

Abstract
Biomarkers for Alzheimer’s disease (AD) are vital for disease detection in the clinical setting. Discovered in our laboratory, activity-dependent neuroprotective protein (ADNP) is essential for brain formation and linked to cognitive functions. Here, we revealed that blood borne expression of ADNP and its paralog ADNP2 is correlated with premorbid intelligence, AD pathology, and clinical stage. Age adjustment showed significant associations between: 1] higher premorbid intelligence and greater serum ADNP, and 2] greater cortical amyloid and lower ADNP and ADNP2 mRNAs. Significant increases in ADNP mRNA levels were observed in patients ranging from mild cognitive impairment (MCI) to AD dementia. ADNP2 transcripts showed high correlation with ADNP transcripts, especially in AD dementia lymphocytes. ADNP plasma/serum and lymphocyte mRNA levels discriminated well between cognitively normal elderly, MCI, and AD dementia participants. Measuring ADNP blood-borne levels could bring us a step closer to effectively screening and tracking AD.
Keywords: Cognitively normal, mild cognitive impairment, Alzheimer’s disease (AD), activity-dependent neuroprotective protein (ADNP), blood-borne biomarkers, amyloid beta, premorbid intelligence
INTRODUCTION
As Alzheimer’s disease (AD) pathologic onset takes place many years prior to clinical manifestation, identification of reliable, non-invasive, and inexpensive biomarkers is imperative toward the goal of the much desired disease modification treatments. Genome-wide association studies (GWAS) linked AD with apolipoprotein E (ApoE, the major susceptibility gene for late onset AD) and others. Further studies addressing biomarkers in AD concentrate on utilizing cerebrospinal fluid (CSF) amyloid-beta 1-42 (Aβ1-42), total tau (t-tau), phosphorylated tau (p-tau181p), p-tau181p/Aβ1-42, and t-tau/Aβ1-42 as circulating biomarkers and molecular imaging of pathology as additional surrogate markers [1–9]. Since CSF sampling and imaging technologies are invasive and expensive, the search for reliable blood-borne biomarkers for AD is vital for widespread detection of AD in the clinical setting.
Activity-dependent neuroprotective protein (ADNP) is a protein/gene discovered in Professor Gozes’ laboratory [10, 11] and found to be essential for brain formation in the mouse [12]. Employing complete gene array, the Gozes laboratory discovered that ADNP regulates >400 genes during brain development and directly interacts with the ApoE promoter region [13]. ADNP deficiency results in marked increases in ApoE expression [13], especially in the female mouse brain [14]. Furthermore, ADNP haploinsufficient mice exhibit aging-related tauopathy, neurodegeneration, and cognitive deficits [15].
In AD mouse models, for example in the PS1(M146L) x APP(751SL) transgenic mice, ADNP messenger ribonucleic acid (mRNA) expression in the hippocampus of 6-month-old PS1xAPP mice is higher than in wild-type (WT) mice, while in the cerebellum, a part of the brain not affected by Aβ deposition, no difference was noted [16]. Malishkevich and Gozes have recently shown similar increases in cortical ADNP preceding tauopathy in the rTg(tau(P301L))4510 mouse expressing the P301L mutation in tau (4R0N) associated with frontotemporal dementia and parkinsonism linked to chromosome 17. These transgenic mice overexpress the mutated tau 4R species in the cerebral cortex but not in the cerebellum and no change in ADNP were found in the cerebellum, as compared to control littermates [17]. However, with aging, cortical ADNP decreases, and a more robust decrease is seen in the in the rTg(tau(P301L))4510 mouse [18]. These results suggest a potential deregulation of ADNP expression in the AD brain [19], raising the question of whether peripheral ADNP can serve as a potential biomarker for AD for initial screening, as well as tracking disease progression.
It should be noted that the Gozes laboratory discovered one ADNP paralog, ADNP2 (33% identity and 46% similarity [11]). ADNP2 is important for cellular protection [20]. In healthy conditions, ADNP mRNA levels correlate well with ADNP2 mRNA levels [21, 22]. Furthermore, peripheral ADNP2 mRNA levels (unlike ADNP mRNA levels) may change as a consequence of drug treatment, as seen in rats subjected to clozapine treatment [23]. Together, these findings further suggest the possibility of blood borne ADNP-ADNP2 mRNA and protein dysregulation in patients with AD dementia and its precursor states.
An independent study by Yang et al., analyzed serum proteins using two-dimensional gel electrophoresis combined with nano-high performance liquid chromatography electrospray ionization tandem mass spectrometry followed by peptide fragmentation patterning, comparing human serum specimens from 45 mild AD dementia patients and 20 cognitively normal (CN) elderly individuals, who did not have any history or evidence of dementia. Human serum samples were collected in sterile glass tubes and no anticoagulant was added. After centrifugation at 1000 g for 10 min at 4 °C, the supernatants were analyzed, showing that the only protein decreasing in the AD dementia patients was ADNP [24].
In the current study, we looked at two groups of participants: A cohort of community-dwelling American CN elderly participating in the Harvard Aging Brain Study (HABS), who had imaging and biomarker data, alongside a cohort of community dwelling Israeli CN elderly and patients with mild cognitive impairment (MCI) and AD dementia. Our objectives were 1] to show that ADNP could be a useful blood-borne AD biomarker and 2] a marker of brain health by relating it to a] established AD imaging and CSF biomarkers, b] different clinical stages of AD, and c] a measure of premorbid intelligence. We hypothesized that different measures of ADNP will be associated with amyloid burden, tau burden, and clinical AD disease severity.
MATERIALS AND METHODS
HABS Participants
Forty community-dwelling CN elderly participating in HABS at the Massachusetts General Hospital (MGH) and Brigham and Women’s Hospital (BWH) in Boston, MA, USA, who had the most complete imaging and biomarker data were included in the current analyses. HABS is a longitudinal cohort study in which CN elderly participants undergo cognitive, behavioral, and functional assessments, multi-model imaging, and serum and CSF biomarker sampling. Participants meet the following inclusion criteria: a Clinical Dementia Rating (CDR)[25] global score of 0, an education-adjusted Mini-Mental State Examination (MMSE)[26] score of 27–30, an education-adjusted performance on the Logical Memory Story Delayed Recall [27] within 1.5 standard deviation, and a 30-item Geriatric Depression Scale [28] score of ≤ 10.
The study was approved by the Partners Healthcare Inc. Institutional Review Board. Prior to initiation of any study procedures, written informed consent was obtained from all participants. Transfer of samples to Tel Aviv University was approved by the University’s Ethical Committee.
Israeli Participants
Fifteen patients with MCI, 17 patients with AD dementia, and 11 community-dwelling CN elderly underwent research assessments at Cognitive Neurology Institute at the Rambam Health Care Campus, Haifa, Israel. MCI patients ranged in age from 60–84 years (mean 71.7±9.8) with a high proportion of men (80%) and mean MMSE score of 27.3±1.4. AD dementia patients ranged in age from 59–89 years (mean 75.6±8.4), with a low proportion of men (37.5%), and mean MMSE score of 20.6±5.4. CN elderly participants ranged in age from 51–72 years (61.7±5.4), with no men (0%).
Blood samples were immediately transferred to Tel Aviv University and subjected to lymphocyte and plasma separation. The study was approved by the Helsinki Committee at the Rambam Health Care Campus (NCT01403519).
HABS Clinical Assessments
In addition to the clinical assessments used for screening, all 40 participants underwent the American National Reading Test (AMNART)[29] intelligence quotient (IQ), a measure of premorbid intelligence (greater scores indicate greater intelligence; the range for the participants in the current study was 97–132).
Isolation of lymphocytes from peripheral blood by Ficoll gradients (Israeli cohort)
10 ml of human peripheral blood/sample were collected by venipuncture into lithium heparin tubes (Vacuette, 455084; Greiner Bio-One International, 4550 Kremsmünster Austria). After gentle homogenization, the whole blood was layered over a Ficoll-Paque gradient in UNI-SEP U-04 tubes (Novamed, Jerusalem, Israel). The tubes were subjected to centrifugation at 3,000 rpm for 20 minutes. Lymphocytes were aspirated at the Ficoll plasma interface where a white band was formed. The lymphocytes were further sedimented through Ficoll-Hypaque and formed a pellet. The resulting pellet was dried and resuspended with TRI Reagent (Sigma, Rehovot, Israel) for further RNA extraction or stored at −80 °C. The plasma fraction from the Ficoll separation was stored at −80 °C for further protein analysis. The red cell pellet was discarded.
RNA extraction by the TRI Reagent (Israeli cohort)
RNA extraction from lymphocytes was performed according to the TRI Reagent protocol. For more details refer to: http://www.sigmaaldrich.com/technical-documents/protocols/biology/tri-reagent.html#sthash.BErG0cJN.dpuf.
Paxgene processing by RNA extraction (HABS cohort)
The RNA extraction from whole blood was performed using PreAnalyix PAXgene kit (Qiagen, Valencia, CA, USA/BD Diagnostic Company, Franklin Lakes, NJ, USA; Cat # 762164). The RNA samples were stored in −80°C.
RNA quantity and quality were analyzed by the Nanodrop ND-1000 UV-Vis spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). Each sample measurement was performed in duplicates.
Reverse Transcription and Quantitative Real-Time polymerase chain reaction (PCR)
Samples containing equal amount of total RNA (1μg RNA/sample) were subjected to reverse transcription (RT) using qScript™ cDNA Synthesis Kit (Quanta Biosciences, Gaithersburg, MD, USA; Cat # 95047-100). Real-Time PCR was performed using the SYBR®Green PCR Master Mix and StepOnePlus™ instrument and software (Applied Biosystems, Foster City, CA, USA) using the default thermocycler program for all genes: 10 minutes of preincubation at 95° C followed by 40 cycles of 15 seconds at 95° C and 1 minute at 60°. Real-Time PCR reactions were carried out in a total volume of 10 μl in a 96-well plate (Applied Biosystems) containing of SYBR®X2 Green PCR Master Mix and 300 nM of each sense and antisense primers. The efficiencies of all primers used were calculated as a precursory step using standard curves, according to the equation: E (efficiency) = [10(−1/slope)−1] × 100 and were near 100% for all primers. All Real-Time PCR reactions were carried out in triplicate. The comparative Ct method was used for quantification of transcripts. Table 1 depicts the various primers used. Results are shown as 2−ΔCT (http://de-de.invitrogen.com/etc/medialib/en/filelibrary/Nucleic-Acid-Amplification-Expression-Profiling/PDFs.Par.83765.File.dat/relative-quant-ct.pdf).
Table 1.
Primers for Real-Time PCR
| Human ADNP (NM_015339) | 5′ acttacgaaaaaccaggactatc 3′ 5′ gacattgcggaaatgact 3′ |
| Human ADNP2 (NM_014913) | 5′ gaaagaaagtgagatatcgaacaaa 3′ 5′ tggtcaatttcatcttcatgg 3′ |
| Human TATA box binding protein (NM_003194) | 5′ ggagccaagagtgaagaacag 3′ 5′ cacagctccccaccatattc 3′ |
ADNP (activity-dependent neuroprotective protein), PCR (polymerase chain reaction).
Serum preparation (HABS cohort)
Human peripheral blood was collected into 4 ml tubes and precipitated in an Eppendorf centrifuge 5810R (25° C), 2,000 rcf for 5 minutes. After centrifugation, 0.5 ml of the liquid phase above the clot was collected and serum samples were aliquoted and stored at −80° C.
ADNP Enzyme-linked Immunosorbent Assay (ELISA)
The levels of ADNP in plasma and serum were detected using an ELISA kit (SEJ67Hu, Cloud-Clone Corporation, Houston, TX, USA) for human ADNP. ELISA assays were performed as described in the manufacture protocol.
Magnetic Resonance Imaging (MRI) Data
All 40 HABS participants underwent MRI scans at the Charlestown Navy Yard research facility of MGH on a Siemens TIM Trio 3T System with a 12-channel head coil. Structural T1-weighted volumetric magnetization-prepared, rapid acquisition gradient echo scans were collected (TR/TE/TI=6400/2.8/900ms, flip angle=8°, 1×1×1.2mm resolution). Region of interest (ROI) labeling was performed using FreeSurfer v5.1 (http://surfer.nmr.mgh.harvard.edu). As previously described [30], hippocampal volume (HV) was summed across hemispheres. A correction for intracranial volume (ICV) was performed by obtaining the residuals from a linear regression model in which HV was the dependent variable and ICV was the independent variable. Residuals were then added to mean bilateral HV for ease of interpretation, yielding an ICV adjusted HV value.
Positron Emission Tomography (PET) Data
All 40 HABS participants underwent PET imaging at the MGH PET facility as previously described [31, 32]. PET images were acquired using a Siemens ECAT EXACT HR+ scanner (3D mode; 63 image planes; 15.2cm axial field of view; 5.6mm transaxial resolution and 2.4mm slice interval; 69 frames: 12×15 seconds, 57×60 seconds).
C11-Pittsburgh Compound B (PiB) PET was used to measure cortical amyloid. PiB was synthesized using a previously published protocol [30, 33, 34]. After injection of 8.5–15 mCi PiB, 60 minutes of dynamic data were acquired in 3D acquisition mode. The Logan graphical analysis method with cerebellar cortex as the reference tissue input function was used to evaluate specific PiB retention expressed as the distribution volume ratio (DVR)[33, 35]. PiB DVR was calculated for an aggregate of cortical regions that typically have elevated PiB retention in AD dementia (FLR, frontal, lateral parietal and temporal, and retrosplenial cortices).
F18-flourodeoxyglucose (FDG) PET was used to measure cerebral metabolism. FDG PET data was acquired according to the Alzheimer’s Disease Neuroimaging Initiative (ADNI) protocol [36]. A bolus of 5 mCi of FDG was injected in a quiet, dimly lit room, with participants in the eyes-open state. FDG acquisition began 30 minutes after injection and lasted 30 minutes. FDG was extracted from a MetaROI reflecting regions known to be vulnerable in AD (posterior cingulate, lateral parietal, lateral inferior temporal cortex) and expressed as the standardized uptake value ratio (SUVR), normalized to pons and cerebellar vermis [36].
CSF Data
Eighteen of the 40 HABS participants underwent lumbar puncture and CSF samples were obtained, processed, and stored according to the ADNI protocol, as previously described [9, 37]. CSF assays of Amyloid-beta 1-42 (Aβ1-42), total tau (t-tau), and tau phosphorylated at the threonine 181 (p-tau181p) were performed using the multiplex xMAP Luminex platform (Luminex Corp, Austin, TX) with Innogenetics (INNO-BIA AlzBio3; Ghent Belgium; for research use-only reagents) immunoassay kit-based reagents [9, 38].
Statistical analyses
Analyses were performed using SPSS Version 22. For initial unadjusted associations between ADNP measures and HABS participant demographics, characteristics, and imaging and CSF biomarkers, we used non-parametric tests (Spearman correlations for continuous variables and the Mann-Whitney U test for categorical variables) because most variables did not have a normal distribution. Marginal (p<0.1) or significant (p<0.05) unadjusted associations were then adjusted for age using partial Spearman correlations since age is associated with the various imaging and CSF biomarkers.
Additional analyses in the Israeli cohort included one or two-way analysis of variance (ANOVA) with the appropriate post hoc test as indicated in the Results section.
RESULTS
HABS CN elderly participants show serum ADNP correlation with higher premorbid intelligence and less ADNP expression was correlated with greater cortical amyloid burden
Table 2 provides the HABS participant demographics and characteristics. The interval between blood sample collection for ADNP and clinical assessments was 7.1 ± 17.4 days, MRI 48.1 ± 63.1 days, PiB PET 122.3 ± 40.2 days, FDG PET 138.6 ± 76.5 days, and CSF 641.8 ± 376.3 days.
Table 2.
Harvard Aging Brain Study demographics and characteristics of participants.
| Mean | SD | Range | |
|---|---|---|---|
| Age (years) | 75.0 | 5.5 | 66–89 |
| Sex (% Male) | 37.5 | ||
| Education (years) | 15.7 | 3.0 | 9–20 |
| AMNART IQ | 121.4 | 9.1 | 97–132 |
| MMSE | 29.2 | 1.2 | 26–30 |
| APOE4 Status (% Carriers) | 28.2 | ||
| ADNP mRNA | 0.50 | 0.27 | 0.02–1.54 |
| ADNP2 mRNA | 0.10 | 0.04 | 0.01–0.20 |
| ADNP/ADNP2 mRNA ratio | 5.15 | 2.07 | 1.93–10.34 |
| ADNP (ng/μl protein) | 0.04 | 0.01 | 0.03–0.06 |
| CSF Aβ1-42 (pg/ml) | 398.05 | 150.87 | 141.00–640.27 |
| CSF t-tau (pg/ml) | 66.55 | 20.42 | 32.71–105.27 |
| CSF p-tau181p (pg/ml) | 34.78 | 12.73 | 21.34–69.24 |
| Cortical amyloid aggregate (PiB PET, DVR) | 1.24 | 0.21 | 1.02–1.73 |
| Cortical metabolism aggregate (FDG PET, SUVR) | 1.31 | 0.13 | 1.01–1.55 |
| Hippocampal volume (adjusted for ICV, mm3) | 7169.02 | 689.75 | 5774.39–8916.76 |
Aβ1-42 (amyloid-beta 1-42), ADNP (activity-dependent neuroprotective protein), AMNART IQ (American National Reading Test intelligence quotient), APOE4 (apolipoprotein E ε4), CSF (cerebrospinal fluid), DVR (distribution volume ratio), FDG (flourodeoxyglucose), ICV (intracranial volume), MMSE (Mini-Mental State Examination), mRNA (messenger ribonucleic acid), PET (positron emission tomography), PiB (Pittsburgh compound B), p-tau181p (tau phosphorylated at the threonine 181), SD (standard deviation), SUVR (standardized uptake value ratio), t-tau (total tau).
One ADNP mRNA and ADNP2 mRNA outlier was excluded from the analyses and 5 ADNP protein outliers were excluded from the analyses (showing inconsistency in the duplicate ELISA assay).
ADNP measures inter-correlations were performed. ADNP mRNA was significantly associated with ADNP2 mRNA (rs=0.61, p<0.0001) and ADNP/ADNP2 mRNA ratio (rs=0.47, p=0.002). ADNP2 mRNA was marginally associated with ADNP/ADNP2 mRNA ratio (rs=−0.27, p=0.09). There were no other significant or marginal associations between ADNP measures.
The unadjusted associations between ADNP measures and HABS participant demographics, characteristics, and imaging and CSF biomarkers are shown in Table 3. Higher premorbid intelligence was marginally associated with greater ADNP (ng/μl protein) (see Figure 1.A). Greater CSF Aβ1-42 was significantly associated with greater ADNP2 mRNA (see Figure 1.B) and marginally associated with greater ADNP mRNA. Greater CSF t-tau was marginally associated with greater ADNP/ADNP2 mRNA ratio. Greater CSF p-tau181p was marginally associated with greater ADNP (ng/μl protein). Greater cortical amyloid aggregate was significantly associated with lower ADNP mRNA and ADNP2 mRNA (see Figures 1.C and 1.D).
Table 3.
Unadjusted associations between ADNP measures and Harvard Aging Brain Study participant demographics, characteristics, and imaging and CSF biomarkers.
| ADNP mRNA | ADNP2 mRNA | ADNP/ADNP2 mRNA ratio | ADNP (ng/μl protein) | |||||
|---|---|---|---|---|---|---|---|---|
| rs | p | rs | P | rs | P | rs | p | |
| Age | −0.17 | 0.32 | −0.07 | 0.65 | −0.21 | 0.19 | −0.17 | 0.34 |
| Sex* | 0.36 | 0.23 | 0.85 | 0.43 | ||||
| Education | 0.04 | 0.82 | 0.08 | 0.64 | −0.13 | 0.44 | 0.12 | 0.51 |
| AMNART IQ | −0.22 | 0.18 | −0.17 | 0.30 | −0.02 | 0.90 | 0.30† | 0.08 |
| APOE4* | 0.51 | 0.78 | 0.92 | 1.00 | ||||
| MMSE | −0.17 | 0.32 | −0.10 | 0.55 | 0.08 | 0.72 | 0.06 | 0.75 |
| CSF Aβ1-42 | 0.43† | 0.09 | 0.53‡ | 0.03 | 0.19 | 0.47 | 0.39 | 0.13 |
| CSF t-tau | 0.35 | 0.17 | −0.08 | 0.76 | 0.45† | 0.07 | 0.20 | 0.46 |
| CSF p-tau181p | 0.17 | 0.52 | −0.21 | 0.41 | 0.33 | 0.20 | 0.43† | 0.09 |
| Cortical amyloid aggregate | −0.37‡ | 0.02 | −0.32‡ | 0.05 | −0.16 | 0.32 | 0.13 | 0.45 |
| Cortical metabolism aggregate | −0.04 | 0.80 | 0.03 | 0.86 | 0.08 | 0.63 | 0.28 | 0.11 |
| Hippocampal volume | −0.13 | 0.45 | −0.05 | 0.75 | 0.03 | 0.85 | −0.05 | 0.80 |
Aβ1-42 (amyloid-beta 1-42), ADNP (activity-dependent neuroprotective protein), AMNART IQ (American National Reading Test intelligence quotient), APOE4 (apolipoprotein E ε4), CSF (cerebrospinal fluid), MMSE (Mini-Mental State Examination), mRNA (messenger ribonucleic acid), p-tau181p (tau phosphorylated at the threonine 181), t-tau (total tau).
Mann-Whitney U test
p<0.1
p≤0.05
Figure 1.
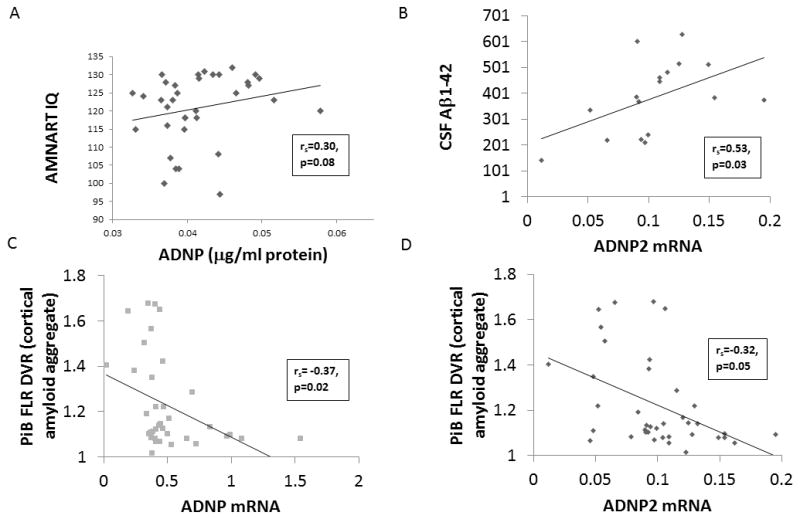
Unadjusted scatter plots depicting the associations in HABS CN elderly participants between ADNP (ng/μl protein) and AMNART IQ (Panel A), ADNP2 mRNA and CSF Aβ1-42 (Panel B), cortical amyloid aggregate (PiB FLR DVR) and ADNP mRNA (Panel C), and cortical amyloid aggregate and ADNP2 mRNA (Panel D). Messenger RNA Results are shown as 2−ΔCT (http://de-de.invitrogen.com/etc/medialib/en/filelibrary/Nucleic-Acid-Amplification-Expression-Profiling/PDFs.Par.83765.File.dat/relative-quant-ct.pdf). Aβ1-42 (amyloid-beta 1-42), ADNP (activity-dependent neuroprotective protein), AMNART IQ (American National Reading Test intelligence quotient), CN (cognitively normal), CSF (cerebrospinal fluid), DVR (distribution volume ratio), FLR (frontal, lateral parietal and temporal, and retrosplenial cortices), mRNA (messenger ribonucleic acid), PiB (Pittsburgh compound B).
After adjusting for age, there was a significant association between higher premorbid intelligence and greater ADNP (ng/μl protein) (prs=0.35, p=0.04); a marginal association between greater CSF Aβ1-42 and greater ADNP2 mRNA (prs=0.44, p=0.09) and no association with ADNP mRNA (prs=0.31, p=0.25); a marginal association between greater CSF t-tau and greater ADNP/ADNP2 mRNA ratio (prs=0.47, p=0.07); a marginal association between greater CSF p-tau181p and greater ADNP (ng/μl protein) (prs=0.50, p=0.06); and a significant association between greater cortical amyloid aggregate and lower ADNP mRNA (prs=−0.33, p=0.04) and ADNP2 mRNA (prs=−0.32, p=0.05).
AD dementia patients show a significant increase in lymphocyte ADNP mRNA expression
Table 4 shows the demographic and characteristics for the Israeli cohort. Figure 2.A shows an 8-fold increase in ADNP mRNA in AD dementia patient lymphocytes when compared to CN participants. The ADNP paralog, ADNP2 mRNA, increased by 2-fold in AD dementia patient lymphocytes when compared to CN participants. Additionally, a significant correlation was seen between ADNP and ADNP2 mRNA in AD dementia patients (see Figure 2.B). A slight increase in ADNP mRNA was also observed in the MCI patients when compared to CN participants. However, this difference was much less prominent than that observed for the AD dementia patients (see Figure 2.A).
Table 4.
Israeli cohort demographics, characteristics, and ADNP measurements of participants.
| Cohort | Age | MMSE | Sex | ||||
|---|---|---|---|---|---|---|---|
| Mean | SD | Range | Mean | SD | Range | %Males | |
| CN | 61.7 | 5.4 | 51–72 | * | * | * | 0 |
| MCI | 71.7 | 9.8 | 60–84 | 27.3 | 1.4 | 25–29 | 80 |
| AD dementia | 75.6 | 8.4 | 59–89 | 20.6 | 5.4 | 11–29 | 37.5 |
| ADNP mRNA | ADNP2 mRNA | n | |||||
| CN | 0.44 | 0.29 | 0.13–0.88 | 0.88 | 0.67 | 0.16–1.82 | 10* |
| MCI | 0.77 | 0.47 | 0.11–1.39 | 0.29 | 0.16 | 0.04–0.53 | 12* |
| AD dementia | 3.64 | 3.0 | 0.57–10.11 | 1.59 | 1.16 | 0.32–3.61 | 17 |
| ADNP/ADNP2 mRNA | ADNP (ng/μl protein) | ||||||
| CN | 0.89 | 0.61 | 0.14–1.77 | 0.047 | 0.002 | 0.035–0.05*** | 7* |
| MCI | 2.82 | 1.36 | 0.29–30** | 0.059 | 0.013 | 0.039–0.085 | 15 |
| AD dementia | 2.32 | 0.79 | 1.14–4.19 | 0.054 | 0.015 | 0.037–0.09 | 16* |
AD (Alzheimer’s disease), ADNP (activity-dependent neuroprotective protein), CN (cognitively normal), MCI (mild cognitive impairment), MMSE (Mini-Mental State Examination), mRNA (messenger ribonucleic acid).
Values were not available for all participants.
A value of 30 was a significant outlier and was removed.
A value of 0.035 was a significant outlier and was removed.
Figure 2.

Panel A depicts the comparison between ADNP and ADNP2 mRNA expression in lymphocytes of CN elderly, MCI, and AD dementia participants in the Israeli cohort. A two-way ANOVA was significant (p<0.001, F (2,77) = 16.176). All pairwise multiple comparison procedures (Fisher LSD Method) indicated a significant difference between ADNP and ADNP2 mRNA values in the AD dementia group, a significant difference for ADNP mRNA between CN and MCI and between MCI and AD dementia (***p<0.001 for all of the above, including also the differences between CN and AD dementia for both ADNP and ADNP2), and a significant difference for ADNP2 mRNA between MCI and AD dementia (#p<0.05). The obvious differences between CN and AD dementia for both ADNP and ADNP2 are not depicted on the graph. Panel B depicts the correlation of ADNP and ADNP2 mRNA expression in lymphocytes across CN, MCI, and AD dementia participants in the Israeli cohort. A highly significant correlation was found in the AD dementia patients (r=0.93).
Panel C compares ADNP plasma protein levels in CN, MCI, and AD dementia Israeli participants.
As we did not find any difference between the serum and the plasma ADNP levels in the HABS vs. Israeli CN cohorts (see Panel D), we grouped the two samples together, increasing the number of tested samples from 7 plasma samples with 35 serum samples (excluding outliers that have shown inconsistency in the duplicate ELISA assay). A one-way ANOVA indicated a significant difference (p<0.001). All Pairwise Multiple Comparison Procedures (Dunn’s Method) Comparison Diff of Ranks indicated a significant difference between CN and MCI and between CN and AD dementia (*p<0.05, in both cases).
All results are depicted with standard error of the mean (SEM).
AD (Alzheimer’s disease), ADNP (activity-dependent neuroprotective protein), ANOVA (analysis of variance), CN (cognitively normal), HABS (Harvard Aging Brain Study), MCI (mild cognitive impairment), mRNA (messenger ribonucleic acid).
ADNP protein increases in MCI patient plasma compared to CN participants were observed, while a small decrease was seen in the AD dementia patient plasma when compared to MCI patients (see Figure 2.C).
It should be noted that the for the AD dementia/MCI/CN comparisons in Figure 2.C, plasma plus serum samples were used, while in Figure 1.A looking at HABS CN participants alone, serum samples were used. Figure 2.D compares the HABS and Israeli CN groups with serum vs. plasma ADNP, showing similarity albeit slight ~10% reduction in serum ADNP vs. plasma ADNP, suggesting potential aggregation of ADNP with coagulated blood.
Lastly, a correlation is seen in AD dementia patient plasma between ADNP protein content and lymphocyte ADNP mRNA. The correlative values differ between AD dementia, MCI, and CN participants (see Figure 3).
Figure 3.
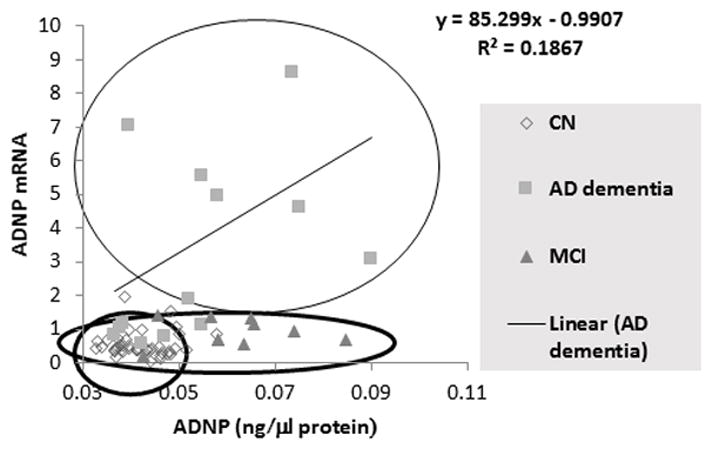
A correlation between ADNP protein and mRNA levels are shown. As no differences were observed between the whole blood ADNP mRNA and lymphocytes ADNP mRNA levels, as well as no differences between plasma and serum ADNP levels among the two CN cohorts (see Figure 2.D), these cohorts were combined for this comparison. The graph shows segregation of the ADNP mRNA/ADNP protein for the three tested diagnostic groups, and a correlation in the AD dementia group (r=0.38).
DISCUSSION
When adjusting for age, higher ADNP serum protein levels are significantly associated with higher premorbid intelligence, while lower ADNP mRNA levels associate with higher cortical amyloid burden in CN elderly. The latter was also found for the ADNP related ADNP2 mRNA. ADNP2 mRNA was also associated with lower CSF Aβ1-42 levels, which, as previously shown, corresponds to greater cortical amyloid burden [39, 40]. In AD dementia patients, the lymphocyte levels of ADNP and ADNP2 mRNA transcripts were significantly increased, in correlation with a modest increase in plasma ADNP protein level. Together, these results suggest that the ADNP measures could potentially be useful surrogate marker for AD disease onset and progression.
This is the first time that ADNP and ADNP2 mRNA are shown to be associated with AD dementia and cortical amyloid burden in human participants. Most previous studies concentrated on tau pathology, given the association of ADNP with microtubules and microtubule-dependent autophagy [15, 23, 41, 42].
The paradoxical decrease in serum ADNP, which is associated with decreased premorbid intelligence in CN elderly, when compared to the increase in plasma ADNP in MCI patients may be related to ADNP association with coagulated blood. This may explain, in part, the previously reported significant decrease in AD dementia patients. Interestingly, ADNP strongly regulates blood coagulation factors, with marked increases in the developing embryo, upon ADNP knockout and with direct interaction with relevant promoters [13]. The fact that plasma and serum do not share the same technical suitability for the protein assays opens another new avenue for future studies. In Figure 2A, an increase of ADNP mRNA from CN to AD dementia is shown, but this did not translate into an increase of the protein. Apart from technical considerations, this reduces the value of the protein as a serum/plasma biomarker because for a clear detection one should extract mRNA and not employ clinically-simplified protein tests. Thus, the development of sophisticated mRNA measurements are warranted for future biomarker detection.
The increased expression in lymphocyte ADNP was also noted in lymphoblastoid cells from cognitively impaired autistic children carrying distinct ADNP mutations [43]. This suggests that there is possibly a compensatory value to ADNP increase in lymphocyte in the face of neurodegeneration, and it should be taken into consideration that ADNP regulates its own expression [13, 44]. A relevant concept in the aging and dementia field is cognitive reserve, which suggests that individuals with higher premorbid intelligence, greater years of education, or more alternative strategies for solving problems may be more resilient to pathologic changes of aging and neurodegeneration later in life [45]. In the current study, we showed an association between higher ADNP serum protein, which is a potential biomarker of AD pathology, and higher premorbid intelligence, which is a proxy of cognitive reserve, in CN elderly from the HABS cohort. We have previously shown that in the HABS cohort cognitive reserve modifies the association between cortical amyloid and cognitive performance such that it may confer protection against cognitive impairment associated with amyloid [46].
ADNP has been studied in many neuropsychiatric conditions other than AD. In contrast to the increase in ADNP mRNA in AD dementia found in the current study, in multiple sclerosis, ADNP was found to decrease in nucleated blood cells alongside with increased expression of inflammatory cytokines. Here, the researchers tried to compensate with the ADNP snippet drug candidate NAP (davunetide) and showed significant reversal of the cytokine profile to an anti-inflammatory profile [47]. NAP has been tested in clinical trials in amnestic MCI patients and showed efficacy in two main measures, delayed match to sample visual short term memory and verbal recall, as well as digit span forward [48]. There was no significant benefit in progressive supranuclear palsy (PSP) patients where the primary outcome was the PSP rating scale, which emphasizes motor abilities [49]. Furthermore, PSP is usually diagnosed in mid-late neurodegenerative state, precluding to a large extent the potential for successful neuroprotective strategies. In contrast to the PSP results, and in agreement with the MCI results, treatment with NAP showed a significant benefit in protecting functional activities of daily living in cognitively impaired schizophrenia patients [50], coupled to brain matter protection [51]. In a follow-up schizophrenia study, Malishkevich and Gozes [23] identified a significantly increased lymphocyte ADNP and ADNP2 mRNA expression when compared to CN matched participants. The increase in ADNP was associated with the initial stages of the disease, possibly reflecting a compensatory effect [23]. However, the schizophrenia study was limited in the number of patients and the available correlates for comparison.
The current study had several limitations. First, the cohort sizes were small making it difficult to observe significant associations, especially in the HABS CN elderly. Future larger studies will strengthen the validation of ADNP as an AD biomarker. Second, the HABS cohort was highly educated and intelligent, which reduces the generalizability of our results. However, this cohort is similar to those of other large observational studies, like ADNI [52], as well as to those participating in clinical trials. Third, several analyses were performed without correction for multiple comparisons as the current study was an early attempt to characterize the potential of ADNP as an AD biomarker and was therefore exploratory in nature. Therefore, the results though encouraging, need to be interpreted with caution. Fourth, the interval between blood sample collection for ADNP and CSF sample collection was long (nearly 2 years on average) and CSF was obtained only in a subset of HABS participants. Thus, these results need to be interpreted with even greater caution. On the other hand, the interval between blood sample collection and imaging biomarkers was relatively short (less than 5 months on average) and imaging biomarkers were obtained in all HABS participants. Fifth, although ADNP distinguished between diagnostic groups (CN, MCI, and AD dementia) as depicted in Figure 3, this was driven by the difference between CN and AD dementia participants. Larger studies will better determine the ability of ADNP to discriminate between CN and MCI and its diagnostic sensitivity and specificity. Finally, the associations were all cross-sectional. Future longitudinal studies will strengthen the results shown in Figure 3 and help determine whether ADNP can predict cognitive decline and disease progression from CN to MCI to AD dementia. Furthermore, an ADNP2 ELISA kit has recently become available, which will allow future studies in larger longitudinal cohorts comparing ADNP and ADNP2.
Based on our results in small cohorts, it is tantalizing to hypothesize that ADNP measures in the blood will serve as good AD biomarkers through which AD will be diagnosed earlier, disease stages will be differentiated, clinical efficacy of drugs in development will be determined, and patient stratification toward neuroprotection in AD will be boosted.
Acknowledgments
FUNDING SOURCES
Professor Gozes laboratory is supported by the AMN Foundation, the Israel Ministry of Science, Technology and Space, Israel Science Foundation, CFTAU Montreal Circle of Friends and the Adams family, Adams Super Center for Brain Studies, the Elton Laboratory for Molecular Neuroendocrinology and the Lily and Avraham Gildor Chair for the Investigation of Growth Factors at Tel Aviv University. This study was further supported by the Harvard Aging Brain Study (P01 AGO36694, R01 AG037497), the Massachusetts Alzheimer’s Disease Research Center (P50 AG005134), the Harvard NeuroDiscovery Center Biomarker Study (HBS). Plasma samples were provided by the Harvard Biomarker Study. The Harvard Biomarker Study is supported by the Harvard NeuroDiscovery Center (HNDC), the Parkinson’s Disease Biomarkers Program (PDBP) grant U01 NS082157 of the NINDS, and the Massachusetts Alzheimer’s Disease Research Center (ADRC) P50 AG005134 grant of the National Institute on Aging
We thank Dr. Eliezer Giladi and Mrs. Michal Levavi for their help in blood collection and coordination of blood transfer. We further thank the Harvard NeuroDiscovery Biomarker Study (HBS) for their invaluable contribution.
Professor Gozes is a Humboldt Award Recipient and a fellow at the Hanse-Wissenschftenkolleg, Germany. This study is in partial fulfilment for the Ph.D. thesis of Mrs. Anna Malishkevich at the Dr. Miriam and Sheldon G. Adelson Graduate School of Medicine, as the Sackler Faculty of Medicine, Tel Aviv University.
Footnotes
DISCLOSURES/CONFLICTS
Dr. Marshall has served as a consultant for Halloran, GliaCure, and Janssen Research & Development. Dr. Sperling has served as a consultant for Merck, Eisai, Janssen, Boehringer-Ingelheim, Isis, Lundbeck, Roche, and Genetech. Professor Gozes is the Chief Scientific Officer of Coronis Partners and is an inventor on patent/patent applications including ADNP and ADNP2 as diagnostic biomarkers. Ms. Malishkevich, Dr. Schultz Professor Aharon-Peretz have no disclosures.
References
- 1.Chung SJ, Lee JH, Kim SY, You S, Kim MJ, Lee JY, Koh J. Association of GWAS Top Hits With Late-onset Alzheimer Disease in Korean Population. Alzheimer Dis Assoc Disord. 2013;27:250–257. doi: 10.1097/WAD.0b013e31826d7281. [DOI] [PubMed] [Google Scholar]
- 2.Swerdlow RH, Corder EH. For Alzheimer disease GWAS, pulling needles from the haystack is just the first step. Neurology. 2012;79:204–205. doi: 10.1212/WNL.0b013e318260581d. [DOI] [PubMed] [Google Scholar]
- 3.Tosto G, Reitz C. Genome-wide Association Studies in Alzheimer’s Disease: A Review. Curr Neurol Neurosci Rep. 2013;13:381. doi: 10.1007/s11910-013-0381-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lehmann S, Delaby C, Touchon J, Hirtz C, Gabelle A. Biomarkers of Alzheimer’s disease: The present and the future. Rev Neurol (Paris) 2013;169:719–723. doi: 10.1016/j.neurol.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 5.Rhinn H, Fujita R, Qiang L, Cheng R, Lee JH, Abeliovich A. Integrative genomics identifies APOE epsilon4 effectors in Alzheimer’s disease. Nature. 2013;500:45–50. doi: 10.1038/nature12415. [DOI] [PubMed] [Google Scholar]
- 6.Chien DT, Bahri S, Szardenings AK, Walsh JC, Mu F, Su MY, Shankle WR, Elizarov A, Kolb HC. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis. 2013;34:457–468. doi: 10.3233/JAD-122059. [DOI] [PubMed] [Google Scholar]
- 7.Jack CR, Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Pankratz VS, Donohue MC, Trojanowski JQ. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 9.Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter W, Lee VM, Trojanowski JQ. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Annals of neurology. 2009;65:403–413. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bassan M, Zamostiano R, Davidson A, Pinhasov A, Giladi E, Perl O, Bassan H, Blat C, Gibney G, Glazner G, Brenneman DE, Gozes I. Complete sequence of a novel protein containing a femtomolar-activity-dependent neuroprotective peptide. J Neurochem. 1999;72:1283–1293. doi: 10.1046/j.1471-4159.1999.0721283.x. [DOI] [PubMed] [Google Scholar]
- 11.Zamostiano R, Pinhasov A, Gelber E, Steingart RA, Seroussi E, Giladi E, Bassan M, Wollman Y, Eyre HJ, Mulley JC, Brenneman DE, Gozes I. Cloning and characterization of the human activity-dependent neuroprotective protein. J Biol Chem. 2001;276:708–714. doi: 10.1074/jbc.M007416200. [DOI] [PubMed] [Google Scholar]
- 12.Pinhasov A, Mandel S, Torchinsky A, Giladi E, Pittel Z, Goldsweig AM, Servoss SJ, Brenneman DE, Gozes I. Activity-dependent neuroprotective protein: a novel gene essential for brain formation. Brain Res Dev Brain Res. 2003;144:83–90. doi: 10.1016/s0165-3806(03)00162-7. [DOI] [PubMed] [Google Scholar]
- 13.Mandel S, Rechavi G, Gozes I. Activity-dependent neuroprotective protein (ADNP) differentially interacts with chromatin to regulate genes essential for embryogenesis. Dev Biol. 2007;303:814–824. doi: 10.1016/j.ydbio.2006.11.039. [DOI] [PubMed] [Google Scholar]
- 14.Malishkevich A, Amram N, Hacohen-Kleiman G, Magen I, Giladi E, Gozes I. Activity-dependent neuroprotective protein (ADNP) exhibits striking sexual dichotomy impacting on autistic and Alzheimer’s pathologies. Transl Psychiatry. 2015;5:e501. doi: 10.1038/tp.2014.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vulih-Shultzman I, Pinhasov A, Mandel S, Grigoriadis N, Touloumi O, Pittel Z, Gozes I. Activity-dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J Pharmacol Exp Ther. 2007;323:438–449. doi: 10.1124/jpet.107.129551. [DOI] [PubMed] [Google Scholar]
- 16.Fernandez-Montesinos R, Torres M, Baglietto-Vargas D, Gutierrez A, Gozes I, Vitorica J, Pozo D. Activity-dependent neuroprotective protein (ADNP) expression in the amyloid precursor protein/presenilin 1 mouse model of Alzheimer’s disease. J Mol Neurosci. 2010;41:114–120. doi: 10.1007/s12031-009-9300-x. [DOI] [PubMed] [Google Scholar]
- 17.Schirer Y, Malishkevich A, Ophir Y, Lewis J, Giladi E, Gozes I. Novel Marker for the Onset of Frontotemporal Dementia: Early Increase in Activity-Dependent Neuroprotective Protein (ADNP) in the Face of Tau Mutation. PLoS One. 2014;9:e87383. doi: 10.1371/journal.pone.0087383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gozes I, Iram T, Maryanovsky E, Arviv C, Rozenberg L, Schirer Y, Giladi E, Furman-Assaf S. Novel Tubulin and Tau Neuroprotective Fragments Sharing Structural Similarities with the Drug Candidate NAP (Davuentide) J Alzheimers Dis. 2014;40(Suppl 1):S23–36. doi: 10.3233/JAD-131664. [DOI] [PubMed] [Google Scholar]
- 19.Gozes I, Yeheskel A, Pasmanik-Chor M. Activity-dependent neuroprotective protein (ADNP): a case study for highly conserved chordata-specific genes shaping the brain and mutated in cancer. J Alzheimers Dis. 2015;45:57–73. doi: 10.3233/JAD-142490. [DOI] [PubMed] [Google Scholar]
- 20.Kushnir M, Dresner E, Mandel S, Gozes I. Silencing of the ADNP-family member, ADNP2, results in changes in cellular viability under oxidative stress. J Neurochem. 2008;105:537–545. doi: 10.1111/j.1471-4159.2007.05173.x. [DOI] [PubMed] [Google Scholar]
- 21.Dresner E, Agam G, Gozes I. Activity-dependent neuroprotective protein (ADNP) expression level is correlated with the expression of the sister protein ADNP2: deregulation in schizophrenia. Eur Neuropsychopharmacol. 2011;21:355–361. doi: 10.1016/j.euroneuro.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 22.Dresner E, Malishkevich A, Arviv C, Leibman Barak S, Alon S, Ofir R, Gothilf Y, Gozes I. Novel evolutionary-conserved role for the activity-dependent neuroprotective protein (ADNP) family that is important for erythropoiesis. J Biol Chem. 2012;287:40173–40185. doi: 10.1074/jbc.M112.387027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merenlender-Wagner A, Malishkevich A, Shemer Z, Udawela M, Gibbons A, Scarr E, Dean B, Levine J, Agam G, Gozes I. Autophagy has a key role in the pathophysiology of schizophrenia. Mol Psychiatry. 2015;20:126–132. doi: 10.1038/mp.2013.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang MH, Yang YH, Lu CY, Jong SB, Chen LJ, Lin YF, Wu SJ, Chu PY, Chung TW, Tyan YC. Activity-dependent neuroprotector homeobox protein: A candidate protein identified in serum as diagnostic biomarker for Alzheimer’s disease. J Proteomics. 2012;75:3617–3629. doi: 10.1016/j.jprot.2012.04.017. [DOI] [PubMed] [Google Scholar]
- 25.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 26.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 27.Wechsler D. Manual for the Wechsler Memory Scale-Revised. The Psychological Corporation; San Antonio, TX: 1987. [Google Scholar]
- 28.Yesavage JA, Brink TL, Rose TL, Lum O, Huang V, Adey M, Leirer VO. Development and validation of a geriatric depression screening scale: a preliminary report. J Psychiatr Res. 1982;17:37–49. doi: 10.1016/0022-3956(82)90033-4. [DOI] [PubMed] [Google Scholar]
- 29.Nelson HE, O’Connell A. Dementia: the estimation of premorbid intelligence levels using the New Adult Reading Test. Cortex. 1978;14:234–244. doi: 10.1016/s0010-9452(78)80049-5. [DOI] [PubMed] [Google Scholar]
- 30.Growdon ME, Schultz AP, Dagley AS, Amariglio RE, Hedden T, Rentz DM, Johnson KA, Sperling RA, Albers MW, Marshall GA. Odor identification and Alzheimer’s disease biomarkers in clinically normal elderly. Neurology. 2015;84:2153–2160. doi: 10.1212/WNL.0000000000001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Donovan NJ, Hsu DC, Dagley AS, Schultz AP, Amariglio RE, Mormino EC, Okereke OI, Rentz DM, Johnson KA, Sperling RA, Marshall GA. Depressive symptoms and biomarkers of Alzheimer’s disease in cognitively normal older adults. Journal of Alzheimer’s Disease. 2015;46:63–73. doi: 10.3233/JAD-142940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mormino E, Betensky R, Hedden T, Schultz A, Ward A, Hujibers W, Rentz D, Johnson K, Sperling R. Amyloid and APOE ε4 interact to influence short-term decline in preclinical Alzheimer disease. Neurology. 2014;82:1760–1767. doi: 10.1212/WNL.0000000000000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Becker J, Hedden T, Carmasin J, Maye J, Reiman E, Rentz D, Putcha D, Fischi B, Greve D, Marshall G, Salloway S, Marks D, Buckner R, Sperling R, Johnson K. Amyloid-β associated cortical thinning in clinically normal elderly. Ann Neurol. 2011;69:1032–1042. doi: 10.1002/ana.22333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gomperts S, Rentz D, Moran E, Becker J, Locascio J, Klunk W, Mathis C, Elmaleh D, Shoup T, Fischman A, Hyman B, Growdon J, Johnson K. Imaging amyloid deposition in Lewy body diseases. Neurology. 2008;71:903–910. doi: 10.1212/01.wnl.0000326146.60732.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Price JC, Klunk WE, Lopresti BJ, Lu X, Hoge JA, Ziolko SK, Holt DP, Meltzer CC, DeKosky ST, Mathis CA. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. J Cereb Blood Flow Metab. 2005;25:1528–1547. doi: 10.1038/sj.jcbfm.9600146. [DOI] [PubMed] [Google Scholar]
- 36.Landau S, Harvey D, Madison C, Koeppe R, Reiman E, Foster N, Weiner M, Jagust W. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging. 2011;32:1207–1218. doi: 10.1016/j.neurobiolaging.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marshall GA, Lorius N, Locascio JJ, Hyman BT, Rentz DM, Johnson KA, Sperling RA Initiative AsDN. Regional cortical thinning and cerebrospinal biomarkers predict worsening daily functioning across the Alzheimer disease spectrum. Journal of Alzheimer’s Disease. 2014;41:719–728. doi: 10.3233/JAD-132768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olsson A, Vanderstichele H, Andreasen N, De Meyer G, Wallin A, Holmberg B, Rosengren L, Vanmechelen E, Blennow K. Simultaneous measurement of beta-amyloid(1-42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clinical chemistry. 2005;51:336–345. doi: 10.1373/clinchem.2004.039347. [DOI] [PubMed] [Google Scholar]
- 39.Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, DeKosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 40.Tapiola T, Alafuzoff I, Herukka SK, Parkkinen L, Hartikainen P, Soininen H, Pirttila T. Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol. 2009;66:382–389. doi: 10.1001/archneurol.2008.596. [DOI] [PubMed] [Google Scholar]
- 41.Oz S, Kapitansky O, Ivashco-Pachima Y, Malishkevich A, Giladi E, Skalka N, Rosin-Arbesfeld R, Mittelman L, Segev O, Hirsch JA, Gozes I. The NAP motif of activity-dependent neuroprotective protein (ADNP) regulates dendritic spines through microtubule end binding proteins. Mol Psychiatry. 2014;19:1115–1124. doi: 10.1038/mp.2014.97. [DOI] [PubMed] [Google Scholar]
- 42.Furman S, Steingart RA, Mandel S, Hauser JM, Brenneman DE, Gozes I. Subcellular localization and secretion of activity-dependent neuroprotective protein in astrocytes. Neuron Glia Biology. 2004;1:193–199. doi: 10.1017/S1740925X05000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vandeweyer G, Helsmoortel C, Van Dijck A, Vulto-van Silfhout AT, Coe BP, Bernier R, Gerdts J, Rooms L, van den Ende J, Bakshi M, Wilson M, Nordgren A, Hendon LG, Abdulrahman OA, Romano C, de Vries BB, Kleefstra T, Eichler EE, Van der Aa N, Kooy RF. The transcriptional regulator ADNP links the BAF (SWI/SNF) complexes with autism. Am J Med Genet C Semin Med Genet. 2014;166C:315–326. doi: 10.1002/ajmg.c.31413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aboonq MS, Vasiliou SA, Haddley K, Quinn JP, Bubb VJ. Activity-dependent neuroprotective protein modulates its own gene expression. J Mol Neurosci. 2012;46:33–39. doi: 10.1007/s12031-011-9562-y. [DOI] [PubMed] [Google Scholar]
- 45.Stern Y. What is cognitive reserve? Theory and research application of the reserve concept. J Int Neuropsychol Soc. 2002;8:448–460. [PubMed] [Google Scholar]
- 46.Rentz DM, Locascio JJ, Becker JA, Moran EK, Eng E, Buckner RL, Sperling RA, Johnson KA. Cognition, reserve, and amyloid deposition in normal aging. Ann Neurol. 2010;67:353–364. doi: 10.1002/ana.21904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Braitch M, Kawabe K, Nyirenda M, Gilles LJ, Robins RA, Gran B, Murphy S, Showe L, Constantinescu CS. Expression of Activity-Dependent Neuroprotective Protein in the Immune System: Possible Functions and Relevance to Multiple Sclerosis. Neuroimmunomodulation. 2009;17:120–125. doi: 10.1159/000258695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gozes I, Stewart A, Morimoto B, Fox A, Sutherland K, Schmechel D. Addressing Alzheimer’s disease tangles: from NAP to AL-108. Curr Alzheimer Res. 2009;6:455–460. doi: 10.2174/156720509789207895. [DOI] [PubMed] [Google Scholar]
- 49.Boxer AL, Lang AE, Grossman M, Knopman DS, Miller BL, Schneider LS, Doody RS, Lees A, Golbe LI, Williams DR, Corvol JC, Ludolph A, Burn D, Lorenzl S, Litvan I, Roberson ED, Hoglinger GU, Koestler M, Jack CR, Jr, Van Deerlin V, Randolph C, Lobach IV, Heuer HW, Gozes I, Parker L, Whitaker S, Hirman J, Stewart AJ, Gold M, Morimoto BH, Investigators AL. Davunetide in patients with progressive supranuclear palsy: a randomised, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol. 2014;13:676–685. doi: 10.1016/S1474-4422(14)70088-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Javitt DC, Buchanan RW, Keefe RS, Kern R, McMahon RP, Green MF, Lieberman J, Goff DC, Csernansky JG, McEvoy JP, Jarskog F, Seidman LJ, Gold JM, Kimhy D, Nolan KS, Barch DS, Ball MP, Robinson J, Marder SR. Effect of the neuroprotective peptide davunetide (AL-108) on cognition and functional capacity in schizophrenia. Schizophr Res. 2012;136:25–31. doi: 10.1016/j.schres.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 51.Jarskog LF, Dong Z, Kangarlu A, Colibazzi T, Girgis RR, Kegeles LS, Barch DM, Buchanan RW, Csernansky JG, Goff DC, Harms MP, Javitt DC, Keefe RS, McEvoy JP, McMahon RP, Marder SR, Peterson BS, Lieberman JA. Effects of davunetide on N-acetylaspartate and choline in dorsolateral prefrontal cortex in patients with schizophrenia. Neuropsychopharmacology. 2013;38:1245–1252. doi: 10.1038/npp.2013.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Green RC, Harvey D, Jack CR, Jagust W, Liu E, Morris JC, Petersen RC, Saykin AJ, Schmidt ME, Shaw L, Siuciak JA, Soares H, Toga AW, Trojanowski JQ. The Alzheimer’s Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers Dement. 2012;8:S1–68. doi: 10.1016/j.jalz.2011.09.172. [DOI] [PMC free article] [PubMed] [Google Scholar]