

Abstract
Prokaryotes are frequently exposed to potentially harmful invasive nucleic acids from phages, plasmids, and transposons. One method of defense is the CRISPR-Cas adaptive immune system. Diverse CRISPR-Cas systems form distinct ribonucleoprotein effector complexes that target and cleave invasive nucleic acids to provide immunity. The Type III-B Cmr effector complex has been found to target the RNA and DNA of the invader in the various bacterial and archaeal organisms where it has been characterized. Interestingly, the gene encoding the Csx1 protein is frequently located in close proximity to the Cmr1-6 genes in many genomes, implicating a role for Csx1 in Cmr function. However, evidence suggests that Csx1 is not a stably associated component of the Cmr effector complex, but is necessary for DNA silencing by the Cmr system in Sulfolobus islandicus. To investigate the function of the Csx1 protein, we characterized the activity of recombinant Pyrococcus furiosus Csx1 against various nucleic acid substrates. We show that Csx1 is a metal-independent, endoribonuclease that acts selectively on single-stranded RNA and cleaves specifically after adenosines. The RNA cleavage activity of Csx1 is dependent upon a conserved HEPN motif located within the C-terminal domain of the protein. This motif is also key for activity in other known ribonucleases. Collectively, the findings indicate that invader silencing by Type III-B CRISPR-Cas systems relies both on RNA and DNA nuclease activities from the Cmr effector complex as well as on the affiliated, trans-acting Csx1 endoribonuclease.
Keywords: CRISPR, Cas, Csx1, CARF, HEPN, ribonuclease
INTRODUCTION
Prokaryotes have evolved a number of ways to defend themselves from viral attack and plasmid invasion. Among these are adaptive and heritable immune systems, known as CRISPR-Cas systems, which are widespread in both bacteria and archaea (Makarova et al. 2006; Terns and Terns 2011; Sorek et al. 2013; van der Oost et al. 2014; Jackson and Wiedenheft 2015). CRISPR (clustered regularly interspaced short palindromic) loci contain repeat sequences that flank short DNA segments (called spacers) shown to originate from phage genomes or other invasive DNA (Bolotin et al. 2005; Mojica et al. 2005; Pourcel et al. 2005; Barrangou et al. 2007). When foreign DNA is introduced, either by phage infection or plasmid uptake, small fragments of the invasive DNA become integrated within the CRISPR locus as a spacer (Fineran and Charpentier 2012; Nuñez et al. 2014). The primary transcript of the CRISPR locus is processed into multiple unit CRISPR RNAs (crRNAs) (Brouns et al. 2008; Carte et al. 2008). Mature crRNAs each form ribonucleoprotein complexes with associated Cas (CRISPR-associated) proteins, and these complexes then recognize and cleave the foreign nucleic acid that is complementary to the crRNA guide element (Terns and Terns 2011; Westra et al. 2012; Sorek et al. 2013; van der Oost et al. 2014; Jackson and Wiedenheft 2015).
CRISPR-Cas systems have been divided into five major types (I, II, III, IV, V) and at least 16 subtypes defined by the identity and arrangement of the associated cas genes and by differences in crRNA processing and invader silencing mechanisms (Makarova et al. 2011, 2015). The hyperthermophilic archaeon Pyrococcus furiosus (Pfu) harbors three coexisting immune effector crRNP complexes: Type I-A (Csa), Type I-G (Cst), and Type III-B (Cmr), along with seven functional CRISPR loci (Hale et al. 2008; Terns and Terns 2013; Majumdar et al. 2015). There is evidence that the Pfu Csa and Cst effector complexes target DNA (Elmore et al. 2015), while the Cmr complex has been shown to target DNA and RNA in vitro and in vivo (Hale et al. 2009, 2012, 2014; Deng et al. 2013; Spilman et al. 2013; Benda et al. 2014; Ramia et al. 2014; J Elmore, N Sheppard, R Terns, and M Terns, unpubl.).
The Pfu Cmr RNA-targeting mechanism and necessary components have recently been characterized. The Cmr complex consists of Cmr1-6 proteins in association with a single crRNA (Hale et al. 2009; Spilman et al. 2013). The interaction of the Cmr complex with target RNA is guided by crRNA/target RNA complementary base-pairing (Hale et al. 2009, 2012, 2014; Ramia et al. 2014). Multiple Cmr4 subunits, which form the backbone of the complex, mediate cleavage of the bound target RNA at regular 6-nt intervals (Staals et al. 2013; Benda et al. 2014; Hale et al. 2014; Ramia et al. 2014; Taylor et al. 2015). Recent data indicate that the Cmr system of Sulfolobus islandicus is capable of transcription-dependent, plasmid silencing in vivo, although this activity has not been recreated with purified components or characterized in detail (Deng et al. 2013). Transcription-dependent plasmid silencing has also been observed with Pfu in vivo, and short DNAs have been cleaved with recombinant Pfu Cmr complexes in vitro (J Elmore, N Sheppard, R Terns, and M Terns, unpubl.).
Notably, the csx1 gene is tightly evolutionarily linked with Type III CRISPR-Cas systems (Garrett et al. 2011; Makarova et al. 2011; Makarova and Koonin 2013). In Pfu, the csx1 (PF1127) gene is located between the cmr3 (PF1128) and cmr4 (PF1126) genes (Terns and Terns 2013). However, data from in vitro and in vivo assays indicate that Pfu Csx1 is not necessary for Cmr-mediated RNA or DNA targeting (Hale et al. 2009, 2012, 2014; Spilman et al. 2013; J Elmore, N Sheppard, R Terns, and M Terns, unpubl.). On the other hand, in S. islandicus, Csx1 was shown to be necessary for Cmr-mediated, transcription-dependent plasmid silencing in vivo, although the specific role of the Csx1 protein is unknown (Deng et al. 2013).
The crystal structure of Pfu Csx1 was determined (Kim et al. 2013), revealing an elongated structure with clearly identifiable N- and C-terminal domains. The N-terminal domain is composed of two Rossmann-like folds, while the C-terminal domain exhibits reported structural similarity to a winged-helix domain (Fig. 1A). Amino acid sequence alignments of Csx1 homologs reveals that the N-terminal domain is relatively well conserved, while there is minimal homology in the C-terminal domain, except for one short motif, R–X4-6–H, that is diagnostic of the HEPN (higher eukaryotes and prokaryotes nucleotide-binding) domain (Fig. 1B; Anantharaman et al. 2013). While the HEPN domain was originally identified as being fused or associated with a nearby nucleotidyl transferase domain (Grynberg et al. 2003), the HEPN protein superfamily was recently expanded to encompass proteins linked to prokaryotic viral defense systems, including the Type III CRISPR-Cas-associated Csx1 and Csm6 proteins (which belong to the COG1517 superfamily), as well as a number of predicted ribonucleases from toxin/antitoxin (T–A) modules and abortive infection (Abi) systems (Makarova et al. 2012, 2014; Anantharaman et al. 2013).
FIGURE 1.
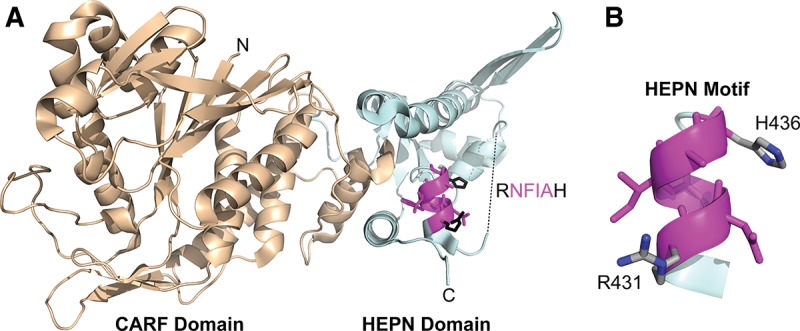
Ribbon diagram of the Pfu Csx1 monomer (PDB 4EOG). (A) (Wheat) The N-terminal modified Rossmannoid fold/CARF domain. (Pale cyan) The C-terminal winged-helix-like domain/HEPN domain. (Magenta) The highly conserved HEPN RxxxxH motif with predicted catalytic residues highlighted in black and middle residues highlighted in magenta. The dashed line represents 17 residues with missing electron density. (B) Isolated HEPN RxxxxH motif with predicted catalytic residues annotated.
The N-terminal Rossmann fold is a unifying feature of a recently proposed family of proteins with largely undefined functions termed CARF (CRISPR-associated Rossmann fold) proteins (Makarova et al. 2014). As Rossmann folds are known (di)nucleotide-binding domains, CARF proteins have been predicted to act as ligand-controlled transcriptional regulators of CRISPR-Cas systems and/or active components of cell defense mechanisms (Lintner et al. 2011; Makarova et al. 2012, 2014; Anantharaman et al. 2013; Liu et al. 2015). Pfu Csx1 was reported to bind double-stranded RNA and DNA in vitro in a sequence-independent manner, although no nucleic acid cleavage activity was reported (Kim et al. 2013). Here, we investigate the activity of Pfu Csx1 in vitro and show that it is a single-strand-specific endoribonuclease that cleaves specifically after adenosines.
RESULTS
Csx1 cleaves single-stranded RNA
CRISPR-Cas systems rely on various nucleases to cleave RNA or DNA targets. To determine if Csx1 is a nuclease, 5′-radiolabeled single-stranded RNA (ssRNA, 37mer A), double-stranded RNA (dsRNA, 37mers A + B), ssDNA (63mer A), dsDNA (63mers A + B), and an RNA/DNA hybrid (45mers A + D) were treated with purified recombinant His-tagged Csx1 (Fig. 2A and see Table 1 for sequences of the nucleic acids used in this and all other experiments). The ssRNA was efficiently cleaved, but none of the other substrates showed significant cleavage, and no cleavage was observed in the absence of Csx1. The small amount of dsRNA and RNA/DNA hybrid cleavage observed is likely due to limited formation of ssRNA in these samples caused by strand separation during incubation at 60°C. The results indicate that recombinant Csx1 has cleavage activity that is specific for ssRNA.
FIGURE 2.
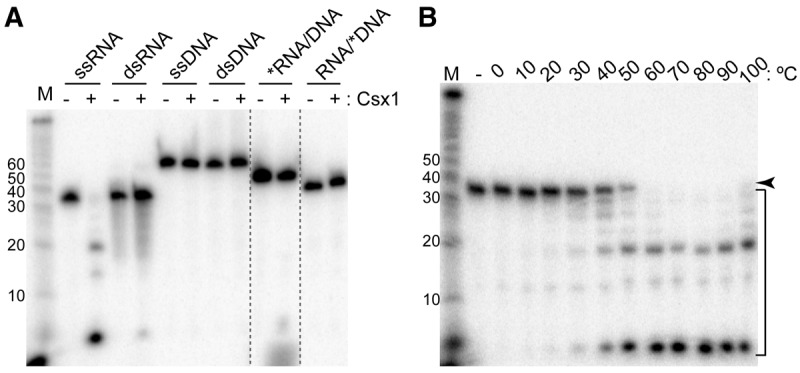
Csx1 is a temperature-dependent, single-strand-specific ribonuclease. (A) Csx1 was tested for nuclease activity (+) on 32P-labeled single-stranded and double-stranded RNA and DNA (37mer A, 63mer A, 37mer A + B, and 63mer A + B, respectively), as well as RNA/DNA hybrids (45mer A + D), which were resolved by denaturing gel electrophoresis alongside no-protein controls (−). See Table 1 for RNA and DNA sequences. Asterisk indicates the labeled oligonucleotide. Size standard (M) is measured in nucleotides. Two lanes not contiguous in the original gel are juxtaposed (dotted lines). (B) 32P-labeled ssRNA was incubated without (−) or with Csx1 across a range of temperatures, then resolved by denaturing gel electrophoresis. The arrow indicates the full-length RNA, while the bracket indicates Csx1 cleavage products.
TABLE 1.
Sequences of RNA and DNA substrates used in this study

Proteins from hyperthermophiles, like Pfu, typically function optimally at elevated temperatures. We determined the optimal temperature for ssRNA cleavage by the Csx1 enzyme by performing the reaction across a wide range of temperatures (Fig. 2B). This analysis showed that Csx1 performs optimally at or above 60°C and was highly active even at 100°C. Under conditions where almost all of the full-length input ssRNA (37mer A) was cleaved, shorter cleavage products persisted, suggesting that Csx1 has a limited substrate specificity. Previous work by others had shown that specific mutations in the conserved HEPN motif (R–X4-6–H, where X is any amino acid) of other known ribonucleases abolished or abrogated the cleavage activity, indicating that this highly conserved motif acted as an RNase active site. Specifically, it was shown that mutation of the conserved histidine eliminates the RNase activity of bacterial antiviral tRNA ribonucleases PrrC (Meineke et al. 2011; Meineke and Shuman 2012) and RloC (Davidov and Kaufmann 2008), as well as eukaryotic Ire1 and antiviral RNase L (Dong et al. 2001; Lee et al. 2008; Han et al. 2014). Mutating the conserved arginine of PrrC (Meineke et al. 2011) or Ire1 (Dong et al. 2001) also blocks catalytic activity.
We tested the prediction that the conserved motif present in the C-terminal domain of Csx1 proteins is responsible for the RNA cleavage activity of Csx1 by mutating the highly conserved residues (R431A and H436A) individually, as well as in combination (Fig. 3A). An equal concentration of wild-type or mutant Csx1 (Fig. 3B) was used in a reaction with ssRNA (37mer A), with time points taken at 1 min and 30 min (Fig. 3A). A similar cleavage pattern was observed for both wild-type and R431A Csx1 mutant; however, the rate of cleavage was significantly reduced for the mutant protein (note that at the 30 min time point, nearly all RNA was cleaved by the wild-type protein, but only a small fraction was cleaved by the mutant protein). In contrast, the activity of the Csx1 protein was abolished by H436A and R431A + H436A mutations. These observations suggest that the conserved HEPN-associated, R–X4-6–H motif found in the C-terminal domain, is critical for the ribonuclease activity of Csx1.
FIGURE 3.
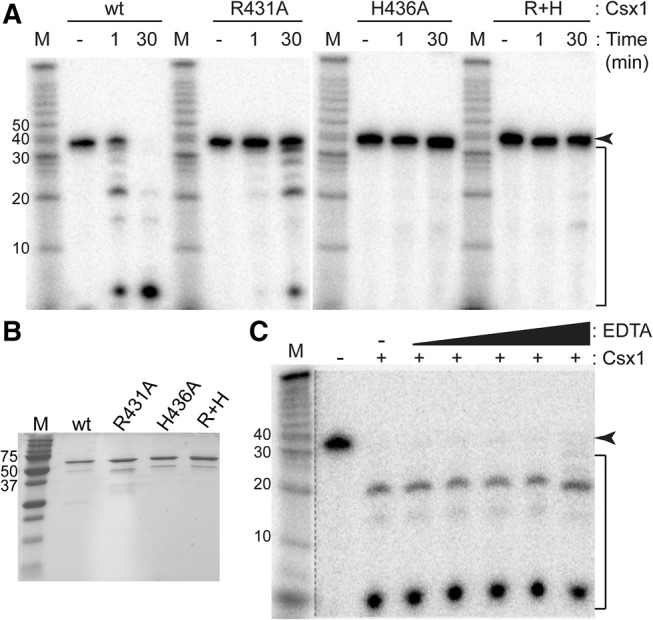
Mutations of highly conserved residues in the HEPN domain affect RNase activity. (A) Radiolabeled ssRNA (37mer A) was incubated with no protein for 30 min (−), with wild-type (wt) or mutant Csx1 for 1 min (1) or for 30 min (30), followed by separation by denaturing gel electrophoresis. The arrow indicates the full-length RNA, while the bracket indicates Csx1 cleavage products. (B) Purified wt and mutant Csx1 proteins were analyzed by SDS-PAGE and Coomassie blue staining. Molecular weight marker is indicated in kilodaltons. (C) Csx1 cleavage activity occurs in the absence of added metal ions (−EDTA) and in the presence of a wide range (0.5, 1, 200, 500, 1000 µM) of EDTA. The dotted line separates data that was subject to longer exposure times to visualize molecular weight markers.
Cleavage mechanism
The ssRNA cleavage activity of Csx1 appears to be metal ion-independent. The metal independence of the reaction is supported by the observation that RNA cleavage by Csx1 occurs in the absence of added metals in the reaction buffer (Fig. 3A,C). Moreover, the RNA cleavage activity of Csx1 is unaffected by the addition of up to millimolar concentrations of the divalent metal ion chelator EDTA (Fig. 3C). Other characterized HEPN RNases employ a metal ion-independent catalytic mechanism (Anantharaman et al. 2013).
To determine whether Csx1 acts as an exo- or endoribonuclease, we tested whether Csx1 could cleave circular RNAs, as would be expected for an endonuclease but not exonuclease (Fig. 4A). 5′-Radiolabeled ssRNA (67mer) was circularized and treated with Csx1, with time points taken at 1 and 30 min. Terminator 5′-phosphate-dependent exonuclease (TEX), which cleaves RNA with a 5′ phosphate, was used to determine the success of circularization. The linear radiolabeled control RNA was cleaved by TEX as expected, while the circular RNA remained intact (Fig. 4A). After 1 min, the circular substrate exhibited a cleavage product the same size as the full-length linear RNA, suggesting a single cleavage by Csx1. Smaller cleavage products were observed in lower abundance. After 30 min, the input RNA was fully cleaved. Due to the radiolabel on the circular RNA becoming internal, different cleavage products are observed with the circular RNA as compared to the linear RNA. These results indicate that Csx1 acts as an endoribonuclease.
FIGURE 4.
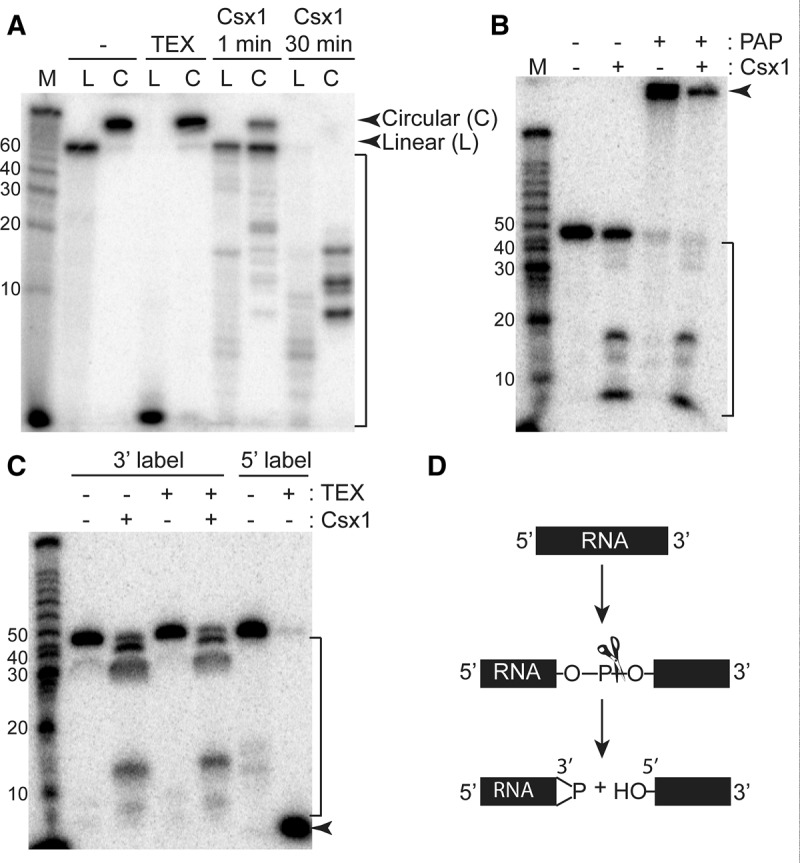
Endonucleolytic cleavage of ssRNA by Csx1. (A) Radiolabeled linear (L) and circular (C) ssRNAs (67mer) were incubated with no protein (−), Terminator 5′-phosphate-dependent exonuclease (TEX), or Csx1 for the indicated time, then resolved by denaturing gel electrophoresis. The full-length linear and circular RNA are indicated by arrows, while the Csx1 cleavage products are indicated by the bracket. (B) 5′-Radiolabeled RNA (45mer A) was treated with no protein (−), Csx1, poly(A) polymerase (PAP), or Csx1 followed by PAP, and resolved by denaturing gel electrophoresis. The arrow indicates RNA elongated by PAP, while the bracket indicates Csx1 cleavage products. (C) 3′-Radiolabeled RNA (45mer A) was treated with no protein (−), with Csx1, with TEX, or with Csx1 followed by Tex, while 5′-radiolabeled RNA was treated with or without TEX. The samples were resolved by denaturing gel electrophoresis. The arrow indicates the expected TEX cleavage product, while the bracket indicates Csx1 cleavage products. (D) A diagram depicting the cleavage method of RNA by Csx1 as suggested by the resistance of the cleavage products to TEX activity and protection from elongation by PAP.
Next, we mapped the 5′ and 3′ end groups of the RNA cleavage products generated by Csx1 cutting (Fig. 4B,C). To this end, 5′-radiolabeled ssRNA (45mer A) was treated with or without Csx1 under reaction conditions that did not go to completion and thus retained some of the uncleaved, full-length RNA species. The RNA products were then treated with poly(A) polymerase (PAP), which adds poly(A) stretches to RNAs with 3′ OH groups (Fig. 4B). In the absence of Csx1 treatment, the full-length RNA was extended by PAP as expected. When incubated in the presence of Csx1, the full-length (uncleaved) RNA in the sample was extended, while the Csx1-generated RNA cleavage products were not extended. This result indicates that the 3′ ends produced by Csx1 cleavage lack a 3′ OH group.
To determine the 5′ end group of Csx1 cleavage products, 3′-radiolabeled ssRNA (45mer A) was treated as described above. The RNA was treated with TEX (5′-3′ exonuclease that selectively digests RNA having a 5′ monophosphate end) to test for the presence of 5′ phosphates on the Csx1 cleavage products (Fig. 4C). Both the full-length RNA and cleavage products were resistant to TEX degradation, while the 5′-radiolabeled control RNA was successfully cleaved as expected. This result indicates that Csx1 cleavage does not result in cleavage products containing 5′ phosphates. Taken together, these data are consistent with Csx1 being a metal-independent endoribonuclease leaving cleavage products with a 5′ OH group and 2′,3′-cyclic phosphate or 3′ phosphate termini (Fig. 4D; Yang 2011).
Sequence specificity
To investigate whether Csx1 cleavage activity had any sequence specificity, we treated all possible RNA homoribopolymers [poly(A), poly(C), poly(G), poly(U)], as well as a poly(C10)/(AUG)3 RNA with Csx1 (Fig. 5 and see Table 1 for sequences of the RNA substrates). We observed robust cleavage of the poly(A) RNA, but no cleavage of the other homoribopolymers. We also observed three products from cleavage of the poly(C10)/(AUG)3 RNA, with sizes consistent with cleavage after each adenosine in the RNA.
FIGURE 5.
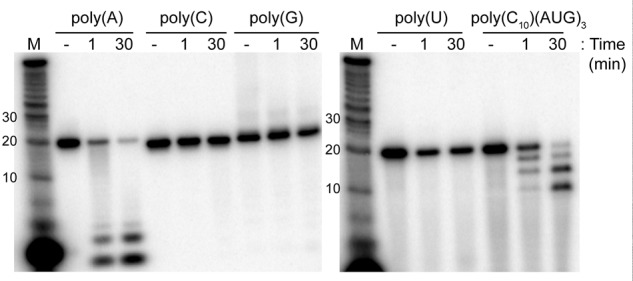
Cleavage of homoribopolymers by Csx1. Radiolabeled RNA homoribopolymers of each ribonucleotide and an RNA composed of 10 cytidylate residues and three repeats of AUG were incubated with no protein (−) or Csx1 for the indicated times, then resolved by denaturing gel electrophoresis.
To get a clearer picture of this apparent base specificity, we treated four “mixed-sequence” RNAs and the poly(C10)/(AUG)3 RNA with Csx1 (Fig. 6A). These were run on sequencing gels, and the cleavage products were mapped at nucleotide resolution to the sequences (Fig. 6B). Alkaline hydrolysis and RNase T1 ladders of each substrate RNA were used in parallel to determine sites of Csx1 cleavage. This mapping revealed that Csx1 cleaved each of the input substrate RNAs after every adenosine in the RNA and not after any other nucleotide.
FIGURE 6.

Csx1 cleaves ssRNA after adenosines. (A) A variety of ssRNAs were treated with no protein (−) or Csx1 for the indicated times, and run alongside 5′-radiolabeled RNA markers (M), RNase T1 ladders (T1), and alkaline hydrolysis ladders (OH). The RNAs were resolved by denaturing sequencing gel electrophoresis. Red arrows indicate Csx1 cleavage products. (B) Cleavage products were mapped back to their respective RNAs. Sites of cleavage are denoted with a red A followed by a dash. No cleavage is mapped after the first A of 45mer B and C because the single nucleotide band was run off the gel. Comparison of the Csx1 ladders with the corresponding T1 ladders confirms that Csx1 cleavage occurs on the 3′ rather than 5′ side of adenosine.
DISCUSSION
Despite its prevalent association with Type III CRISPR-Cas systems (Haft et al. 2005; Garrett et al. 2011; Makarova et al. 2011), the function and activity of Csx1 proteins have remained largely uncharacterized. Here we have experimentally determined that Pfu Csx1 functions as a metal-independent, single-strand-specific endoribonuclease that relies on an HEPN active site found in other characterized RNases (Dong et al. 2001; Davidov and Kaufmann 2008; Lee et al. 2008; Meineke et al. 2011; Meineke and Shuman 2012; Anantharaman et al. 2013). The RNase activity of Csx1 was previously anticipated based on the occurrence of the highly conserved HEPN motif in Csx1 homologs by sequence analysis (Makarova et al. 2012; Anantharaman et al. 2013).
Interestingly, we found that Pfu Csx1 cleaves specifically after adenosines (Figs. 5, 6). An RNase with complete specificity for adenosines has not been reported. While the RNases T2 and U2 have been shown to have a preference for adenosines, they have also been found to cleave after other nucleotides, and U2 cleavage is highly dependent on the adjacent nucleotides (Rogg and Staehelin 1972; Yasuda and Inoue 1982; Deshpande and Shankar 2002; MacIntosh 2011). In contrast, Pf Csx1 shows remarkable specificity for cleaving diverse RNA substrates at sites containing an adenosine in several sequence contexts (Fig. 6). The novel specificity of Pf Csx1 as an adenosine-specific RNA cleaving enzyme has the potential to be leveraged as a useful molecular tool. Analogous to the commonly used RNase T1 enzyme that specifically cleaves RNAs after guanine (Sato and Egami 1957), Csx1 has the potential to be used in determining RNA sequence, mapping cleavage sites of other ribonucleases, and leaving RNAs with 3′-terminal adenosines, among other potentially useful applications.
Our mutational analysis of the HEPN R–X4-6–H motif of Csx1 confirms that the highly conserved arginine and histidine are important for RNase activity (as shown with other studied HEPN RNases) and provides insight into the possible catalytic mechanism of the enzyme (Fig. 3; Dong et al. 2001; Davidov and Kaufmann 2008; Lee et al. 2008; Meineke et al. 2011; Meineke and Shuman 2012; Anantharaman et al. 2013). Consistent with findings for other HEPN RNases (Anantharaman et al. 2013), our results support a metal ion-independent cleavage mechanism for Csx1, generating RNA fragments with 5′ hydroxyl and 2′,3′-cyclic phosphate termini (Figs. 3, 4). Based on the proposed general acid–base catalytic mechanism of other HEPN RNases (Anantharaman et al. 2013), the predicted Csx1 active site His436 likely functions as a general base to deprotonate the nucleophilic 2′-hydroxyl of the ribose ring leading to an attack of the 2′ oxygen on the phosphate backbone. Alternatively or additionally, His436 may act as a general acid to protonate the 5′ oxyanion leaving group to facilitate cleavage of the scissile phosphate. We found that mutation of Csx1 His436 abolished activity, while mutation of the predicted active site Arg431 residue significantly impaired, but did not prevent, RNA cleavage by the Csx1 enzyme (Fig. 3). The role of the arginine may be charge stabilization of the predicted pentavalent transition state during the cleavage reaction or interaction with the backbone of the RNA substrate. A Csx1-specific HEPN motif consensus motif was determined as R–N–X–θ–A–H (Kim et al. 2013), suggesting that the identity of the residues flanking the broadly conserved R and H residues may also be important for Csx1 activity.
Csx1 is structurally related to the Csm6 protein, and, by inference, our results make a strong prediction that Csm6 also exhibits single-strand-specific RNase activity. Indeed, the many shared features of Csx1 and Csm6 indicate that these proteins perform similar or identical functional roles. Csx1 and Csm6 are each CARF proteins that harbor N-terminal Rossman fold domains and C-terminal domains containing the R–X4-6–H HEPN RNase active site (Makarova et al. 2012, 2014; Anantharaman et al. 2013). The csx1 and csm6 genes are evolutionarily linked to Type III-B (Cmr) and Type III-A (Csm) CRISPR-Cas systems, respectively (Garrett et al. 2011; Makarova and Koonin 2013), indicating these two protein families cofunction with Type III CRISPR-Cas systems, which are known to cleave both target (e.g., viral) RNA as well as target DNA in a transcription-dependent manner (Hale et al. 2009; Marraffini and Sontheimer 2010; Zhang et al. 2012; Deng et al. 2013; Staals et al. 2013, 2014; Hale et al. 2014; Hatoum-Aslan et al. 2014; Ramia et al. 2014; Tamulaitis et al. 2014; Samai et al. 2015).
The function of Csx1 and Csm6 Cas proteins remains enigmatic. Intriguingly, evidence has emerged that both csx1 and csm6 genes are vital for transcription-dependent plasmid interference in vivo (Deng et al. 2013; Hatoum-Aslan et al. 2014), despite clear evidence in vitro that both Csx1 and Csm6 proteins are dispensible for target RNA cleavage (Hale et al. 2009, 2014; Zhang et al. 2012; Staals et al. 2013, 2014; Ramia et al. 2014; Tamulaitis et al. 2014; Samai et al. 2015) as well as for transcription-dependent target DNA cleavage (Samai et al. 2015). Furthermore, Csx1 and Csm6 are not required for the proper processing or maturation of crRNAs (Hatoum-Aslan et al. 2014; J Elmore, N Sheppard, R Terns, and M Terns, unpubl.), and neither protein is stably associated with its affiliated multisubunit Cmr or Csm crRNP effector complex, respectively (Hale et al. 2009; Hatoum-Aslan et al. 2014). These observations indicate that Csx1 and Csm6 may play a role in antiviral defense that is auxiliary to that of the evolutionarily linked Cmr and Csm effector crRNPs.
Our results indicate a possible key role for RNase activity in the functioning of Csx1 and Csm6 CARF proteins. Conceivably, Csx1 and Csm6 are regulated to selectively destroy invasive RNAs (e.g., viral mRNAs) either in addition to, or in conjunction with, the crRNP-guided Type III effector complexes. Another intriguing proposal is that these CARF proteins may cleave (certain) host RNAs to act as dormancy/suicide inducers in the event the CRISPR defense mechanism fails to dispel the invader in a timely manner (Makarova et al. 2012; Anantharaman et al. 2013). It is not clear how Csx1 or Csm6 RNase activity might affect transcription-dependent DNA silencing activity of Cmr and Csm effector complexes or whether the observed adenosine-specific cleavage by Csx1 (Figs. 5, 6) is significant for its physiological function.
Understanding how Csx1 (and related Csm6) activity is regulated remains an important challenge. In general, the activity of cellular ribonucleases is tightly controlled such that they cleave only their intended substrates. We have found that Csx1 protein is constitutively expressed in Pfu cells (N Sheppard, M Ellis, R Terns, and M Terns, unpubl.), suggesting that Csx1 activity may be post-translationally controlled in vivo. Indeed, the N-terminal CARF domain of Csx1 (Kim et al. 2013) is predicted to interact with a yet-to-be-determined (di)nucleotide that may allosterically regulate Csx1 cleavage activity, perhaps in response to viral infection and associated nucleotide metabolites that might be triggered in response to the invasion (Lintner et al. 2011; Makarova et al. 2012, 2014; Anantharaman et al. 2013). The oligomeric state of the protein may represent an additional point of control for the activity of Csx1 (and Csm6). Monomeric Pfu Csx1 was found to homodimerize following binding to dsDNA, bringing the HEPN RNase active sites in close proximity to one another (Kim et al. 2013). This raises the possibility that there is a nucleic acid regulator of Csx1 function.
Additional studies are required to define the detailed mechanism of action of Csx1 in prokaryotic cell defense mechanisms and to determine how Csx1 activity is regulated.
MATERIALS AND METHODS
Purification of Csx1
The gene encoding P. furiosus Csx1 (PF1127) was amplified by PCR from genomic DNA and cloned into a modified form of pET24d. N-terminal, 6x-histidine-tagged Csx1 protein was expressed in Escherichia coli BL21-RIPL cells (DE3, Stratagene). Cells (1 L culture) were grown to an OD600 of 0.7, and protein expression was induced overnight at room temperature by the addition of isopropylthio-β-D galactoside (IPTG) to a final concentration of 1 mM. The cells were resuspended in native binding buffer (NBB; 50 mM sodium phosphate [pH 7.6], 500 mM NaCl, and 0.1 mM phenylmethylsulfonyl fluoride) and were disrupted by sonication (Misonix Sonicator 3000). The lysate was cleared by centrifugation at 6000 rpm for 10 min, followed by incubation at 70°C for 20 min. The sample was centrifuged at 9000 rpm for 10 min, syringe-filtered (Corning Incorporated, 0.80 μm), and applied to a HisTrap HP column (GE Healthcare) that had been equilibrated with NBB. The protein was eluted from the column using NBB containing increasing concentrations of imidazole (50, 100, 200, and 500 mM). Fractions were evaluated by SDS-PAGE and staining with Coomassie blue. The peak fraction of Csx1 was further purified by gel filtration using an XK26 HiLoad 26/60 Superdex 200 gel filtration column (GE Healthcare) that had been equilibrated with 2× assay buffer (40 mM Tris–HCl [pH 7.5] and 200 mM NaCl).
Generation of RNA and DNA substrates
Synthetic RNAs were purchased from Integrated DNA Technologies (IDT), DNA oligos from Eurofins MWG Operon, and the RNA size standards (Decade Markers) from Life Technologies. The sequences of the RNAs used in this study are given in Table 1. The oligonucleotides were 5′ end-labeled with T4 polynucleotide kinase (New England Biolabs [NEB]) in a 20 μL reaction containing 20 pmol oligonucleotide, 150 μCi of [γ-32P] ATP (6000 Ci/mmol; Perkin Elmer), 1× T4 PNK buffer, and 10 U of T4 kinase (NEB). RNAs were 3′ end-labeled with T4 RNA ligase (NEB) in a 20 μL reaction containing 20 pmol RNA, 10 μCi of [α-32P] pCp (3000 Ci/mmol; Perkin Elmer), 20 U of T4 ligase, 10 U of SUPERase-IN RNase inhibitor (Ambion), 1× T4 RNA ligase buffer (NEB), and 20% polyethylene glycol M.W. 8000 (NEB). The oligonucleotides were then run on a denaturing (7 M urea) 15% polyacrylamide gel containing 1× TBE (89 mM Tris base, 89 mM Boric acid, 2 mM EDTA, pH 8.0), followed by autoradiographic exposure to guide excision of the appropriate bands. The oligonucleotides were eluted by end-over-end rotation for 12–14 h at 4°C in 500 μL of 2× assay buffer. This was followed by phenol/chloroform/isoamyl alcohol (PCI, 25:24:1 at pH 5.2; Fisher Biosciences) extraction, then precipitation with 2.5 volumes of 100% ethanol, 0.3 M sodium acetate, and 20 μg glycogen after incubation for 30 min at −80°C.
Double-stranded oligonucleotides were created by mixing labeled oligonucleotides with a twofold molar excess of nonlabeled complement in 30 mM HEPES (pH 7.4), 100 mM potassium acetate, 2 mM magnesium acetate and incubating for 1 min at 95°C, followed by temperatures decreasing by 1° each minute, down to 23°C. Annealing was confirmed and substrates were purified following electrophoresis on nondenaturing 15% polyacrylamide gels. Double-stranded substrates were then removed, eluted, extracted, and precipitated as described above, but PCI of pH 8.0 was used.
Circular RNAs were created using 5′ end-labeled RNA (67mer A), as described above, in a 20 μL reaction containing ∼10 pmol RNA, 20 μg BSA, 1 mM ATP, 20 U of T4 ligase, 10 U of SUPERase-IN RNase inhibitor, and 1× T4 RNA ligase buffer. Circularization was confirmed and circular RNA was purified with denaturing (8.3 M urea) 20% polyacrylamide gels in TBE. The circular RNA was then removed, eluted, extracted, and precipitated as described above.
Nuclease assays
Assays were carried out in 20 μL reactions made up of 1× assay buffer (20 mM Tris–HCl [pH 7.5 at room temperature] and 100 mM NaCl) with 500 nM Csx1, as determined by Qubit 2.0 Fluorometer (Life Technologies) quantification, and 5000 cpm (∼15–20 fmol) of oligonucleotide at 70°C for 30 min, unless otherwise noted in Results. Assays involving double-stranded nucleic acids were incubated at 60°C to reduce heat-induced strand separation. Reactions were stopped by placing tubes on ice and adding an equal volume of Gel Loading Buffer II (95% formamide, 18 mM EDTA, and 0.025% SDS, Xylene Cyanol, and Bromophenol Blue; Life Technologies). The reaction products were separated by electrophoresis on either 15% (7.0 M urea, linear substrates) or 20% (8.3 M urea, circular RNAs) denaturing polyacrylamide gels. Radiolabeled Decade Markers (Life Technologies) were used to determine the sizes of observed products. For sequencing gels, partial alkaline hydrolysis (cleaves phosphodiester linkages) and RNase T1 (cleaves after guanylate residues) ladders (Ambion) were generated using single-hit conditions, as described by the manufacturer. Gels were dried, and radiolabeled substrates were visualized by phosphorimaging.
Creation of Csx1 mutants
QuikChange site-directed mutagenesis (Stratagene) was used to create site-specific mutations in the csx1 gene. The R431A mutant was generated using the primers 5′-gacaatagaatctccaaatgttgttgctaactttatagcacattctggattt-3′ and 5′-aaatccagaatgtgctataaagttagcaacaacatttggagattctattgtc-3′. The H436A mutant was generated using the primers 5′ caaatgttgttcgtaactttatagcagcttctggatttgagtataacattgtct-3′ and 5′-agacaatgttatactcaaatccagaagctgctataaagttacgaacaacatttg-3′. The R431A + H436A double mutant was generated using primers 5′-gacaatagaatctccaaatgttgttgctaactttatagcagcttctggattt-3′ and 5′-aaatccagaagctgctataaagttagcaacaacatttggagattctattgtc-3′ using the plasmid encoding the R431A csx1 mutant gene. Mutations were confirmed by sequencing. The mutant proteins were expressed as described above and purified using a Ni-NTA agarose column (Qiagen).
End-group analysis for cleaved RNA
Circular, 5′ end-labeled, and 3′ end-labeled RNAs were treated with Csx1, as described above. Products of circular and 3′ end-labeled RNA were treated with 1 U Terminator Exonuclease (TEX; EpiBio), 1× terminator reaction buffer B (EpiBio), and 10 U of SUPERase-IN RNase inhibitor and incubated at 42°C for 30 min. Products of 5′ end-labeled RNA were treated with 5 U E. coli poly(A) polymerase (PAP; NEB), 1× PAP reaction buffer (NEB), and 10 U of SUPERase-IN RNase inhibitor and incubated at 37°C for 20 min. Reactions were stopped by placing on ice and adding an equal volume of Gel Loading Buffer II (Life Technologies). The reaction products were separated by electrophoresis on denaturing 15% or 20% polyacrylamide as described above.
ACKNOWLEDGMENTS
We thank the members of the Terns laboratory and Sidney Kushner for meaningful input, discussion, and feedback. This work was supported by National Institutes of Health grant GM54682 to M.P.T. and R.M.T.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.039842.113.
REFERENCES
- Anantharaman V, Makarova KS, Burroughs AM, Koonin EV, Aravind L. 2013. Comprehensive analysis of the HEPN superfamily: identification of novel roles in intra-genomic conflicts, defense, pathogenesis and RNA processing. Biol Direct 8: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315: 1709–1712. [DOI] [PubMed] [Google Scholar]
- Benda C, Ebert J, Scheltema RA, Schiller HB, Baumgartner M, Bonneau F, Mann M, Conti E. 2014. Structural model of a CRISPR RNA-silencing complex reveals the RNA-target cleavage activity in Cmr4. Mol Cell 56: 43–54. [DOI] [PubMed] [Google Scholar]
- Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. 2005. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151: 2551–2561. [DOI] [PubMed] [Google Scholar]
- Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. 2008. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321: 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carte J, Wang R, Li H, Terns RM, Terns MP. 2008. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev 22: 3489–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidov E, Kaufmann G. 2008. RloC: a wobble nucleotide-excising and zinc-responsive bacterial tRNase. Mol Microbiol 69: 1560–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Garrett RA, Shah SA, Peng X, She Q. 2013. A novel interference mechanism by a type IIIB CRISPR-Cmr module in Sulfolobus. Mol Microbiol 87: 1088–1099. [DOI] [PubMed] [Google Scholar]
- Deshpande RA, Shankar V. 2002. Ribonucleases from T2 family. Crit Rev Microbiol 28: 79–122. [DOI] [PubMed] [Google Scholar]
- Dong B, Niwa M, Walter P, Silverman RH. 2001. Basis for regulated RNA cleavage by functional analysis of RNase L and Ire1p. RNA 7: 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore J, Deighan T, Westpheling J, Terns RM, Terns MP. 2015. DNA targeting by the type I-G and type I-A CRISPR-Cas systems of Pyrococcus furiosus. Nucleic Acids Res 10.1093/nar/gkv1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineran PC, Charpentier E. 2012. Memory of viral infections by CRISPR-Cas adaptive immune systems: acquisition of new information. Virology 434: 202–209. [DOI] [PubMed] [Google Scholar]
- Garrett RA, Vestergaard G, Shah SA. 2011. Archaeal CRISPR-based immune systems: exchangeable functional modules. Trends Microbiol 19: 549–556. [DOI] [PubMed] [Google Scholar]
- Grynberg M, Erlandsen H, Godzik A. 2003. HEPN: a common domain in bacterial drug resistance and human neurodegenerative proteins. Trends Biochem Sci 28: 224–226. [DOI] [PubMed] [Google Scholar]
- Haft DH, Selengut J, Mongodin EF, Nelson KE. 2005. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol 1: e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale C, Kleppe K, Terns RM, Terns MP. 2008. Prokaryotic silencing (psi)RNAs in Pyrococcus furiosus. RNA 14: 2572–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, Terns MP. 2009. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139: 945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CR, Majumdar S, Elmore J, Pfister N, Compton M, Olson S, Resch AM, Glover CV III, Graveley BR, Terns RM, et al. 2012. Essential features and rational design of CRISPR RNAs that function with the Cas RAMP module complex to cleave RNAs. Mol Cell 45: 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CR, Cocozaki A, Li H, Terns RM, Terns MP. 2014. Target RNA capture and cleavage by the Cmr type III-B CRISPR-Cas effector complex. Genes Dev 28: 2432–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Donovan J, Rath S, Whitney G, Chitrakar A, Korennykh A. 2014. Structure of human RNase L reveals the basis for regulated RNA decay in the IFN response. Science 343: 1244–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatoum-Aslan A, Maniv I, Samai P, Marraffini LA. 2014. Genetic characterization of antiplasmid immunity through a type III-A CRISPR-Cas system. J Bacteriol 196: 310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RN, Wiedenheft B. 2015. A conserved structural chassis for mounting versatile CRISPR RNA-guided immune responses. Mol Cell 58: 722–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Kim YG, Oh BH. 2013. Crystal structure and nucleic acid-binding activity of the CRISPR-associated protein Csx1 of Pyrococcus furiosus. Proteins 81: 261–270. [DOI] [PubMed] [Google Scholar]
- Lee KP, Dey M, Neculai D, Cao C, Dever TE, Sicheri F. 2008. Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell 132: 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lintner NG, Frankel KA, Tsutakawa SE, Alsbury DL, Copie V, Young MJ, Tainer JA, Lawrence CM. 2011. The structure of the CRISPR-associated protein Csa3 provides insight into the regulation of the CRISPR/Cas system. J Mol Biol 405: 939–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Li Y, Wang X, Ye Q, Li H, Liang Y, She Q, Peng N. 2015. Transcriptional regulator-mediated activation of adaptation genes triggers CRISPR de novo spacer acquisition. Nucleic Acids Res 43: 1044–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIntosh GC. 2011. RNase T2 family: enzymatic properties, functional diversity, and evolution of ancient ribonucleases. In Ribonucleases (ed. Nicholson AW), pp. 89–114. Springer, Berlin/Heidelberg. [Google Scholar]
- Majumdar S, Zhao P, Pfister NT, Compton M, Olson S, Glover CV III, Wells L, Graveley BR, Terns RM, Terns MP. 2015. Three CRISPR-Cas immune effector complexes coexist in Pyrococcus furiosus. RNA 21: 1147–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Koonin EV. 2013. Evolution and classification of CRISPR-Cas systems and cas protein families. In CRISPR-Cas systems (ed. Barrangou R, van der Oost J), pp. 61–91. Springer, Berlin/Heidelberg. [Google Scholar]
- Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. 2006. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct 1: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, et al. 2011. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol 9: 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Anantharaman V, Aravind L, Koonin EV. 2012. Live virus-free or die: coupling of antivirus immunity and programmed suicide or dormancy in prokaryotes. Biol Direct 7: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Anantharaman V, Grishin NV, Koonin EV, Aravind L. 2014. CARF and WYL domains: ligand-binding regulators of prokaryotic defense systems. Front Genet 5: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, et al. 2015. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol 13: 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, Sontheimer EJ. 2010. Self vs. non-self discrimination during CRISPR RNA-directed immunity. Nature 463: 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meineke B, Shuman S. 2012. Structure-function relations in the NTPase domain of the antiviral tRNA ribotoxin Escherichia coli PrrC. Virology 427: 144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meineke B, Schwer B, Schaffrath R, Shuman S. 2011. Determinants of eukaryal cell killing by the bacterial ribotoxin PrrC. Nucleic Acids Res 39: 687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJ, Diez-Villaseñor C, Garcia-Martínez J, Soria E. 2005. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60: 174–182. [DOI] [PubMed] [Google Scholar]
- Nuñez JK, Kranzusch PJ, Noeske J, Wright AV, Davies C, Doudna JA. 2014. Cas1-Cas2 complex formation mediates spacer acquisition during CRISPR-Cas adaptive immunity. Nat Struct Mol Biol 21: 528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourcel C, Salvignol G, Vergnaud G. 2005. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151: 653–663. [DOI] [PubMed] [Google Scholar]
- Ramia NF, Spilman M, Tang L, Shao Y, Elmore J, Hale C, Cocozaki A, Bhattacharya N, Terns RM, Terns MP, et al. 2014. Essential structural and functional roles of the Cmr4 subunit in RNA cleavage by the Cmr CRISPR-Cas complex. Cell Rep 9: 1610–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogg H, Staehelin M. 1972. On the specificity of ribonuclease U2. Biochim Biophys Acta 262: 314–319. [DOI] [PubMed] [Google Scholar]
- Samai P, Pyenson N, Jiang W, Goldberg GW, Hatoum-Aslan A, Marraffini LA. 2015. Co-transcriptional DNA and RNA cleavage during type III CRISPR-Cas immunity. Cell 161: 1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Egami F. 1957. Studies on ribonucleases in takadiastase. J Biochem 44: 753–767. [Google Scholar]
- Sorek R, Lawrence CM, Wiedenheft B. 2013. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu Rev Biochem 82: 237–266. [DOI] [PubMed] [Google Scholar]
- Spilman M, Cocozaki A, Hale C, Shao Y, Ramia N, Terns R, Terns M, Li H, Stagg S. 2013. Structure of an RNA silencing complex of the CRISPR-Cas immune system. Mol Cell 52: 146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staals RH, Agari Y, Maki-Yonekura S, Zhu Y, Taylor DW, van Duijn E, Barendregt A, Vlot M, Koehorst JJ, Sakamoto K, et al. 2013. Structure and activity of the RNA-targeting type III-B CRISPR-Cas complex of Thermus thermophilus. Mol Cell 52: 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staals RH, Zhu Y, Taylor DW, Kornfeld JE, Sharma K, Barendregt A, Koehorst JJ, Vlot M, Neupane N, Varossieau K, et al. 2014. RNA targeting by the type III-A CRISPR-Cas Csm complex of Thermus thermophilus. Mol Cell 56: 518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamulaitis G, Kazlauskiene M, Manakova E, Venclovas C, Nwokeoji AO, Dickman MJ, Horvath P, Siksnys V. 2014. Programmable RNA shredding by the type III-A CRISPR-Cas system of Streptococcus thermophilus. Mol Cell 56: 506–517. [DOI] [PubMed] [Google Scholar]
- Taylor DW, Zhu Y, Staals RH, Kornfeld JE, Shinkai A, van der Oost J, Nogales E, Doudna JA. 2015. Structures of the CRISPR-Cmr complex reveal mode of RNA target positioning. Science 348: 581–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terns MP, Terns RM. 2011. CRISPR-based adaptive immune systems. Curr Opin Microbiol 14: 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terns RM, Terns MP. 2013. The RNA- and DNA-targeting CRISPR-Cas immune systems of Pyrococcus furiosus. Biochem Soc Trans 41: 1416–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Oost J, Westra ER, Jackson RN, Wiedenheft B. 2014. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat Rev Microbiol 12: 479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra ER, Swarts DC, Staals RH, Jore MM, Brouns SJ, van der Oost J. 2012. The CRISPRs, they are a-changin’: how prokaryotes generate adaptive immunity. Annu Rev Genet 46: 311–339. [DOI] [PubMed] [Google Scholar]
- Yang W. 2011. Nucleases: diversity of structure, function and mechanism. Q Rev Biophys 44: 1–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Inoue Y. 1982. Studies of catalysis by ribonuclease U2. Steady-state kinetics for transphosphorylation of oligonucleotide and synthetic substrates. Biochemistry 21: 364–369. [DOI] [PubMed] [Google Scholar]
- Zhang J, Rouillon C, Kerou M, Reeks J, Brugger K, Graham S, Reimann J, Cannone G, Liu H, Albers SV, et al. 2012. Structure and mechanism of the CMR complex for CRISPR-mediated antiviral immunity. Mol Cell 45: 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]