HCl, EtOH, 100 °C, 6–24 h, 75–95 %. (d) mCPBA, CH2Cl2, r.t., 24–48 h 70–90%. (e) POBr3, CH2Cl2, 60 °C, 3h, 40–60 %. (f) Method A: R2B(OH)2 or 4,4,5,5-tetrametyl-2-R2-1,3,2-dioxaborolane, PdCl2(dppf) (cat.), Cs2CO3, dioxane, 80–100 °C, 18h, 15–97 %; method B: R2B(OH)2, Pd2(dba)3 (cat.), S-Phos, K3PO4, toluene, 100 °C, 18– 72 h, 25–50 %; method C: 1,3-difluorobenzene or 1,3-difluoro-5-methoxybenzene, nBuLi, THF, −78 °C, 30 min, then ZnCl2, THF, −50 °C, 20 min, and then Pd (PPh3)4 (cat.), THF, 40 °C, 48–72 h, 21–93 %. (g) mCPBA, CH2Cl2, r.t., 18 h, 40–100 %.
The naphthyridine N-oxides, on the other hand, were synthesized from the corresponding aminopyridines, starting with a modified Friedlander cyclization with acetaldehyde or the corresponding ketone under acid catalysis (Figure 36).
Figure 36.

Synthetic route to 1,7-naphthyridine N-oxide p38α MAP kinase inhibitors. [133]
Reaction conditions: (a) method A: CH3C(O)R3, (R3 = H), AcOH, microwave, 100 °C, 32–85 %; method B: (R3≠ H), H2SO4, MgSO4, PhMe, 115 °C, 32–73 %. (b) mCPBA.
For instance, the condensation of acetaldehyde with aminopyridine 143 under acetic acid catalysis and microwave irradiation afforded the corresponding naphthyridine 144 (Figure 37). N-oxidation of the pyridine ring gave rise to desired naphthyridine N-oxide 145. For substituted naphthyridines, the aminopyridine and the corresponding methyl ketones were cyclized under sulfuric acid catalysis and with MgSO4 as a desiccant.
Figure 37.

Alternative synthetic route to 1,7-naphthyridine N-oxide-based p38 α MAP kinase inhibitors. [133]
Reaction conditions: (a) pivaloyl chloride, DIEA, dioxane, 110 °C. (b) LDA, AcOtBu, THF, −78 °C. (c) HCl, 100 °C. (d) mCPBA, CH2Cl2, r.t. (e) POCl3, 110 °C; (f) R4NH2, base, 110–130 °C.
When there is a heteroatom in the 7-position of the naphthyridine-2-oxide system, the corresponding aminopyridine is first acylated with pivaloyl chloride and then treated with the enolate of t-butyl acetate followed by cyclization under acidic conditions to give 146. The oxidation of pyridone 146 afforded the corresponding N-oxide 147, which was converted to 7-chloronaphthyridine-2-oxide by reaction with phosphoryl chloride. Nucleophilic displacement of the chloride afforded various substituted naphthyridine inhibitors 148.
3.8. Emerging bioactive furoxans (1,2,5-oxadiazole N-oxides)
3.8.1. Introduction
The furoxan (1,2,5-oxadiazole N-oxide) skeleton is a simple heterocyclic N-oxide scaffold with a wealth of fascinating chemistry (Figure 38). [135] A variety of compounds of this class have been implicated with a wide range of biological activities. [27, 136] An interesting aspect of the furoxan chemistry is their ability to react selectively with thiols. This has been linked to their varied biological activities. Some furoxan derivatives have been shown to produce highly reactive free-radicals via a bioreductive pathway in hypoxia. [137] Thus, the thiophilicity and/or bioreduction are the underlying factors responsible for the biological activity of furoxan and related benzofuroxan derivatives. [138] There is growing chemical evidence in support of both mechanisms. The thiophilicity of furoxans has been shown to lead to the release of nitric oxide (NO).
Figure 38.

Furoxan and benzofuroxan core structures.
The NO produced by these compounds is responsible for the procongnitive activity, [25] as well as cytotoxicity. [139] Thus, the ability of some furoxans to be bioreductively activated has been shown to elicit cytotoxicity. In addition, some benzofuroxan derivatives have antichagasic acitivity. [140]
3.8.2. Furoxans as nitric oxide mimetic neuroprotective and procognitive agents
The studies of furoxans as thiol-mediated nitric oxide (NO) release agents were initiated by Gasco and coworkers in the early 1980s. More recently, a number of reports have focused on highly reactive furoxans capable of producing reasonable amounts of NO that is necessary for cytotoxicity. While NO possess inherent cytotoxic effects at high concentrations, low levels of NO have been linked to protective activity particularly in the CNS. [141] In an effort to synthesize highly reactive furoxans, the Thatcher group postulated that “the identity and pattern of furoxan substitution can potentially dictate reactivity and propensity for NO release.” Thus manipulation of these substituents can be used to produce furoxans with attenuated reactivity and with a potential as neuroprotective agents. It has been shown that nitric oxide (NO) signaling in combination with secondary messenger molecule (cGMP) aid in the maintenance of normal brain function. [26] This happens via activation of soluble guanylyl cyclase (sGC) by cytosolic NO, and the activated cGC in turn catalyzes the conversion of GMP to cGMP. The cGMP potentiates a cascade of events through the protein kinase pathway leading to activation of memory-related transcription factor, cyclic-AMP response element binding protein (CREB) and serum response factor (SRF) [142] The activation of CREB and/or SRF can lead to the expression of plasticity-related genes, which in turn provide the functional and morphological changes necessary for neuronal plasticity to occur. [143] There is evidence that also supports the notion that NO elicits its neuronal effect by direct action in the presynaptic neurons [144] Thus increase in NO or NO-sGC signaling can be used as a potential treatment for neurological disorders, such as Alzheimer’s and Parkinson’s disease. Furthermore, a secreted neurotrophin growth factor known as BDNF (brain derived neurotrophic growth factor), a downstream target of CREB is known to nourish neurons, which in turn leads to increase in neurogenesis and enhanced synaptic plasticity. [145] The impairment of BDNF has been observed in Alzheimer’s disease brain. Amyloid β, the major component of amyloid plaques has been shown to inhibit NO/scGC/CREB signaling cascade, leading to synaptic impairment. [146] Thus, development of NO mimetic agents, is necessary to provide some modulation in synaptic plasticity and enhancement of learning or memory, all of which may be useful for the treatment of Alzheimer’s and Parkinson’s diseases.
An important aspect in the design of a neuroprotective and procognitive NO mimetic agent is the ability to produce lower fluxes of NO over a specified period of time. Towards this goal, Thatcher and coworkers designed and synthesized a variety of furoxans embedded in the backbone of peptidomimetic scaffolds (Figure 39). These furoxan derivatives were tested for the ability to react with excess cysteine and produce inorganic nitrite. Inorganic nitrite formation has been demonstrated as a direct measure of NO production. [147]
Figure 39.

Based on the reaction with cysteine, compound 158 showed the highest reactivity followed by other analogues with electron-withdrawing groups, such as 157. The high reactivity of compound 158 compared to the other analogues despite the lack of electron withdrawing groups has been attributed to the anchimeric assistance of the amide hydrogen through stabilization of the negative charge, thereby increasing the electrophilicity of the attacking carbon atom. [148] The closely related furazan derivative 159 that lacked the N-oxide moiety did not produce nitrite in the reaction with excess cysteine. All other analogues showed some ability to produce NO after reaction with excess cysteine. Analogues 155, 156 and 157 produced more NOx compared to their corresponding furoxan tautomers (not shown). The highest NO production was observed for 157.
The neuroprotection by the furoxan derivatives has been accessed based on their ability to restore normal neuronal function in primary rat neuronal cell cultures after being subjected to oxygen glucose deprivation (OGD). [149] OGD is a model used to mimic neuronal loss in neurodegenerative diseases such as stroke and Alzheimer’s disease. [150] Results from these experiments showed that several of the furoxan compounds have considerable neuroprotective activity. These activities have been shown to be in accordance with the reactivity profile of the furoxans. The neuroprotective activity elicited by electron-rich analogues suggests that even low levels of generated NOx are capable of producing a biological response. Some of these compounds were also found to be cytotoxic, e.g. carbazole 153. The cytotoxicity could be correlated with the overproduction of NO, as some highly reactive furoxans showed beneficial effects. The furazan 159 was also shown to be cytotoxic. In this case, the activity is only limited to thiophilicity without any release of nitric oxide. Experiments with 1H-[1,2,4] oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), a potent selective inhibitor for sGC, demonstrated a complete shutdown of the neuroprotective activity of compound 158. This experiment supports active involvement of NO-sGC pathway via a cGMP-dependent activation of the required transcription factors in the furoxan activity profiles, thus leading to neurogenesis. In addition, furoxan 158 (used as a prototype) was also found to provide restoration of synaptic plasticity and reversal of cognitive deficits in hippocampal slices that were treated with oligomeric Aβ. This observation suggests activation of the NO/sGC/CREB cascade as the mechanism of furoxan-elicited neuroprotection and synaptic plasticity.
3.8.3. Mechanism of thiol-mediated nitric oxide release by the furoxans
The mechanism that outlines the degradation of furoxan leading to the release of NO has not been clearly elucidated. However, Thatcher and coworkers reported intermediates from an associated mass spectrometric experiment, which indicate that the reaction of cysteine with furoxans leads to the formation of a keto-oxime intermediate 167 (Figure 40). [27] The formation of this intermediate requires release of some form of nitrogen oxide (NOx). Formation of this intermediate implies that the cysteine may be regenerated after a ring opening, leading to the formation of 165. The initial ring opening may also result in the oxidation of cysteine to its disulfide and concomitant formation of 164. Indeed, it was shown that the amount of cysteine consumed by this process did not correlate with the production of NOx, due to the oxidation of concomitantly formed cysteine from the reaction.
Figure 40.

Proposed mechanism of cysteine-mediated NO release from furoxans. [26]
3.8.4. Synthesis of nitric oxide-producing furoxan derivatives
The furoxan derivatives were synthesized by a route that was designed to increase structural diversity (Figure 41). Furoxan core 170 was assembled by reaction of arylcinnamyl alcohols 169 with sodium nitrite in acetic acid. Furoxan 170 was converted to azide 171 by way of tosylation of the hydroxy group followed by substitution with sodium azide in DMF.
Figure 41.

Synthetic route to peptidomimetic furoxan analogues. [152]
Reaction conditions: (a) NaNO2, AcOH, < 20 °C. (b) TsCl, NEt3, CH2Cl2, 0 °C. (c) NaN3, DMF, 60 °C. (d) CuI, DIEA, toluene, R2C≡CH. (e) toluene, 120 °C, 3 days. (f) PPh3, 35% NH4OH, DMF, r.t. (g) EDCl, DMAP, (S)-4-methyl-2-(phenylsulfonamido)pentanoic acid, CH2Cl2, 0 °C. (h) Zn, HCO2NH4, MeOH, 60 °C.
Azide 171 was then employed in a click reaction with a variety of alkynes to give a library of furoxans of type 172. [152] The regioisomeric furoxan 174 was obtained by thermal isomerization of 173 in toluene. The peptidomimetic scaffolds 177 were assembled by initial conversion of the azide to amine function via the Staudinger reduction. The resulting amines 176 were coupled to the corresponding protected amino acid using DMAP as catalyst. The furazan analogue 178 was synthesized by reduction of furoxan 177 by a combination of activated zinc and ammonium formate under reflux.
3.8.5. Furoxan-amodiaquine hybrid as an antiparasitic agent
Parasitic diseases caused by protozoans or helminths continue to pose a significant threat to humans. It has been estimated by the WHO that one in every four person is infected with parasite and over 400 million people are infected with malaria, hookworm or schistosomiasis. Most of this population is concentrated in sub-Saharan Africa. A lot of research has been devoted to provide remedy to this problem. Some of these efforts include the development of vector controls, vaccination and direct therapeutic intervention. For instance, chloroquine (CQ) and amodiaquine have been used for malaria parasite. However, both compounds have shown inherent limitations as anti-malaria drugs, such as resistance that has been reported in every region, where malaria is prevalent. Although amodiaquine is still widely used, it exhibits significant toxicity, which has been linked to the presence of the aminophenol moiety. [151] It has been shown that the quinine core common to amodiaquine and chloroquine possess activity against other parasitic diseases, most notably schistosomiasis. Thus, Mott and coworkers [152] applied the concept of drug hybridization in the development of a new therapy by combining the beneficial features of furoxans (known NO donor) and that of the quinine core (Figure 42).
Figure 42.

Amodiaquine-furoxan hybrid drug design.
The system eliminates the phenolic moiety in amodiaquine and introduces a potent NO donor on the meta-position of the aryl core. SAR studies aimed at combining the furoxan and amodiaquine resulted in the hybrid 182, which lacks phenolic functions on the para position of amodiaquine but is connected to furoxan at the meta-position and contains a t-butylamino group instead of diethylamino group. [153] Biological testing of this compound revealed noticeable activity against three parasites: Plasmodium falciparum, Schistosoma mansoni and Ancylostoma ceylanicum.
3.8.6. Synthesis of furoxan-amodiaquine hybrid agent
The synthesis of this hybrid agent starts with the reaction of 2, 7-dichloroquinoline 183 with (3-amino-5-bromophenyl) methanol (184) in the presence of hydrochloric acid to afford a 2-substituted amodiaquinoline derivative (Figure 43). The benzyl alcohol was converted to amine function by treatment of the alcohol with HCl followed by reaction with t-butylamine in the presence of pyridine to give 185. Heck reaction of bromide 185 with ethyl acrylate afforded 186, which upon reaction with two equivalents of DIBAL afforded the primary alcohol. The reaction of this precursor with sodium nitrite and acetic acid afforded the furoxan-amodiaquine analogue 187 with concomitant nitrosylation of the secondary amine. Since the preceding intermediate possessed several nitrogens, the oxidation of the alcohol to the aldehyde before reaction with hydroxylamine was problematic. The alcohol was instead reacted with N-(tert-butyldimethylsilyloxy)benzenesulfonamide (TBSONHTs) under Mitsunobu conditions. Deprotection of TBS group with cesium fluoride followed by treatment with thionyl chloride yielded oxime 188. The oxime was then converted to nitrile 182 by treatment with HCl in dioxane.
Figure 43.

Synthesis of amodiaquine-furoxan hybrid antiparasitic agent. [152]
Reaction conditions: (a) HCl, MeOH, 89%. (b) conc. HCl, reflux in sealed tube, 6 h, 77%. (c) tBuNH2, Py, DMF, 80 °C, 80%. (d) ethyl acrylate, K2CO3, Pd(OAc)2, dppp, DMF, MW, 130 °C, 7 h, 81%. (e) DIBAL, CH2Cl2, −78 °C, 74%. (f) NaNO2, AcOH, 57%. (g) TBSONHTs, DEAD, PPh3, THF, 62%. (h)CsF, MeCN, 48%. (i) SOCl2, 0 °C, DMF, 99%. (j) 4M HCl in dioxane, 99%.
3.8.7. Benzofuroxans as bioreductively activated anti-chagas prodrugs
Chagas disease also known as American trypanosomiasis is a tropical disease that affects about 15 million people worldwide, and an additional 25 million people are at risk of infection. [154] This disease, caused by a protozoan known as Trypanosoma cruzi (T. cruzi) leads to symptoms progressing from mild swelling to intestinal disease and ultimately chronic cases that can lead to heart failure. The current treatment includes the use nifurtimox, (Nfx, Lampit) or benznidazole (Bnz, Rochagan) and has some limitations, such as incomplete elimination of the parasite in chronic cases, and potentially harmful side effects. [155] In addition, to these limitations, more resistant strains of the parasite have recently emerged. Thus, there is an urgent need for the development of alternative therapies. An important approach adopted to combat this disease includes the use of multitarget-directed ligands (MTDLs), which involves the combination of two or more pharmacophores in a single chemical entity, also known as a hybrid drug.
Merlino and coworkers reported the implementation of this strategy by employing entities capable of producing free-radicals that are linked to: (a) DNA-interacting agents, [156] sterol-biosynthesis inhibitors [157] and endoproteinase (cruzipain) CP inhibitors. [158] The latter strategy involving CP inhibitors appears most promising since cruzipain (CP) is a cysteine protease that is heavily expressed by the causative agent (Trypanosoma cruzi) of Chagas disease. With the intricate knowledge and understanding of the active site of this enzyme, Merlino and coworkers designed the benzofuroxans of type 190 and benzimidazole-1,3-dioxides of type 191 based on in silico docking and NMR experiments (Figure 44). These compounds were evaluated, and the results show excellent trypanosomicidal activities with good selectivity indices, although without improved cruzipain-inhibitory activity compared to a reference inhibitor. [159]
Figure 44.

Schematic of the design strategy of anti-Chagas prodrugs.
3.9. Quinoxaline N-oxides, benzofuroxans and furoxans as agricultural agents
3.9.1. Introduction
The quinoxaline, benzofuroxan and furoxans have been endowed with a wide range of biological activities. Compared to their reduced analogues, these compounds show superior potency, most of which is related to the presence of the N-oxide moiety. The underlying factor responsible for this activity, as discussed in the preceeding sections, is related to the ability to undergo bioreduction in selective microenvironments leading to the formation of highly reactive radicals. Furthermore, compounds such as furoxans and benzofuroxans have been shown to be very thiophilic. The thiophilicity is thought to be responsible for the inhibition of cysteine protease and also for the nitric oxide production. The release of large fluxes of NOx has been to shown to produce cytotoxicity in a variety of cell lines.
3.9.2. Quinoxaline N-oxides, benzofuroxans and furoxans as herbicidal agents
Over the years, the agricultural industry has seen some applications of these compounds both as herbicidal and as anti-bacterial growth promoters in animals. With the aim of developing highly effective herbicides, Cerecetto and coworkers [15] synthesized and conducted herbicidal evaluations of a series of quinoxaline N-oxides, benzofuroxans and furoxans (Figure 45). These analogues were based on the combination of the toxophoric features of some of the existing herbicidal classes. The various herbicidal classes used in this design included: (1) the pyrazole phenyl ethers 192, which act by inhibiting the protoporphyrinogen oxidase, [160] (2) sulfonylureas 193 and imidazolinones 195 that are known to inhibit amino acid biosynthesis, [161] and (3) bipyridinium herbicides 194 that elicit their effects by competing for electrons with the natural substrate ferredoxin [162] Heterocyclic N-oxide-based herbicides 196, 197 and 198 combine the toxophores from the above classes of compounds. For instance, they contain quaternary nitrogen (as in the bipyridinium family) and a heterocyclic system, which is structurally related to the other families (pyrazole phenyl ethers, sulfonylureas and imidazolinones).
Figure 45.

Design of heterocyclic N-oxide-based herbicides.
The herbicidal profiles of these heterocyclic N-oxides were evaluated by Cerecetto and coworkers in pre-emergence (ability to prevent germination) assay using Triticum aestivum L. as the biological model species. [15] Dose-response profiles were established and the active compounds were carried to post-emergence assay. The pre-emergence assay identified a number of compounds with some herbicidal activity. Compounds 199, 200, 201 and 202 showed the highest potency among all the compounds synthesized (Figure 46). All four compounds showed a dose-dependent response in the herbicidal activity.
Figure 46.

Structures of bioactive herbicides of the furoxan, benzofuroxan and quinoxaline di-N-oxide family.
The most potent compound 199 needed 4.18 g/ha to completely inhibit 100% growth of the model weed, while compound 200 and 201 needed 4.27 and 4.07 g/ha to inhibit approximately 50% growth. [15] Control experiments with the analogous non-N-oxides showed no growth inhibition, which led to the conclusion that the N-oxide moiety in these compounds is the active unit. The mechanism of this herbicidal activity has not been evaluated. However, it is likely that the heterocyclic N-oxide undergoes selective bioreduction producing highly reactive radicals, and these radicals are responsible for the observed activity. Also, production of nitric oxide by the furoxan under thiol mediation could not be ruled out.
3.9.3. Quinoxaline N-oxides as growth promoters in animal husbandry
The antibacterial properties of some of these compounds have been used intensely in animal husbandry. The heterocyclic N-oxides of the quinoxaline class for example, have been reported as growth promoters in animals, due to the ability to affect both gram positive and negative strains of bacteria. [163] Some of these compounds have been used for the prevention of porcine infectious diseases caused by a wide range of bacteria especially salmonella, Escherichia coli, and Treponema hyodysenteriae (Figure 47). Carbadox (205), olaquidox (204), and cyadox (206) for example, are commercially available products that are usually added to pig or cattle feed as growth promoters agents. Quindoxin (203), which is also an N-oxide, has been reported to possess some side effects in humans, such as eczema, and hepatic toxicity in rodents. As a result, it has been withdrawn from market.
Figure 47.

Structures of bioactive quinoxaline di-N-oxide-based animal growth promoters.
3.10. Purine N-oxides and their biological activities
3.10.1. Introduction
The isolation of purine N-oxides from Streptomyces species by three groups in the mid-1980s [164] revealed compounds with a wide range of biological activities. [165] Guanine 7-oxide for example, was shown to have antitumor, antimicrobial, and antiviral activities. [166] On the other hand, this compound and related purine N-oxides were shown to be oncogenic in mice models in the late 1960s. [167] Significant efforts were, thus, directed towards the synthesis and re-evaluation of diverse analogues of these heterocyclic N-oxides with defined oxidation patterns on the purine backbone. [168]
3.10.2. Guanine 7-N-oxides, hypoxanthine 7-N-oxide, and adenine 1-N- and 7-N-oxides
In the 1985, Watanabe and coworkers reported on the antitumor activities of some purine N-oxides. Notably, guanine-N-oxide (207) was shown to possess excellent activities in mice that were inoculated with L1210 leukemia cells (Figure 48). [169] In these studies, compound 207 caused significant growth inhibition of murine and human cancer cell lines in vitro. The compound had a dose-dependent inhibition of the growth of Ehrlich solid carcinoma in mice after oral administration.
Figure 48.

Structures of selected bioactive purine N-oxides. [169]
The compound was tested across a host of bacteria lines, and compound 207 did not show any activity against Staphylococcus aureus 209P, Bacillus subtilis PCI 219, Escherichia coli NIHJ, Pseudomonas aeruginosa IFO 3445, Micrococcus luteus PCI 1001, Candida albilcans 3147, and Saccharomyces cerevisiae. Other reports, e.g. by Jackson and coworkers, also demonstrated that compound 207 was converted into guanosine 7-oxide 5′-triphosphate within sensitive cells and this led to the inhibition of cellular protein synthesis. [173] Nishii and coworkers reported a weak antimicrobial activity of the nucleoside analogue 208. Nucleoside N-oxide 208 also caused apparent inhibition of L5178Y mouse leukemia cell in culture with IC50 = 0.60 mg/mL. Life-prolongation effect on mice with P388 leukemia was observed with intraperitoneal administration of 208. [170] In addition, 208 also demonstrated a dose dependent inhibition of the growth of Ehrlich solid carcinoma in mice. Kitahara and coworkers also studied the antitumor properties of a range of purine-N-oxides and reported that compound 208 and its 2′-deoxy analogue 209 is active against mouse leukemia L1210 cells. N-oxides 210 and 211 on the other hand have been shown to be weakly cytotoxic and have not be shown to possess any antimicrobial activity. [171] Studies by Parra and coworkers [172] showed that hypoxanthine 3-N-oxide 213 is the main chemical that elicits alarm reactions in zebrafish. These results are based on the observation of an efficaciously induced fear reactions and increase in the number of erratic movement episodes and jumps upon administration of this compound to zebrafish.
3.10.3. Synthesis of bioactive purine N-oxides
(a) Guanine 7-N-oxides
The synthesis of guanine-7-oxide was first accomplished by Fujio and coworkers (Figure 49). [173] This strategy commenced with the reaction of the phenacylamines 214 with 2-amino-6-chloro-5-nitro-4(3H)-pyrimidinone (215) in the presence of NaOH to afford compound 216, Further treatment with NaOH yielded protected guanine 7-oxide 217, which could be deprotected with sulfuric acid to afford the desired N-oxide 207. It should be noted that direct oxidation of guanine did not lead to this compound, but rather afforded guanine-3N- oxide.
Figure 49.

Synthesis of guanine 7-N-oxides and hypoxanthine N-oxides. [169]
Reaction conditions: (a) NaOH, EtOH. (b) 2N NaOH, r.t., 1 h. (c) H2SO4, toluene, r.t., 1–3 h.
(b) Hypoxanthine 7-N-oxide
Hypoxanthine 7-N-oxide (210) was unknown until its discovery in 1988 by Fujio and coworkers. It was synthesized using the same sequence of reactions as for the guanine-7-N-oxides. [173] The isomeric hypoxanthine-3-N-oxide could be obtained by direct oxidation of hypoxanthine with peracids.
(c) Adenine N-oxides: 1-N- and 7-N-oxides
The direct oxidation of adenine with hydrogen peroxide or peracids affords exclusively adenine-1-N-oxide 212. Fujio and coworkers examined strategies to selectively synthesize potential antitumor adenine-7-N-oxides (Figure 50). They started by directly oxidizing adenine under different buffer conditions and found no evidence of the desired 7-N-oxides. Based on these unsuccessful attempts, they developed a 3-step route that yielded the desired compound. The synthesis started with N-oxidation of 3-N-benzyl adenine 224 with a peracid followed by debenzylation of the resulting compound 224 to afford the adenine 7-N-oxide (211). The reaction of 211 with sodium nitrite in the presence of HCl also affords hypoxanthine 7-N-oxide (210). Upon hydrogenation of adenine-7-N-oxide over nickel followed by N-oxidation with hydrogen peroxide in acetic acid, the corresponding adenine-1-N-oxide (213) was made in a good yield.
Figure 50.

Synthetic route to adenine-N-oxides. [169]
Reaction conditions: (a) mCPBA or MMPP, MeOH. (b) H2SO4, toluene. (c) NaNO2, HCl. (d) H2, Ni. (e) 30% H2O2 in AcOH, r.t.
4. CONCLUSION
The heterocyclic N-oxide motif has proven to be a very productive emerging scaffold in drug discovery. The four modes of action outlined in this review encompass a large therapeutic arena extending into areas, where nitric oxide has been observed to have excellent therapeutic value, to areas dealing with the bioisosteric replacement of carbonyl with N-oxide units for stronger binding within enzyme active sites. In addition, the ability of these compounds to produce highly reactive radicals selectively under hypoxic conditions has a significant potential in the field of anti-cancer drug development. It is our hope that new mechanisms will be discovered which may explore the intrinsic electrophilicity of the C2 carbon in pyridine N-oxides, quinoline N-oxides and quinoxaline mono and di-N-oxides, and the C1 of isoquinoline N-oxide. [174] Thus, a new mode of action can be envisioned, whereby a nucleophilic group in an enzyme’s active site reacts with the electrophilic C2 or C1 site in a heterocyclic N-oxide, resulting in an inhibition or modulation of the enzyme’s activity, similar to the proteasomal inhibitory activity of β-lactones. [175] Another potential mode of action may involve complexation of metal ions by the oxygen atom of heterocyclic N-oxides. [176] These potential modes of action in addition to the known ones, and in conjunction with the ease of synthesis [177] and rapid structural diversification [178] of the heterocyclic N-oxide scaffold make it a very promising structural motif for drug discovery. It is expected that the review will attract further attention to this topic and lead to the discovery of new potent medicinal N-oxide heterocycles.
Figure 10.

Initial lead investigation towards otamixaban.
Table 2.
Optimization of the P1-subunit. [76]

| ||||
|---|---|---|---|---|
| Compound | X | P1 | Ki (nM) | 2× APPT (μM) |
| 54 | CN |

|
150 | __ |
| 55 | Cl |

|
2.60 | 0.46 |
| 56 | CN |

|
<0.01 | 0.09 |
| 57 | Cl |
|
0.40 | 0.13 |
| 58 | Cl |

|
0.08 | 0.20 |
| 59 | CN |

|
0.05 | 0.23 |
| 60 | Cl |

|
<0.01 | 0.15 |
| 61 | CN |
|
0.001 | 0.07 |
Acknowledgments
Financial support by the Welch Foundation (AX-1788), NIGMS (SC3GM105579), the Voelcker Fund and the University of Texas at San Antonio is gratefully acknowledged.
LIST OF ABBREBIATIONS
- cGMP
Cyclic guanosine monophosphate
- dba
Dibenzylideneacetone
- DCE
1,2-Dichloroethane
- DIEA
Diisopropylethylamine
- DIBAL
Diisobutylaluminum hydride
- DMAP
4-Dimethylaminopyridine
- DMF
N,N-Dimethylformamide
- dppf
1,1′-Bis(diphenylphosphino)ferrocene
- EDCl
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide
- FXa
Activated factor X also known as Stuart-Prower factor or as prothrombinase, thrombokinase or thromboplastin
- GMP
Guanosine monophosphate
- HOAt
1-Hydroxy-7-azabenzotriazole
- HOBt
Hydroxybenzotriazole
- LDA
Lithium diisopropylamide
- LiHMDS
Lithium hexamethyldisilazide
- mCPBA
m-Chloroperbenzoic acid
- MMPP
Magnesium monoperoxyphthalate
- MW
Microwave irradiation
- NCS
N-Chlorosuccinimide
- NMM
N-Methylmorpholine Pyr, Pyridine
- TBTU
N,N,N′,N′-Tetramethyl-O-(benzotriazol-1-yl)-uronium tetrafluoroborate TFA, Trifluoroacetic acid
- TFAA
Trifluoroacetic anhydride
- THF
Tetrahydrofuran
- TMEDA
Tetramethylethylenediamine
- Ts
p-Toluenesulfonyl
Footnotes
CONFLICT OF INTEREST
The authors declare no actual or potential conflicts of interest.
References
- 1.(a) Schreiber SL. Organic synthesis toward small-molecule probes and drugs. Proc Nat Acad Sci USA. 2011;108:6699–6702. doi: 10.1073/pnas.1103205108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stockwell BR. Exploring biology with small organic molecules. Nature. 2004;432(7019):846–854. doi: 10.1038/nature03196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ray RS, Corcoran AE, Brus RD, Kim JC, Richerson GB, Nattie E, Dymecki SM. Impaired Respiratory and body temperature control upon acute serotonergic neuron inhibition. Science. 2011;333:637–642. doi: 10.1126/science.1205295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Welsch ME, Snyder SA, Stockwell BR. Privileged scaffolds for library design and drug discovery. Curr Opin Chem Biol. 2010;14(3):347–361. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) DeSimone RW, Currie KS, Mitchell SA, Darrow JW, Pippin DA. Privileged Structures: applications in drug discovery. Comb Chem High Throughput Screening. 2004;7(3):473–493. doi: 10.2174/1386207043328544. [DOI] [PubMed] [Google Scholar]; (c) Dai W-M. Generation of Molecular Shape Diversity. From Privileged Scaffolds to Diverted Total Synthesis. Diversity-Oriented Synth. 2012;1:11–20. [Google Scholar]
- 4.Fishman MC, Porter JA. Pharmaceuticals: a new grammar for drug discovery. Nature. 2005;437(7058):491–493. doi: 10.1038/437491a. [DOI] [PubMed] [Google Scholar]
- 5.Showell GA, Mills JS. Chemistry challenges in lead optimization: silicon isosteres in drug discovery. Drug Discov Today. 2003;8(12):551–556. doi: 10.1016/s1359-6446(03)02726-0. [DOI] [PubMed] [Google Scholar]
- 6.(a) Vagner J, Qu H, Hruby VJ. Peptidomimetics, a synthetic tool of drug discovery. Curr Opin Chem Biol. 2008;12(3):292–296. doi: 10.1016/j.cbpa.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Meanwel NA. Synopsis of some recent tactical application of bioisosteres in drug design. J Med Chem. 2011;54(8):2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 7.Lackey KE. Lessons from the drug discovery of lapatinib, a dual ErbB1/2 tyrosine kinase inhibitor. Curr Top Med Chem. 2006;6(5):435–460. doi: 10.2174/156802606776743156. [DOI] [PubMed] [Google Scholar]
- 8.Guertin KR, Choi YM. The discovery of the factor Xa inhibitor otamixaban: from lead identification to clinical development. Curr Med Chem. 2007;14(23):2471–2481. doi: 10.2174/092986707782023659. [DOI] [PubMed] [Google Scholar]
- 9.Chaykin S, Bloch K. The metabolism of nicotinamide-N-oxide. Biochim Biophys Acta. 1959;31(1):1213–1216. doi: 10.1016/0006-3002(59)90458-5. [DOI] [PubMed] [Google Scholar]
- 10.Ihsan A, Wang X, Tu H-G, Zhang W, Dai M-H, Peng D-P, Wang Yu-L, Huang L-L, Chen D-M, Mannan S, Tao Y-F, Liu Z-L, Yuan Z-H. Genotoxicity evaluation of mequindox in different short-term tests. Food Chem Toxicol. 2013;51:330–336. doi: 10.1016/j.fct.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 11.(a) Palani A, Tagat JR. Discovery and development of small-molecule chemokine coreceptor CCR5 antagonists. J Med Chem. 2006;49:2851–2856. doi: 10.1021/jm060009x. [DOI] [PubMed] [Google Scholar]; (b) Debnath AK. Generation of predictive pharmacophore models for CCR5 antagonists: study with piperidine- and piperazine-based compounds as a new class of HIV-1 entry inhibitors. J Med Chem. 2003;46:4501–4515. doi: 10.1021/jm030265z. [DOI] [PubMed] [Google Scholar]; (c) Tagat JR, Steensm RW, McCombie SW, Dennis V, Nazaren DV, Lin SL, Neustadt BR, Cox K, Xu S, Wojcik L, Murray MG, Vantuno N, Baroudy BM, Strizki JM. Piperazine-based CCR5 antagonists as HIV-1 Inhibitors. II. Discovery of 1-[(2,4-dimethyl-3-pyridinyl)carbonyl]-4-methyl-4-[3(S)-methyl-4-[1(S)-[4-(trifluorome-thyl)phenyl]ethyl]-1-piperazinyl]-piperidine N1-oxide (Sch-350634), an orally bioavailable, potent CCR5 antagonist. J Med Chem. 2001;44:3343–3346. doi: 10.1021/jm0155401. [DOI] [PubMed] [Google Scholar]
- 12.Ganley B, Chowdhury G, Bhansali J, Daniels JS, Gates KS. Redox-activated, hypoxia-selective DNA cleavage by quinoxaline 1,4-di-N-oxide. Bioorg Med Chem. 2001;9:2395–2401. doi: 10.1016/s0968-0896(01)00163-8. [DOI] [PubMed] [Google Scholar]
- 13.Miroshnikova OV, Hudson TH, Gerena L, Kyle DE, Lin AJ. Synthesis and antimalarial activity of new isotebuquine analogues. J Med Chem. 2007;50:889–896. doi: 10.1021/jm061232x. [DOI] [PubMed] [Google Scholar]
- 14.Ibrahim H, Furiga A, Najahi E, Pigasse Hénocq C, Nallet JP, Roques C, Aubouy A, Sauvain M, Constant P, Daffé M, Nepveu F. Antibacterial, antifungal and antileishmanial activities of indolone-N-oxide derivatives. J Antibiot. 2012;65(10):499–504. doi: 10.1038/ja.2012.60. [DOI] [PubMed] [Google Scholar]
- 15.Cerecetto H, Dias E, Di Maio R, Gonzalez M, Pacce S, Saenz P, Seoane G. Synthesis and herbicidal activity of N-oxide derivatives. J Agric Food Chem. 2000;48:2995–3002. doi: 10.1021/jf9904766. [DOI] [PubMed] [Google Scholar]
- 16.(a) Messenger AG, Rundegren J. Minoxidil: mechanisms of action on hair growth. Br J Dermatol. 2004;150:186–194. doi: 10.1111/j.1365-2133.2004.05785.x. [DOI] [PubMed] [Google Scholar]; (b) Padois K, Cantien C, Bertholle V. Solid lipid nanoparticles suspension versus commercial solutions for dermal delivery of minoxidil. Int J Pharm. 2011;416:300–304. doi: 10.1016/j.ijpharm.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 17.(a) Rand MJ, Li CG. Nitric oxide as a neurotransmitter in peripheral nerves: nature of transmitter and mechanism of transmission. Ann Rev Physiol. 1995;57:659–682. doi: 10.1146/annurev.ph.57.030195.003303. [DOI] [PubMed] [Google Scholar]; (b) Bult H, Boeckxstaens GE, Pelckmans PA, Jordaens FH, Van Maercke YM, Herman AG. Nitric oxide as an inhibitory non-adrenergic non-cholinergic neurotransmitter. Nature. 1990;345:346–347. doi: 10.1038/345346a0. [DOI] [PubMed] [Google Scholar]
- 18.(a) Li M, Marubayashi A, Nakaya Y, Fukui K, Arase S. Minoxidil-induced hair growth is mediated by adenosine in cultured dermal papilla cells: possible involvement of sulfonylurea receptor 2B as a target of minoxidil. J Invest Dermatol. 2001;117:1594–1600. doi: 10.1046/j.0022-202x.2001.01570.x. [DOI] [PubMed] [Google Scholar]; (b) Johnson GA, Barsuhn KJ, McCall JM. Sulfation of minoxidil by liver sulfotransferase. Biochem Pharmacol. 1982;31:2949–2954. doi: 10.1016/0006-2952(82)90268-4. [DOI] [PubMed] [Google Scholar]
- 19.Davies GC, Thornton MJ, Jenner TJ, Chen YJ, Hansen JB, Carr RD, Randall VA. Novel and established potassium channel openers stimulate hair growth in vitro: implications for their modes of action in hair follicles. J Invest Dermatol. 2005;124(4):686–694. doi: 10.1111/j.0022-202X.2005.23643.x. [DOI] [PubMed] [Google Scholar]
- 20.Weisman M, Furst D, Schiff M, Kauffman R, Merica E, Martin-Munley S. A double-blind, placebo-controlled trial of VX-745, an oral p38 mitogen activated protein kinase (MAPK) inhibitor, in patients with rheumatoid arthritis (RA). Ann Rheum Dis; Present at EULAR 2002; Stockholm, Sweden. 2002. p. 166. Abstract FRI0018. [Google Scholar]
- 21.Burgey CS, Robinson KA, Lyle TA, Sanderson PEJ, Lewis SD, Lucas BJ, Krueger JA, Singh R, Miller-Stein C, White RB, Wong B, Lyle EA, Williams PD, Coburn CA, Dorsey BD, Barrow JC, Stranieri MT, Holahan MA, Sitko GR, Cook JJ, McMasters DR, McDonough CM, Sanders WM, Wallace AA, Clayton FC, Bohn D, Leonard YM, Jr, Detwiler TJ, Jr, Lynch JJ, Jr, Yan Y, Chen Z, Kuo L, Gardell SJ, Shafer JA, Vacca JP. Metabolism-directed optimization of 3-aminopyrazinone acetamide thrombin inhibitors. Development of an orally bioavailable series containing P1 and P3 pyridines. J Med Chem. 2003;46:461–473. doi: 10.1021/jm020311f. [DOI] [PubMed] [Google Scholar]
- 22.(a) Anderson RF, Shinde SS, Hay MP, Gamage SA, Denny WA. Potentiation of the cytotoxicity of the anticancer agent tirapazamine by benzotriazine N-oxides: the role of redox equilibria. J Am Chem Soc. 2003;125:748–756. doi: 10.1021/ja0559101. [DOI] [PubMed] [Google Scholar]; (b) Shinde SS, Anderson RF, Hay MP, Gamage SA, Denny WA. Oxidation of 2-deoxyribose by benzotriazinyl radicals of antitumor 3-amino-1,2,4-benzotriazine 1,4-dioxides. J Am Chem Soc. 2004;126:7853–7864. doi: 10.1021/ja048740l. [DOI] [PubMed] [Google Scholar]
- 23.(a) Wang J, Biedermann KA, Brown JM. Repair of DNA and chromosome breaks in cells exposed to SR 4233 under hypoxia or to ionizing radiation. Cancer Res. 1992;52:4473–4477. [PubMed] [Google Scholar]; (b) Siim BG, van Zijl PL, Brown JM. Tirapazamine-induced DNA damage measured using the comet assay correlates with cytotoxicity towards hypoxic tumour cells in vitro. Br J Cancer. 1996;73:952–960. doi: 10.1038/bjc.1996.187. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Siim BG, Menke DR, Dorie MJ, Brown JM. Tirapazamine-induced cytotoxicity and DNA damage in transplanted tumors: relationship to tumor hypoxia. Cancer Res. 1997;57:2922–2928. [PubMed] [Google Scholar]; (d) Peters KB, Brown JM. Tirapazamine. Cancer Res. 2002;62:5248–5253. [PubMed] [Google Scholar]; (e) Evans JW, Chernikova SB, Kachnic LA, Banath JP, Sordet O, Delahoussaye YM, Treszezamsky A, Chon BH, Feng Z, Gu Y, Wilson WR, Pommier Y, Olive PL, Powell SN, Brown JM. Repair of DNA and chromosome breaks in cells exposed to SR 4233 under hypoxia or to ionizing radiation. Cancer Res. 2008;68:257–265. [Google Scholar]
- 24.(a) Daniels JS, Gates KS. DNA cleavage by the antitumor agent 3-amino-1,2,4-benzotriazine 1,4-dioxide (SR4233): Evidence for Involvement of Hydroxyl Radical. J Am Chem Soc. 1996;118:3380–3385. [Google Scholar]; (b) Kotandeniya D, Ganley B, Gates K. Oxidative DNA base damage by the antitumor agent 3-amino-1,2,4-benzotriazine 1,4-dioxide (Tirapazamine) Bioorg Med Chem Lett. 2002;12:2325–2329. doi: 10.1016/s0960-894x(02)00468-7. [DOI] [PubMed] [Google Scholar]; (c) Zagorevskii D, Song M, Breneman C, Yuan Y, Fuchs T, Gates KS, Greenlief CM. A mass spectrometry study of tirapazamine and its metabolites: insights into the mechanism of metabolic transformations and characterization of reaction intermediates. J Am Soc Mass Spectrom. 2003;14:881–892. doi: 10.1016/S1044-0305(03)00334-9. [DOI] [PubMed] [Google Scholar]
- 25.Schiefer IT, VandeVrede L, Fa’ M, Arancio O, Thatcher GRJ. Furoxans (1,2,5-oxadiazole-N-oxides) as novel NO mimetic neuroprotective and procognitive agents. J Med Chem. 2012;55:3076–3087. doi: 10.1021/jm201504s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thatcher GRJ, Bennett BM, Reynolds JN. NO chimeras as therapeutic agents in Alzheimer’s disease. Curr Alzheimer Res. 2006;3:237–245. doi: 10.2174/156720506777632925. [DOI] [PubMed] [Google Scholar]
- 27.Cena C, Boschi D, Tron GC, Chegaev K, Lazzarato L, Di Stilo A, Aragno M, Fruttero R, Gasco A. Development of a new class of potential antiatherosclerosis agents: NO-donor antioxidants. Bioorg Med Chem Lett. 2004;14:5971–5974. doi: 10.1016/j.bmcl.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 28.Zins GR. The history of the development of minoxidil. Clin Dermatol. 1988;6:132–147. doi: 10.1016/0738-081x(88)90078-8. [DOI] [PubMed] [Google Scholar]
- 29.Buhl AE, Waldon DJ, Baker CA, Johnson GA. Minoxidil sulfate is the active metabolite that stimulates hair follicles. J Invest Dermatol. 1990;95:553–557. doi: 10.1111/1523-1747.ep12504905. [DOI] [PubMed] [Google Scholar]
- 30.(a) Harmon CS, Ducote LD. Potassium channel openers stimulate DNA synthesis in mouse epidermal keratinocyte and whole hair follicle cultures. J Skin Pharmacol. 1993;6:170–178. doi: 10.1159/000211131. [DOI] [PubMed] [Google Scholar]; (b) Johnson GA, Barsuhn KJ, McCall JM. Sulfation of minoxidil by liver sulfotransferase. Biochem Pharmacol. 1982;31:2949–2954. doi: 10.1016/0006-2952(82)90268-4. [DOI] [PubMed] [Google Scholar]
- 31.Fleishaker JC, Andreadis NA, Welshma IR, Wright CE. The pharmacokinetics of 2.5- to 10-mg oral doses of minoxidil in healthy volunteers. J Clin Pharmacol. 1989;29:162–167. doi: 10.1002/j.1552-4604.1989.tb03307.x. [DOI] [PubMed] [Google Scholar]
- 32.Thoma RC, Harpootlian H. Metabolism of minoxidil, a new hypotensive agent I: Absorption, distribution, and excretion following administration to rats, dogs, and monkeys. J Pharm Sci. 1975;64:1366–1371. doi: 10.1002/jps.2600640822. [DOI] [PubMed] [Google Scholar]
- 33.(a) Johnson GA, Barsuhn KJ, McCall JM. Sulfation of minoxidil by liver sulfotransferase. Biochem Pharmacol. 1982;31:2949–2954. doi: 10.1016/0006-2952(82)90268-4. [DOI] [PubMed] [Google Scholar]; (b) Meisheri KD, Johnson GA, Puddington L. Enzymatic and non-enzymatic sulfation mechanisms in the biological actions of minoxidil. Biochem Pharmacol. 1993;45:271–279. doi: 10.1016/0006-2952(93)90061-z. [DOI] [PubMed] [Google Scholar]
- 34.Falany CN, Kerl EA. Sulfation of minoxidil by human liver phenol sulfotransferase. Biochem Pharmacol. 1990;40:1027–1032. doi: 10.1016/0006-2952(90)90489-8. [DOI] [PubMed] [Google Scholar]
- 35.Johnson GA, Baker CA. Sulfation of minoxidil by human platelet sulfotransferase. Clin Chim Acta. 1987;169:217–227. doi: 10.1016/0009-8981(87)90322-6. [DOI] [PubMed] [Google Scholar]
- 36.Johnson GA, Baker CA, Knight KA. Minoxidil sulfotransferase, a marker of human keratinocyte differentiation. J Invest Dermatol. 1992;98:730–733. doi: 10.1111/1523-1747.ep12499930. [DOI] [PubMed] [Google Scholar]
- 37.Anderson R, Kudlacek PE, Clemens DL. Sulfation of minoxidil by multiple human cytosolic sulfotransferases. Chem Biol Interact. 1998;109:53–67. doi: 10.1016/s0009-2797(97)00120-8. [DOI] [PubMed] [Google Scholar]
- 38.Dooley TP. Molecular biology of the human cytosolic sulfotransferase gene superfamily implicated in the bioactivation of minoxidil and cholesterol in skin. Exp Dermatol. 1999;8:328–329. doi: 10.1159/000026112. [DOI] [PubMed] [Google Scholar]
- 39.Buhl AE, Baker CA, Dietz AJ. Minoxidil sulfotransferase activity influences the efficacy of rogaine topical solution (TS): enzyme studies using scalp and platelets. J Invest Dermatol. 1994;102:534. [Google Scholar]
- 40.(a) Babenko AP, Aguilar-Bryan L, Bryan J. A view of sur/KIR6.X, KATP channels. Annu Rev Physiol. 1998;60:667–687. doi: 10.1146/annurev.physiol.60.1.667. [DOI] [PubMed] [Google Scholar]; (b) Ashcroft FM, Gribble FM. J Diabetes Complicat. 2000;14:192–196. doi: 10.1016/s1056-8727(00)00081-7. [DOI] [PubMed] [Google Scholar]
- 41.McCall JM, TenBrink RE, Ursprung JJ. New approach to triaminopyrimidine N-oxides. J Org Chem. 1975;40(22):3304–3306. doi: 10.1021/jo00910a040. [DOI] [PubMed] [Google Scholar]
- 42.Anthony WC, Ursprung JJ. 6-Amino-1,2-dihydro-1-hydroxy-2-imino-4-phenoxypirimidines. 3,382,247. US Patent. 1965 Nov 1;
- 43.Anthony WC. Compounds and process. 3,644,364. US Patent. 1970 Mar 31;
- 44.Skerritt JH, Johnston GA. Enhancement of GABA binding by benzodiazepines and related anxiolytics. Eur J Pharmacol. 1983;89:193–198. doi: 10.1016/0014-2999(83)90494-6. [DOI] [PubMed] [Google Scholar]
- 45.Ban TA. The role of serendipity in drug discovery. Dialogues Clin Neurosci. 2006;8:335–344. doi: 10.31887/DCNS.2006.8.3/tban. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sternbach L, Stempe RA, Rachlin A. Quinazolines and 1,4-benzodiazepines. XIV. Synthesis and transformation of 5-phenyl-1,4-benzodiazepine-2-thiones. J Org Chem. 1964;29:3744–3744. [Google Scholar]
- 47.Greenblatt DJ, Shader RI, MacLeod SM, Sellers EM. Benzodiazepines: A summary of pharmacokinetic properties. Clin Pharmacokinet. 1978;3:381–394. doi: 10.2165/00003088-197803050-00004. [DOI] [PubMed] [Google Scholar]
- 48.Campo-Soria C, Chang Y, Weiss DS. Mechanism of action of benzodiazepines on GABAA receptors. Br J Pharmacol. 2006;148:984–990. doi: 10.1038/sj.bjp.0706796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Derry JM, Dunn SM, Davies M. Identification of a residue in the gamma-aminobutyric acid type A receptor alpha subunit that differentially affects diazepam-sensitive and -insensitive benzodiazepine site binding. J Neurochem. 2004;88:1431–1438. doi: 10.1046/j.1471-4159.2003.02264.x. [DOI] [PubMed] [Google Scholar]
- 50.Seymour VA, Curmi JP, Howitt SM, Casarotto MG, Laver DR, Tierney ML. Selective modulation of different GABAA receptor isoforms by diazepam and etomidate in hippocampal neurons. Int J Biochem Cell Biol. 2012;44:1491–1500. doi: 10.1016/j.biocel.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 51.Möhler H. Role of GABAA receptors in cognition. Biochem Soc Trans. 2009;37:1328–1333. doi: 10.1042/BST0371328. [DOI] [PubMed] [Google Scholar]
- 52.Sigel E. Mapping of the benzodiazepine recognition site on GABAA receptors. Curr Top Med Chem. 2002;2:833–839. doi: 10.2174/1568026023393444. [DOI] [PubMed] [Google Scholar]
- 53.Jacob TC, Moss SJ, Jurd R. GABAA receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat Rev Neurosci. 2008;9:331–343. doi: 10.1038/nrn2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Akabas MH. GABAA receptor structure-function studies: a reexamination in light of new acetylcholine receptor structures. Int Rev Neurobiol. 2004;62:1–43. doi: 10.1016/S0074-7742(04)62001-0. [DOI] [PubMed] [Google Scholar]
- 55.Hanson SM, Czajkowski C. Structural mechanisms underlying benzodiazepine modulation of the GABAA receptor. J Neurosci. 2008;28(13):3490–3499. doi: 10.1523/JNEUROSCI.5727-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.(a) Sternbach LH, Reeder E, Stempel A, Rachlin A. Quinazolines and 1,4-benzodiazepines. XVIII. The acetylation of chlordiazepoxide and its transformation into 6-chloro-4-phenyl-2-quinazoline-carboxaldehyde. J Org Chem. 1964;29:332–336. [Google Scholar]; (b) Sternbach LH, Reeder E, Stempel A, Rachlin A. Additions and corrections - quinazolines and 1,4-benzodiazepines. XVIII. The acetylation of chlordiazepoxide and its transformation into 6-chloro-4-phenyl-2-quinazolinecarboxaldehyde. J Org Chem. 1964;29:3744–3744. [Google Scholar]
- 57.(a) Nutescu EA, Pater K. Drug evaluation: the directly activated Factor Xa inhibitor otamixaban. Idrugs. 2006;9:854–865. [PubMed] [Google Scholar]; (b) Hinder M, Frick A, Jordaan P, Hesse G, Gebauer A, Maas J, Paccaly A. Direct and rapid inhibition of factor Xa by otamixaban: a pharmacokinetic and pharmacodynamic investigation in patients with coronary artery disease. Clin Pharmacol Ther. 2006;80:691–702. doi: 10.1016/j.clpt.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 58.Gould WR, McClanahan TB, Welch KM, Baxi SM, Saiya-Cork K, Chi L, Johnson TR, Leadley RJ. Inhibitors of blood coagulation factors Xa and IIa synergize to reduce thrombus weight and thrombin generation in vivo and in vitro. J Thromb Haemostasis. 2006;4:834–841. doi: 10.1111/j.1538-7836.2006.01830.x. [DOI] [PubMed] [Google Scholar]
- 59.Lillicrap D, Key N, Makris M, O’Shaughnessy D. Practical Hemostasis and Thrombosis. Wiley-Blackwell; 2009. pp. 1–5. [Google Scholar]
- 60.Michiels C. Endothelial cell functions. J Cell Physiol. 2003;196:430–443. doi: 10.1002/jcp.10333. [DOI] [PubMed] [Google Scholar]
- 61.Catella F, Healy D, Lawson JA, FitzGerald GA. 11-Dehydrothromboxane B2: a quantitative index of thromboxane A2 formation in the human circulation. Proc Nat Acad Sci USA. 1986;83:5861–5865. doi: 10.1073/pnas.83.16.5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gomez K, McVey JH. Tissue factor initiated blood coagulation. Front Biosci. 2006;11:1349–1359. doi: 10.2741/1888. [DOI] [PubMed] [Google Scholar]
- 63.Bajaj MS, Birktoft JJ, Steer SA, Bajaj SP. Structure and biology of tissue factor pathway inhibitor. Thromb Haemostasis. 2001;86:959–972. [PubMed] [Google Scholar]
- 64.(a) Nesheim ME, Taswell JB, Mann KG. The contribution of bovine Factor V and Factor Va to the activity of prothrombinase. J Biol Chem. 1979;254:10952–10962. [PubMed] [Google Scholar]; (b) van Rijn JL, Govers-Riemslag JW, Zwaal RF, Rosing J. Kinetic studies of prothrombin activation: effect of factor Va and phospholipids on the formation of the enzyme-substrate complex. Biochemistry. 1984;23:4557–4564. doi: 10.1021/bi00315a008. [DOI] [PubMed] [Google Scholar]
- 65.Mosesson MW. Fibrinogen and fibrin structure and functions. J Thromb Haemostasis. 2005;3:1894–1904. doi: 10.1111/j.1538-7836.2005.01365.x. [DOI] [PubMed] [Google Scholar]
- 66.(a) Laki K, Lóránd L. On the solubility of fibrin clots. Science. 1948;108(2802):280. doi: 10.1126/science.108.2802.280. [DOI] [PubMed] [Google Scholar]; (b) Ariëns RAS, Lai TS, Weisel JW, Greenberg CS, Grant PG. Role of factor XIII in fibrin clot formation and effects of genetic polymorphisms. Blood. 2002;100:743–754. doi: 10.1182/blood.v100.3.743. [DOI] [PubMed] [Google Scholar]
- 67.Cesarman-Maus G, Hajjar KA. Molecular mechanisms of fibrinolysis. Brit J Haematol. 2005;129:307–321. doi: 10.1111/j.1365-2141.2005.05444.x. [DOI] [PubMed] [Google Scholar]
- 68.Fabre JE, Gurney ME. Limitations of current therapies to prevent thrombosis: a need for novel strategies. Mol BioSyst. 2010;6:305–315. doi: 10.1039/b914375k. [DOI] [PubMed] [Google Scholar]
- 69.(a) Firestone RA, Barker PL, Pisano JM, Asher BM, Dahlgren ME. Monocyclic β-lactam inhibitors of human leukocyte elastase. Tetrahedron. 1990;46(7):2255–2262. [Google Scholar]; (b) Knight WB, Maycock AL, Green BG, Asher BM, Gale P, Weston H, Finke PE, Hagmann WK, Shah SK, Doherty JB. Mechanism of inhibition of human leukocyte elastase by two cephalosporin derivatives. Biochemistry. 1992;31(21):4980–4986. doi: 10.1021/bi00136a010. [DOI] [PubMed] [Google Scholar]
- 70.Klein SI, Czekaj M, Gardner CJ, Guertin KR, Cheney DL, Spada AP, Bolton SA, Brown K, Colussi D, Heran CL, Morgan SR, Leadley RJ, Dunwiddie CT, Perrone MH, Chu V. Identification and initial structure-activity relationships of a novel class of nonpeptide inhibitors of blood coagulation factor Xa. J Med Chem. 1998;41:437–450. doi: 10.1021/jm970482y. [DOI] [PubMed] [Google Scholar]
- 71.Czekaj M, Klein S, Guertin K, Gardner C, Zulli A, Pauls H, Spada A, Cheney D, Brown K, Colussi D, Chu V, Leadley R, Dunwiddie C. Optimization of the β-aminoester class of factor Xa inhibitors. Part 1: P(4) and side-chain modifications for improved in vitro potency. Bioorg Med Chem Lett. 2002;12:1667–1670. doi: 10.1016/s0960-894x(02)00212-3. [DOI] [PubMed] [Google Scholar]
- 72.Guertin KR, Gardner CJ, Klein SI, Zulli AL, Czekaj M, Gong Y, Spada AP, Cheney DL, Maignan S, Guilloteau J-P, Brown KD, Colussi DJ, Chu V, Heran CL, Morgan SR, Bentley RG, Dunwiddie CT, Leadley RJ, Pauls HW. Optimization of the β-aminoester class of factor Xa inhibitors. Part 2: Identification of FXV673 as a potent and selective inhibitor with excellent in vivo anticoagulant activity. Bioorg Med Chem Lett. 2002;12:1671–1674. doi: 10.1016/s0960-894x(02)00213-5. [DOI] [PubMed] [Google Scholar]
- 73.Jarvis LM. AstraZeneca, Sanofi cut programs. Chem Eng News. 2013;91(23):17. [Google Scholar]
- 74.Chu V, Brown K, Colussi D, Gao J, Bostwick J, Kasiewski C, Bentley R, Morgan S, Guertin K, Pauls HW, Gong Y, Zulli A, Perrone MH, Dunwiddie CT, Leadley RJ. Pharmacological characterization of a novel factor Xa inhibitor, FXV673. Thromb Res. 2001;103:309–324. doi: 10.1016/s0049-3848(01)00328-0. [DOI] [PubMed] [Google Scholar]
- 75.(a) Singh R, Silva Elipe MV, Pearson PG, Arison BH, Wong BK, White R, Yu X, Burgey CS, Lin JH, Baillie TA. Metabolic activation of a pyrazinone-containing thrombin inhibitor. Evidence for novel biotransformation involving pyrazinone ring oxidation, rearrangement, and covalent binding to proteins. Chem Res Toxicol. 2003;16:198–207. doi: 10.1021/tx025635l. [DOI] [PubMed] [Google Scholar]; (b) Subramanian R, Lin CC, Ho JZ, Pitzenberger SM, Silva-Elipe MV, Gibson CR, Braun MP, Yu X, Yergey JL, Singh R. Bioactivation of the 3-amino-6-chloropyrazinone ring in a thrombin inhibitor leads to novel dihydro-imidazole and imidazolidine derivatives: structures and mechanism using 13C-labels, mass spectrometry, and NMR. Drug Metab Disposition. 2003;31:1437–14347. doi: 10.1124/dmd.31.11.1437. [DOI] [PubMed] [Google Scholar]
- 76.Nantermet PG, Burgey CS, Robinson KA, Pellicore JM, Newton CL, Deng JZ, Selnick HG, Lewis SD, Lucas BJ, Krueger JA, Miller-Stein C, White RB, Wong B, McMasters DR, Wallace AA, Lynch JJ, Yan Y, Chen Z, Kuo L, Gardell SJ, Shafer JA, Vacca JP, Lyle TA. P2 pyridine N-oxide thrombin inhibitors: a novel peptidomimetic scaffold. Bioorg Med Chem Lett. 2005;15:2771–2775. doi: 10.1016/j.bmcl.2005.03.110. [DOI] [PubMed] [Google Scholar]
- 77.(a) Lewis SD, Ng AS, Lyle EA, Mellott MJ, Appelby SD, Brady SF, Stauffer KS, Sisko JT, Mao S-S, Veber DF, Nutt RF, Lynch JJ, Cook JJ, Gardell SJ, Shafer JA. Inhibition of thrombin by peptides containing lysyl-alpha-keto carbonyl derivatives. Thromb Haemostasis. 1995;74:1107–1112. [PubMed] [Google Scholar]; (b) Piccione G, Casella S, Giannetto1 C, Giudice E. Effect of storage conditions on prothrombin time, activated partial thromboplastin time and fibrinogen concentration on canine plasma samples. J Vet Sci. 2010;11:121–124. doi: 10.4142/jvs.2010.11.2.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.(a) Lumma WC, Witherup KM, Tucker TJ, Brady SF, Sisko JT, Naylor-Olsen AM, Lewis SD, Lucas BJ, Vacca JP. Design of novel, potent, noncovalent inhibitors of thrombin with nonbasic P-1 substructures: rapid structure-activity studies by solid-phase synthesis. J Med Chem. 1998;41:1011–1013. doi: 10.1021/jm9706933. [DOI] [PubMed] [Google Scholar]; (b) Tucker TJ, Lumma WC, Lewis SD, Gardell SJ, Lucas BJ, Baskin EP, Woltmann R, Lynch JJ, Lyle EA, Appleby SD, Chen I-W, Dancheck KB, Vacca JP. Potent noncovalent thrombin inhibitors that utilize the unique amino acid d-dicyclohexylalanine in the P3 position. Implications on oral bioavailability and antithrombotic efficacy. J Med Chem. 1997;40:1565–1569. doi: 10.1021/jm970140s. [DOI] [PubMed] [Google Scholar]
- 79.Boden D, Hurley A, Zhang L, Cao Y, Guo Y, Jones E, Tsay J, Ip J, Farthing C, Limoli K, Parkin N, Markowitz M. HIV-1 drug resistance in newly infected individuals. J Am Med Assoc. 1999;282:1135–1141. doi: 10.1001/jama.282.12.1135. [DOI] [PubMed] [Google Scholar]
- 80.(a) Littman DR. Chemokine receptors: keys to AIDS pathogenesis? Cell. 1998;93:677–680. doi: 10.1016/s0092-8674(00)81429-4. [DOI] [PubMed] [Google Scholar]; (b) Murphy PM. Viral exploitation and subversion of the immune system through chemokine mimicry. Nat Immun. 2001;2:116–122. doi: 10.1038/84214. [DOI] [PubMed] [Google Scholar]; (c) Hunt SW, III, LaRosa GJ. Chemokine Receptors As HIV Co-Receptors: Targets for Therapeutic Intervention in AIDS. Annu Rep Med Chem. 1998;33:263–270. [Google Scholar]; (d) Saunders J, Tarby CM. Opportunities for novel therapeutic agents acting at chemokine receptors. Drug Discov Today. 1999;4:80–92. doi: 10.1016/s1359-6446(98)01280-x. [DOI] [PubMed] [Google Scholar]
- 81.NIH, National Institute of Allergy and Infectious Diseases. ( http://www.niaid.nih.gov/daids/dtpdb/attach.asp)
- 82.(a) Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science. 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]; (b) Michael NL, Moore JP. HIV-1 entry inhibitors: evading the issue. Nat Med. 1999;5(7):740–743. doi: 10.1038/10462. [DOI] [PubMed] [Google Scholar]
- 83.Michael NL, Chang G, Louie LG, Mascola JR, Dondero D, Birx DL, Sheppard HW. The role of viral phenotype and CCR-5 gene defects in HIV-1 transmission and disease progression. Nat Med. 1997;3(3):338–340. doi: 10.1038/nm0397-338. [DOI] [PubMed] [Google Scholar]
- 84.(a) Maeda K, Yoshimura K, Shibayama S, Habashita H, Tada H, Sagawa K, Miyakawa T, Aoki M, Fukushima D. Novel low molecular weight spirodiketopiperazine derivatives potently inhibit R5 HIV-1 infection through their antagonistic effects on CCR5. J Biol Chem. 2001;276(37):35194–35200. doi: 10.1074/jbc.M105670200. [DOI] [PubMed] [Google Scholar]; (b) Mastrolorenzo A, Scozzafava A, Supuran CT. Update on the development of HIV entry inhibitors. Expert Opin Ther Pat. 2001;11:1245–1252. [Google Scholar]
- 85.Palani A, Shapiro S, Josien H, Bara T, Clader JW, Greenlee WJ, Cox K, Strizki JM, Baroudy BM. Synthesis, SAR, and biological evaluation of oximino-piperidino-piperidine amides. 1. Orally bioavailable CCR5 receptor antagonists with potent anti-HIV activity. J Med Chem. 2002;45:3143–3160. doi: 10.1021/jm0200815. [DOI] [PubMed] [Google Scholar]
- 86.Tagat JR, McCombie SW, Steensma RW, Lin S-I, Nazareno DV, Baroudy B, Vantuno N, Xu S, Liu J. Piperazine-based CCR5 antagonists as HIV-1 inhibitors I: 2(S)-methyl piperazine as a key pharmacophore element. Bioorg Med Chem Lett. 2001;11:2143–2146. doi: 10.1016/s0960-894x(01)00381-x. [DOI] [PubMed] [Google Scholar]
- 87.Palani A, Shapiro S, Clader JW, Greenlee WJ, Cox K, Strizki J, Endres M, Baroudy BM. Discovery of 4-[(Z)-(4-bromophenyl)-(ethoxyimino)methyl]-1′-[(2,4-dimethyl-3-pyridinyl)carbonyl]-4′-methyl-1,4′-bipiperidine N-oxide (SCH 351125): an orally bioavailable human CCR5 antagonist for the treatment of HIV infection. J Med Chem. 2001;44(21):3339–3342. doi: 10.1021/jm015526o. [DOI] [PubMed] [Google Scholar]
- 88.Strizki JM, Xu S, Wagner NE, Wojcik L, Liu J, Hou Y, Endres M, Palani A, Shapiro S, Clader JW, Greenlee WJ, Tagat JR, McCombie S, Cox K, Fawzi AB, Chou CC, Pugliese-Sivo C, Davies L, Moreno ME, Ho DD, Trkola A, Stoddart CA, Moore JP, Reyes GR, Baroudy BM. SCH-C (SCH 351125), an orally bioavailable, small molecule antagonist of the chemokine receptor CCR5, is a potent inhibitor of HIV-1 infection in vitro and in vivo. Proc Natl Acad Sci USA. 2001;98(22):12718–12723. doi: 10.1073/pnas.221375398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Strizki JM, Tremblay C, Xu S, Wojcik L, Wagner N, Gonsiorek W, Hipkin RW, Chou CC, Pugliese-Sivo C, Xiao Y, Tagat JR, Cox K, Priestley T, Sorota S, Huang W, Hirsch M, Reyes GR, Baroudy BM. Discovery and characterization of vicriviroc (SCH 417690), a CCR5 antagonist with potent activity against human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2005;49:4911–4919. doi: 10.1128/AAC.49.12.4911-4919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.(a) Fermini B, Fossa AA. The impact of drug-induced QT interval prolongation on drug discovery and development. Nat Rev Drug Discov. 2003;2(6):439–447. doi: 10.1038/nrd1108. [DOI] [PubMed] [Google Scholar]; (b) Möller C. Keeping the rhythm: hERG and beyond in cardiovascular safety pharmacology. Expert Rev Clin Pharmacol. 2010;3:321–329. doi: 10.1586/ecp.10.24. [DOI] [PubMed] [Google Scholar]
- 91.Vermeire K, Schols D, Bell TW. CD4 down-modulating compounds with potent anti-HIV activity. Curr Pharm Des. 2004;10:1795–1803. doi: 10.2174/1381612043384547. [DOI] [PubMed] [Google Scholar]
- 92.Patterson AV, Saunders MP, Chinje EC, Patterson LH, Stratford IJ. Enzymology of tirapazamine metabolism: a review. Anti-Cancer Drug Des. 1998;13:541–573. [PubMed] [Google Scholar]
- 93.Zeman EM, Brown JM, Lemon MJ, Hirst VK, Lee WW. SR-4233: a new bioreductive agent with high selective toxicity for hypoxic mammalian cells. Int J Radiat Oncol Biol Phys. 1986;12:1239–1242. doi: 10.1016/0360-3016(86)90267-1. [DOI] [PubMed] [Google Scholar]
- 94.Robbins RF, Schofield K. Polyazabicyclic compounds. Part II. Further derivatives of benzo-1,2,4-triazine. J Chem Soc. 1957:3186–3194. [Google Scholar]
- 95.Ke Q, Costa M. Hypoxia-Inducible Factor-1 (HIF-1) Mol Pharmacol. 2006;70:1469–1480. doi: 10.1124/mol.106.027029. [DOI] [PubMed] [Google Scholar]
- 96.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 97.Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology. 2004;19:176–182. doi: 10.1152/physiol.00001.2004. [DOI] [PubMed] [Google Scholar]
- 98.Thomlinson RH, Gray LH. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer. 1955;9:539–549. doi: 10.1038/bjc.1955.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 100.Marcu L, Bezak E, Olver I. Scheduling cisplatin and radiotherapy in the treatment of squamous cell carcinomas of the head and neck: a modelling approach. Phys Med Biol. 2006;51:3625–3637. doi: 10.1088/0031-9155/51/15/002. [DOI] [PubMed] [Google Scholar]
- 101.Brown JM, Lemmon MJ. Potentiation by the hypoxic cytotoxin SR-4233 of cell killing produced by fractionated irradiation of mouse tumors. Cancer Res. 1990;50:7745–1149. [PubMed] [Google Scholar]
- 102.Hay MP, Gamage SA, Kovacs MS, Pruijn FB, Anderson RF, Patterson AV, Wilson WR, Brown JM, Denny WA. Structure activity relationships of 1,2,4-benzotriazine 1,4-dioxides as hypoxia-selective analogues of tirapazamine. J Med Chem. 2003;46:169–182. doi: 10.1021/jm020367+. [DOI] [PubMed] [Google Scholar]
- 103.Hicks KO, Siim BG, Jaiswal JK, Pruijn FB, Fraser AM, Patel R, Hogg A, Sarath Liyanage HD, Dorie MJ, Brown JM, Denny WA, Hay MP, Wilson WR. Pharmacokinetic/pharmacodynamic modeling identifies SN30000 and SN29751 as tirapazamine analogs with improved tissue penetration and hypoxic cell killing in tumors. Clin Cancer Res. 2010;16:4946–4957. doi: 10.1158/1078-0432.CCR-10-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Solano B, Junnotula V, Marín A, Villar R, Burguete A, Vicente E, Pérez-Silanes S, Aldana I, Monge A, Dutta S. Synthesis and biological evaluation of new 2-arylcarbonyl-3-trifluoromethyl-quinoxaline 1,4-di-N-oxide derivatives and their reduced analogues. J Med Chem. 2007;50:5485–5492. doi: 10.1021/jm0703993. [DOI] [PubMed] [Google Scholar]
- 105.(a) Jiang FQ, Yang B, Fan LL, He QJ, Hu YZ. Synthesis and hypoxic-cytotoxic activity of some 3-amino-1,2,4-benzotriazine-1,4-dioxide derivatives. Bioorg Med Chem Lett. 2006;16:4209–4213. doi: 10.1016/j.bmcl.2006.05.095. [DOI] [PubMed] [Google Scholar]; (b) Xia Q, Zhang L, Zhang J, Sheng R, Yang B, He QJ, Hu YZ. Synthesis, hypoxia-selective cytotoxicity of new 3-amino-1,2,4-benzotriazine-1,4-dioxide derivatives. Eur J Med Chem. 2011;46:919–926. doi: 10.1016/j.ejmech.2011.01.007. [DOI] [PubMed] [Google Scholar]; (c) Jiang FQ, Weng QJ, Sheng R, Xia Q, He QJ, Yang B, Hu YZ. Synthesis, structure and hypoxic cytotoxicity of 3-amino-1,2,4-benzotriazine-1,4-dioxide derivatives. Arch Pharm. 2007;340:258–263. doi: 10.1002/ardp.200600201. [DOI] [PubMed] [Google Scholar]
- 106.(a) Sheng R, Xu Y, Weng QJ, Xia Q, He QJ, Yang B, Hu YZ. Synthesis and cytotoxic activity of 3-phenyl-2-thioquinoxaline 1,4-dioxide derivatives in hypoxia and in normoxia. Drug Discov Ther. 2007;1:119–123. [PubMed] [Google Scholar]; (b) Weng QJ, Wang DD, Guo P, Fang L, Hu YZ, He QJ, Yang B. Q39, a novel synthetic quinoxaline 1,4-di-N-oxide compound with anti-cancer activity in hypoxia. Eur J Pharmacol. 2008;581:262–269. doi: 10.1016/j.ejphar.2007.12.006. [DOI] [PubMed] [Google Scholar]; (c) Weng QJ, Zhang J, Cao J, Xia Q, Wang DD, Hu YZ, Sheng R, Wu HH, Zhu DF, Zhu H, He Q, Yang B. Q39, a quinoxaline 1,4-di-N-oxide derivative, inhibits hypoxia-inducible factor-1α expression and the Akt/mTOR/4E-BP1 signaling pathway in human hepatoma cells. Invest New Drugs. 2011;29:1177–1187. doi: 10.1007/s10637-010-9462-y. [DOI] [PubMed] [Google Scholar]
- 107.Hu Y, Xia Q, Shangguan S, Liu X, Hu Y, Sheng R. Synthesis and biological evaluation of 3-aryl-quinoxaline-2-carbonitrile 1,4-diN-oxide derivatives as hypoxic selective anti-tumor agents. Molecules. 2012;17:9683–9696. doi: 10.3390/molecules17089683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nakashima H, Ikkyu K, Nakashima K, Sano K, Uto Y, Nakata E, Nagasawa H, Sugimoto H, Shiro Y, Nakagawa Y, Hori H. Design of novel hypoxia-targeting IDO hybrid inhibitors conjugated with an unsubstituted L-TRP as an IDO affinity moiety. Adv Exp Med Biol. 2010;662:415–421. doi: 10.1007/978-1-4419-1241-1_60. [DOI] [PubMed] [Google Scholar]
- 109.Nakashima H, Uto Y, Nakata E, Nagasawa H, Ikkyu K, Hiraoka N, Nakashima K, Sasaki Y, Sugimoto H, Shiro Y, Hashimoto T, Okamoto Y, Asakawa Y, Hori H. Synthesis and biological activity of 1-methyl-tryptophan-tirapazamine hybrids as hypoxia-targeting indoleamine 2,3-dioxygenase inhibitors. Bioorg Med Chem. 2008;16:8661–8669. doi: 10.1016/j.bmc.2008.07.087. [DOI] [PubMed] [Google Scholar]
- 110.Adams Wilson JR, Morandi A, Girard TD, Thompson JL, Boomershine CS, Shintani AK, Ely EW, Pandharipande PP. The association of the kynurenine pathway of tryptophan metabolism with acute brain dysfunction during critical illness. Crit Care Med. 2012;40:835–841. doi: 10.1097/CCM.0b013e318236f62d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 112.Peters KB, Wang H, Brown JM, Iliakis G. Inhibition of DNA replication by tirapazamine. Cancer Res. 2001;61:5425–5431. [PubMed] [Google Scholar]
- 113.Peters KB, Brown JM. Tirapazamine: a hypoxia-activated topoisomerase II poison. Cancer Res. 2002;62:5248–5253. [PubMed] [Google Scholar]
- 114.Delahoussaye YM, Hay MP, Pruijn FB, Denny WA, Brown JM. Improved potency of the hypoxic cytotoxin tirapazamine by DNA-targeting. Biochem Pharmacol. 2003;65:1807–1815. doi: 10.1016/s0006-2952(03)00199-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.(a) Delahoussaye YM, Evans JW, Brown JM. Metabolism of tirapazamine by multiple reductases in the nucleus. Biochem Pharmacol. 2001;62:1201–1209. doi: 10.1016/s0006-2952(01)00784-5. [DOI] [PubMed] [Google Scholar]; (b) Biedermann KA, Wang J, Graham RP, Brown JM. SR 4233 cytotoxicity and metabolism in DNA repair-competent and repair-deficient cell cultures. Br J Cancer. 1991;63:358–362. doi: 10.1038/bjc.1991.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jones GD, Weinfeld M. Dual action of tirapazamine in the induction of DNA strand breaks. Cancer Res. 1996;56:1584–1590. [PubMed] [Google Scholar]
- 117.Daniels JS, Gates KS, Tronche C, Greenberg MM. Direct evidence for bimodal DNA damage induced by tirapazamine. Chem Res Toxicol. 1998;11:1254–1257. doi: 10.1021/tx980184j. [DOI] [PubMed] [Google Scholar]
- 118.Hwang JT, Greenberg MM, Fuchs T, Gates KS. Reaction of the hypoxia-selective antitumor agent tirapazamine with a C1′-radical in single-stranded and double-stranded DNA: the drug and its metabolites can serve as surrogates for molecular oxygen in radical-mediated DNA-damage reactions. Biochemistry. 1999;38:14248–14255. doi: 10.1021/bi991488n. [DOI] [PubMed] [Google Scholar]
- 119.Siim BG, Pruijn FB, Sturman JR, Hogg A, Hay MP, Brown JM, Wilson WR. Selective potentiation of the hypoxic cytotoxicity of tirapazamine by its 1-N-oxide metabolite SR 4317. Cancer Res. 2004;64:736–742. doi: 10.1158/0008-5472.can-03-2488. [DOI] [PubMed] [Google Scholar]
- 120.(a) Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838–2849. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]; (b) Yang DP, Kim J, Syed N, Tung YJ, Bhaskaran A, Mindos T, Mirsky R, Jessen KR, Maurel P, Parkinson DB, Kim HA. p38 MAPK activation promotes denervated Schwann cell phenotype and functions as a negative regulator of Schwann cell differentiation and myelination. J Neurosci. 2012;32:7158–7168. doi: 10.1523/JNEUROSCI.5812-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fernandes DJ, Ravenhall CE, Harris T, Tran T, Vlahos R, Stewart AG. Contribution of the p38MAPK signalling pathway to proliferation in human cultured airway smooth muscle cells is mitogen-specific. Br J Pharmacol. 2004;142:1182–1190. doi: 10.1038/sj.bjp.0705809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Svensson C, Part K, Künnis-Beres K, Kaldmäe M, Fernaeus SZ, Land T. Pro-survival effects of JNK and p38 MAPK pathways in LPS-induced activation of BV-2 cells. Biochem Biophys Res Commun. 2011;406:488–492. doi: 10.1016/j.bbrc.2011.02.083. [DOI] [PubMed] [Google Scholar]
- 122.Sinfield JK, Das A, O’Regan DJ, Ball SG, Porter KE, Turner NA. p38 MAPK alpha mediates cytokine-induced IL-6 and MMP-3 expression in human cardiac fibroblasts. Biochem Biophys Res Commun. 2013;430:419–424. doi: 10.1016/j.bbrc.2012.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yong HY, Koh MS, Moon A. The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin Invest Drugs. 2009;18:1893–1905. doi: 10.1517/13543780903321490. [DOI] [PubMed] [Google Scholar]
- 124.Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heyes JR, Landvatter SW, Strickler JE, McLaughlin MM, Siemens IR, Fisher SM, Livi GP, White JR, Adams JL, Young PR. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 125.Fitzgerald CE, Patel SB, Becker JW, Cameron PM, Zaller D, Pikounis VB, O’Keefe SJ, Scapin G. Structural basis for p38α MAP kinase quinazolinone and pyridol-pyrimidine inhibitor specificity. Nat Struct Biol. 2003;10:764–769. doi: 10.1038/nsb949. [DOI] [PubMed] [Google Scholar]
- 126.(a) Ferraciola GF. VX-745 Vertex Pharmaceutical. Curr Opin Anti-Inflam Immunomodul Invest Drugs. 2000;2:74–77. [Google Scholar]; (b) Haddad JJ. VX-745 (Vertex Pharmaceuticals) Curr Opin Invest Drugs. 2001;2:1070–1076. [PubMed] [Google Scholar]
- 127.(a) Alonso-Alija C, Michels M, Schirok H, Schlemmer K-H, Bell J, Fitzgerald MF, Dodd S, Gill A. Monocyclic aroylpyr-idinones as antiinflammatory agents. 076405. WO Patent. 2003 Sep 4;; (b) Schirok H, Alonso-Alija C, Benet-Buchholz J, Goller AH, Grosser R, Michels M, Paulsen H. Efficient regioselective synthesis of 6-amino-5-benzoyl-1-substituted 2(1H)-pyridinones. J Org Chem. 2005;70:9463–9469. doi: 10.1021/jo0515428. [DOI] [PubMed] [Google Scholar]
- 128.(a) Zimmiti CS, Schwartz R, Torcellini CA, Pargellis CA, Madwed JB, Weldon SM. Suppression of p38 activity in vitro and THF alpha production in vivo with BIRB-796 BS, a novel p38 kinase inhibitor. Arthritis Rheum. 2001;44:S114. [Google Scholar]; (b) Regan J, Capolino A, Cirillo PF, Gilmore T, Graham AG, Hickey E, Kroe RR, Madwed J, Moriak M, Nelson R, Pargellis CA, Swinamer A, Torcellini C, Tsang M, Moss N. Structure-activity relationships of the p38R MAP kinase inhibitor 1-(5-tert-Butyl-2-p-tolyl-2H-pyrazol-3-yl)-3-[4-(2-morpholin-4-yl-ethoxy)napthalen-1-yl]urea (BIRB-796) J Med Chem. 2003;46:4676–4686. doi: 10.1021/jm030121k. [DOI] [PubMed] [Google Scholar]
- 129.Devraj RV. Discovery and development of orally active p38 kinase inhibitors as anti-TNF agents. Abstracts of Papers of the 229th national meeting of the American Chemical Society; San Diego, CA. March 13–17, 2005; Washington, DC: American Chemical Society; 2005. p. MEDI-296. [Google Scholar]
- 130.(a) Zack DJ, Campagnuolo G, Middleton S, Koch A, Zhu L, Bolon B, Feige U. Efficacy of AMG 548, a novel p38 kinase inhibitor, in rats with adjuvant-induced or collagen-induced arthritis. Abstracts of the 67th Annual Scientific Meeting of the American College of Rheumatology; Orlando, FL. October 23–28, 2003; p. Abstract 834. [Google Scholar]; (b) Domınguez C, Liu L, Zhang D, Tamayo N, Powers D, Min W, Feige F, Harris R, Wild S, Neervannan S, Rashid S, Harvey T. p38 MAP kinase: an exciting target for the treatment of inflammatory diseases. Abstracts of Papers of the 228th National Meeting of the American Chemical Society; Philadelphia, PA. Washington, DC: American Chemical Society; 2004. p. MEDI-189. [Google Scholar]
- 131.Nikas SN, Drossos AA. SCIOS-469 (Scios Inc.) Curr Opin Investig Drugs. 2004;5:1205–1212. [PubMed] [Google Scholar]
- 132.Lumeras W, Caturla F, Vidal L, Esteve C, Balague C, Orellana A, Domınguez M, Roca R, Huerta JM, Godessart N, Vidal B. Design, synthesis, and structure-activity relationships of aminopyridine N-oxides, a novel scaffold for the potent and selective inhibition of p38 mitogen activated protein kinase. J Med Chem. 2009;52:5531–5545. doi: 10.1021/jm9008604. [DOI] [PubMed] [Google Scholar]
- 133.Lumeras W, Vidal L, Vidal B, Balague C, Orellana A, Maldonado M, Domińguez M, Segarra V, Caturla F. 1,7-Naphthyridine 1-oxides as novel potent and selective inhibitors of p38 mitogen activated protein kinase. J Med Chem. 2011;54:7899–7910. doi: 10.1021/jm200975u. [DOI] [PubMed] [Google Scholar]
- 134.(a) Walker SD, Barder TE, Martinelli JR, Buchwald SL. A rationally designed universal catalyst for Suzuki-Miyaura coupling processes. Angew Chem, Int Ed. 2004;43:1871–1876. doi: 10.1002/anie.200353615. [DOI] [PubMed] [Google Scholar]; (b) Negishi E-I, King AO, Okukado N. Selective carbon-carbon bond formation via transition-metal catalysis. 3. Highly selective synthesis of unsymmetrical biaryls and diarylmethanes by nickel-catalyzed or palladium-catalyzed reaction of aryl derivatives and benzylzinc derivatives with aryl. J Org Chem. 1977;42:1821–1823. [Google Scholar]; (c) Negishi E-I, Luo F-T, Frisbee R, Matsushita HA. Regiospecific synthesis of carbosubstituted heteroaromatic derivatives via Pd-catalyzed cross coupling. Heterocycles. 1982;18:117–122. [Google Scholar]
- 135.Gasco A, Boulton AJ. Furoxans and benzofuroxans. Adv Heterocycl Chem. 1981;29:251–340. [Google Scholar]
- 136.Fang L, Zhang Y, Lehmann J, Wang Y, Ji H, Ding D. Design and synthesis of furoxan-based nitric oxide-releasing glucocorticoid derivatives with potent anti-inflammatory activity and improved safety. Bioorg Med Chem Lett. 2007;17:1062–1066. doi: 10.1016/j.bmcl.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 137.Olea-Azar C, Rigo C, Mendizábal F, Cerecetto H, Di Maio R, González M, Porcal W, Morello A, Repetto Y, Maya JD. Novel benzo [1,2-c]1,2,5-oxadiazole N-oxide derivatives as antichagasic agents: chemical and biological studies. Lett Drug Des Discovery. 2005;4:294–301. [Google Scholar]
- 138.(a) Ghosh PB, Whitehouse MW. 7-Chloro-4-nitrobenzo-2-oxa-1,3-diazole: a new fluorigenic reagent for amino acids and other amines. J Med Chem. 1968;11:305–311. doi: 10.1042/bj1080155. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Whitehouse MW, Ghosh PB. 4-Nitrobenzofurazans and 4-nitrobenzofuroxans: a new class of thiol-neutralising agents and potent inhibitors of nucleic acid synthesis in leucocytes. Biochem Pharmacol. 1968;17:158–161. doi: 10.1016/0006-2952(68)90169-x. [DOI] [PubMed] [Google Scholar]
- 139.Aguirre G, Boiani M, Cerecetto H, Fernández M, González M, León E, Pintos C, Raymondo S, Arredondo C, Pacheco JP, Basombrío M. Furoxan derivatives as cytotoxic agents: preliminary in vivo antitumoral activity studies. Pharmazie. 2006;61:54–59. [PubMed] [Google Scholar]
- 140.Boiani L, Aguirre G, González M, Cerecetto H, Chidichimo A, Cazzulo JJ, Bertinaria M, Guglielmo S. Furoxan-, alkylnitrate-derivatives and related compounds as anti-trypanosomatid agents: mechanism of action studies. Bioorg Med Chem. 2008;16:7900–7907. doi: 10.1016/j.bmc.2008.07.077. [DOI] [PubMed] [Google Scholar]
- 141.Thatcher GRJ, Nicolescu AC, Bennett BM, Toader V. Nitrates and NO release: contemporary aspects in biological and medicinal chemistry. Free Radical Biol Med. 2004;37:1122–1143. doi: 10.1016/j.freeradbiomed.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 142.(a) Zhuo M, Hu Y, Schultz C, Kandel ER, Hawkins RD. Role of guanylyl cyclase and cGMP-dependent protein kinase in long-term potentiation. Nature. 1994;368:635–639. doi: 10.1038/368635a0. [DOI] [PubMed] [Google Scholar]; (b) Lu YF, Kandel ER, Hawkins RD. Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J Neurosci. 1999;19:10250–10261. doi: 10.1523/JNEUROSCI.19-23-10250.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bon CL, Garthwaite J. On the role of nitric oxide in hippocampal long-term potentiation. J Neurosci. 2003;23:1941–1948. doi: 10.1523/JNEUROSCI.23-05-01941.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Haley JE, Wilcox GL, Chapman PF. The role of nitric oxide in hippocampal long-term potentiation. Neuron. 1992;8:211–216. doi: 10.1016/0896-6273(92)90288-o. [DOI] [PubMed] [Google Scholar]
- 143.Cirelli C, Tonon G. Differential expression of plasticity-related genes in waking and sleep and their regulation by the noradrenergic system. J Neurosci. 2000;20:9187–9194. doi: 10.1523/JNEUROSCI.20-24-09187.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Arancio O, Kiebler M, Lee CJ, Lev-Ram V, Tsien RY, Kandel ER, Hawkins RD. Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell. 1996;87:1025–1035. doi: 10.1016/s0092-8674(00)81797-3. [DOI] [PubMed] [Google Scholar]
- 145.Gass P, Riva MA. CREB, neurogenesis and depression. Bioessays. 2007;29:957–961. doi: 10.1002/bies.20658. [DOI] [PubMed] [Google Scholar]
- 146.Peng S, Wu J, Mufson EJ, Fahnestock M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J Neurochem. 2005;93:1412–1421. doi: 10.1111/j.1471-4159.2005.03135.x. [DOI] [PubMed] [Google Scholar]
- 147.Rai G, Sayed AA, Lea WA, Luecke HF, Chakrapani H, Prast-Nielsen S, Jadhav A, Leister W, Shen M, Inglese J, Austin CP, Keefer L, Arner ES, Simeonov A, Maloney DJ, Williams DL, Thomas CJ. Structure mechanism insights and the role of nitric oxide donation guide the development of oxadiazole-2-oxides as therapeutic agents against schistosomiasis. J Med Chem. 2009;52:6474–6483. doi: 10.1021/jm901021k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.(a) Sorba G, Medana C, Fruttero R, Cena C, Di Stilo A, Galli U, Gasco A. Water soluble furoxan derivatives as NO prodrugs. J Med Chem. 1997;40:463–469. doi: 10.1021/jm960379t. [DOI] [PubMed] [Google Scholar]; (b) Ferioli R, Folco GC, Ferretti C, Gasco AM, Medana C, Fruttero R, Civelli M, Gasco A. A new class of furoxan derivatives as NO donors: mechanism of action and biological activity. Br J Pharmacol. 1995;114:816–820. doi: 10.1111/j.1476-5381.1995.tb13277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Goldberg MP, Strasser U, Dugan LL. Techniques for assessing neuroprotective drugs in vitro. Int Rev Neurobiol. 1997;40:69–93. doi: 10.1016/s0074-7742(08)60716-3. [DOI] [PubMed] [Google Scholar]
- 150.Lee KS, Frank S, Vanderklish P, Arai A, Lynch G. Inhibition of proteolysis protects hippocampal neurons from ischemia. Proc Natl Acad Sci USA. 1991;88:7233–7237. doi: 10.1073/pnas.88.16.7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bisby RH. Reactions of a free radical intermediate in the oxidation of amodiaquine. Biochem Pharmacol. 1990;39:2051–2055. doi: 10.1016/0006-2952(90)90628-x. [DOI] [PubMed] [Google Scholar]
- 152.Mott BT, Cheng KC, Guha R, Kommer VP, Williams DL, Vermeire JJ, Cappello M, Maloney DJ, Rai G, Jadhav A, Simeonov A, Inglese J, Posner GH, Thomas CJ. A furoxan-amodiaquine hybrid as a potential therapeutic for three parasitic diseases. MedChemComm. 2012;12:1505–1511. doi: 10.1039/C2MD20238G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.(a) Egan TJ. Quinoline antimalarials. Expert Opin Ther Pat. 2001;11:185–209. [Google Scholar]; (b) Egan TJ. Structure-function relationships in chloroquine and related 4-aminoquinoline antimalarials. Mini-Rev Med Chem. 2001;1:113–123. doi: 10.2174/1389557013407188. [DOI] [PubMed] [Google Scholar]
- 154.Schmunis GA, Yadon ZE. Chagas disease: a Latin American health problem becoming a world health problem. Acta Trop. 2010;115(1–2):14–21. doi: 10.1016/j.actatropica.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 155.Cerecetto H, Gonzalez M. Synthetic medicinal chemistry in Chagas’ disease: compounds at the final stage of “Hit-To-Lead” phase. Pharmaceuticals. 2010;3:810–838. doi: 10.3390/ph3040810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Otero L, Vieites M, Boiani L, Denicola A, Rigol C, Opazo L, Olea-Azar C, Maya JD, Morello A, Krauth-Siegel RL, Piro OE, Castellano E, Gonzalez M, Gambino D, Cerecetto H. Novel antitrypanosomal agents based on palladium nitrofuryl-thiosemicarbazone complexes: DNA and redox metabolism as potential therapeutic targets. J Med Chem. 2006;49:3322–3331. doi: 10.1021/jm0512241. [DOI] [PubMed] [Google Scholar]
- 157.(a) Gerpe A, Odreman-Nunez I, Draper P, Boiani l, Urbina JA, Gonzalez M, Cerecetto H. Heteroallyl-containing 5-nitrofuranes as new anti-Trypanosoma cruzi agents with a dual mechanism of action. Bioorg Med Chem. 2008;16:569–577. doi: 10.1016/j.bmc.2007.07.031. [DOI] [PubMed] [Google Scholar]; (b) Gerpe A, Alvarez G, Benıtez D, Boiani L, Quiroga M, Hernandez P, Sortino M, Zacchino S, Gonzalez M, Cerecetto H. 5-Nitrofuranes and 5-nitrothiophenes with anti-Trypanosoma cruzi activity and ability to accumulate squalene. Bioorg Med Chem. 2009;17:7500–7509. doi: 10.1016/j.bmc.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 158.(a) Porcal W, Hernandez P, Boiani M, Aguirre G, Boiani L, Chidichimo A, Cazzulo JJ, Campillo NE, Paez JA, Castro A, Krauth Siegel RL, Davies C, Basombr MA, Gonzalez M, Cerecetto H. In Vivo Anti-Chagas Vinylthio-, Vinylsulfinyl-, and Vinylsulfonylbenzofuroxan Derivatives. J Med Chem. 2007;50:6004–6015. doi: 10.1021/jm070604e. [DOI] [PubMed] [Google Scholar]; (b) Porcal W, Hernandez P, Boiani L, Boiani M, Ferreira A, Chidichimo A, Cazzulo JJ, Olea-Azar C, Gonzalez M, Cerecetto H. New trypanocidal hybrid compounds from the association of hydrazone moieties and benzofuroxan heterocycle. Bioorg Med Chem. 2008;16:6995–7004. doi: 10.1016/j.bmc.2008.05.038. [DOI] [PubMed] [Google Scholar]; (c) Merlino A, Benitez D, Chavez S, Da Cunha J, Hernandez P, Tinoco LW, Campillo NE, Paez JA, Cerecetto H, Gonzalez M. Development of second generation amidinohydrazones, thio- and semicarbazones as Trypanosoma cruzi-inhibitors bearing benzofuroxan and benzimidazole 1,3-dioxide core scaffolds. MedChemComm. 2010;1:216–228. [Google Scholar]
- 159.Merlino A, Benitez D, Campillo NE, Paez JA, Tinoco LW, Gonzalez M, Cerecetto H. Amidines bearing benzofuroxan or benzimidazole 1,3-dioxide core scaffolds as Trypanosoma cruzi-inhibitors: structural basis for their interactions with cruzipain. MedChemComm. 2012;3:90–101. [Google Scholar]
- 160.(a) Clark RD. Synthesis and QSAR of herbicidal 3-pyrazolyl α,α,α-trifluorotolyl ethers. J Agric Food Chem. 1996;44:3643–3652. [Google Scholar]; (b) Nandihalli UB, Duke MV, Ashmore JA, Musco VA, Clark RD, Duke OS. Enantioselectivity of protoporphyrinogen oxidase-inhibiting herbicides. Pestic Sci. 1994;40:265–277. [Google Scholar]
- 161.(a) Kleschick WA, Costales MJ, Dunbar JE, Meikle RW, Monte WT, Pearson NR, Snider SW, Vinogradoff AP. New herbicidal derivatives of 1,2,4-triazolo [1,5-a] pyrimidine. Pestic Sci. 1990;29:341–355. [Google Scholar]; (b) Wepplo P. Imidazolinone herbicides: synthesis and novel chemistry. Pestic Sci. 1990;39:293–315. [Google Scholar]
- 162.Baker NR, Percival MP. Herbicides and photosynthesis. In: Baker NR, Percival MP, Barber J, editors. Herbicides Topics in Photosynthesis. Vol. 10. Elsevier Science Publishers; Amsterdam, The Netherlands: 1990. pp. 1–26. Chapter 9.4. [Google Scholar]
- 163.(a) English AR, Dunegan CM. Quinoxaline-1, 4-di-N-oxides. I. Inhibition of deoxyribonucleic acid synthesis in Escherichia coli by 2,3-dihydroxymethyl-quinoxaline-1, 4-di-N-oxide. Proc Soc Exp Biol Med. 1970;133:398–400. doi: 10.3181/00379727-133-34481. [DOI] [PubMed] [Google Scholar]; (b) Suter W, Rosselet A, Knusel F. Mode of action of quindoxin and substituted quinoxaline-di-N-oxides on Escherichia coli. Antimicrob Agents Chemother. 1978;13:770–783. doi: 10.1128/aac.13.5.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.(a) Kern DL, Hokanson GC, French JC, Dally NK. Guanine-7-oxide, a novel antitumor antibiotic. J Antibiot. 1985;38:572–574. doi: 10.7164/antibiotics.38.572. [DOI] [PubMed] [Google Scholar]; (b) Kitahara M, Ishii K, Kumada Y, Shiraishi T, Furuta T, Miwa T, Kawaharada H, Watanabe K. 7-Hydroxyguanine, a novel anti-metabolite from a strain of Streptomyces purpurascens. I. Taxonomy of producing organism, fermentation, isolation and biological activity. J Antibiot. 1985;38:972–976. doi: 10.7164/antibiotics.38.972. [DOI] [PubMed] [Google Scholar]; (c) Kitahara M, Ishii K, Kawaharada H, Watanabe K, Suga T, Hirata T, Nakamura S. 7-Hydroxyguanine, a novel antimetabolite from a strain of Streptomyces purpurascens. II. Physico-chemical properties and structure determination. J Antibiot. 1985;38:977–980. doi: 10.7164/antibiotics.38.977. [DOI] [PubMed] [Google Scholar]; (d) Nishii M, Inagaki J, Nohara F, Isono K, Kusakabe H, Kobayashi K, Sakurai T, Koshimura S, Sethi SK, McCloskey JA. A new antitumor antibiotic, guanine 7-N-oxide produced by Streptomyces sp. J Antibiot. 1985;38:1440–1443. doi: 10.7164/antibiotics.38.1440. [DOI] [PubMed] [Google Scholar]
- 165.Jackson RC, Boritzki TJ, Besserer JA, Hamelehle KL, Shillis JL, Leopold WR, Fry DW. Biochemical pharmacology and experimental chemotherapy studies with guanine-7-oxide, a novel purine antibiotic. Adv Enzyme Regul. 1987;26:301–316. doi: 10.1016/0065-2571(87)90020-3. [DOI] [PubMed] [Google Scholar]
- 166.Hasobe M, Saneyoshi M, Isono K. Antiviral activity and its mechanism of guanine 7-N-oxide on DNA and RNA viruses derived from salmonid. J Antibiot. 1985;38:1581–1587. doi: 10.7164/antibiotics.38.1581. [DOI] [PubMed] [Google Scholar]
- 167.Kanematsu S, Brown GB. Purine N-oxides. XIX. On the oncogenic N-oxide derivatives of guanine and xanthine and a nononcogenic isomer of xanthine N-oxide. Cancer Res. 1967;27:925–931. [PubMed] [Google Scholar]
- 168.Fujii T, Itaya T, Ogawa K. The 7-N-oxides of purines related to nucleic acid: Their chemistry, synthesis and biological evaluation. Heterocycles. 1997;44:573–592. [Google Scholar]
- 169.Kitahara M, Ishii K, Kumada Y, Shiraishi T, Furuta T, Miwa T, Kawaharada H, Watanabe K. 7-Hydroxyguanine, a novel antimetabolite from a strain of Streptomyces purpurascens. I. Taxonomy of producing organism, fermentation, isolation and biological activity. J Antibiot. 1985;38:972–976. doi: 10.7164/antibiotics.38.972. [DOI] [PubMed] [Google Scholar]
- 170.Kitahara M, Shinjyo K, Fukae M, Hosoe K, Shiraishi T, Ishii K, Watanabe K, Kawaharada H. Preparation of new 7-hydroxyguanine derivatives and their biological activities. J Antibiot. 1990;43:352–356. doi: 10.7164/antibiotics.43.352. [DOI] [PubMed] [Google Scholar]
- 171.(a) Ogawa K, Nishii M, Nohara F, Saito T, Itaya T, Fujii T. Purines. Part 51. Synthesis and biological activity of hypoxanthine 7-N-oxide and related compounds. Chem Pharm Bull. 1992;40:612–616. doi: 10.1248/cpb.40.612. [DOI] [PubMed] [Google Scholar]; (b) Fujii T, Ogawa K, Itaya T, Date T, Inagaki J, Nohara F. Purines. LXX. An Extension of the “Phenacylamine Route” to the Syntheses of the 7-N-Oxides of 6-Mercaptopurine and 6-Methylthiopurine, and Antileukemic Activity of Some Purine N-Oxides. Chem Pharm Bull. 1995;43:408–413. doi: 10.1248/cpb.43.408. [DOI] [PubMed] [Google Scholar]
- 172.Parra KV, Adrian JC, Gerlai R. The synthetic substance hypoxanthine 3-N-oxide elicits alarm reactions in zebrafish (Danio rerio) Behav Brain Res. 2009;205:336–341. doi: 10.1016/j.bbr.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Nohara F, Nishii M, Ogawa K, Isono K, Ubukata M, Fujii T, Itaya T, Saito T. Synthesis of guanine 7-oxide, an antitumor antibiotic from Streptomyces species. Tetrahedron Lett. 1987;28:1287–1290. [Google Scholar]
- 174.Albini A. Heterocyclic N-oxides. chapter 4 CRC Press; 1991. [Google Scholar]
- 175.(a) Groll M, Larionov OV, Huber R, de Meijere A. Inhibitor-binding mode of homobelactosin C to proteasomes: new insights into class I MHC ligand generation. Proc Natl Acad Sci USA. 2006;103(12):4576–4579. doi: 10.1073/pnas.0600647103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) de Meijere A, Korotkov VS, Lygin AV, Larionov OV, Sokolov VV, Graef T, Es-Sayed M. Synthesis and biological activity of simplified belactosin C analogues. Org Biomol Chem. 2012;10(31):6363–6374. doi: 10.1039/c2ob25586c. [DOI] [PubMed] [Google Scholar]
- 176.(a) Carlin RL. Transition metal complexes of pyridine N-oxide. J Am Chem Soc. 1961;83(18):3773–3775. [Google Scholar]; (b) Popp CJ, Garlough GD. Transition metal complexes of diazine N-oxides. J Inorg Nucl Chem. 1981;43(3):501–507. [Google Scholar]; (c) Sarma R, Karmakar A, Baruah JB. Synthesis and characterization of pyridine N-oxide complexes of manganese, copper and zinc. Inorg Chim Acta. 2008;361:2081–2086. [Google Scholar]
- 177.(a) Coperet C, Adolfsson H, Khuong T-AW, Yudin AK, Sharpless KB. A simple and efficient method for the preparation of pyridine N-oxides. J Org Chem. 1998;63:1740–1741. [Google Scholar]; (b) Larionov OV, Stephens D, Mfuh AM, Arman HD, Naumova AS, Chavez G, Skenderi B. Insights into the mechanistic and synthetic aspects of the Mo/P-catalyzed oxidation of N-heterocycles. Org Biomol Chem. 2014;12(19):3026–3036. doi: 10.1039/c4ob00115j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.(a) Larionov OV, Stephens D, Mfuh A, Chavez G. Direct, catalytic, and regioselective synthesis of 2-alkyl-, aryl-, and alkenyl-substituted N-heterocycles from N-oxides. Org Lett. 2014;16(3):864–867. doi: 10.1021/ol403631k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stephens DE, Chavez G, Valdes M, Dovalina M, Arman HD, Larionov OV. Synthetic and mechanistic aspects of the regioselective base-mediated reaction of perfluoroalkyl- and perfluoroarylsilanes with heterocyclic N-oxides. Org Biomol Chem. 2014;12(32):6190–6199. doi: 10.1039/c4ob01088d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Stephens DE, Lakey-Beitia J, Atesin AC, Ateşin TA, Chavez G, Arman HD, Larionov OV. Palladium-catalyzed C8-selective C–H arylation of quinoline N-oxides: insights into the electronic, steric and solvation effects on the site-selectivity by mechanistic and DFT computational studies. ACS Catal. 2015;5(1):167–175. doi: 10.1021/cs501813v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Stephens DE, Lakey-Beitia J, Chavez G, Ilie C, Arman HD, Larionov OV. Experimental and mechanistic analysis of the palladium-catalyzed oxidative C8-selective C–H homocoupling of quinoline N-oxides. Chem Commun. 2015;51:9507–9510. doi: 10.1039/c5cc02227d. [DOI] [PMC free article] [PubMed] [Google Scholar]