

Abstract
Study Objectives:
Patients with Huntington's disease (HD) show a high prevalence of sleep disorders that typically occur prior to the onset of motoric symptoms and neurodegeneration. Our understanding of the pathophysiological alterations in premanifest HD is limited, hindering the ability to measure disease modification in response to treatment. We used a full-length knock-in HD model to determine early changes in the electroencephalogram (EEG) and sleep that may predict the onset and progression of the disease.
Methods:
A 10-month longitudinal study was designed to determine the effect of the HD mutation on the EEG and sleep/wake changes in heterozygous (HET) and homozygous (HOM) zQ175 mice and wild-type (WT) littermates from 8 to 48 w of age. Mice were instrumented with tethered headmounts to record EEG/electromyography signals. Telemeters were implanted to continuously measure locomotor activity (LMA) and body temperature (Tb). Sleep deprivation (SDep) was performed at 8, 12, 16, 24, 32, and 48 w of age.
Results:
The HD mutation disrupted the EEG field potential from 8–12 w in an age- and mutant huntington dose-dependent manner, prior to changes in sleep/wake states, LMA, and Tb. Prominent effects of the HD mutation on the EEG included a progressive reduction in low frequency power, a slowing of rapid eye movement peak theta frequency, and the emergence of state-dependent beta/gamma oscillations. There was no effect of genotype on the relative increase in nonrapid eye movement delta power or sleep time in response to SDep.
Conclusions:
The expression of the Huntington's disease (HD) mutation results in complex EEG alterations that occur prior to deficits in behavioral measures and are one of the earliest phenotypes uncovered in this mouse model. Despite these EEG changes, homeostatic responses to sleep loss were preserved in HET and HOM zQ175 mice. Greater insight into the localization and response of these EEG alterations to novel therapies may enable early intervention and improve outcomes for patients with HD.
Citation:
Fisher SP, Schwartz MD, Wurts-Black S, Thomas AM, Chen TM, Miller MA, Palmerston JB, Kilduff TS, Morairty SR. Quantitative electroencephalographic analysis provides an early-stage indicator of disease onset and progression in the zQ175 knock-in mouse model of Huntington's disease. SLEEP 2016;39(2):379–391.
Keywords: biomarker, EEG, Huntington's disease, sleep, zQ175 mouse
Significance.
The presence of mutant huntingtin underlies the development of HD, a late-onset neurodegenerative disorder. However, reliable biomarkers that indicate disease onset and progression in either HD patients or preclinical models are currently lacking. Here, we employ a longitudinal analysis of EEG and sleep/wake behavior in knock-in mice heterozygous and homozygous for the HD mutation. Significantly, these mice show early-stage and progressive alterations in qEEG measurements that precede changes in sleep/wake states, LMA, and Tb. These results support our initial hypothesis that EEG parameters can serve as highly sensitive biomarkers indicating the presence and progression of HD long before the onset of major disease symptoms. Further studies are needed to determine if these results are translational to HD patients.
INTRODUCTION
Huntington's disease (HD) is a dominantly inherited neuro-degenerative disorder with only partial symptomatic relief available for the motor, cognitive, and psychiatric deficits that characterize the disease. One of the main challenges facing late-onset neurodegenerative conditions is determination of the initiation and progression of the disease prior to the onset of major symptoms. Due to the monogenic nature and full penetrance of the abnormal CAG expansion in the huntingtin (HTT) gene, individuals in whom HD will inevitably develop can be identified prior to symptom onset, thereby enabling the evaluation of early-stage biomarkers of the disease.1,2 Recent collaborative multidisciplinary investigations, including TRACK-HD, IMAGE-HD, and PREDICT-HD, have employed longitudinal assessments in premanifest HD subjects and identified alterations in a range of imaging, motor, cognitive, functional, and psychiatric parameters.3–6 These findings highlight the consensus view that HD gene-positive individuals exhibit neurobiological changes years, if not decades, before the onset of major symptoms and that early intervention is necessary to delay disease progression before the onset of neurodegeneration and loss of function.7,8 Promising disease-modifying strategies on the horizon include silencing of the mutant HTT (mHTT) gene,9–11 histone deacetylase inhibition,12,13 and stem cell therapy.14 However, validation of the efficacy of novel therapies remains difficult without sensitive biomarkers of pre-manifest HD that can ultimately be used as outcome measures in preclinical and clinical trials.
Despite its translational potential, relatively little attention has focused on the electroencephalogram (EEG) as a source of early-stage HD biomarkers. The EEG measures the summation of excitatory and inhibitory currents generated by cortical and subcortical neurons. It provides high temporal resolution and, when analyzed in terms of its frequency components by quantitative EEG (qEEG) analysis, results in objective measures of neuronal functioning.15 Compared to other electrophysiological techniques, EEG recordings are easily obtainable in both patients and animal models. Sleep and circadian abnormalities that are common early features in patients with HD include insomnia, sleep fragmentation, excessive daytime sleepiness, delayed sleep phase, and rapid eye movement (REM) sleep behavior disorders.16–19 EEG abnormalities are also prevalent with a reduction in alpha activity and a slowing of EEG frequencies.20–22 Few studies have assessed EEG changes in pre-manifest subjects with sufficient power across different sleep/ wake states.23–25 In the most recent of these studies, a loss of the anterior-posterior gradient in relative alpha power (8–12 Hz) was identified during wakefulness that distinguished pre-manifest HD subjects from healthy age-matched controls.25 Importantly, the extent of this gradient loss increased in patients exhibiting more advanced disease severity, indicating that qEEG parameters may serve as valuable biomarkers of HD progression.
Expression of the HD mutation in transgenic animals replicates many of the phenotypic features of the human disease26 and results in valuable models to understand the influence of mHtt on behavioral, molecular, and cellular abnormalities of the disorder.27 Altered electrophysiology is a salient feature of HD models, including synaptic dysfunction and deficits in corticostriatal processing preferentially in striatal medium-sized spiny and cortical pyramidal neurons.28–33 Our previous work in the R6/2 HD mouse found a gradual disruption of sleep/ wake patterns with extensive abnormalities in the EEG.34 These abnormalities included large increases in beta/gamma EEG activity evident in all sleep/wake states from our earliest recording at 9 w of age, indicating a highly symptomatic electrophysiological phenotype that precluded evaluation of the very early stages of the disease. Analogous findings were replicated independently in both R6/235 and R6/1 transgenic mouse strains.36 Increasingly, attention is focusing on HD knock-in (KI) models with chimeric human/mouse expanded CAG constructs inserted into the native Htt gene homologue37 that exhibit slower disease progression and relatively normal lifespans.38 Heterozygous (HET) KI models are thought to have greater translational relevance because they more precisely replicate the genetic context of patients with HD, although such strains have not been extensively studied due to their subtle phenotypes.39 This problem is circumvented in the novel zQ175 KI HD model, derived from a spontaneous CAG expansion in the CAG140 line27 that exhibits robust behavioral, histopathological and molecular alterations40,41 and age- and gene dosage-dependent circadian disruption.42 Based on motor abnormalities, homozygous (HOM) and HET zQ175 mice are considered to be in the manifest stage of the disease from approximately 8 and 20 w, respectively.41 It will be important to determine the effect of mHtt expression on the EEG to determine whether the abnormalities previously identified in the R6/2 and R6/1 mice are also evident in KI HD models. Consequently, we characterized longitudinal changes in the EEG, sleep/wake behavior, locomotor activity (LMA), and core body temperature (Tb) in HET, HOM, and wild-type (WT) zQ175 mice. In an attempt to identify other biomarkers of disease onset or progression, we also assessed the integrity of the sleep homeostatic system in zQ175 mice by performing short-term sleep deprivation throughout the study. We found gene dosage-and age-dependent changes in qEEG parameters that occurred early in the disease, thereby identifying these EEG alterations as candidate biomarkers for HD progression.
METHODS
Animals
A cohort of 5-w-old male HET (mean CAG repeat length, 186 ± 1.54 nucleotides), HOM (mean CAG repeat length, 188 ± 1.61), and wild-type (WT) zQ175 KI mice on a C57BL/6J background were obtained from the CHDI colony (Jackson Laboratories, B6J.129S1-Htttm1Mfc/190ChdiJ: Stock Number: 027410, Bar Harbor, ME). HOM, HET, and WT mice were generated by crossing HET zQ175 mice on a congenic C57BL/6J background. The zQ175 line was originally derived from a spontaneous CAG repeat expansion in the CAG140 KI line.41 Genotyping and CAG repeat sizing were conducted on tail biopsies by Laragen (Los Angeles, CA) before the start and after completion of the study. For core Tb and LMA data analysis, group sizes were WT = 7–10, HET = 12, and HOM = 7–9. Group sizes were variable for WT and HOM groups due to telemetry transmitter failures at 32 and 48 w. Group sizes for EEG data analysis were WT = 8, HET = 9, and HOM = 7. All mice were housed in light-tight, ventilated, sound-isolated chambers under a standard 12:12 h light/dark cycle (lights on at 09:00, Zeitgeber time [ZT]0) with food and water available ad libitum. Room temperature and relative humidity were maintained at 24 ± 2°C and 50 ± 20% respectively. Animals were inspected daily in accordance with Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) and SRI International guidelines. All experimental procedures involving animals were approved by SRI's Institutional Animal Care and Use Committee and were in accordance with US National Institutes of Health guidelines and EU Directive 2010/63/EU.
Experimental Protocol
WT, HET, and HOM zQ175 mice were evaluated longitudinally for changes in the EEG, sleep/wake parameters, Tb, LMA, and body weight from 8 to 48 w of age. Whereas Tb and LMA were recorded continuously across the study, EEG/EMG recording sessions occurred at 8, 12, 16, 24, 32, and 48 w of age (Figure 1A). Each experimental period/week consisted of a 48-h baseline recording (days 1–2) followed by three sleep deprivations (SDep) of increasing duration (1, 3, 6 h) with a minimum of 42 h recovery between each SDep. Body weight was determined approximately every 2 w (Figure 1B).
Figure 1.
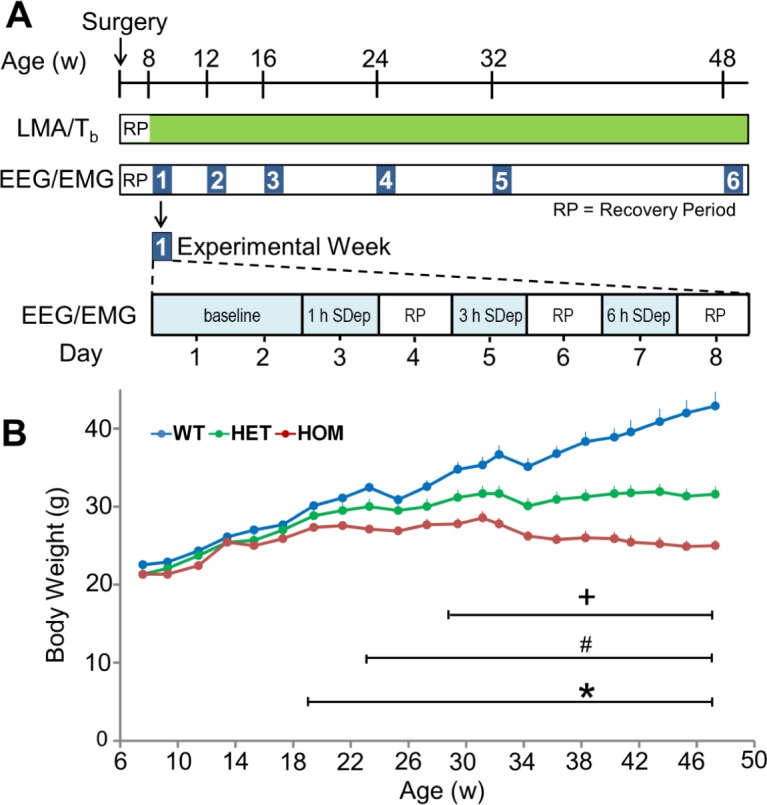
Overview of the longitudinal experimental protocol and body weight assessments determined from zQ175 HET, HOM, and WT mice across the study. (A) Mice were surgically implanted at 5 w of age with telemetry transmitters and EEG/ EMG head mounts and allowed to recover for 2 w. Locomotor activity (LMA) and core body temperature (Tb) were sampled at 1-min intervals continuously throughout the study (green bar). EEG/EMG data were collected continuously during each experimental week (blue numbered squares, expanded on bottom timeline) at 8, 12, 16, 24, 32, and 48 w of age. The experimental week consisted of a 48-h baseline recording followed by sleep deprivations (SDep) of increasing duration (1, 3, and 6 h) interspersed with recovery periods (RP) of at least 42 h. (B) Body weight was determined for WT, HET, and HOM male zQ175 mice from 8–48 w of age. *Significant differences between HOM and WT mice (P < 0.05). +Significant differences between HET and WT mice (P < 0.05). #Significant differences between HET and HOM mice. EEG, electroencephalogram; EMG, electromyography; HET, heterozygous; HOM, homozygous; WT, wild-type.
Surgical Procedures
All surgeries were performed aseptically under isoflurane anesthesia. Mice (age 5–6 w, 21.4 ± 0.3 g) were implanted with Mini-Mitter telemetry devices (G2-E-Mitter, Starr Lite Sciences) to record Tb and LMA. During the same surgical procedure, mice were implanted with custom prefabricated headmounts (8201-C, Pinnacle Technologies) to enable tethered EEG and EMG recordings as described previously.34 The front edge of the headmount was placed 3.0 mm anterior to bregma aligned centrally along the sagittal suture. In this configuration, EEG electrodes were positioned 1.5 mm lateral to the sagittal suture, 2.0 mm anterior of bregma and −4.0 mm posterior to bregma. EMG electrodes were inserted and sutured into the trapezius muscle. Mice were administered postoperative hydration (0.9% NaCl), analgesics (buprenorphine, 0.05–0.1 mg/kg and ketoprofen, 2–5 mg/kg), thermal support, and soft chow. During the 14 d postsurgery recovery and for the remainder of the study, mice were individually housed in cages (280 × 175 × 130 mm) with raised vertical sides to prevent escape during tethered recordings.
Activity and Body Temperature Recordings and Data Analyses
LMA and Tb were collected continuously from ∼8 to 49 w of age using inductive telemetry (Mini-Mitter, Starr Life Sciences, Oakmont, PA) as described previously.34 Data were sampled every minute using Vitalview software (version 5.0). Hourly average values were then calculated for all individual animals and experimental groups. Activity data were analyzed as actograms using Actiview software (version 1.3, Starr Life Sciences, Oakmont, PA) in which raw LMA counts were plotted as vertical bars in a double-plot format. Tb data were similarly analyzed in ClockLab (Actimetrics, Wilmette, IL) on a percentile distribution basis. To determine the amplitude of circadian periodicity of LMA and Tb rhythms, chi-square periodograms (period lengths: 10–36 h) were calculated using ClockLab on data collected over the 14 d interval preceding the EEG/EMG recordings at 10–12, 14–16, 22–24, 30–32, and 46–48 w of age. The first time point (7–9 w) was excluded from the analysis because it overlapped with the postsurgical recovery period.
EEG/EMG Data Collection
Electrophysiological signals (EEG/EMG) were collected using iox2 software (Version 2.8.0.11, EMKA Technologies, Falls Church, VA) using tethered recordings.34 Mice were connected to swivel commutators (8204, Pinnacle Technologies, Lawrence, KS) mounted above the cage, allowing them to move freely at all times and were habituated to the recording cable for 4–5 d before data collection. Commutators were connected to an amplifier system (M15 biopolar amplifier, Grass Instruments, Warwick, RI). Due to limitations on the number of channels, we recorded a single EEG derivative (frontal - parietal electrode configuration) and EMG signal from each mouse. EEG and EMG signals were amplified 10,000×, routed to iox2 interface boxes (EMKA Technologies, Falls Church, VA) and sampled at 500 Hz. The EEG was low-pass filtered at 300 Hz and a high-pass filtered at 0.3 Hz. EMG was low-pass filtered at 6 KHz and high-pass filtered at 3 Hz.
Signal Analysis of EEG/EMG Data
The electrophysiological data presented here are derived from 3,456 h of recordings from 8 WT, 9 HET, and 7 HOM zQ175 mice during baseline conditions and 5,184 h of recordings from SDep and recovery periods. EEG/EMG data were analyzed offline using ecgAUTO (Version 3.3.0.15 EEG Module, EMKA Technologies, Falls Church, VA). 10-sec epochs were examined visually and sleep/wake states were determined by experienced scorers (blinded to genotype) as either wakefulness (Wake) rapid eye movement (REM) sleep or non-REM (NREM) sleep as defined previously.34 Data were analyzed and processed using MatLab (Mathworks, Natick, MA) scripts to calculate sleep/wake amounts and sleep consolidation measures. As indices of behavioral state consolidation, the average duration and number of bouts for each state were calculated. A bout was defined as a minimum of two consecutive 10-sec epochs of waking or NREM sleep or a single epoch of REM sleep and ended with any single state change epoch. For spectral analysis of the EEG data, a fast Fourier transform (range: 0.5–100 Hz, Hanning window function, resolution 0.1 Hz) was performed on artifact-free epochs (10 sec) for each behavioral state. Baseline spectral data for all mice are either presented as raw EEG spectra (average log values) or normalized to the age-matched WT condition (average group value). During the 6-h recovery period from SDep, normalized NREM delta power (0.5–4.5 Hz, normalized to 12 h of an undisturbed age-matched baseline recording for each individual mouse from ZT0–12) and the recovery of sleep time lost (recovery sleep time minus baseline NREM and REM sleep) were calculated.
Sleep Deprivation Procedures
To probe the integrity of the sleep homeostat, mice were sleep deprived on alternate days for 1, 3, or 6 h at 8, 12, 16, 24, 32, and 48 w of age. Animals were sleep deprived by light tapping of the cage and movement of bedding materials as detailed previously.34
Statistical Analyses
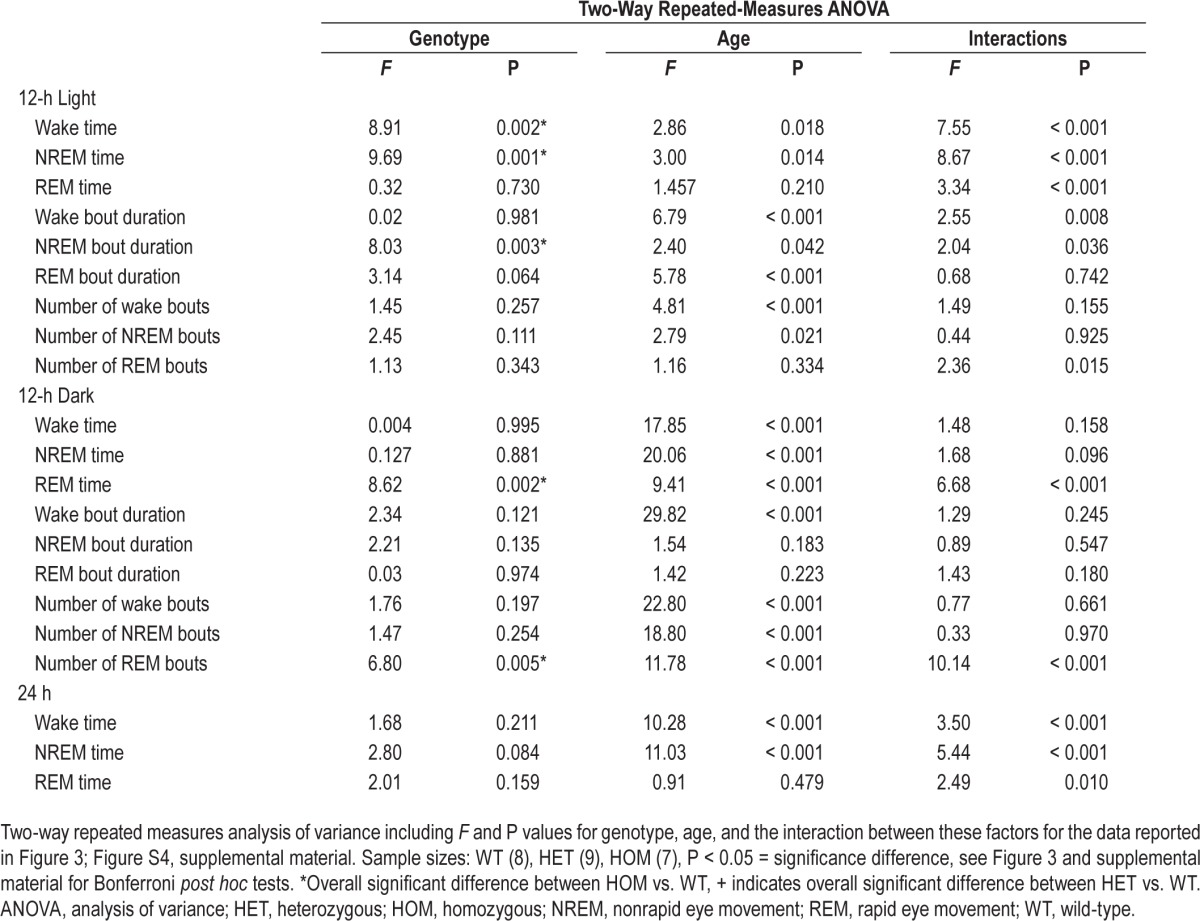
Statistical analyses were performed using SigmaPlot 12.0 (Systat Software Inc., San Jose, CA). Differences among group means were assessed using two-way repeated-measures analysis of variance (ANOVA). For LMA, Tb, sleep/wake and EEG data, comparisons were made between “genotype” and “age” or between “genotype” and “time of day.” For the EEG spectral data, statistical analysis was performed on total EEG power data for wake, NREM, and REM sleep. If an ANOVA test indicated significance, multiple comparisons were made using post hoc Bonferroni t tests in which differences were considered statistically significant if P < 0.05. For all periodo-gram analyses, the significance level was set at P < 0.01.
RESULTS
Gene Dosage- and Age-dependent Reduction of Body Weight Gain in zQ175 Mice
As indicated in Figure 1B, there was a significant effect of genotype (F2,607 = 36.22, P < 0.001) and age (F21,607 = 149.54, P < 0.001) on body weight assessed across the duration of the study, as well as a significant interaction between these factors (F42,607 = 28.87, P < 0.001). Post hoc analysis revealed that HOM mice stopped gaining weight and were significantly lighter than WT and HET mice from 19 and 23 w of age, respectively, a trend that continued across the study. HET mice weight plateaued at ∼23 w and were significantly lighter compared to WT mice from 29 w.
Disruption of Locomotor Activity and Body Temperature Evident in HOM zQ175 Mice
There was an overall effect of age on LMA during the 12 h light (main effect: F2,120 = 10.71, P < 0.001) and dark phases (main effect: F2,120 = 23.24, P < 0.001). However, there was no effect of genotype on these parameters nor was there an interaction between these factors (Figures 2A and 2B). For the 12 h dark phase, there was a trend for HOM mice to be less active than WT or HET mice from 12 to 48 w of age, although this difference was not statistically significant (Figure 2B) likely due to the decline in activity observed across all genotypes with age. Chi square periodogram analysis indicated that all genotypes exhibited robust LMA rhythms entrained to the 12:12 LD cycle across the study (Figure S1A, supplemental material), although the amplitude of the 24-h rhythm was consistently lower in HOM mice (Figure S1B). Inspection of 24-h activity profiles indicated that the first signs of reduced activity in HOM mice occurred at 16 w in the early dark phase (Figure S1C). When LMA was expressed as activity per hour spent awake, the trend of reduced activity apparent in the HOM mice persisted during the 12 h dark phase (Figure S1D) and was significantly different compared to HET mice at 48 w (P < 0.05). Therefore, the reduction in activity in HOM mice occurred largely independent of total waking time, which may be an indication of motor deficits in these animals. Despite maintaining diurnal organization of LMA rhythms, HOM mice exhibited decreases in activity during the dark phase of the LD cycle.
Figure 2.

Locomotor activity and core body temperature across the 12 h light and dark phase during baseline recordings in HET, HOM and WT zQ175 mice from 8–48 w of age. (A,B) Total activity determined from implanted telemeters (data points represent total activity counts across 6 d prior to the first sleep deprivation in each experimental week) during the light (ZT0–12) and dark phase (ZT13–24) from 8–48 w of age. (C) Representative “actogram-style” double-plots of body temperature for WT, HET and HOM zQ175 mice recorded continuously from ∼8–48 w of age maintained under a 12:12 light/ dark cycle (indicated by black and white bars at the top of the plot). Each horizontal line represents two consecutive 24-h periods in which the second half of each line is repeated on the first half of the following line. (D,E) Average core body temperature (average values calculated during 6 d prior to the first sleep deprivation in each experimental week) during the light (ZT0–12) and dark phase (ZT13–24) from 8–48 w of age. *Significant differences between HOM and WT mice (P < 0.05). +Significant differences between HET and WT mice (P < 0.05). #Significant differences between HET and HOM mice. HET, heterozygous; HOM, homozygous; WT, wild-type; ZT, Zeitgeber time.
Tb was measured in mice continuously throughout the study using implanted telemeters. An overall effect of age on average T b was confirmed by two-way ANOVA comparing all three genotypes across 8–48 w separately for the 12-h light and dark phases (Figures 2D and 2E). A significant effect of genotype was apparent only during the dark period (Figure 2E, genotype F 2,120 = 4.18 P = 0.028; age F5,120 = 26.17, P < 0.001; interaction F 10,120 = 2.99, P = 0.002) and post hoc comparisons revealed a significant difference between WT and HOM mice at 32 w (P = 0.002) and 48 w (P = 0.005) of age. Average Tb was indistinguishable between HET zQ175 and WT mice throughout the duration of the study in either phase of the LD cycle. The reduction in Tb in HOM mice occurred predominantly during the middle of the 12-h dark phase, as shown by the reduced contrast in the temperature-actogram style plots beyond 32 w of age compared to WT and HET (Figure 2C). All genotypes exhibited robust 24-h rhythms of Tb across the study but the amplitude of the diurnal rhythm in HOM mice was reduced by ∼50% at 48 w of age (Figure S2A, supplemental material). Differences in hourly Tb between WT and HOM mice were apparent from 16 to 48 w of age (Figure S2B). However, as with LMA, Tb decreased with age in the WT group (particularly evident at 48 w) with no overall effect of genotype (P = 0.913) on the 24-h profile.
Because Tb would be predicted to increase when animals are more active, we performed correlational analyses between these two parameters across the three genotypes from 8 to 48 w of age (Figure S3, supplemental material). We identified robust positive correlations between Tb and LMA across all ages and genotypes. The correlation plots at 32 and 48 w clearly demonstrate the reduction in LMA and Tb with age in all mice shown by the compression of the data points to a narrower range. There was also a trend toward a weaker correlation (R2 = 0.683) between these parameters in HOM mice at 48 w.
Disruption of Sleep/Wake Amount and Architecture Evident Only in HOM zQ175 Mice
From 24 w of age, HOM zQ175 mice exhibited progressive increases in wakefulness and a parallel decrease in NREM sleep during the light phase due to a reduction in NREM bout duration (Figure 3A, Table 1, Figure S4A). There were no differences in Wake or NREM sleep amount or architecture between the genotypes during the 12-h dark phase. However, REM sleep amount increased in HOM mice from 24 w, was twofold greater than WT and HET mice by 48 w, and was largely confined to the mid-dark phase (ZT15–20, Figure 3B; Figure S5, supplemental material). The increased REM sleep was due to an increase in the number of REM bouts with no change in REM bout duration (Figures S4A and S4B, supplemental material). Total time spent (24 h) in each state was more mildly affected in zQ175 HOM mice, with NREM decreasing from 32 w and Wake and REM increasing relative to WT mice from 48 w (Figure 3C). Hourly sleep/wake profiles indicated progressive changes in Wake and NREM from 24 to 48 w in HOM mice, predominantly in the first 6 h of the light phase (Figure S5).
Figure 3.

HOM zQ175 mice exhibit progressive deficits in NREM sleep in the light phase and increases in REM sleep in the dark phase. (A) Time spent (min) in wake, NREM and REM in the 12-h light period. (B) Time spent (min) in wake, NREM and REM in the 12 h dark period. (C) Time spent (min) in wake, NREM and REM across the entire 24 h period. *Significant differences between HOM and WT mice (P < 0.05). +Significant differences between HET and WT mice (P < 0.05). #Significant differences between HET and HOM mice. For hourly sleep/wake data, see Figure S5 in the supplemental material. HET, heterozygous; HOM, homozygous; NREM, nonrapid eye movement; REM, rapid eye movement; WT, wild-type.
Table 1.
Longitudinal assessment of the effects of genotype and age on baseline sleep/wake parameters in zQ175 mice from 8 to 48 w.
Early EEG Alterations and Late-stage Aberrant Beta/Gamma Oscillations in zQ175 Mice
Gene-dosage and age-dependent alterations in the EEG were apparent in the raw EEG spectra, particularly the overall decrease in EEG power and emergence of the beta/ gamma activity in HOM zQ175 mice (Figures 4A and 4B; Figures S6–S8, supplemental material). To enable visualization of the changes at higher EEG frequencies, spectral EEG power was normalized to the age-matched WT condition (average group value) within each vigilance state from 8 to 48 w (Figure 4C). Genotypic differences in the EEG were evident from our first recording at 8 w of age, particularly across the 0–20 Hz range in the HOM zQ175 mice. For HET mice, EEG power was similar across the states at 8 w but then progressively declined in the low frequency range. The normalized EEG power curves for HET and HOM mice, particularly during Wake in the 0–30 Hz range, are remarkably parallel with peaks and troughs closely mirrored, indicating the specificity of the gene-dosage effect of mHtt on the EEG.
Figure 4.
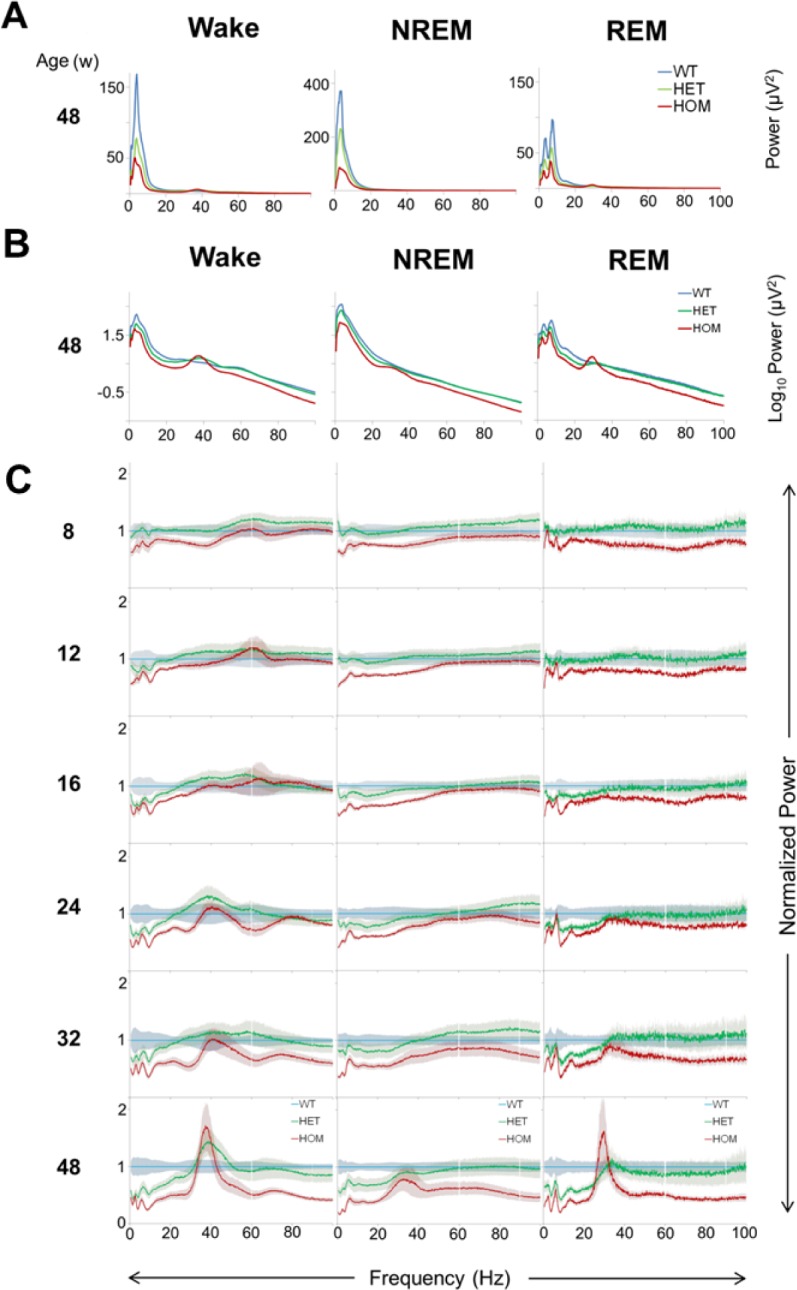
Early quantitative EEG abnormalities and late stage development of beta/gamma oscillations in zQ175 mice. (A) Raw EEG spectra (0.5–100 Hz, 0.1 Hz resolution, mean values) during Wake and NREM and REM sleep in WT (blue), HET (green), and HOM (red) zQ175 mice from 24-h baseline recordings at 48 w of age. (B) The same data as in panel A but plotted as mean log values for better visualization of high frequencies. (C) Average EEG spectra (0.5–100 Hz, 0.1 Hz resolution, normalized to the age-matched WT group average value, which is depicted as the blue line, mean ± SEM) for HET (green line) and HOM (red line) during Wake, and NREM and REM sleep for 24-h baseline recordings at 8–48 w of age. EEG, electroencephalograph; HET, heterozygous; HOM, homozygous; NREM, nonrapid eye movement; REM, rapid eye movement; SEM, standard error of the mean; WT, wild-type.
Analysis of total EEG power (0.5–100 Hz) across the three sleep/wake states revealed significant overall genotype effects (Figure 5). Post hoc analysis indicated that absolute EEG power was lower in HOM mice from 12 w in NREM sleep and 16 w of age in wake and REM sleep. In addition, a significant difference emerged in absolute EEG power between WT and HET mice at 48 w for Wake and REM sleep. The earliest significant frequency changes in HOM mice were a decrease in NREM delta power at 8 w and, for HET mice, a decrease in waking delta power and REM alpha power at 24 w (data not shown for individual frequency bands). Interestingly, despite the dramatic attenuation of total EEG power during all sleep/ wake states in HOM zQ175 mice, a large peak in beta/gamma power became evident predominantly in Wake (38 Hz peak frequency) and REM sleep (30 Hz peak frequency) at 48 w of age. This beta/gamma peak was similar in magnitude during Wake and REM but was largely attenuated during NREM sleep. In HET zQ175 mice, a beta/gamma peak was only apparent in the waking EEG.
Figure 5.

Age- and gene dosage-dependent changes in total EEG power (0.5–100 Hz) in wake, NREM and REM sleep in WT, HET and HOM zQ175 mice from 8–48 w of age. Data represent baseline 24-h mean values ± SEM, age- and genotype-matched. *Significant differences between HOM and WT mice (P < 0.05). +Significant differences between HET and WT mice (P < 0.05). #Significant differences between HET and HOM mice. EEG, electroencephalograph; HET, heterozygous; HOM, homozygous; NREM, nonrapid eye movement; REM, rapid eye movement; SEM, standard error of the mean; WT, wild-type.
Gene Dosage- and Age-dependent Slowing of Theta Peak Frequency in zQ175 Mice
A decrease in theta peak frequency (TPF) in REM sleep was a consistent EEG alteration identified previously in R6/234,35 and R6/1 mice.36 Therefore, we were interested to determine whether a slowing of the EEG was a consistent feature across multiple HD mouse models. Visual inspection of log raw EEG frequencies (4–40 Hz) in REM sleep showed a clear theta peak in all genotypes at all ages studied and a beta/gamma peak evident only in 48-w-old zQ175 HOM mice (Figures 6A–6C). Two-way ANOVA revealed an effect of genotype (F2,105 = 21.65, P < 0.001), age (F5,105 = 8.86 P < 0.001) and an interaction (F10,105 = 4.46 P < 0.001) between these factors on TPF in zQ175 mice (Figure 6D). Post hoc analysis identified that TPF was reduced in HOM Q175 from 12 w and from 24 w in HET mice compared to WT mice.
Figure 6.

Age- and gene dosage-dependent slowing of REM theta peak frequency in zQ175 mice. Average EEG spectra in REM sleep across 24-h baseline recordings (4–40 Hz, log scale) for (A) WT, (B) HET, and (C) HOM zQ175 mice from 8–48 w of age. (D) Average theta peak frequency in REM sleep determined from individual HET, HOM, and WT zQ175 mice from 8–48 w of age. Theta peak frequency was defined as the frequency bin (0.1 Hz resolution) within the theta range (5–9 Hz) with the highest EEG power value derived from the average 24-h REM spectra (averaged within individuals and then between individuals, age- and genotype-matched) *Significant differences between HOM and WT mice (P < 0.05). +Significant differences between HET and WT mice (P < 0.05). #Significant differences between HET and HOM mice. EEG, electroencephalograph; HET, heterozygous; HOM, homozygous; REM, rapid eye movement; WT, wild-type.
Homeostatic Sleep Regulation is Preserved in HET and HOM zQ175 Mice
To compare the functioning of the sleep homeostat in HET, HOM, and WT zQ175 mice across the course of the disease (8–48 w), we performed SDep for 1, 3, and 6 h on alternate days. Although absolute delta power levels in NREM sleep differed between genotypes and across age (Figure 4A and 4B), there is no evidence to indicate that these alterations are associated with changes in homeostatic sleep need. Therefore, to examine sleep/wake history-dependent changes in NREM delta power (0.5–4 Hz), we normalized hourly NREM delta power values during recovery from SDep (ZT7–12) against each individual animal's age-matched average 12 h light phase baseline value (Figure 7A). This analysis revealed that there were no differences between the genotypes from 8–48 w, indicating that, although absolute delta power values were generally lower in HET and HOM zQ175 mice compared to WT across the study duration, the relative increase from baseline levels in response to sleep loss were comparable.
Figure 7.

Age and genotype do not affect homeostatic responses to sleep deprivation in zQ175 mice. (A) NREM delta power (normalized to the 12-h light period (ZT0–12) average NREM delta power in each week's baseline recording) for 6 h following 1, 3, and 6 h SDep for WT, HET, and HOM zQ175 mice from 8–48 w of age. (B,C) Recovery of sleep time (min) lost for NREM (B) and REM sleep (C) in response to 1, 3 and 6 h SDep for WT, HET and HOM zQ175 mice. Recovery was calculated as total recovery (REC) sleep (6 h following SDep) minus baseline (BL) sleep for the same time period (ZT7–12), age- and genotype-matched. EEG, electroencephalograph; EMG, electromyography; HET, heterozygous; HOM, homozygous; NREM, nonrapid eye movement; REM, rapid eye movement; SDep, sleep deprivation; WT, wild-type; ZT, Zeitgeber time.
We then determined whether there were changes in the amount of sleep time lost in response to the different SDep durations. Because sleep amount varied with genotype and age, we subtracted baseline sleep (time-of-day-, genotype-, and age-matched hourly values) from the durations of NREM and REM sleep during the 6-h recovery period (Figures 7B and 7C). The 3- and 6-h SDep resulted in fairly large increases in sleep time, particularly for NREM sleep (dotted line on plots denotes no overall change in hourly sleep time). However, no significant differences were evident among the three genotypes. Thus, despite exhibiting changes in absolute NREM EEG delta power and baseline sleep amount, zQ175 HET and HOM mice maintained a homeostatic response to sleep loss across the progression of the disease.
DISCUSSION
Despite identification of the genetic basis of HD more than two decades ago, our understanding of the neuropathology in the premanifest stage remains largely incomplete. There is a compelling need to identify biomarkers that correlate with HD initiation and development and which can serve as objective endpoints in preventive preclinical and clinical pharmacological trials. Consequently, over a 40-w period, we examined EEG and sleep/wake changes that accompany the behavioral, histopathological, and neurophysiological deficits previously identified in the novel zQ175 HD KI model.40,41 Expression of mHtt occurs in the appropriate genetic and protein context in this strain, which develops a slowly progressing phenotype, thereby permitting examination of the presymptomatic phase. We identified age- and genotype-dependent EEG alterations in zQ175 mice prior to deficits in sleep/wake behavior, LMA, or Tb. Early and progressive changes in qEEG parameters included a decrease in low frequency power and a slowing of REM TPF while, later in the disease, state-dependent beta/gamma oscillations developed. These findings demonstrate the specificity of early EEG alterations in relation to mHTT load in this model and establish a basis for future preclinical therapeutic trials.
Early Gene Dosage- and Age-Related EEG Alterations in zQ175 HD Mice
EEG abnormalities and sleep/wake disturbances are highly prevalent in patients with HD16–19 and are also a common feature of transgenic HD mouse models.34–36 Here, we confirm that sleep and EEG alterations are also found in KI HD mice that represent a more precise genetic model of the human condition. Similar to the R6/2 model, HOM zQ175 mice exhibited early changes in the EEG spectra compared to age-matched WT mice. A number of these EEG differences relative to WT mice were already present in the HOM zQ175 mice from our earliest recording and, thus, may not be indicative of a premanifest EEG pattern. These EEG alterations could represent early developmental changes caused by mHtt expression (discussed later). TPF in REM sleep decreased in zQ175 mice, a finding consistent with R6/234,35 and R6/136 mice and which has also been reported in patients with HD.21 This observation could indicate a slowing of the entire EEG spectra, as implied by the decrease in beta/gamma peak frequency in R6/2 mice.34 However, this feature did not fully develop in zQ175 mice until the final recording at 48 w, an age when cognitive deficits first appear in these mice.40 In contrast, beta/gamma activity was greatly elevated in R6/2 mice from 8 w of age,34 consistent with the view that this model is a more aggressive juvenile form of HD.43 In comparison, zQ175 mice exhibit a less symptomatic EEG phenotype, signified by the late development of beta/ gamma oscillations.
The source of the observed increase in beta/gamma activity is unknown but electrophysiological studies in striatal slices indicate that medium spiny neurons (MSNs) become intrinsically more excitable with age in zQ175 mice and have impaired corticostriatal communication.40 The beta/gamma oscillations in HOM zQ175 mice exhibited some degree of state-dependency (increasing in Wake and REM sleep) but were largely attenuated in NREM sleep and thus preferentially occurred during activated brain states. Interestingly, an identical pattern of EEG beta/gamma activity is observed in Parkinson disease (PD) in relation to sleep/wake state44,45 and has been associated with motor deficits and tremor,46,47 suggesting common dysfunction across corticobasal ganglia networks. In addition, striatal levels of dopamine and its metabolite homovanillic acid decrease in zQ175 HET mice48 from 12 mo of age when beta/gamma activity is elevated during Wake. A different pattern of beta/gamma activity in relation to behavioral state has been found in transgenic R6/1 and R6/2 HD mice.36,49 In these models, beta/gamma oscillations were lower during active Wake and exploration and higher during quiet rest, NREM, and REM sleep, suggesting key differences exist among murine HD models. For HET mice, EEG power was similar across the states at 8 w but then progressively declined in the low frequency range, with an increase in beta/gamma activity evident during Wake at 48 w.
Sleep/wake amount and architecture were indistinguishable between HET and WT zQ175 mice and differences were only evident in HOM mice from 24 w, confirming that EEG alterations precede arousal state changes. Future studies will be needed to verify whether sleep disruption becomes evident in HET zQ175 mice beyond 48 w of age. A previous study assessed immobility-defined sleep using video tracking and found similar late-stage changes exclusively in HOM mice.42 R6/2 mice also exhibit increased REM sleep propensity34,35; however, this is not observed in patients with HD. We challenged the compensatory response to sleep loss across disease progression in the zQ175 mouse in a duration-dependent manner (1, 3, 6 h SDep). Following SDep, sleep homeostatic responses, such as relative delta power and recovery of sleep time lost, were preserved in the HET and HOM zQ175 mice. These observations contrast with our previous findings in the R6/2 model in which attenuated sleep homeostatic responses were evident from 13 w of age.34 Preservation of a functional sleep homeostatic system in HOM mice indicates that mHtt expression in this strain does not affect the ability of the brain to regulate sleep intensity or sleep amount in response to sleep loss, at least up to age 48 w.
Mutant HTT Expression in Relation to Neuronal Dysfunction and Atrophy
The normal physiological function of the HTT protein is unknown but it appears to play a role in neuronal development and survival.50,51 Compared to other neurodegenerative diseases, the monogenic basis and full penetrance of the abnormal CAG expansion in HD uniquely enables the identification and study of prodromal biomarkers prior to classic symptom development. Controversy remains over whether the CAG expansion leads to neurodegeneration from a toxic gain of function and/or a loss of WT HTT function.52 The mutated form of HTT is expressed ubiquitously in all tissues; however, neurodegeneration occurs preferentially in striatal MSNs and cortical pyramidal neurons. We hypothesized that mHTT accumulation in the zQ175 mouse alters neuronal functioning and leads to neuronal and synaptic dysfunction, including deficits in striatal MSNs and cortical pyramidal neurons,28–33 culminating in specific “signature” changes detectable in the global EEG. Aggregates of mHTT are absent at 1 mo but are present in the striatum, motor cortex, prefrontal cortex, and hippocampus in the HET zQ175 mice from 6 mo,48 an age similar to when we detected robust EEG alterations in these mice. HTT associates with a number of proteins involved in synaptic functioning53 and, in HET zQ175 mice with only one mutated allele, synaptic proteins such as SNAP-25, Rab3A, and PSD-95 decrease from 6 mo of age.48 Modifications in the activity of these critical brain proteins could affect excitatory and inhibitory currents generated by cortical and subcortical neurons and, cumulatively, such changes could lead to alterations in EEG patterns. For example, SNAP-25, a component of the SNARE complex involved in the regulation of synaptic vesicle exocytosis, is known to negatively regulate neuronal calcium responsiveness to depolarization.54 Reduction in SNAP-25 protein levels could alter neuronal and network excitability, as recently reported in SNAP-25 heterozygous mice.55 In terms of neuronal loss, HOM zQ175 mice exhibit an early decrease in striatal neurons and in striatal and cortical volumes from 3–4.5 mo,40 progressing to a 21% and 14% reduction in striatal and cortical volumes, respectively, by 8 mo. In HET zQ175 mice, striatal and cortical volume decreased from 4 mo when assessed using MRI40 but were not apparent until 12 mo of age when measured from Nissl-stained sections.48 The reduction in total EEG power we identified across the progression of the disease (Figure 5) may be a direct result of such cortical atrophy. Similar volumetric changes and cortical thinning are also evident in patients with early-stage HD.56
Disturbances in rest/activity rhythms are common in patients with HD.57 We measured intrinsic home-cage LMA and detected minor decreases in activity in HOM zQ175 mice, but HET mice were largely unaffected. A decrease in activity has been reported previously in these mice using open field and PhenoCube activity monitoring40,41 with an age-related decline also shown in running wheel activity.42 Motor deficits previously identified in zQ175 mice could be one factor that accounts for this decrease in activity. Smith and colleagues48 reported motor deficits in zQ175 HET mice at 6 mo of age using slow-motion video analysis at a time when striatal gamma-aminobutyric acid levels increase and mHTT inclusions are present in the brain. The EEG abnormalities we found in the current study occurred in advance of disrupted Tb and also before the cognitive deficits that have been reported in HOM mice at 10–12 mo40 and in HET mice at 16 mo of age.48 Excessive weight loss is not a feature of the zQ175 model; instead, we confirmed the previously reported failure of these mice to gain weight in a gene-dosage dependent manner.40,41
In addition to the well-characterized dysfunction of striatal MSNs and cortical pyramidal neurons in HD,30–32 changes in glial and/or immune function could also contribute to the pathophysiology of the disease. Microglia activation, by exposure of cells to abnormal mHTT aggregates, has been reported to occur in the striatum of premanifest HD subjects and may be an early event in HD pathogenesis.58,59 Aberrant immune responses, including an increase in glial markers of neuroinflammation60 and secretion of proinflammatory cytokines, have also been found in presymptomatic HdhQ150 and R6/2 mice.61 Activation of microglia in this manner within the striatum and cortex is associated with neuronal loss,62 although the exact mechanisms involved and the effects on global cortical electrophysiology are not fully understood and warrant further investigation. Furthermore, recent evidence suggests astrocyte physiology is altered in HD mouse models, with expression of mHtt in astrocytes leading to age-dependent neurological symptoms in mice63 and decreased Kir4.1 potassium ion channel expression.64 The resultant loss of Kir4.1 channels elevates striatal extracellular K+ levels and subsequently depolarizes and increases the firing of MSNs. It is tempting to speculate that such changes in striatal neuronal excitability may contribute to the aberrant beta/gamma oscillations identified in the zQ175 model and previously in the R6/2 mouse.34–36
The EEG as a Tool to Identify Candidate HD Biomarkers: Translational Relevance of the zQ175 Model
To fully realize the value of the EEG to identify early stage biomarkers of HD, future studies will need to determine its predictive validity against effective treatments. Ideally, these therapies should inhibit specific early-stage pathogenic mechanisms such as mHTT expression, particularly in cortical and striatal neurons recently found to account for the behavioral deficits and brain atrophy in the BACHD model.65 Intrinsic differences between mouse models and the human condition also need to be considered. The CAG repeat length in the zQ175 mouse (186–188) is significantly larger than human HD alleles (37–84 repeats); however, there is a delicate balance between a disease-relevant repeat size and the occurrence of a robust enough phenotype for preclinical testing. Animals with greater construct validity and lower CAG repeats sizes are available,66,67 including the novel Hu97/18 mouse that fully mimics HD genetics by expressing human HTT HET for a CAG expansion (97 repeats) in the absence of endogenous murine Htt.68 These mice exhibit synaptic alterations in the striatum and hippocampus from 6 mo of age69 and would be an interesting model to further validate use of the EEG to reveal translational HD biomarkers. zQ175 HOM mice exhibit very early neuropathological changes, including EEG alterations, raising the possibility that mHtt expression causes developmental changes in these mice. The EEG was more mildly affected in HET mice in which the presence of normal and mutant Htt make it a more viable translational preclinical model. Interestingly, the effects on the EEG appeared earlier and were larger in HOM compared to HET mice, indicating a gene dosage effect of mHtt as was previously reported in this model.40–42 Similarly, the onset of the phenotype was exacerbated in the YAC128 mouse model by increasing levels of mHtt.70 In patients with HD, disease progression is more aggressive in patients homozygous for the mutation, although small numbers of subjects were studied and age of clinical onset was comparable with HET patients.71 Although analysis of the EEG in patients with HD is not a new concept, a comprehensive longitudinal evaluation of the qEEG parameters in premanifest subjects is still lacking (after submission of the final revised manuscript, Lasar et al.72 was published and the authors would like to direct the readers' attention to this publication). Identification of parallel changes in animal models and humans may reveal key insights into the early events in HD onset and development and may help accelerate drug development for HD.
In summary, we performed a comprehensive longitudinal evaluation of EEG and sleep/wake alterations in the zQ175 KI HD mouse. We found that the EEG, an easily obtainable measurement in human subjects, uncovered one of the earliest neurophysiological phenotypes uncovered in this model. These data suggest that qEEG measures may be valuable early indicators of HD onset and progression and that further validation against novel therapeutics may provide greater insight into their utility for preclinical and clinical drug development.
DISCLOSURE STATEMENT
This was not an industry supported study. This work was supported by the CHDI Foundation, Inc., National Institutes of Health (R01 NS057464) and internal research funds from SRI International. Dr. Morairty has received research support from Sunovion, F. Hoffmann LaRoche, and Merck.
Dr. Thomas has received research support from F. Hoffmann LaRoche.
Dr. Schwartz has received research support from F. Hoffmann LaRoche and Merck. Dr. Black has received research support from F. Hoffmann LaRoche. Dr. Kilduff has received research support from F. Hoffmann LaRoche, Sunovion, Dr. Palmerston has received research support from F. Hoffmann LaRoche, Sunovion, and Merck. The other authors have indicated no financial conflicts of interest. The work was performed at the Center for Neuroscience, Bioscience Division, SRI International, Menlo Park, CA. Dr. Fisher's current affiliation is with the Department of Physiology, Anatomy and Genetics, University of Oxford, Sherrington Building, Parks Road, Oxford, OX1 3PT, United Kingdom.
ACKNOWLEDGMENTS
The authors thank David Howland for advice on the zQ175 mouse model and members of the Center for Neuroscience and Mammalian Toxicology at SRI International for technical assistance during sleep deprivations.
ABBREVIATIONS
- ANOVA
analysis of variance
- EEG
electroencephalogram
- EMG
eclectromyogram
- HD
Huntington's disease
- HET
heterozygous
- HOM
homozygous
- Htt
mouse Huntingtin protein
- Htt
mouse Huntingtin gene or RNA
- HTT
human Huntingtin protein
- HTT
human Huntingtin gene or RNA
- mHtt
mutant huntingtin protein (mouse)
- mHTT
mutant huntingtin protein (human)
- LMA
locomotor activity
- KI
knock-in
- NREM
non-rapid eye movement sleep
- qEEG
quantitative electroencephalogram
- REM
rapid-eye movement sleep
- SDep
sleep deprivation
- Tb
body temperature
- TPF
theta peak frequency
- WT
wildtype
REFERENCES
- 1.Gusella JF, MacDonald ME. Huntington's disease: seeing the pathogenic process through a genetic lens. Trends Biochem Sci. 2006;31:533–40. doi: 10.1016/j.tibs.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 2.Ross CA, Tabrizi SJ. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 3.Tabrizi SJ, Reilmann R, Roos RA, et al. Potential endpoints for clinical trials in premanifest and early Huntington's disease in the TRACK-HD study: analysis of 24 month observational data. Lancet Neurol. 2012;11:42–53. doi: 10.1016/S1474-4422(11)70263-0. [DOI] [PubMed] [Google Scholar]
- 4.Tabrizi SJ, Scahill RI, Owen G, et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington's disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol. 2013;12:637–49. doi: 10.1016/S1474-4422(13)70088-7. [DOI] [PubMed] [Google Scholar]
- 5.Paulsen JS, Long JD, Johnson HJ, et al. Clinical and biomarker changes in premanifest Huntington disease show trial feasibility: a decade of the PREDICT-HD study. Front Aging Neurosci. 2014;6:78. doi: 10.3389/fnagi.2014.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dominguez DJ, Egan GF, Gray MA, et al. Multi-modal neuroimaging in premanifest and early Huntington's disease: 18 month longitudinal data from the IMAGE-HD study. PloS One. 2013;8:e74131. doi: 10.1371/journal.pone.0074131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paulsen JS. Biomarkers to predict and track diseases. Lancet Neurol. 2009;8:776–7. doi: 10.1016/S1474-4422(09)70203-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ross CA, Aylward EH, Wild EJ, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10:204–16. doi: 10.1038/nrneurol.2014.24. [DOI] [PubMed] [Google Scholar]
- 9.Carroll JB, Warby SC, Southwell AL, et al. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene / allele-specific silencing of mutant huntingtin. Mol Ther. 2011;19:2178–85. doi: 10.1038/mt.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ostergaard ME, Southwell AL, Kordasiewicz H, et al. Rational design of antisense oligonucleotides targeting single nucleotide polymorphisms for potent and allele selective suppression of mutant Huntingtin in the CNS. Nucleic Acids Res. 2013;41:9634–50. doi: 10.1093/nar/gkt725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garriga-Canut M, Agustin-Pavon C, Herrmann F, et al. Synthetic zinc finger repressors reduce mutant huntingtin expression in the brain of R6/2 mice. Proc Natl Acad Sci U S A. 2012;109:E3136–45. doi: 10.1073/pnas.1206506109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sadri-Vakili G, Cha JH. Histone deacetylase inhibitors: a novel therapeutic approach to Huntington's disease (complex mechanism of neuronal death) Curr Alzheimer Res. 2006;3:403–8. doi: 10.2174/156720506778249407. [DOI] [PubMed] [Google Scholar]
- 13.Mielcarek M, Landles C, Weiss A, et al. HDAC4 reduction: a novel therapeutic strategy to target cytoplasmic huntingtin and ameliorate neurodegeneration. PLoS Biol. 2013;11:e1001717. doi: 10.1371/journal.pbio.1001717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fink KD, Crane AT, Leveque X, et al. Intrastriatal transplantation of adenovirus-generated induced pluripotent stem cells for treating neuropathological and functional deficits in a rodent model of Huntington's disease. Stem Cells Transl Med. 2014;3:620–31. doi: 10.5966/sctm.2013-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen L, Bradshaw JL, Stout JC, Croft RJ, Georgiou-Karistianis N. Electrophysiological measures as potential biomarkers in Huntington's disease: review and future directions. Brain Res Rev. 2010;64:177–94. doi: 10.1016/j.brainresrev.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 16.Arnulf I, Nielsen J, Lohmann E, et al. Rapid eye movement sleep disturbances in Huntington disease. Arch Neurol. 2008;65:482–8. doi: 10.1001/archneur.65.4.482. [DOI] [PubMed] [Google Scholar]
- 17.Aziz NA, Anguelova GV, Marinus J, Lammers GJ, Roos RA. Sleep and circadian rhythm alterations correlate with depression and cognitive impairment in Huntington's disease. Parkinsonism Relat Dis. 2010;16:345–50. doi: 10.1016/j.parkreldis.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 18.Goodman AO, Barker RA. How vital is sleep in Huntington's disease? J Neurol. 2010;257:882–97. doi: 10.1007/s00415-010-5517-4. [DOI] [PubMed] [Google Scholar]
- 19.Morton AJ. Circadian and sleep disorder in Huntington's disease. Exper Neurol. 2013;243:34–44. doi: 10.1016/j.expneurol.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 20.Scott DF, Heathfield KW, Toone B, Margerison JH. The EEG in Huntington's chorea: a clinical and neuropathological study. J Neurol Neurosurg Psychiatry. 1972;35:97–102. doi: 10.1136/jnnp.35.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bylsma FW, Peyser CE, Folstein SE, Folstein MF, Ross C, Brandt J. EEG power spectra in Huntington's disease: clinical and neuropsychological correlates. Neuropsychologia. 1994;32:137–50. doi: 10.1016/0028-3932(94)90001-9. [DOI] [PubMed] [Google Scholar]
- 22.Painold A, Anderer P, Holl AK, et al. Comparative EEG mapping studies in Huntington's disease patients and controls. J Neural Transm. 2010;117:1307–18. doi: 10.1007/s00702-010-0491-7. [DOI] [PubMed] [Google Scholar]
- 23.de Tommaso M, Sciruicchio V, Specchio N, et al. Early modifications of auditory event-related potentials in carriers of the Huntington's disease gene. Acta Neurologica Belgica. 2003;103:192–8. [PubMed] [Google Scholar]
- 24.van der Hiele K, Jurgens CK, Vein AA, et al. Memory activation reveals abnormal EEG in preclinical Huntington's disease. Mov Dis. 2007;22:690–5. doi: 10.1002/mds.21390. [DOI] [PubMed] [Google Scholar]
- 25.Hunter A, Bordelon Y, Cook I, Leuchter A. QEEG measures in Huntington's disease: a pilot study. PLoS Curr. 2010;2:RRN1192. doi: 10.1371/currents.RRN1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pouladi MA, Morton AJ, Hayden MR. Choosing an animal model for the study of Huntington's disease. Nat Rev Neurosci. 2013;14:708–21. doi: 10.1038/nrn3570. [DOI] [PubMed] [Google Scholar]
- 27.Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington's disease with 140 CAG repeats. J Comp Neurol. 2003;465:11–26. doi: 10.1002/cne.10776. [DOI] [PubMed] [Google Scholar]
- 28.Milnerwood AJ, Raymond LA. Corticostriatal synaptic function in mouse models of Huntington's disease: early effects of huntingtin repeat length and protein load. J Physiol. 2007;585:817–31. doi: 10.1113/jphysiol.2007.142448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walker AG, Miller BR, Fritsch JN, Barton SJ, Rebec GV. Altered information processing in the prefrontal cortex of Huntington's disease mouse models. J Neurosci. 2008;28:8973–82. doi: 10.1523/JNEUROSCI.2804-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cummings DM, Andre VM, Uzgil BO, et al. Alterations in cortical excitation and inhibition in genetic mouse models of Huntington's disease. J Neurosci. 2009;29:10371–86. doi: 10.1523/JNEUROSCI.1592-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cepeda C, Cummings DM, Andre VM, Holley SM, Levine MS. Genetic mouse models of Huntington's disease: focus on electrophysiological mechanisms. ASN Neuro. 2010;2:e00033. doi: 10.1042/AN20090058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller BR, Walker AG, Barton SJ, Rebec GV. Dysregulated neuronal activity patterns implicate corticostriatal circuit dysfunction in multiple rodent models of Huntington's disease. Front Syst Neurosci. 2011;5:26. doi: 10.3389/fnsys.2011.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raymond LA, Andre VM, Cepeda C, Gladding CM, Milnerwood AJ, Levine MS. Pathophysiology of Huntington's disease: time-dependent alterations in synaptic and receptor function. Neuroscience. 2011;198:252–73. doi: 10.1016/j.neuroscience.2011.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fisher SP, Black SW, Schwartz MD, et al. Longitudinal analysis of the electroencephalogram and sleep phenotype in the R6/2 mouse model of Huntington's disease. Brain. 2013;136:2159–72. doi: 10.1093/brain/awt132. [DOI] [PubMed] [Google Scholar]
- 35.Kantor S, Szabo L, Varga J, Cuesta M, Morton AJ. Progressive sleep and electroencephalogram changes in mice carrying the Huntington's disease mutation. Brain. 2013;136:2147–58. doi: 10.1093/brain/awt128. [DOI] [PubMed] [Google Scholar]
- 36.Jeantet Y, Cayzac S, Cho YH. Beta oscillation during slow wave sleep and rapid eye movement sleep in the electroencephalogram of a transgenic mouse model of Huntington's disease. PloS One. 2013;8:e79509. doi: 10.1371/journal.pone.0079509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shelbourne PF, Killeen N, Hevner RF, et al. A Huntington's disease CAG expansion at the murine Hdh locus is unstable and associated with behavioural abnormalities in mice. Hum Mol Genet. 1999;8:763–74. doi: 10.1093/hmg/8.5.763. [DOI] [PubMed] [Google Scholar]
- 38.Menalled LB. Knock-in mouse models of Huntington's disease. NeuroRx. 2005;2:465–70. doi: 10.1602/neurorx.2.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rising AC, Xu J, Carlson A, Napoli VV, Denovan-Wright EM, Mandel RJ. Longitudinal behavioral, cross-sectional transcriptional and histopathological characterization of a knock-in mouse model of Huntington's disease with 140 CAG repeats. Exper Neurol. 2011;228:173–82. doi: 10.1016/j.expneurol.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heikkinen T, Lehtimaki K, Vartiainen N, et al. Characterization of neurophysiological and behavioral changes, MRI brain volumetry and 1H MRS in zQ175 knock-in mouse model of Huntington's disease. PloS One. 2012;7:e50717. doi: 10.1371/journal.pone.0050717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Menalled LB, Kudwa AE, Miller S, et al. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington's disease: zQ175. PloS One. 2012;7:e49838. doi: 10.1371/journal.pone.0049838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loh DH, Kudo T, Truong D, Wu Y, Colwell CS. The Q175 mouse model of Huntington's disease shows gene dosage- and age-related decline in circadian rhythms of activity and sleep. PloS One. 2013;8:e69993. doi: 10.1371/journal.pone.0069993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woodman B, Butler R, Landles C, et al. The Hdh(Q150/Q150) knock-in mouse model of HD and the R6/2 exon 1 model develop comparable and widespread molecular phenotypes. Brain Res Bull. 2007;72:83–97. doi: 10.1016/j.brainresbull.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 44.Degos B, Deniau JM, Chavez M, Maurice N. Chronic but not acute dopaminergic transmission interruption promotes a progressive increase in cortical beta frequency synchronization: relationships to vigilance state and akinesia. Cereb Cortex. 2009;19:1616–30. doi: 10.1093/cercor/bhn199. [DOI] [PubMed] [Google Scholar]
- 45.Urrestarazu E, Iriarte J, Alegre M, et al. Beta activity in the subthalamic nucleus during sleep in patients with Parkinson's disease. Mov Dis. 2009;24:254–60. doi: 10.1002/mds.22351. [DOI] [PubMed] [Google Scholar]
- 46.Weinberger M, Hutchison WD, Lozano AM, Hodaie M, Dostrovsky JO. Increased gamma oscillatory activity in the subthalamic nucleus during tremor in Parkinson's disease patients. J Neurophysiol. 2009;101:789–802. doi: 10.1152/jn.90837.2008. [DOI] [PubMed] [Google Scholar]
- 47.Mallet N, Pogosyan A, Marton LF, Bolam JP, Brown P, Magill PJ. Parkinsonian beta oscillations in the external globus pallidus and their relationship with subthalamic nucleus activity. J Neurosci. 2008;28:14245–58. doi: 10.1523/JNEUROSCI.4199-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith GA, Rocha EM, McLean JR, et al. Progressive axonal transport and synaptic protein changes correlate with behavioral and neuropathological abnormalities in the heterozygous Q175 KI mouse model of Huntington's disease. Hum Mol Genet. 2014;23:4510–27. doi: 10.1093/hmg/ddu166. [DOI] [PubMed] [Google Scholar]
- 49.Hong SL, Cossyleon D, Hussain WA, Walker LJ, Barton SJ, Rebec GV. Dysfunctional behavioral modulation of corticostriatal communication in the R6/2 mouse model of Huntington's disease. PloS One. 2012;7:e47026. doi: 10.1371/journal.pone.0047026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nasir J, Floresco SB, O'Kusky JR, et al. Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–23. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- 51.Reiner A, Dragatsis I, Zeitlin S, Goldowitz D. Wild-type huntingtin plays a role in brain development and neuronal survival. Mol Neurobiol. 2003;28:259–76. doi: 10.1385/MN:28:3:259. [DOI] [PubMed] [Google Scholar]
- 52.Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. 2000;26:300–6. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- 53.Wood JD, MacMillan JC, Harper PS, Lowenstein PR, Jones AL. Partial characterisation of murine huntingtin and apparent variations in the subcellular localisation of huntingtin in human, mouse and rat brain. Hum Mol Genet. 1996;5:481–7. doi: 10.1093/hmg/5.4.481. [DOI] [PubMed] [Google Scholar]
- 54.Verderio C, Pozzi D, Pravettoni E, et al. SNAP-25 modulation of calcium dynamics underlies differences in GABAergic and glutamatergic responsiveness to depolarization. Neuron. 2004;41:599–610. doi: 10.1016/s0896-6273(04)00077-7. [DOI] [PubMed] [Google Scholar]
- 55.Corradini I, Donzelli A, Antonucci F, et al. Epileptiform activity and cognitive deficits in SNAP-25(+/-) mice are normalized by antiepileptic drugs. Cereb Cortex. 2014;24:364–76. doi: 10.1093/cercor/bhs316. [DOI] [PubMed] [Google Scholar]
- 56.Rosas HD, Koroshetz WJ, Chen YI, et al. Evidence for more widespread cerebral pathology in early HD: an MRI-based morphometric analysis. Neurology. 2003;60:1615–20. doi: 10.1212/01.wnl.0000065888.88988.6e. [DOI] [PubMed] [Google Scholar]
- 57.Morton AJ, Wood NI, Hastings MH, Hurelbrink C, Barker RA, Maywood ES. Disintegration of the sleep-wake cycle and circadian timing in Huntington's disease. J Neurosci. 2005;25:157–63. doi: 10.1523/JNEUROSCI.3842-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tai YF, Pavese N, Gerhard A, et al. Microglial activation in presymptomatic Huntington's disease gene carriers. Brain. 2007;130:1759–66. doi: 10.1093/brain/awm044. [DOI] [PubMed] [Google Scholar]
- 59.Politis M, Pavese N, Tai YF, et al. Microglial activation in regions related to cognitive function predicts disease onset in Huntington's disease: a multimodal imaging study. Hum Brain Mapp. 2011;32:258–70. doi: 10.1002/hbm.21008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Young D, Mayer F, Vidotto N, et al. Mutant huntingtin gene-dose impacts on aggregate deposition, DARPP32 expression and neuroinflammation in HdhQ150 mice. PloS One. 2013;8:e75108. doi: 10.1371/journal.pone.0075108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bjorkqvist M, Wild EJ, Thiele J, et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. J Exper Med. 2008;205:1869–77. doi: 10.1084/jem.20080178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sapp E, Kegel KB, Aronin N, et al. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exper Neurol. 2001;60:161–72. doi: 10.1093/jnen/60.2.161. [DOI] [PubMed] [Google Scholar]
- 63.Bradford J, Shin JY, Roberts M, Wang CE, Li XJ, Li S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc Natl Acad Sci U S A. 2009;106:22480–5. doi: 10.1073/pnas.0911503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tong X, Ao Y, Faas GC, et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington's disease model mice. Nat Neurosci. 2014;17:694–703. doi: 10.1038/nn.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang N, Gray M, Lu XH, et al. Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington's disease. Nat Med. 2014;20:536–41. doi: 10.1038/nm.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jacobsen JC, Gregory GC, Woda JM, et al. HD CAG-correlated gene expression changes support a simple dominant gain of function. Hum Mol Genet. 2011;20:2846–60. doi: 10.1093/hmg/ddr195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Holter SM, Stromberg M, Kovalenko M, et al. A broad phenotypic screen identifies novel phenotypes driven by a single mutant allele in Huntington's disease CAG knock-in mice. PloS One. 2013;8:e80923. doi: 10.1371/journal.pone.0080923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Southwell AL, Warby SC, Carroll JB, et al. A fully humanized transgenic mouse model of Huntington disease. Hum Mol Genet. 2013;22:18–34. doi: 10.1093/hmg/dds397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kolodziejczyk K, Parsons MP, Southwell AL, Hayden MR, Raymond LA. Striatal synaptic dysfunction and hippocampal plasticity deficits in the hu97/18 mouse model of huntington disease. PloS One. 2014;9:e94562. doi: 10.1371/journal.pone.0094562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Graham RK, Slow EJ, Deng Y, et al. Levels of mutant huntingtin influence the phenotypic severity of Huntington disease in YAC128 mouse models. Neurobiol Dis. 2006;21:444–55. doi: 10.1016/j.nbd.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 71.Squitieri F, Gellera C, Cannella M, et al. Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain. 2003;126:946–55. doi: 10.1093/brain/awg077. [DOI] [PubMed] [Google Scholar]
- 72.Lazar AS, Panin F, Goodman AO, et al. Sleep deficits but no metabolic deficits in premanifest Huntington's disease. Ann Neurol. 2015;78:630–48. doi: 10.1002/ana.24495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.