

Abstract
Background
Breakdown of renal endothelial, tubular and glomerular matrix collagen plays a major role in acute kidney injury (AKI) development. Such collagen breakdown releases endostatin into the circulation. The aim of this study was to compare the AKI predictive value of plasma endostatin with two previously suggested biomarkers of AKI, cystatin C and neutrophil gelatinase-associated lipocalin (NGAL).
Methods
We studied 93 patients without kidney disease who had a first plasma sample obtained within 48 h of ICU admission. We identified risk factors for AKI within the population and designed a predictive model. The individual ability and net contribution of endostatin, cystatin C and NGAL to predict AKI were evaluated by the area under the receiver operating characteristics curve (AUC), likelihood-ratio test, net reclassification improvement (NRI) and integrated discrimination improvement (IDI).
Results
In total, 21 (23 %) patients experienced AKI within 72 h. A three-parameter model (age, illness severity score and early oliguria) predicted AKI with an AUC of 0.759 (95 % CI 0.646–0.872). Adding endostatin to the predictive model significantly (P = 0.04) improved the AUC to 0.839 (95 % CI 0.752–0.925). In addition, endostatin significantly improved risk prediction using the likelihood-ratio test (P = 0.005), NRI analysis (0.27; P = 0.04) and IDI analysis (0.07; P = 0.04). In contrast, adding cystatin C or NGAL to the three-parameter model did not improve risk prediction in any of the four analyses.
Conclusions
In this cohort of critically ill patients, plasma endostatin improved AKI prediction based on clinical risk factors, while cystatin C and NGAL did not.
Electronic supplementary material
The online version of this article (doi:10.1186/s13613-016-0108-x) contains supplementary material, which is available to authorized users.
Keywords: Endostatin, Cystatin C, NGAL, Acute kidney injury, Sepsis
Background
Acute kidney injury (AKI) is common in the critically ill and associated with a high mortality rate [1]. Biomarkers may allow earlier detection of patients at risk of AKI and enable earlier intervention. Endostatin, the C-terminal fragment of collagen XVIII, is released into the circulation as a consequence of accelerated turnover of collagen XVIII in the basement membranes of the renal tubular epithelium, Bowman’s capsule, mesangium and renal capillaries, and may be one such biomarker [2].
In animal AKI models, upregulated renal endostatin expression preceded deteriorating kidney function by several hours [3, 4]. In addition, elevated serum endostatin has been associated with the degree of renal dysfunction in elderly patients and independently predicts the subsequent development of chronic kidney disease (CKD) in this population [5]. Endostatin has also been associated with mortality in several patient groups [6–8]. However, the potential of endostatin to identify critically ill patients at a higher risk of AKI remains unexplored.
Accordingly, we conducted a prospective, exploratory observational study to investigate the value of plasma endostatin as an early biomarker of AKI by comparing it with traditional clinical assessments of renal function and with plasma neutrophil gelatinase-associated lipocalin (NGAL) and plasma cystatin C. We hypothesized that, in patients with normal renal function on ICU admission, plasma endostatin levels would be higher among those patients who subsequently develop AKI compared with those who do not. In addition, we hypothesized that admission plasma endostatin would improve the AKI predictive ability of a clinical risk model.
Methods
This study was approved by the regional ethical review board in Stockholm and has therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Written informed consent was obtained from patients or their next of kin.
Patient selection and operational definitions
We screened patients admitted to the general intensive care unit (ICU) at the Karolinska University Hospital, Solna, Sweden, from August 2007 to November 2010. We enrolled patients with an expected length of stay of more than 24 h and an estimated glomerular filtration rate (eGFR) of more than 60 mL/min/1.73 m2 (modification of diet in renal disease [MDRD] equation) on ICU admission.
We defined AKI as a ≥50 % increase in plasma creatinine from baseline or an increase in plasma creatinine by ≥26.5 µmol/L within 48 h and/or a urine output less than 0.5 mL/kg/h for at least 6 consecutive hours according to the Kidney Disease: Improving Global Outcomes (KDIGO) criteria [9]. We used the lowest creatinine level obtained within 3 months before ICU admission as baseline for the KDIGO classification. Missing baseline creatinine was imputed using the MDRD formula and an eGFR of 75 mL/min/1.73 m2 [9]. We decided a priori to exclude patients having their first study sample obtained >48 h after ICU admission and patients with AKI on the day of their first study sample collection. Our primary outcome was development of AKI within 72 h of first study sample collection. Accordingly, we recorded AKI status until a maximum of 5 days following ICU admission.
We defined the systemic inflammatory response syndrome (SIRS) using three or more SIRS criteria [10]. Sepsis was defined as a suspected or confirmed infection together with SIRS.
Plasma sampling and biomarker analyses
We collected blood samples on study inclusion and twice daily thereafter until ICU discharge or start of renal replacement therapy. After centrifugation at 2000 rpm at 4 °C for 10 min, the supernatant plasma was stored at −80 °C.
Endostatin and NGAL were analyzed during 2013 using a commercially available enzyme-linked immunosorbent assay (ELISA) kit [DY1098 (endostatin) and DY1757 (NGAL), R&D Systems, Minneapolis, MN]. The assays had a total coefficient of variation (CV) of approximately 6 %. Cystatin C was measured with a particle-enhanced turbidimetric immunoassay on the Architect Ci8200 analyzer (Abbott Laboratories, Abbott Park, IL) with cystatin C reagents from Gentian (Moss, Norway).
Statistical analysis
Data were analyzed using STATA® version 11.2 software (Stata Corporation, College Station, TX, USA). Data are presented as medians and interquartile ranges (IQR) or as numbers and percentages. The Mann–Whitney test and Fisher’s exact test were used to test for differences between groups. The change over time for endostatin was tested by a repeated-measure analysis of variance (ANOVA). To compare the change over time between groups (AKI versus no AKI), we introduced an interaction variable between group and time to the ANOVA model. We assessed correlation using Spearman’s rank correlation coefficient (rho). The association of clinical variables with AKI development was assessed by multivariate logistic regression analysis. The following clinical predictor variables were considered: age, sex, acute physiology and chronic health evaluation (APACHE) II score, baseline creatinine, delta creatinine, early oliguria (urine output <0.5 mL/kg/h for >2 h but <6 h), presence of SIRS, presence of sepsis and noradrenaline dose. Clinical predictor variables were included in the multivariate models if they were statistically significant at P < 0.10 in the univariate analyses.
We assessed whether the addition of the measured biomarkers to the clinical model improved the predictive power for AKI by using the likelihood-ratio test. In addition, we calculated the area under the receiver operating characteristics curve (AUC) for the clinical model with and without inclusion of the measured biomarkers. The equality of AUCs was assessed by the method of DeLong et al. [11]. We described AUCs using the following values: 0.90–1.0 excellent, 0.80–0.89 good, 0.70–0.79 fair, 0.60–0.69 poor and 0.50–0.59 no useful performance [12]. The contributions of the biomarkers to risk prediction were further assessed by the net reclassification improvement (NRI) and the integrated discrimination improvement (IDI) methods. Log-transformed (base 10) biomarker values were used in the statistical analyses. Two-sided P values below 0.05 were considered statistically significant.
Results
Study patients
We enrolled 138 patients with eGFR > 60 mL/min/1.73 m2 and an expected length of stay >24 h (Fig. 1). We excluded 16 patients who were enrolled after >48 h of ICU admission and 29 patients with AKI on the day of their first study sample. We therefore studied 786 plasma samples in 93 patients; of these, 21 [22.6 % (95 % CI 14.6–32.4 %)] patients developed AKI within 72 h. Of the 21 AKI patients, 16 [76.2 % (52.8–91.8 %)] developed stage 1 AKI, 3 [14.3 % (3.0–36.3 %)] developed stage 2 AKI, 2 [9.5 % (1.2–30.4 %)] developed stage 3 AKI and 2 [9.5 % (1.2–30.4 %)] received renal replacement therapy.
Fig. 1.
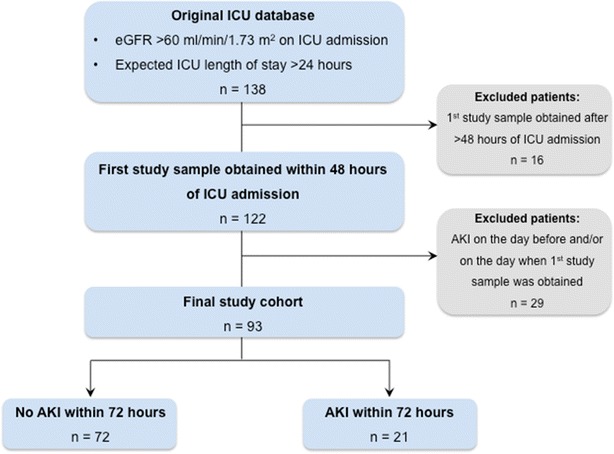
Selection of study patients
Compared to non-AKI patients, AKI patients were older and had greater illness severity on presentation and more comorbidities (Table 1) but did not have significantly worse renal function at baseline. By 30 days after ICU admission, 4 (19 %) of 21 patients developing AKI had died compared to 8 (11 %) of the 72 non-AKI patients (P = 0.46).
Table 1.
Patient characteristics and outcomes
| Variable | No AKI (n = 72) |
AKI (n = 21) |
P value |
|---|---|---|---|
| Age (years) | 50 (28, 65) | 66 (57, 71) | 0.002 |
| Female gender | 22 (31 %) | 5 (24 %) | 0.79 |
| APACHE II score | 15 (11, 19) | 19 (14, 24) | 0.01 |
| Body weight (kg) | 78 (70, 90) | 88 (79, 93) | 0.06 |
| Baseline creatinine (µmol/l) | 82 (69, 91) | 83 (71, 88) | 0.69 |
| True baseline creatinine available | 52 (72 %) | 12 (57 %) | 0.28 |
| Time admission–enrolled (h) | 12 (5, 23) | 8 (3, 14) | 0.10 |
| Time from first biomarker analysis to AKI diagnosis (days) | N/A | 1.0 (0.5, 1.5) | |
| Comorbidity | |||
| Diabetes | 6 (8 %) | 5 (24 %) | 0.12 |
| Cardiovascular disease | 20 (28 %) | 11 (52 %) | 0.06 |
| COPD/asthma | 5 (7 %) | 2 (10 %) | 0.65 |
| Gastrointestinal/liver disease | 2 (3 %) | 3 (14 %) | 0.07 |
| Any malignancy | 11 (15 %) | 3 (14 %) | 1.0 |
| Admission diagnosis | |||
| Neurologic | 4 (6 %) | 1 (5 %) | 0.03 |
| Respiratory | 14 (19 %) | 6 (29 %) | |
| Cardiovascular | 2 (3 %) | 5 (24 %) | |
| Trauma | 37 (51 %) | 7 (33 %) | |
| Gastrointestinal | 3 (4 %) | 1 (5 %) | |
| Sepsis | 12 (17 %) | 1 (5 %) | |
| Outcome | |||
| ICU length of stay, days | 5 (3, 9) | 4 (3, 8) | 0.72 |
| ICU mortality | 4 (6 %) | 2 (10 %) | 0.62 |
| 30-day mortality | 8 (11 %) | 4 (19 %) | 0.46 |
Values are median (interquartile range) or n (%)
Clinical renal characteristics at study inclusion
At inclusion, plasma creatinine levels (P = 0.20) and the changes in plasma creatinine from estimated or known baseline creatinine (P = 0.07) were similar (Table 2). Nine AKI patients (42.9 %) had early oliguria at inclusion, compared to 6 (8.3 %) non-AKI patients (P = 0.001). There was no difference in the prevalence of SIRS or sepsis at inclusion between groups (Table 2).
Table 2.
Illness severity, organ function and inflammatory response at study inclusion
| No AKI (n = 72) | AKI (n = 21) | P value | |
|---|---|---|---|
| Vasopressors and inotropes | |||
| Noradrenaline dose (µg/kg/min) | 0 (0, 0.03) | 0.01 (0, 0.04) | 0.43 |
| Adrenaline [n (%)] | 0 (0) | 1 (4.8 %) | 0.23 |
| Dobutamine [n (%)] | 1 (1.4 %) | 3 (14.3 %) | 0.04 |
| Kidney function | |||
| Plasma creatinine [µmol/l] | 82 (69, 96) | 91 (74, 93) | 0.20 |
| Δ creatinine [%] | 0 (−12, 18) | 8 (1, 25) | 0.07 |
| Early oliguriaa [n (%)] | 6 (8.3 %) | 9 (42.9 %) | 0.001 |
| Furosemide dose, mg during previous 24 h | 6 (0, 23) | 0 (0, 35) | 0.93 |
| Lung function | |||
| Mechanical ventilation [n (%)] | 56 (77.8 %) | 16 (76.2 %) | 1.0 |
| FiO2 | 0.35 (0.26, 0.47) | 0.40 (0.30, 0.50) | 0.15 |
| PaO2 (kPa) | 10 (9, 11) | 10 (9, 11) | 0.95 |
| PaO2/FiO2 ratio | 29 (20, 42) | 26 (18, 35) | 0.31 |
| PaCO2 (kPa) | 4.8 (4.3, 5.4) | 4.5 (4.3, 5.0) | 0.12 |
| Metabolic | |||
| pH | 7.4 (7.3, 7.4) | 7.4 (7.3, 7.4) | 0.64 |
| Base excess (mmol/L) | 0.2 (−2.4, 2.8) | −0.3 (−4.1, 1.0) | 0.18 |
| Lactate (mmol/L) | 1.6 (1.2, 2.6) | 2.1 (1.2, 2.8) | 0.50 |
| Inflammatory response | |||
| SIRS [n (%)] | 52 (72.2 %) | 14 (66.7 %) | 0.60 |
| SIRS + suspected or confirmed infection [n (%)] | 38 (52.8 %) | 13 (61.9 %) | 0.62 |
| White cell count (×109/L) | 11 (8, 15) | 10 (8, 13) | 0.45 |
| C-reactive protein (mg/dL) | 74 (26, 181) | 45 (15, 193) | 0.56 |
| Plasma biomarkers levels on inclusion | |||
| Endostatin (ng/mL) | 31 (23, 40) | 42 (35, 54) | 0.002 |
| Cystatin C (mg/dL) | 0.75 (0.64, 1.00) | 1.10 (0.82, 1.40) | 0.02 |
| NGAL (ng/mL) | 97 (66, 149) | 133 (67, 180) | 0.29 |
Values are reported as median (interquartile range) or as n (%)
aUrine output <0.5 mL/kg/h during >2 h but <6 h
Biomarker characteristics at study inclusion
Plasma endostatin levels were significantly higher in patients who developed AKI (P = 0.002; Table 2) and remained higher during the first four study days (P = 0.01; Fig. 2). Inclusion cystatin C concentrations were also significantly greater in the AKI cohort at inclusion (P = 0.02; Table 2) and during the four study days (P = 0.002; Additional file 1: Fig. S1). However, we found no significant difference in plasma NGAL between the groups at inclusion (P = 0.29; Table 2) or over time (P = 0.06; Additional file 1: Fig. S2). Septic patients, as compared to non-septic patients, had higher plasma NGAL at inclusion (P < 0.001), whereas endostatin (P = 0.09) and cystatin C (P = 0.08) levels were similar in patients with and without sepsis (Fig. 3). We found a significant correlation between age and endostatin (Spearman’s rho 0.28, P = 0.006) and between age and cystatin C (Spearman’s rho 0.50, P < 0.001) but not between age and NGAL (Spearman’s rho 0.13, P = 0.17).
Fig. 2.
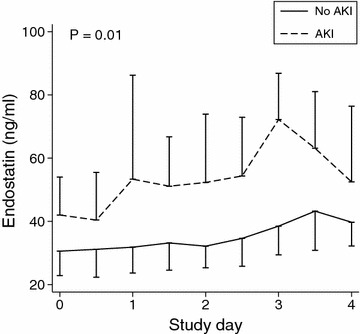
Plasma endostatin levels during the first five study days in AKI and non-AKI patients. Values are median and interquartile range. P value is for the repeated-measure ANOVA between groups
Fig. 3.
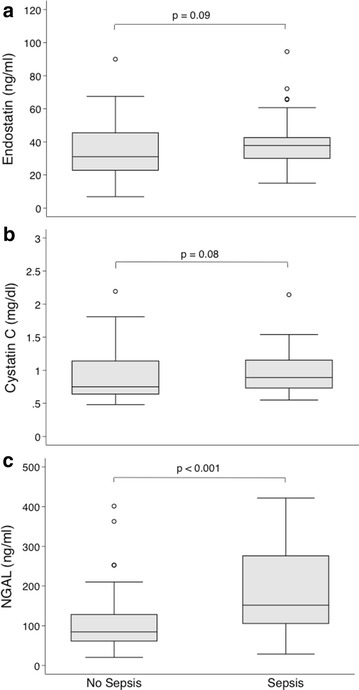
Plasma endostatin (a), cystatin C (b) and NGAL (c) at study inclusion in patients with and without sepsis
Predicting the development of AKI
On univariate analysis, age, APACHE II score and early oliguria were associated with the development of AKI within 72 h and were included in a three-parameter clinical risk prediction model (Additional file 1: Table S1). This model predicted AKI with an AUC of 0.759 (95 % CI 0.646–0.872; Table 3).
Table 3.
Values for prediction of AKI within 72 h
| Statistic | Estimate (95 % CI) | P valuea |
|---|---|---|
| AUC endostatin alone | 0.726 (0.603 to 0.848) | <0.001 |
| AUC cystatin C alone | 0.674 (0.535 to 0.812) | 0.01 |
| AUC NGAL alone | 0.577 (0.430 to 0.723) | 0.22 |
| AUC early oliguria | 0.618 (0.509 to 0.727) | 0.04 |
| AUC clinical model | 0.759 (0.646 to 0.872) | <0.001 |
| AUC clinical model + endostatin | 0.839 (0.752 to 0.925) | <0.001 |
| AUC clinical model + cystatin C | 0.776 (0.662 to 0.890) | <0.001 |
| AUC clinical model + NGAL | 0.766 (0.657 to 0.874) | <0.001 |
| NRI (endostatin) | 0.268 (0.010 to 0.526) | 0.04 |
| IDI (endostatin) | 0.073 (0.038 to 0.142) | 0.04 |
| NRI (cystatin C) | 0.06 (−0.16 to 0.28) | 0.63 |
| IDI (cystatin C) | 0.012 (−0.013 to 0.037) | 0.34 |
| NRI (NGAL) | 0.035 (−0.15 to 0.22) | 0.71 |
| IDI (NGAL) | 0.0061 (−0.013 to 0.025) | 0.53 |
AUC area under the receiver operating characteristics curve, NRI net reclassification improvement, IDI integrated discrimination improvement
a P values for AUC assess the difference from 0.5
Endostatin alone predicted AKI with a similar AUC of 0.726 (0.603–0.848). Its optimal cutoff value of 37 ng/mL predicted AKI with a sensitivity of 71 % and a specificity of 65 % (Additional file 1: Table S2).
The addition of endostatin significantly improved the clinical three-parameter regression model, as confirmed by the likelihood-ratio test (P = 0.005; Additional file 1: Table S3). This endostatin-enhanced model was significantly better at predicting subsequent AKI than the clinical model alone, with an AUC of 0.839 (95 % CI 0.752–0.925; Table 3; Fig. 4), supported by both the net reclassification (P = 0.04; Table 3) and the integrated discrimination improvement indices (P = 0.04; Table 3). Cystatin C and NGAL were both poorly individually predictive of subsequent AKI (Table 3). Furthermore, the addition of either cystatin C or NGAL to the clinical predictive model failed to achieve any significant improvement in risk prediction (Table 3; Fig. 4; Additional file 1: Tables S4 and S5).
Fig. 4.
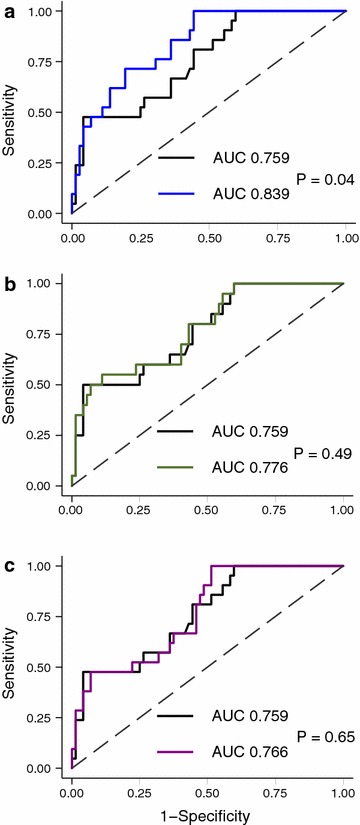
Receiver operating characteristics curves for prediction of AKI within 72 h using a clinical model (black line) and a clinical model together with endostatin (blue line, a), cystatin C (green line, b) or NGAL (purple line, c) at study inclusion. AUC area under the receiver operating characteristics curve
Sensitivity analysis
We repeated the risk prediction analyses after removing age from the clinical model since age is a component of the APACHE II score. Both APACHE II and early oliguria were associated with AKI in this bivariate model. Adding endostatin to the bivariate model improved AKI prediction on the likelihood-ratio test (P < 0.001), the net reclassification improvement test (P = 0.02) and the integrated discrimination improvement test (P = 0.01). In contrast, adding cystatin C or NGAL did not improve risk prediction based on the bivariate model in any of the analyses (Additional file 1: Tables S6–S9 and Additional file 1: Fig. S3–S5).
Discussion
Key findings
We conducted a prospective, exploratory study assessing the relationship between a novel potential AKI biomarker (plasma endostatin) and AKI in critically ill patients. In this cohort, we found that plasma endostatin levels were higher in patients who developed AKI than in those who did not and that elevated plasma endostatin levels were independently associated with an increased risk of AKI. Moreover, the addition of plasma endostatin to a clinical prediction model significantly improved risk prediction performance for subsequent AKI. Finally, plasma endostatin concentration achieved greater utility in the prediction of subsequent AKI than either plasma NGAL or plasma cystatin C concentration.
Relationship to previous studies
To our knowledge, this is the first investigation of plasma endostatin in critically ill humans as a predictor of AKI. However, in a cohort of elderly patients, serum endostatin levels independently predicted the future development of CKD [5]. In this population, endostatin also correlated strongly with eGFR. Moreover, up to fivefold higher endostatin levels have been observed in patients with CKD or ESRD [13, 14].
In the present study, inclusion endostatin levels were low and comparable to those observed in community-based cohorts (the PIVUS and ULSAM cohorts) [5]. Compared to these cohorts, our patients were markedly younger (median age 50–66 years versus a mean age >75 years in PIVUS and ULSAM) and did not have AKI or CKD on study inception. However, the majority of our critically ill patients had SIRS or sepsis at inclusion, which could be expected to affect endostatin levels in the same way it affects other biomarkers of AKI [15, 16]. For example, NGAL, which is released both by kidney epithelial cells and by activated neutrophils during systemic inflammation [17, 18], was elevated in our septic subgroup.
Similarly, collagen turnover is enhanced in sepsis, and this is reflected in elevated levels of circulating collagen degradation products [19]. Theoretically, matrix breakdown in non-renal tissues during sepsis could contribute to circulating endostatin levels. However, when comparing septic and non-septic patients we observed similar levels in both groups, suggesting minimal release of non-renal endostatin into the circulation in our patients.
Kidney extracellular matrix remodeling and angiogenesis appear to play an important role in the initiation, maintenance and progression of AKI [20, 21]. In experimental animal models of AKI, endothelial activation and damage lead to both disruption of renal endothelial cell integrity and endothelial matrix breakdown [3, 22]. This appears to be mediated via proteinase activation and cleavage of matrix collagens and leads to vascular leak, renal parenchymal edema and organ dysfunction [23]. Similar mechanisms have been demonstrated to contribute to the development of CKD [21]. Our finding that elevated circulating endostatin levels preceded AKI, as evident by a subsequent rise in creatinine, and remained elevated until such AKI was established supports the pathophysiological role of matrix breakdown and endothelial injury/dysfunction in AKI development. Additionally, we observed a slight increase in median endostatin levels in non-AKI patients reaching the optimal AKI predictive cutoff level (37 ng/mL) on day 3 (Fig. 2). Whether this delayed endostatin rise was triggered by some degree of “subclinical” AKI, by non-renal matrix breakdown or by both remains, however, uncertain.
Finally, as endostatin is a middle-sized molecule (20 kDa), which is freely filtered by the glomeruli, its plasma concentration may reflect glomerular filtration rate (GFR). In keeping with this hypothesis, an inverse correlation between plasma endostatin and GFR has indeed been established [5, 13]. Hence, impaired clearance of middle-sized molecules but not yet affecting smaller molecules such as creatinine (0.11 kDa) could potentially explain the observed early rise in endostatin. However, cystatin C (13 kDa) and NGAL (25 kDa) are both middle-sized molecules, but their patterns of release in our cohort differed from that of endostatin, and neither improved predictive modeling. In their aggregate, these observations suggest that increased production/release rather than impaired clearance of endostatin likely caused its early rise in plasma.
Implications of study findings
Our findings may have implications for patient management. They support the emerging view that microcirculatory dysfunction is a key step in the establishment of AKI [24]. Endostatin may, in conjunction with other biomarkers of renal stress or injury, be a useful biomarker of the natural history of AKI and the transition from AKI to CKD. Within the limitations of this small cohort, our data suggest that elevated plasma endostatin may be an earlier and more specific signal of kidney stress or injury, especially in conjunction with clinical risk factors, than two previously proposed AKI markers: NGAL and cystatin C. Whether this signal is a useful tool to detect patients “at risk” of AKI, however, needs to be assessed in combination with validated clinical risk models in independent cohorts of critically ill patients. In particular, biomarker performance may be different in patients with sepsis. In fact, in a recent cohort of 112 septic patients, the ability to predict AKI within 24 h was fair using plasma cystatin C [AUC 0.737 (95 % CI 0.633–0.841)] and good using plasma NGAL [AUC 0.830 (95 % CI 0.741–0.919)] [25].
In contrast, cystatin C and NGAL demonstrated poor predictive performance in our study. This may be explained by the fact that we included both septic and non-septic patients and that we measured the occurrence of AKI up to 72 h after biomarker measurement. Based on these conflicting findings, future studies need to compare the AKI predictive performances of endostatin, cystatin C and NGAL in cohorts of septic patients and within different time-points of AKI diagnosis.
Promising therapies against acute renal microcirculatory changes have been demonstrated in animal AKI models but failed to improve kidney function or other patient-centered outcomes in clinical studies [24, 26]. Future studies should explore the role of plasma endostatin as a trigger of therapies targeting the renal microcirculation. Finally, endostatin may be particularly attractive in conjunction with novel imaging techniques of the renal microcirculation such as contrast-enhanced ultrasound [27, 28] and blood-oxygen level-dependent contrast imaging [29] to monitor therapeutic effects.
Strengths and limitations
Our study has several strengths. It includes a well-defined cohort of critically ill patients without renal disease on ICU admission, yet at significant risk of developing AKI. Secondly, detailed data collection allowed us to develop a risk model using meaningful clinical and physiological parameters and to compare endostatin to established renal biomarkers like cystatin C and NGAL. Thirdly, data were prospectively collected and are therefore unlikely to be biased. Finally, despite a limited number of AKI cases, the added value of endostatin in risk prediction was consistently found in multiple analyses, which increases the robustness of our findings.
Our study has, however, limitations. It was a single-center study, which limits the generalizability of the study findings. It was, however, performed in a tertiary hospital suggesting some degree of external validity to similar hospitals in the developed world. CKD patients were excluded and the value of endostatin to predict acute-on-chronic kidney injury cannot be extrapolated from our findings. Illness severity (APACHE II) was generally low in our cohort. This was, however, expected since we excluded patients admitted with AKI, a population well known to be more severely ill. Furthermore, given our small study sample the robustness of our clinical risk model may be limited. However, our model includes clinically relevant and established risk factors (early oliguria, illness severity and age) of AKI. Additionally, we developed and validated the clinical risk model on the same cohort. This approach may inflate its predictive value. However, such potential inflation applied to all other biomarkers as well, and yet the addition of endostatin proved superior and significantly improved risk prediction. Finally, plasma samples were collected between 2007 and 2010 and analyzed for endostatin and NGAL during 2013. Such an extended storage period may impact biomarker concentrations. However, samples were stored at −80 °C, which preserve NGAL concentrations during long-term storage [30]. It is unlikely that identical storage conditions would affect endostatin levels differently.
Conclusions
Our results suggest that plasma endostatin may be a fair predictor of AKI developing within 72 h of ICU admission in patients without preexisting renal disease. Combining endostatin with clinical variables may further enhance AKI risk prediction in such patients. In addition, within the limitations of an exploratory investigation, our findings suggest that endostatin may outperform two previously proposed AKI markers, cystatin C and NGAL. Our results provide the basis for further evaluation of endostatin as a biomarker of early AKI in independent cohorts of critically ill patients.
Authors’ contributions
JM and CRM designed and coordinated the study. AL conducted biomarker analyses. JM, NJ and MB collected study data. JM, NJG and RB analyzed and interpreted the data. JM performed the statistical analysis. All authors participated in writing the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interest.
Abbreviations
- AKI
acute kidney injury
- APACHE
acute physiology and chronic health evaluation
- AUC
area under the receiver operating characteristics curve
- CKD
chronic kidney disease
- ELISA
enzyme-linked immunosorbent assay
- ESRD
end-stage renal disease
- GFR
glomerular filtration rate
- ICU
intensive care unit
- IDI
integrated discrimination improvement
- KDIGO
Kidney Disease: Improving Global Outcomes
- MDRD
modification of diet in renal disease
- NGAL
neutrophil gelatinase-associated lipocalin
- NRI
net reclassification improvement
- SIRS
systemic inflammatory response syndrome
Additional file
10.1186/s13613-016-0108-x Tables S1–S5. Clinical model and biomarker performances for AKI prediction. Figures S1, S2. Cystain C and NGAL kinetics. Tables S6–S9 and Figures S3–S5. Sensitivity analyses excluding age from clinical model.
Contributor Information
Johan Mårtensson, Phone: +61 3 94965992, Email: johan.martensson@austin.org.au.
Niklas Jonsson, Email: niklas.jonsson@karolinska.se.
Neil J. Glassford, Email: neil.glassford@austin.org.au
Max Bell, Email: max.bell@karolinska.se.
Claes-Roland Martling, Email: claes-roland.martling@karolinska.se.
Rinaldo Bellomo, Email: rinaldo.bellomo@austin.org.au.
Anders Larsson, Email: anders.larsson@medsci.uu.se.
References
- 1.Nisula S, Kaukonen KM, Vaara ST, Korhonen AM, Poukkanen M, Karlsson S, et al. Incidence, risk factors and 90-day mortality of patients with acute kidney injury in Finnish intensive care units: the FINNAKI study. Intensive Care Med. 2013;39(3):420–428. doi: 10.1007/s00134-012-2796-5. [DOI] [PubMed] [Google Scholar]
- 2.Marneros AG, Olsen BR. Physiological role of collagen XVIII and endostatin. FASEB J. 2005;19(7):716–728. doi: 10.1096/fj.04-2134rev. [DOI] [PubMed] [Google Scholar]
- 3.Bellini MH, Coutinho EL, Filgueiras TC, Maciel TT, Schor N. Endostatin expression in the murine model of ischaemia/reperfusion-induced acute renal failure. Nephrology. 2007;12(5):459–465. doi: 10.1111/j.1440-1797.2007.00850.x. [DOI] [PubMed] [Google Scholar]
- 4.Bellini MH, Malpighi TF, Calvo FB, Miranda AR, Spencer PJ, Cichy MC, et al. Immobilized kidney 28-kDa endostatin-related (KES28 kDa) fragment promotes endothelial cell survival. Am J Nephrol. 2010;31(3):255–261. doi: 10.1159/000278756. [DOI] [PubMed] [Google Scholar]
- 5.Ruge T, Carlsson AC, Larsson TE, Carrero JJ, Larsson A, Lind L, et al. Endostatin level is associated with kidney injury in the elderly: findings from two community-based cohorts. Am J Nephrol. 2014;40(5):417–424. doi: 10.1159/000369076. [DOI] [PubMed] [Google Scholar]
- 6.Gouya G, Siller-Matula JM, Fritzer-Szekeres M, Neuhold S, Storka A, Neuhofer LM, et al. Association of endostatin with mortality in patients with chronic heart failure. Eur J Clin Invest. 2014;44(2):125–135. doi: 10.1111/eci.12197. [DOI] [PubMed] [Google Scholar]
- 7.Damico R, Kolb TM, Valera L, Wang L, Housten T, Tedford RJ, et al. Serum endostatin is a genetically determined predictor of survival in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;191(2):208–218. doi: 10.1164/rccm.201409-1742OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arnlov J, Ruge T, Ingelsson E, Larsson A, Sundstrom J, Lind L. Serum endostatin and risk of mortality in the elderly: findings from 2 community-based cohorts. Arterioscler Thromb Vac Biol. 2013;33(11):2689–2695. doi: 10.1161/ATVBAHA.113.301704. [DOI] [PubMed] [Google Scholar]
- 9.Kellum JA, Lameire N. Diagnosis, evaluation, and management of acute kidney injury: a KDIGO summary (Part 1) Crit Care. 2013;17(1):204. doi: 10.1186/cc11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344(10):699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 11.DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44(3):837–845. doi: 10.2307/2531595. [DOI] [PubMed] [Google Scholar]
- 12.Glassford NJ, Schneider AG, Xu S, Eastwood GM, Young H, Peck L, et al. The nature and discriminatory value of urinary neutrophil gelatinase-associated lipocalin in critically ill patients at risk of acute kidney injury. Intensive Care Med. 2013;39(10):1714–1724. doi: 10.1007/s00134-013-3040-7. [DOI] [PubMed] [Google Scholar]
- 13.Chen J, Hamm LL, Kleinpeter MA, Husserl F, Khan IE, Chen CS, et al. Elevated plasma levels of endostatin are associated with chronic kidney disease. Am J Nephrol. 2012;35(4):335–340. doi: 10.1159/000336109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carlsson AC, Carrero JJ, Stenvinkel P, Bottai M, Barany P, Larsson A, et al. Endostatin, Cathepsin S, and Cathepsin L, and their association with inflammatory markers and mortality in patients undergoing hemodialysis. Blood Purif. 2015;39(4):259–265. doi: 10.1159/000381664. [DOI] [PubMed] [Google Scholar]
- 15.Martensson J, Bellomo R. The Rise and Fall of NGAL in Acute Kidney Injury. Blood Purif. 2014;37(4):304–310. doi: 10.1159/000364937. [DOI] [PubMed] [Google Scholar]
- 16.Bell M, Larsson A, Venge P, Bellomo R, Martensson J. Assessment of cell-cycle arrest biomarkers to predict early and delayed acute kidney injury. Dis Markers. 2015;2015:158658. doi: 10.1155/2015/158658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martensson J, Bell M, Oldner A, Xu S, Venge P, Martling CR. Neutrophil gelatinase-associated lipocalin in adult septic patients with and without acute kidney injury. Intensive Care Med. 2010;36(8):1333–1340. doi: 10.1007/s00134-010-1887-4. [DOI] [PubMed] [Google Scholar]
- 18.Martensson J, Bell M, Xu S, Bottai M, Ravn B, Venge P, et al. Association of plasma neutrophil gelatinase-associated lipocalin (NGAL) with sepsis and acute kidney dysfunction. Biomarkers. 2013;18(4):349–356. doi: 10.3109/1354750X.2013.787460. [DOI] [PubMed] [Google Scholar]
- 19.Gaddnas F, Koskela M, Koivukangas V, Risteli J, Oikarinen A, Laurila J, et al. Markers of collagen synthesis and degradation are increased in serum in severe sepsis: a longitudinal study of 44 patients. Crit Care. 2009;13(2):R53. doi: 10.1186/cc7780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ergin B, Kapucu A, Demirci-Tansel C, Ince C. The renal microcirculation in sepsis. Nephrol Dial Transplant. 2015;30(2):169–177. doi: 10.1093/ndt/gfu105. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka T, Nangaku M. Angiogenesis and hypoxia in the kidney. Nat Rev Nephrol. 2013;9(4):211–222. doi: 10.1038/nrneph.2013.35. [DOI] [PubMed] [Google Scholar]
- 22.Basile DP. The endothelial cell in ischemic acute kidney injury: implications for acute and chronic function. Kidney Int. 2007;72(2):151–156. doi: 10.1038/sj.ki.5002312. [DOI] [PubMed] [Google Scholar]
- 23.Horbelt M, Lee SY, Mang HE, Knipe NL, Sado Y, Kribben A, et al. Acute and chronic microvascular alterations in a mouse model of ischemic acute kidney injury. Am J Physiol Renal Physiol. 2007;293(3):F688–F695. doi: 10.1152/ajprenal.00452.2006. [DOI] [PubMed] [Google Scholar]
- 24.Zafrani L, Ince C. Microcirculation in acute and chronic kidney diseases. Am J Kidney Dis. 2015;66(6):1083–1094. doi: 10.1053/j.ajkd.2015.06.019. [DOI] [PubMed] [Google Scholar]
- 25.Dai X, Zeng Z, Fu C, Zhang S, Cai Y, Chen Z. Diagnostic value of neutrophil gelatinase-associated lipocalin, cystatin C, and soluble triggering receptor expressed on myeloid cells-1 in critically ill patients with sepsis-associated acute kidney injury. Crit Care. 2015;19:223. doi: 10.1186/s13054-015-0941-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duke GJ. Renal protective agents: a review. Crit Care Resusc. 1999;1(3):265–275. [PubMed] [Google Scholar]
- 27.Schneider A, Johnson L, Goodwin M, Schelleman A, Bellomo R. Bench-to-bedside review: contrast enhanced ultrasonography—a promising technique to assess renal perfusion in the ICU. Crit Care. 2011;15(3):157. doi: 10.1186/cc10058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schneider AG, Goodwin MD, Schelleman A, Bailey M, Johnson L, Bellomo R. Contrast-enhanced ultrasound to evaluate changes in renal cortical perfusion around cardiac surgery: a pilot study. Crit Care. 2013;17(4):R138. doi: 10.1186/cc12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inoue T, Kozawa E, Okada H, Inukai K, Watanabe S, Kikuta T, et al. Noninvasive evaluation of kidney hypoxia and fibrosis using magnetic resonance imaging. J Am Soc Nephrol. 2011;22(8):1429–1434. doi: 10.1681/ASN.2010111143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haase-Fielitz A, Haase M, Bellomo R. Instability of urinary NGAL during long-term storage. Am J Kidney Dis. 2009;53(3):564–565. doi: 10.1053/j.ajkd.2009.01.009. [DOI] [PubMed] [Google Scholar]