

Abstract
Sclerosing and spindle cell rhabdomyosarcoma (SRMS) have been recently re-classified as a stand-alone pathologic entity, separate from embryonal RMS (ERMS). Genetically a subset of the congenital cases display NCOA2 gene rearrangements, while tumors occurring in older children or adults harbor MYOD1 gene mutations with or without coexisting PIK3CA mutations. Despite these recent advances, a significant number of tumors lack known genetic alterations. In this study we sought to investigate a large group of pediatric spindle cell/sclerosing RMS, spanning a diverse clinical and pathologic spectrum, by using a combined FISH, targeted DNA and whole transcriptome sequencing methodology for a more definitive molecular classification. A total of 26 spindle and sclerosing RMS cases were selected from the two participating Institutions for the molecular analysis. Ten of the 11 congenital/infantile SRMS showed recurrent fusion genes: with novel VGLL2 rearrangements seen in 7 (63%), including VGLL2-CITED2 fusion in 4 and VGLL2-NCOA2 in 2 cases. Three (27%) cases harbored the previously described NCOA2 gene fusions, including TEAD1-NCOA2 in 2, and SRF-NCOA2 in 1. All fusion positive congenital/infantile SRMS patients with available long term follow-up were alive and well, none developing distant metastases. Among the remaining 15 SRMS patients older than age of one, 10 (67%) showed MYOD1 L122R mutations, most of them following a fatal outcome despite an aggressive multi-modality treatment. All 4 cases harboring co-existing MYOD1/PIK3CA mutations shared sclerosing morphology. All 5 fusion/mutation-negative SRMS cases presented as intra-abdominal or para-testicular lesions.
Keywords: rhabdomyosarcoma, spindle cell, congenital, VGLL2, SRF, TEAD1, MYOD1, NCOA2, CITED2
INTRODUCTION
Rhabdomyosarcomas (RMS) are the most common soft tissue sarcomas in children accounting for 5–8% of all pediatric malignancies. Traditionally, RMS have been divided into two clinicopathologic groups: embryonal RMS (ERMS), occurring in younger patients, histologically reminiscent to fetal skeletal muscle, and alveolar RMS (ARMS), often diagnosed in older children, being characterized by a monomorphic round cell cytology, with a variable alveolar pattern and carrying recurrent translocations, a t(2;13)(q35;q14) or t(1;13)(p36;q14) in about 80% of cases 1. While ARMS follow a highly aggressive course, the prognosis of ERMS has significantly improved in recent years with an overall survival of 70% at 5 years for patients presenting with localized disease 2, 3.
The spindle cell variant of rhabdomyosarcoma (SRMS) is an uncommon subtype of RMS that was initially grouped under ERMS, having predilection for paratesticular and head and neck sites and being associated with a more favorable behavior in children 4, 5. A subset of SRMSs display areas of prominent hyaline sclerosis and pseudo-vascular growth pattern, suggesting morphologic overlap with the even less common sclerosing RMS (ScRMS). ScRMS is characterized by cords or micro-alveolar patterns, embedded in a prominent sclerotic or hyalinized stroma 6, 7. As both spindle cell and sclerosing RMS have similar clinical presentations and overlapping histologic features, it was suggested that they might represent a morphologic spectrum, with possibly shared pathogenesis. 6, 8, 9. Subsequently, the latest WHO classification merged these two entities into a single pathologic entity, distinct from ERMS and ARMS 10. The recent evidence of recurrent MYOD1 gene mutations present in both spindle cell or sclerosing RMS support the unifying concept proposed by WHO 2013 by morphologic grounds alone 11. However, despite these genetic advances and refinement in classification, the heterogeneity of this subgroup of RMS even within the pediatric population has become apparent, as evidenced by important genetic and clinical characteristics being age-dependent. Notably, recurrent NCOA2 gene rearrangements have been identified in a subset of congenital/infantile spindle cell RMS associated with a favorable clinical course 12. In contrast, all 4 pediatric patients with spindle/sclerosing RMS carrying MYOD1 mutations, followed a highly aggressive course, similar to the adult patients 11. In this study we further expand our investigation of pediatric spindle cell and sclerosing RMS in a large cohort of different clinical presentations, using a combined molecular approach, including next generation paired-end RNA sequencing for novel fusion discovery, mutation analysis and FISH, for a better molecular subclassification and risk factor stratification in the pediatric age group.
MATERIAL AND METHODS
Patient Selection
Archival material from pediatric patients with diagnosis of spindle cell or sclerosing RMS was retrieved from the Institutional and consultation files of the Departments of Pathology of the University of Padua and Memorial Sloan-Kettering Cancer Center. Twenty-six cases were identified and the diagnosis was confirmed based on histological features and positive immunohistochemical reactivity for desmin and myogenin. All cases were screened at diagnosis for PAX3/7-FOXO1 gene fusions either by RT-PCR or FISH and were negative. All cases had archival formalin-fixed paraffin-embedded material for further molecular testing. In addition 6 cases had adequate frozen tissue material, 4 of these being subjected to paired-end RNA sequencing and 2 were previously analyzed by 5’RACE 12. Three cases were previously included in the study by Mosquera et al. 12 and 4 cases by Agaram et al. 11, see Table 1. The study was approved by the Institutional Review Board at each institution.
Table 1.
Clinicopathologic and Molecular Features of Pediatric Spindle cell/Sclerosing RMS
| RMS # |
Age/ Sex |
Site | Diagnosis | Molecular Results | FU (y) |
IRS | Status |
|---|---|---|---|---|---|---|---|
| 1* | 0.7/M | Post-neck | SRMS | SRF-NCOA2¥ | 3y | I | NED, CR2 |
| 2* | 4W/M | Chest wall | SRMS | TEAD1-NCOA2 | 3y | IIa | NED |
| 3*α | 0.3/F | Chest wall | SRMS | VGLL2-NCOA2 | 13y | III | NED, CR4 |
| 4 | 0/M | Calf | SRMS | TEAD1-NCOA2 | NA | NA | NA |
| 5 | 0.2/F | Back | SRMS | VGLL2-NCOA2 | NA | NA | NA |
| 6 | 0/F | Back | SRMS | VGLL2-CITED2¥ | 9y | III | NED, CR |
| 7 | 0/F | Back | SRMS | VGLL2-CITED2¥ | 6y | III | NED, CR |
| 8 | 0.7/F | Back | SRMS | VGLL2-CITED2 | 8y | II | NED, CR |
| 9 | 0/F | Lower neck/back | SRMS | VGLL2-CITED2 | Recent case | I | In therapy |
| 10 | 0.8/M | Chest wall | SRMS | VGLL2 | NA | NA | NA |
| 11 | 0.4/M | Paravertebral | SRMS | - | NA | NA | NA |
| 12€ | 10/F | Paraspinal | SRMS | MyoD1 (L122R)β | 3y | III | DOD |
| 13€ | 2/F | Buttock | SRMS | MyoD1 (L122R) | 1y | IIa | DOD |
| 14 | 17/M | Paravertebral | ScRMS | MyoD1 (L122R)β | 2y | III | DOD |
| 15€ | 15/F | Infratemporal | ScRMS | MyoD1 (L122R)∞β | 1y | III | DOD |
| 16€ | 13/F | Back | ScRMS | MyoD1 (L122R)∞ β | 2y | III | DOD |
| 17 | 10/F | Buttock | ScRMS | MyoD1 (L122R)∞
¥ FGFR4 (V548M) |
0.5y | III | DOD |
| 18 | 8/M | Thigh | ScRMS | MyoD1 (L122R)β | 1y | III | NED, CR1 |
| 19 | 11/F | H&N | ScRMS | MyoD1 (L122R)∞ β | Recent case | I | In therapy |
| 20 | 9/M | H&N | SRMS | MyoD1 (L122R)β | 3y | III | AWD (2nd LR) |
| 21 | 9/F | H&N | SRMS | MyoD1 (L122R)β | 1y | III | DOD |
| 22 | 3/M | Intra-abd | SRMS | - | 13y | I | NED, CR2 |
| 23 | 16/M | Paratesticular | SRMS | - | 1y | I | NED, CR |
| 24 | 5/F | Intra-abd | SRMS | FGFR4 (V550L) | NA | NA | NA |
| 25 | 2.8/F | Ovary/salpinx | SRMS | - | 2y | III | DOD |
| 26 | 11/M | Paratesticular | ScRMS | - | 1y | II | NED, CR |
included in Mosquera et al12,
included in Agaram et al11;
cases tested by RNAseq;
case tested by 5’-RACE;
co-existing PIK3CA mutations;
homozygous MyoD1 (L122R) mutations;
M, male; F, female; FU, follow-up; Tx, therapy; y, years; H&N, head and neck; intra-abd, intra-abdominal; CR, complete remission; NED, no evidence of disease; AWD, alive with disease; DOD, dead of disease; LR, local recurrence; NA, not available; IRS, according to the Intergroup Rhabdomyosarcoma Study Group pretreatment staging classification.
Clinicopathologic Features
The clinical data of the 26 patients (11 males and 15 females) with an overall age range of 0–17 years (median 3, mean 5.5) are summarized in Table 1. Eleven of these patients were diagnosed at birth (congenital) or within one year of age (infantile), with equal gender distribution. All except one of the congenital/infantile cases were located in the trunk: back/paravertebral areas, 5; chest wall, 3; posterior neck/paraspinal, 2. Only one occurred in the limb soft tissues (calf). The remaining 15 patients, 9 males and 6 females, were diagnosed in older children, with a mean age of 9.4 years (range 2–17, median 9.5). The anatomic distribution for this latter subgroup was more variable, with 5 intra-abdominal/paratesticular, 3 cases in the trunk (paraspinal, paravertebral, back), 4 in the head and neck (infratemporal, cheek, orbit), 2 in the buttock and 1 in the thigh.
Hematoxylin and eosin (H&E) stained slides from all cases were reviewed by two sarcoma pathologists (RA, CRA). The diagnosis of SRMS was defined according to the current criteria proposed by WHO 2013 classification, when there was a predominant spindle cell morphology, with only focal if any rhabdomyoblastic differentiation and absent or minimal nuclear pleomorphism. Sc-RMS was defined as tumors with variable cellularity, composed of spindle cells or round cells arranged in a pseudovascular/pseudoalveolar or tubular-like pattern, embedded in a hyalinizing stroma. Positive immunostaining for desmin and, at least focally, for myogenin were required for diagnosis.
A detailed morphologic examination was carried out in each case, recording the predominant growth pattern (herring-bone, fascicular, fibromatosis-like), amount and quality of cytoplasm, degree of cytologic atypia (nuclear hyperchromasia, type of chromatin degree of nuclear pleomorphism), mitotic rate, and type of stromal component. Based on some of these features the tumors were also defined as resembling other mesenchymal tumors, such as fibrosarcoma-like, leiomyosarcoma-like, and fibromatosis-like. Clinicopathologic features, including size, location, management and follow-up, were obtained from the medical records for all cases. Subsequently, the histological features were compared to the results of cytogenetic and molecular characterization.
RNA Sequencing
Four cases were analyzed by RNA sequencing (SRMS#1, 6, 7, 17). Total RNA was prepared for RNA sequencing in accordance with the standard Illumina mRNA sample preparation protocol (Illumina). Briefly, mRNA was isolated with oligo(dT) magnetic beads from total RNA (2µg). The mRNA was fragmented by incubation at 94°C for 2.5 min in fragmentation buffer (Illumina). To reduce the inclusion of artifact chimeric transcripts into the sequencing library, an additional gel size-selection step was introduced prior to the adapter ligation step (Quail et al., 2008). The adaptor-ligated library was then enriched by PCR for 15 cycles and purified. The library was sized and quantified using DNA1000 kit (Agilent) on an Agilent 2100 Bioanalyzer according to the manufacturer’s instructions. Paired-end RNA- sequencing at read lengths of 50 or 51 bp was per- formed with the HiSeq 2000 (Illumina). A total of about 60.9 million paired-end reads were generated, corresponding to about 3.1 billion bases.
Analysis of RNA Sequencing Results with FusionSeq and TopHat
All reads were independently aligned with the STAR (ver 2.3) 13 and TopHat2 (ver 2.0.14) 14 against the human reference genome (hg19), followed by FusionSeq 15 and TopHat-Fusion (ver 0.1.0) 16 respectively to identify fusion candidates. In the first analysis mapped reads were converted into Mapped Read Format 17 and analyzed with FusionSeq to identify potential fusion transcripts. An SRF-NCOA2 fusion candidate was identified by FusionSeq in SRMS1 and experimentally validated as previously reported 12. However, for the additional 3 new cases (SRMS# 6, 7, 16) no fusion candidates were identified with this pipeline. We then applied TopHat-fusion analysis tool to detect potential gene fusion events with spanning reads > 20 bp. In contrast to FusionSeq, TopHat-fusion aligns reads without relying on existing annotation, and directly detects individual reads that span the fusion event. This tool proved to be more sensitive to detect small intra-chromosomal inversions, which most likely were filtered out by the Fusion Seq algorithm. In addition, RNA seq data was analyzed for gene mutation calls. BAM files were generated by STAR alignment, followed by PicardTools (ver 1.130) standard preprocessing, which include marking of duplicate reads, recalibrate of base quality scores and local realignment. MuTect (var 1.15) (Cibulskis et al., 2013) and VarScan (var 2.3.8) (Koboldt et al., 2012) variant callers were both applied for mutation detection, followed by vcf2maf (https://github.com/ckandoth/vcf2maf) for converting VCF into MAF files, with the annotation added by Variant Effect Predictor tool provided by Ensembl. (http://useast.ensembl.org/info/docs/tools/vep/index.html). Variants with missense and frame shift mutation in the 340 genes from the IMPACT panel were considered, and potential mutation locations were compared to NCBI dbSNP (http://www.ncbi.nlm.nih.gov/snp), cBioPortal (http://www.cbioportal.org/), and COSMIC (http://cancer.sanger.ac.uk/cosmic). Sanger PCR validation was performed subsequently.
Fluorescence in situ hybridization (FISH)
FISH on interphase nuclei from paraffin embedded 4-micron sections was performed applying custom probes using bacterial artificial chromosomes (BAC), covering and flanking VGLL2, CITED2, NCOA2, SRF and TEAD1 (Supporting Information Table 1). BAC clones were chosen according to UCSC genome browser (http://genome.ucsc.edu) and obtained from BAC-PAC sources of Children’s Hospital of Oakland Research Institute (CHORI; Oakland, CA; http://bacpac.chori.org). DNA from individual BACs was isolated according to the manufacturer’s instructions, labeled with different fluorochromes in a nick translation reaction, denatured, and hybridized to pretreated slides. Slides were then incubated, washed, and mounted with DAPI in an antifade solution. The genomic location of each BAC set was verified by hybridizing them to normal metaphase chromosomes. Two hundred successive nuclei were examined using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberko- chen, Germany), controlled by Isis 5 software (Metasystems). A positive score was interpreted when at least 20% of the nuclei showed a break-apart signal. Nuclei with incomplete set of signals were omitted from the score.
Targeted PCR and Sanger Sequencing
Genomic DNA was isolated either from fresh-frozen or archival paraffin tissue, as described previously (Antonescu et al., 2003) in all fusion negative samples. Targeted PCR was performed for the known MYOD1 exon 1 hot spot mutation and PIK3CA exons 9 and 20 mutations as previously reported 11. Direct sequencing of PCR products was performed and compared to the NCBI human MYOD1 and PIK3CA gene sequences. For the FGFR4 kinase domain validation we used the following primers: FGFR4 exon 13 Fwd: 5'-CAATGTCCCGACTTCTCCCTCTC-3’ and FGFR4 exon 13 Rev 5'-CAGGTTGATGATGTTCTTGTGTCG-3.
RT-PCR validation and 5’RACE
The VGLL2-CITED2 fusion candidate identified by TopHAT fusion was validated by RT-PCR using 1 µg of total RNA and cDNA synthesis by SuperScript ® III First-Strand Synthesis Kit (Invitrogen; Carlsbad, CA). RT-PCR was performed using the advantage 2 PCR kit (Clonetech; Mountain View, CA) for 33 cycles at a 65°C annealing temperature, using the following primers: VGLL2 Intron3 fwd 5’-GTCTGCCCACTGCCCCTTTC-3’; CITED2 Ex2 rev 5’-GGTTGAAATACTGGTTGTTGAGC-3’. The PCR product was confirmed by agarose gel electrophoresis and then sequenced using the Sanger method.
5’RACE was performed in SRMS3 using the SMARTer™ RACE cDNA Amplification Kit (Clonetech). Reverse transcribed mRNA was initiated by SMARTer II A Oligonucleotide with appropriate 5’-RACE CDS Primer A in a 10 μl reaction volume according to the manufacturer's protocol. Primary PCR was performed by Advantage® 2 polymerase chain reaction (Clonetech) with the SMARTer™ RACE Universal Primer A Mix and reverse primer on NCOA2 exon 15 (5'-TCCTCCAAATCAGACTGCCCCAT-3'). Nested PCR was performed with the SMARTer™ RACE Nested Universal Primer A and reverse primer on NCOA2 exon 14 (5'- GGAACCCAGCAGCCAGCATC -3'). Amplified PCR products were being cloned by TOPO® TA Cloning® Kit for Sequencing with One Shot® TOP10 Chemically Competent E. coli (Invitrogen). The constructed plasmid DNA was sequenced using Sanger’s method.
Western blotting
Frozen tissue from the 2 samples with VGLL2-CITED1 fusions (SRMS6, SPRMS7) and one control sample (normal skeletal muscle) were homogenized in Tissue Protein Extraction Reagent (Thermo Scientific, Cat# 78510) supplemented with protease inhibitors. Electrophoresis and immunoblotting were performed on the protein extracts using 30 μg of protein per sample. VGLL2 and β-Actin expression were detected by anti-VGLL2 mouse polyclonal antibody (Abcam, Cat# ab169247) with 1:500 dilution and anti-Actin mouse monoclonal antibody (Cell Signaling, Cat#3700) with 1:1,000 dilution, respectively. Following hybridization with the secondary rabbit anti-mouse antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA, #sc-358923) with 1:5,000 dilution, the blots were incubated with Immun-Star horseradish peroxidase luminal/enhancer (Bio-Rad, Hercules, CA, USA) and exposed onto Kodak Biomax MR Film (Eastman Kodak Co., Rochester, NY, USA).
RESULTS
Congenital/infantile SRMSs show recurrent gene fusions, involving VGLL2, SRF and TEAD1, critical transcriptional regulators of skeletal muscle function
Two of the 3 infantile SRMS tested by TopHat Fusion algorithm showed similar VGLL2-CITED2 gene fusion candidates (SRMS6, SRMS7), which resulted through an intra-chromosomal 6 inversion, as the 2 genes have opposites directions of transcription (Fig. 1 A). RT-PCR confirmed the fusion of VGLL2 intron 3 with CITED2 exon 2 (Fig. 1B). FISH studies further validated this result in both cases (Fig. 1C). All the remaining cases were further tested by FISH for VGLL2 gene abnormalities and 5 additional cases were found to have VGLL2 break/inversion. These 5 cases were further investigated for potential partners and confirmed two additional cases with VGLL2-CITED2 fusion (SRMS8, SRMS9), two with VGLL2-NCOA2 (SRMS3, SRMS5) and one without an identifiable partner (SRMS10). SRMS3 was previously reported by our group as having a NCOA2 gene rearrangement without a proven gene partner 12. Repeat 5’RACE analysis on this case showed the fusion of VGLL2 exon 2 with NCOA2 exon 14 (Fig. 2), result being further confirmed by FISH (Fig. 2). The remaining 3 cases showed SRF-NCOA2 fusions in one case (SRMS1) and TEAD1-NCOA2 (SRMS2, SRMS4) in 2 cases 12. Only one tumor (SRMS11) in this group was not associated with gene fusion abnormalities.
Figure 1. Novel VGLL2-CITED2 fusion gene discovery in congenital/infantile SRMS by RNA sequencing and TopHat-seq data analysis.

(A) Diagrammatic representation of the 6q22-q23 region breaks at the VGLL2 and CITED2 loci, followed by an inversion and fusion of the 3’CITED2 to the 5’VGLL2 portion. (B) Fusion candidates were validated by RT-PCR showing fusion of the 1020 bp of VGLL2 intron 3 to CITED2 exon 2 (SRMS6). (C) FISH validation of VGLL2-CITED2 fusion, first by the VGLL2 break-apart assay (left panel)(arrows show constant split signal, in keeping with an intra-chromosomal inversion/break; red, centromeric; green, telomeric) and followed by the VGLL2-CITED2 three-color fusion assay (right panel)(arrows show red-yellow fused signals; red centromeric of CITED2; yellow VGLL2). (D) Western blotting showing lack of the 28 kDa wild-type VGLL2 band in the 2 VGLL2-CITED1 fusion positive cases (SRMS6&SRMS7) compared to the control skeletal muscle. Instead they show a lower band, possibly truncated protein at 22 kDa molecular weight.
Figure 2. Novel VGLL2-NCOA2 complex fusion in congenital/infantile SRMS by 5’RACE.

As VGLL2 and NCOA2 genes have opposite directions of transcription a functional fusion requires a break/inversion of one of the partners. (A) Schematic view of the intragenic VGLL2 break and inversion, confirmed by VGLL2 break-apart FISH with constant split signals (yellow, centromeric; green, telomeric). (B) Schematic diagram of the t(6;8) translocation, fusing the 5’VGLL2 portion to the 3’NCOA2; subsequent FISH validation using 3-color VGLL2-NCOA2 fusion assay (arrow showing fused red-yellow signals; yellow, centromeric of VGLL2; red, centromeric of NCOA2); (C) 5’RACE showing the fusion of VGLL2 exon 2 to NCOA2 exon 14 (SRMS3).
Histologically all 10 fusion-positive infantile SRMS showed a striking resemblance with infantile fibrosarcoma (Fig. 3), with no morphologic differences among the different fusion types. The lesional cells were monomorphic, with delicate, scant eosinophilic cytoplasm and ovale to wavy nuclei, with finely dispersed chromatin, and occasional, inconspicuous nucleoli (Fig. 3 A–C). Hyperchromasia was seen in 2 cases as focal and in one case as more diffuse (Fig. 3 D–H). Alternating hypocellular areas were present in 4 cases, with collagen bundles interspersed between cells, reminiscent of a desmoid-type fibromatosis (Fig. 3I). There was no evidence of necrosis. Mitotic rate was typically < 5 mitoses/10HPFs, except for SRMS2 with >10/10 HPFs. One case (SRMS10) displayed unusual myoid features with a vaguely perivascular pattern of arrangement, mimicking a myofibromatosis. The only fusion-negative tumor, occurring in a 4 month-old baby (SRMS11) showed a more conventional spindle cell RMS morphology, characterized by elongated cells with more abundant eosinophilic cytoplasm, scattered large hyperchromatic nuclei and >10/10 HPFs. All tumors showed patchy to diffuse desmin reactivity, and scattered, multifocal myogenin and MyoD1 reactivity.
Figure 3. Pathologic features of congenital/infantile fusion positive SRMS.
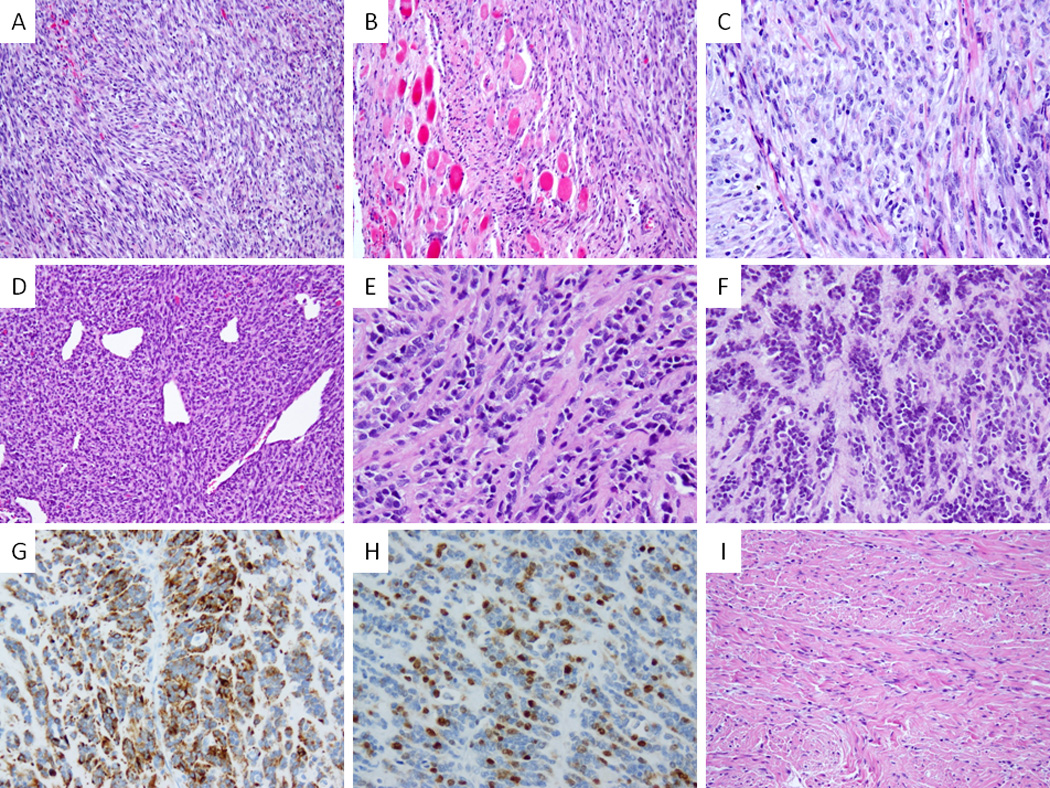
VGLL2-CITED2 fusion positive tumors shared similar morphology (A–C) with monomorphic spindle cells arranged in short intersecting fascicles (A, SRMS6), infiltrating within skeletal muscle (B, SRMS6) and showing a more plump ovale cells with pale eosinophilic to clear cytoplasm, fine chromatin and scattered mitotic figures (C, SRMS7). VGLL2-NCOA2 fusion positive tumor (SRMS3) showed a highly cellular and hyperchromatic appearance reminiscent of infantile fibrosarcoma with a distinctive hemangiopericytoma-like vascular pattern in the primary tumor (D); in the subsequent two local recurrences, 2 years (E) and 5 years (F) later, it had a more sclerosing appearance; latter recurrence showed reactivity for desmin (G) and myogenin (H). In contrast a VGLL2 rearranged tumor with no identifiable partner showed a more sclerotic background mimicking fibromatosis (SRMS10, I).
MYOD1-positive S/ScRMS occur in older children with predilection in the trunk and head and neck
Among the 15 spindle cell RMS occurring in older children, 10 (67%) had MYOD1 L122R exon 1 mutations, as detected by Sanger sequencing. In all except 2 cases there were MYOD1 L122R homozygous mutations (Table 1); both heterozygous MYOD1 mutant cases occurred in females and buttock location (2 year-old and 10 year-old girls). Four of these MYOD1-mutant cases harbored co-existing PIK3CA mutations, three being in the exon 10 helical domain (E542V, E545K, and Q546R) and one in the exon 21 kinase domain (G1049R). The latter case had available frozen tissue (RMS17) and was studied by RNAseq as initially being diagnosed as an unclassified rhabdomyosarcoma (possibly representing a fusion-negative solid ARMS). As the TopHat algorithm did not identify any fusion candidates, it was further subjected to mutation detection algorithms, which identified MYOD1 L122R, PIK3CA G1049R and FGFR4 V548M mutations. These mutations were then confirmed by Sanger sequencing. Upon morphologic re-review, the tumor showed focal areas of sclerosis intermixed within the mostly solid round cell component (Fig. 4); thus the case was reclassified as a cellular variant of sclerosing RMS supported by the above mutational profile. In fact all cases with co-existing MYOD1 and PIK3CA mutations were associated with sclerosing histology, being composed of variably cellular, but monomorphic, round to spindle cells embedded in a distinctive hyalinized stroma. The remaining MYOD1-mutant SRMSs displayed a classic morphology, composed of intersecting long fascicles of elongated cells with scant to moderate amount of fibrillary eosinophilic cytoplasms and uniform ovale nuclei, reminiscent of leiomyosarcoma. Rhabdomyoblastic differentiation, although very focal, was often present in the form of scattered rhabdomyoblasts intermixed among the spindle cells. Patchy hyperchromasia was also present, with mostly scattered cytologic atypia. Three tumors showed increased mitotic activity of >10MF/10 HPFs and geographic necrosis.
Figure 4. Novel FGFR4 kinase domain mutation in a poorly differentiated ScRMS with co-existing MYOD1 and PIK3CA mutations.

(A) IGV (integrated genomic viewer) tool using RNAseq data showing FGFR4 kinase domain mutation at V548 position (exon 13) with a 49% allelic frequency and read coverage of 717 in SRMS17, compared with other RMS cases; (B) Bar chart showing high FGFR4 mRNA expression in ScRMS17, compared to other tumor types and RMS cases (including SRMS6 and SRMS7 with VGLL2-CITED2 fusions; 2 ectomesenchymomas [ECTM], 1 infantile myofibroma [MYO1] and other subtypes of RMS). Of note, similarly high FGFR4 expression was seen in an adult SRMS, lacking MYOD1, PIK3CA and FGFR4 mutation (adSRMS1). (C) IGV showing MYOD1 L122R mutation with a 42% allele fraction and read coverage of 883 and further confirmed by Sanger sequencing as a heterozygous mutation. (D) Bar chart showing overall high expression of MYOD1 mRNA across different RMS and ectomesenchymomas compared to other tumor types, with highest levels in the 2 cases harboring MYOD1 mutations (adSRMS2, SRMS17).
The remaining 5 cases lacking both fusions and MYOD1 mutations showed overlapping morphologic features with the MYOD1-mutant SRMS (Supplementary Fig. 1). In addition 2 of the cases showed focal myxoid areas. Two cases showed increased mitotic rate of >10 MF/10 HPFs. All cases showed mostly diffuse positivity for desmin, while myogenin nuclear staining was found ranging from 5–50% of the cells. Additional targeted DNA PCR for the FGFR4 hot spots mutations showed the presence of a FGFR4 exon 13 V550L in SRMS24 (Table 1).
Fusion-positive infantile SRMS follow a favorable clinical course compared to the highly aggressive behavior of older children with SRMS harboring MYOD1 mutations
All patients have been treated with pre-operative chemotherapy after the diagnosis of spindle cell or sclerosing RMS was rendered on the biopsy, except for the 2 paratesticular tumors, treated with surgery and subsequent chemotherapy. All 6 fusion positive congenital/infantile SRMS patients with available long term follow-up (FU) are presently alive and well after a median of 7 years (range 3–13 years). Two patients developed local recurrences, but none developed distant metastases. In contrast, 7 of the 9 MYOD1-mutant RMS patients died of disease, including the 3 cases with sclerosing morphology and co-existing MYOD1/PIK3CA mutations, with a median of 1.5 years (range 1–3 years). Only one patient in this group is alive and well after only one year follow-up and an additional patient is alive with disease after developing a 2nd local recurrence. Among the fusion/MYOD1 mutation negative subset, 3 of the 4 cases with available follow-up are with no evidence of disease, including the 2 patients with para-testicular and one with para-ovarian tumors.
DISCUSSION
Despite a relatively homogeneous morphologic phenotype, SRMS has emerged as a diverse group of tumors, with different molecular genetic profiles and variable clinical behaviors depending on the age at presentation. In the pediatric population, SRMS was first described by Cavazzana et al. in 1992 4 and subsequently by Leuschner a year later 5 as a variant of ERMS, typically occurring in the paratesticular or head and neck region, with histologic features mimicking a leiomyosarcoma. Their clinical behavior appeared to be more favorable compared to classic ERMS. In contrast, Lundgren et al. described a small series of 3 SRMS occurring in children aged 1–3 years old and displaying a morphology reminiscent of infantile fibrosarcomas (‘infantile rhabdomyofibrosarcomas’), but followed a fatal course in 2 cases 18. Moreover, in adults, SRMSs also have a predilection for the head and neck region, but unlike the pediatric tumors, seem to follow a more aggressive clinical course 8, 19, 20. As the evidence for SRMS having distinct clinicopathologic features from ERMS and sharing morphologic features with ScRMS became more widely accepted, it lead to its reclassification in the latest WHO classification as a stand-alone pathologic entity 10.
From the genetic viewpoint, we have previously described recurrent NCOA2 gene rearrangements in a small subset of SRMS occurring in the infantile/congenital setting 12. Since NCOA2-related fusions were only seen in infants, we speculated that spindle cell/sclerosing RMS represents a heterogenous genetic group of tumors among different age groups. Notably, all 3 NCOA2-rearranged SRMS pursued a favorable clinical course and lacked metastatic potential. The additional 7 fusion-positive infantile SRMS reported here reinforce our initial results of a very favorable outcome, all patients being without evidence of disease after long term follow-up.
Herein we identify novel VGLL2 gene rearrangements located at 6q22 locus as the most common (64%) genetic abnormality in congenital/infantile SRMS, being fused to either NCOA2 or CITED1 gene partners. This result is not so surprising since VGLL2 is expressed in differentiating somites and branchial arches during embryogenesis and is exclusively expressed in skeletal muscle in the adult 21. VGLL2 (Vestigial-like 2, a.k.a. VGL2 or VITO1) was identified as a human homolog of a Drosophila protein, Vestigial, a transcriptional coactivator (without DNA binding sequence) of Scalloped, that contains the TEA domain and is required for wing formation 22, 23. VGLL2 is a key cofactor of TEF-1 (transcription enhancer factor-1, a.k.a. TEAD1) and MEF2 (myocyte enhancer factor-2) family members regulating muscle-specific gene transcription in skeletal muscle 21,24 (Supplementary Fig 2). In Drosophila, Vgl-2 plays a key role in the development and patterning of the wing: its loss results in a failure of wing development, while its overexpression leads to the development of ectopic wings 25. In humans, upon muscle differentiation VGLL2 is up-regulated and translocates from the cytoplasm to the nucleus, where it interacts with MEF2 and co-activates TEF-1 and MEF2-dependent muscle-specific promoters. Although VGLL2 alone is not sufficient to initiate myogenic conversion of 10T1/2 fibroblasts in vitro, it enhanced MyoD-mediated myogenic conversion, suggesting that it is a crucial cofactor of the muscle regulatory program 26.
Our results thus implicate a number of critical transcription factors or co-activators involved in skeletal muscle function in the pathogenesis of congenital/infantile SRMS. As most of these cases do not show overt rhabdomyoblastic differentiation morphologically and sometimes display only patchy desmin reactivity, it is intriguing to speculate that their oncogenic role in this tumor is to block skeletal muscle differentiation and maintain a primitive phenotype. Both SRF (serum response factor) and TEAD1 (TEA domain, a.k.a. TEF-1) have been shown to be involved in the control of muscle-specific gene transcription. SRF is highly expressed in skeletal muscle and controls the expression of genes specifically expressed in skeletal muscle (dystrophin, muscle creatine kinase, myoD), including several genes encoding sarcomeric proteins (such as α skeletal actin, myosin light chain, tropomyosin) 27. TEAD1 is constitutively expressed in cardiac and skeletal muscle, acting as a key molecule of muscle development, transactivating multiple target genes involved in cell proliferation and differentiation pathways 28.
In the context of SRMS, these muscle specific factors (VGLL2, SRF, TEAD1) participate as 5’partners in the recurrent translocations and maintain most of their key functional domains within the fusion protein. Thus VGLL2 retains its vestigial motif (Vg), SRF preserves the MADS box and TEAD1 its TEA domain (Supplementary Fig 2 diagram). Interestingly, our RNA sequencing showed that the VGLL2-CITED2 fusion product contained a large intronic sequence of VGLL2 intron 3, which most likely results in a truncated protein. This result is further confirmed by our Western blotting, showing lack of a wild-type VGLL2 protein, compared to normal skeletal muscle, in keeping with a loss of function effect (Fig 1). VGLL2 3’fusion partners are either NCOA2 on 8q13.3 or CITED2 on 6q23.3. The exact role of these genes within the fusion remains unclear, since no upregulation of either VGLL2 or CITED2, were noted in the 2 cases with available RNAseq data, suggesting a different role of the fusion transcript other than gene overexpression. CITED2 (Cbp/p300-interacting transactivator with Glu-Asp-rich carboxy-terminal domain, 2) is a cardiac transcription factor which is essential for heart development 29. Cited2-deficient mice show cardiac malformations, adrenal agenesis and neural crest defects. Mutations in this gene were reported to cause cardiac septal defects 30. Of interest, fusions involving CITED2 gene have been described recently in a case of high grade undifferentiated pleomorphic sarcoma, however being the 5’ partner and fused to PRDM10 on 11q24 31. CITED2 is a nuclear protein which binds closely to the CH1 region of p300 and CBP. In contrast, NCOA2 (nuclear receptor coactivator) is a member of the p160 steroid receptor co-activator gene family. In the COOH-terminal part of NCOA2 there are two intrinsic transcriptional activation domains, TAD1 and TAD2 32, 33, which are responsible for interaction with general transcriptional co-integrators such as CBP and p300. This is intriguing since both CITED2 and NCOA2 maintain their carboxy-terminal in the projected fusion protein structure, thus sharing a CBP/p300 interaction domain which may be involved in the pathogenesis of SRMS (see Supplementary Fig 2).
SRMSs harboring MYOD1 mutations have been first described in the adult population, in about 44% of cases tested 34. Our group subsequently reported their occurrence in both pediatric age group and in sclerosing morphology 11. In our initial study, all the 4 pediatric MYOD1-mutated RMS patients died of the disease at 2-year median follow-up. Interestingly 2 of the 4 cases showing co-existing PIK3CA helical mutations were associated with sclerosing morphology 11. Of interest MYOD1 were also reported in 10% of ERMS 35, albeit described as having mostly a spindle cell morphology. However, MYOD1 mutations were not identified in a large genomic study encompassing both fusion-positive ARMS and fusion negative ERMS 36. In our expanded cohort of 9 MYOD1-mutant SRMS we confirm the initial observations that this genotype is seen in older children (mean of 10 years old), presenting with predilection in the trunk and head and neck area. The presence of MYOD1 mutations is associated with a highly aggressive course despite multimodality treatment, with 7/9 children with available follow-up dying of disease within one year of diagnosis. Once again the co-existing PIK3CA mutation was associated with sclerosing morphology. Of note one of the Sc-RMS tested by RNAseq showed 3 coexisting mutations: MYOD1 L122R, kinase domain PIK3CA G1049R and kinase domain FGFR4 V548M. Although previous kinase domain mutations in FGFR4 have been reported in about 6–7% of RMS 36, 37, mostly in ERMS 38, this case appears to be the first report of FGFR4 mutations in a spindle/sclerosing RMS. The RNAseq in this case showed high up-regulation of FGRF4 mRNA (Fig 4). Furthermore, the association of these 3 activating mutations in one single case might explain the highly undifferentiated phenotype noted, reminiscent of a fusion-negative solid ARMS. Only in retrospect, after the mutation profile became available, the case was reclassified as a cellular variant of sclerosing RMS due to the presence of focal areas of sclerosis. This case raises the question if a subset of the so-called fusion-negative solid ARMS represent in fact cellular/solid-variants of ScRMS. Indeed a recent re-review of the ARMS in patients enrolled in the Children Oncology Group (COG) studies from 1999–2005 resulted in the reclassification of 30% of cases, either into ERMS with so called ‘dense pattern’ or sclerosing RMS 39. It also raises the possibility that accumulating secondary oncogenic mutations, possibly synergistic to the MYOD1 initiating event, are responsible for blocking differentiation and associated fatal outcome. SRMS with MYOD1 mutations showed significantly higher upregulation of MYOD1 mRNA, based on 2 cases with available RNAseq data, either occurring in an adult (adSRMS2) or in a child (SRMS17), compared to other RMS subtypes (Fig 4).
Despite our detailed molecular investigation, there was a remaining third group of tumors, lacking recurrent genetic abnormalities and presenting with predilection in the genito-urinary or intra-abdominal location. Although the data is quite limited for definitive conclusions, these patients appeared to follow a more favorable course compared to the MYOD1 mutant cases, and might either represent a third genetic group of SRMS or more closely related to the ERMS. Only one of these 5 cases showed FGFR4 V550L mutations, as previously documented in ERMS37.
In conclusion, despite a relatively similar histomorphology, pediatric SRMS is a heterogeneous disease genetically as well as clinically. Our study identifies three distinct molecular subsets of spindle cell/sclerosing RMS within the pediatric-age group. First, tumors presenting at birth or within one year of age occur with predilection in the trunk and are associated with recurrent gene fusions, involving critical transcriptional activators of muscle-specific genes, such as VGLL2, TEAD1 and SRF. Most importantly these patients follow a favorable clinical outcome, lacking metastatic potential, all being alive and well at long term follow-up. These fusion-positive infantile SRMS group appears closely reminiscent to the behavior of ETV6-NTRK3-positive infantile fibrosarcomas and militate against their classification as a ‘high grade neoplasm’. Second, our findings further show that pediatric MYOD1-mutant SRMSs, with or without accompanying PIK3CA mutations, follow a highly aggressive course with high mortality despite multimodality therapy. In fact, MYOD1 mutations was the most common genetic abnormality in pediatric SRMS, occurring in 67% of children beyond one year of age, and can be used as molecular diagnostic test to stratify these high risk patients. Third, despite our detailed molecular investigation, there is a remaining group lacking gene fusions or MYOD1 mutations often presenting intra-abdominally or in the genito-urinary area. This latter subset seems to follow a favorable clinical course, however the data remains limited to determine if it represents a separate molecular group or has a closer relationship to the more common ERMS in this location.
Supplementary Material
SRMS showing monotonous spindle cells arranged in intersecting fascicles (A, RMS25) and moderate amount of mostly clear cytoplasm (B, RMS23); ScRMS with densely sclerotic background and encasing testicular tubules (C, RMS26) and a more distinctive micro-alveolar growth pattern (D, RMS26).
Figure 5. Wide morphologic spectrum of MYOD1-mutant pediatric SRMS.
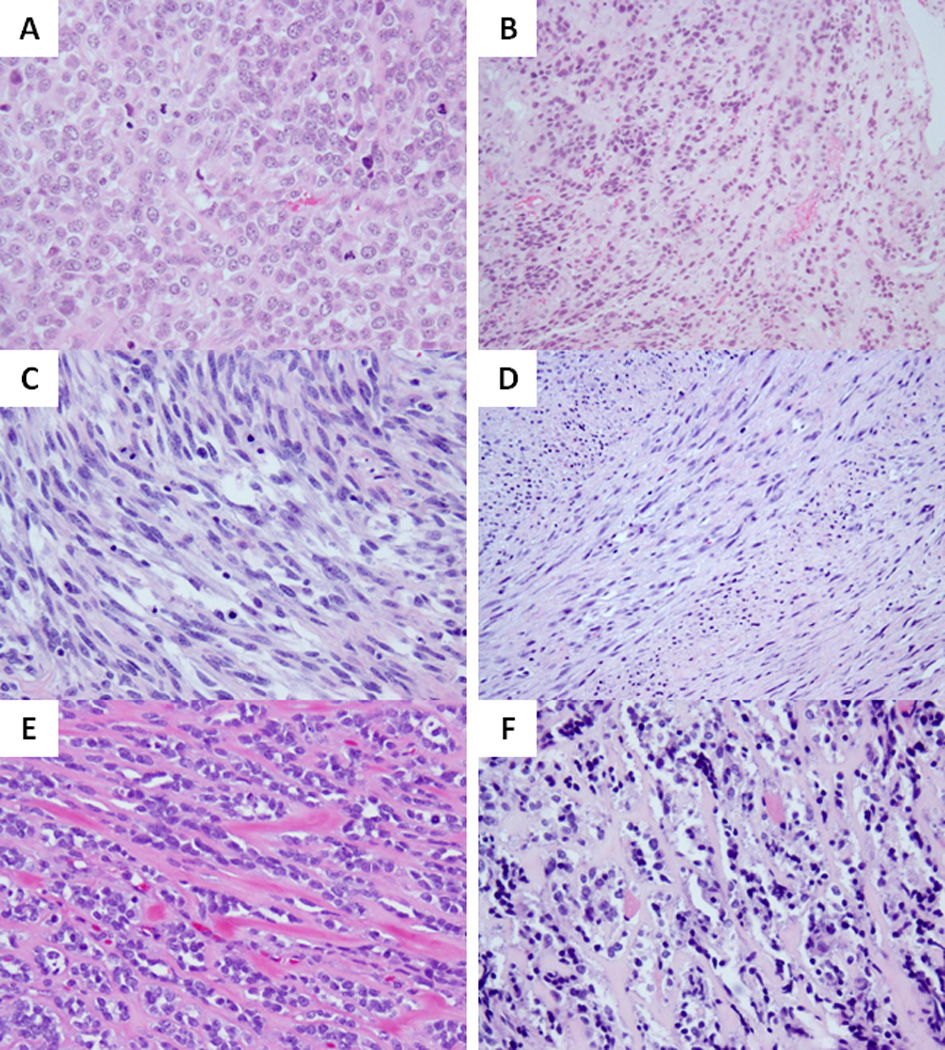
Co-existing MYOD1, PIK3CA and FGFR4 mutations were associated with a highly primitive and solid growth of undifferentiated round cells with high mitotic activity and necrosis (A; RMS17). Focal areas of dense sclerosis were noted in keeping with a cellular variant of ScRMS (B). MYOD1-mutant SRMSs from the H&N area showing monomorphic spindle cells with fibrillary pale eosinophilic cytoplasm, fine chromatin and scattered mitotic figures, in a loose edematous stroma (C, RMS20) or in a more sclerotic background and arranged in long sweeping fascicles (D, RMS21); Co-existing MYOD1 and PIK3CA helical mutations in 2 Sc-RMSs showing distinctive micro-alveolar growth pattern and alternating refractile collagen bundles (E, RMS15; F, RMS16).
Acknowledgements
Supported in part by: P50 CA 140146-01 (CRA), Cycle for Survival ‘Luke’s team’ (CRA, LW), Kristen Ann Carr Fund (CRA).
The authors thank Milagros Soto for editorial assistance and the efforts of Agnès Viale and the Genomics Core Laboratory at MSKCC where RNA sequencing was performed.
Footnotes
Conflict of interest: none
REFERENCES
- 1.Missiaglia E, Williamson D, Chisholm J, et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol. 2012;30:1670–1677. doi: 10.1200/JCO.2011.38.5591. [DOI] [PubMed] [Google Scholar]
- 2.Carli M, Colombatti R, Oberlin O, et al. European intergroup studies (MMT4-89 and MMT4-91) on childhood metastatic rhabdomyosarcoma: final results and analysis of prognostic factors. J Clin Oncol. 2004;22:4787–4794. doi: 10.1200/JCO.2004.04.083. [DOI] [PubMed] [Google Scholar]
- 3.Oberlin O, Rey A, Lyden E, et al. Prognostic factors in metastatic rhabdomyosarcomas: results of a pooled analysis from United States and European cooperative groups. J Clin Oncol. 2008;26:2384–2389. doi: 10.1200/JCO.2007.14.7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavazzana AO, Schmidt D, Ninfo V, et al. Spindle cell rhabdomyosarcoma. A prognostically favorable variant of rhabdomyosarcoma. Am J Surg Pathol. 1992;16:229–235. doi: 10.1097/00000478-199203000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Leuschner I, Newton WA, Jr, Schmidt D, et al. Spindle cell variants of embryonal rhabdomyosarcoma in the paratesticular region. A report of the Intergroup Rhabdomyosarcoma Study. Am J Surg Pathol. 1993;17:221–230. doi: 10.1097/00000478-199303000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Mentzel T, Katenkamp D. Sclerosing, pseudovascular rhabdomyosarcoma in adults. Clinicopathological and immunohistochemical analysis of three cases. Virchows Arch. 2000;436:305–311. doi: 10.1007/s004280050451. [DOI] [PubMed] [Google Scholar]
- 7.Folpe AL, McKenney JK, Bridge JA, et al. Sclerosing rhabdomyosarcoma in adults: report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. Am J Surg Pathol. 2002;26:1175–1183. doi: 10.1097/00000478-200209000-00008. [DOI] [PubMed] [Google Scholar]
- 8.Mentzel T, Kuhnen C. Spindle cell rhabdomyosarcoma in adults: clinicopathological and immunohistochemical analysis of seven new cases. Virchows Arch. 2006;449:554–560. doi: 10.1007/s00428-006-0284-4. [DOI] [PubMed] [Google Scholar]
- 9.Mentzel T. Spindle cell rhabdomyosarcoma in adults: a new entity in the spectrum of malignant mesenchymal tumors of soft tissues. Pathologe. 2010;31:91–96. doi: 10.1007/s00292-009-1249-6. [DOI] [PubMed] [Google Scholar]
- 10.Fletcher C, Bridge JA, Hogendoorn PC, et al. WHO Classification of Tumours of Soft Tissue and Bone. Lyon: IARC; 2013. [Google Scholar]
- 11.Agaram NP, Chen CL, Zhang L, et al. Recurrent MYOD1 mutations in pediatric and adult sclerosing and spindle cell rhabdomyosarcomas: evidence for a common pathogenesis. Genes Chromosomes Cancer. 2014;53:779–787. doi: 10.1002/gcc.22187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mosquera JM, Sboner A, Zhang L, et al. Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes Chromosomes Cancer. 2013;52:538–550. doi: 10.1002/gcc.22050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim D, Pertea G, Trapnell C, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sboner A, Habegger L, Pflueger D, et al. FusionSeq: a modular framework for finding gene fusions by analyzing paired-end RNA-sequencing data. Genome Biol. 2010;11:R104. doi: 10.1186/gb-2010-11-10-r104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Habegger L, Sboner A, Gianoulis TA, et al. RSEQtools: a modular framework to analyze RNA-Seq data using compact, anonymized data summaries. Bioinformatics. 2011;27:281–283. doi: 10.1093/bioinformatics/btq643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lundgren L, Angervall L, Stenman G, et al. Infantile rhabdomyofibrosarcoma: a high-grade sarcoma distinguishable from infantile fibrosarcoma and rhabdomyosarcoma. Hum Pathol. 1993;24:785–795. doi: 10.1016/0046-8177(93)90017-b. [DOI] [PubMed] [Google Scholar]
- 19.Rubin BP, Hasserjian RP, Singer S, et al. Spindle cell rhabdomyosarcoma (so-called) in adults: report of two cases with emphasis on differential diagnosis. Am J Surg Pathol. 1998;22:459–464. doi: 10.1097/00000478-199804000-00011. [DOI] [PubMed] [Google Scholar]
- 20.Nascimento AF, Fletcher CD. Spindle cell rhabdomyosarcoma in adults. Am J Surg Pathol. 2005;29:1106–1113. [PubMed] [Google Scholar]
- 21.Maeda T, Chapman DL, Stewart AF. Mammalian vestigial-like 2, a cofactor of TEF-1 and MEF2 transcription factors that promotes skeletal muscle differentiation. J Biol Chem. 2002;277:48889–48898. doi: 10.1074/jbc.M206858200. [DOI] [PubMed] [Google Scholar]
- 22.Paumard-Rigal S, Zider A, Vaudin P, et al. Specific interactions between vestigial and scalloped are required to promote wing tissue proliferation in Drosophila melanogaster. Dev Genes Evol. 1998;208:440–446. doi: 10.1007/s004270050201. [DOI] [PubMed] [Google Scholar]
- 23.Vaudin P, Delanoue R, Davidson I, et al. TONDU (TDU), a novel human protein related to the product of vestigial (vg) gene of Drosophila melanogaster interacts with vertebrate TEF factors and substitutes for Vg function in wing formation. Development. 1999;126:4807–4816. doi: 10.1242/dev.126.21.4807. [DOI] [PubMed] [Google Scholar]
- 24.Yoshida T. MCAT elements and the TEF-1 family of transcription factors in muscle development and disease. Arterioscler Thromb Vasc Biol. 2008;28:8–17. doi: 10.1161/ATVBAHA.107.155788. [DOI] [PubMed] [Google Scholar]
- 25.Kim J, Sebring A, Esch JJ, et al. Integration of positional signals and regulation of wing formation and identity by Drosophila vestigial gene. Nature. 1996;382:133–138. doi: 10.1038/382133a0. [DOI] [PubMed] [Google Scholar]
- 26.Gunther S, Mielcarek M, Kruger M, et al. VITO-1 is an essential cofactor of TEF1-dependent muscle-specific gene regulation. Nucleic Acids Res. 2004;32:791–802. doi: 10.1093/nar/gkh248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pipes GC, Creemers EE, Olson EN. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 2006;20:1545–1556. doi: 10.1101/gad.1428006. [DOI] [PubMed] [Google Scholar]
- 28.Qiu H, Wang F, Liu C, et al. TEAD1-dependent expression of the FoxO3a gene in mouse skeletal muscle. BMC Mol Biol. 2011;12:1. doi: 10.1186/1471-2199-12-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, Wang F, Wu Y, et al. Variations of CITED2 are associated with congenital heart disease (CHD) in Chinese population. PLoS One. 2014;9:e98157. doi: 10.1371/journal.pone.0098157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu M, Wu X, Li Y, et al. CITED2 mutation and methylation in children with congenital heart disease. J Biomed Sci. 2014;21:7. doi: 10.1186/1423-0127-21-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hofvander J, Tayebwa J, Nilsson J, et al. Recurrent PRDM10 gene fusions in undifferentiated pleomorphic sarcoma. Clin Cancer Res. 2015;21:864–869. doi: 10.1158/1078-0432.CCR-14-2399. [DOI] [PubMed] [Google Scholar]
- 32.Chen D, Ma H, Hong H, et al. Regulation of transcription by a protein methyltransferase. Science. 1999;284:2174–2177. doi: 10.1126/science.284.5423.2174. [DOI] [PubMed] [Google Scholar]
- 33.Koh SS, Chen D, Lee YH, et al. Synergistic enhancement of nuclear receptor function by p160 coactivators and two coactivators with protein methyltransferase activities. J Biol Chem. 2001;276:1089–1098. doi: 10.1074/jbc.M004228200. [DOI] [PubMed] [Google Scholar]
- 34.Szuhai K, de Jong D, Leung WY, et al. Transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma. J Pathol. 2014;232:300–307. doi: 10.1002/path.4307. [DOI] [PubMed] [Google Scholar]
- 35.Kohsaka S, Shukla N, Ameur N, et al. A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat Genet. 2014;46:595–600. doi: 10.1038/ng.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shern JF, Chen L, Chmielecki J, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4:216–231. doi: 10.1158/2159-8290.CD-13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taylor JGt, Cheuk AT, Tsang PS, et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J Clin Invest. 2009;119:3395–3407. doi: 10.1172/JCI39703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shukla N, Ameur N, Yilmaz I, et al. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin Cancer Res. 2012;18:748–757. doi: 10.1158/1078-0432.CCR-11-2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rudzinski ER, Anderson JR, Hawkins DS, et al. The World Health Organization Classification of Skeletal Muscle Tumors in Pediatric Rhabdomyosarcoma: A Report From the Children's Oncology Group. Arch Pathol Lab Med. 2015 doi: 10.5858/arpa.2014-0475-OA. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SRMS showing monotonous spindle cells arranged in intersecting fascicles (A, RMS25) and moderate amount of mostly clear cytoplasm (B, RMS23); ScRMS with densely sclerotic background and encasing testicular tubules (C, RMS26) and a more distinctive micro-alveolar growth pattern (D, RMS26).