

Abstract
Approximately 75% of the human genome is transcribed and many of these spliced transcripts contain primate-specific Alu elements, the most abundant mobile element in the human genome. The majority of exonized Alu elements are located in long noncoding RNAs (lncRNAs) and the untranslated regions of mRNA, with some performing molecular functions. To further assess the potential for Alu elements to be repurposed as functional RNA domains, we investigated the distribution and evolution of Alu elements in spliced transcripts. Our analysis revealed that Alu elements are underrepresented in mRNAs and lncRNAs, suggesting that most exonized Alu elements arising in the population are rare or deleterious to RNA function. When mRNAs and lncRNAs retain exonized Alu elements, they have a clear preference for Alu dimers, left monomers, and right monomers. mRNAs often acquire Alu elements when their genes are duplicated within Alu-rich regions. In lncRNAs, reverse-oriented Alu elements are significantly enriched and are not restricted to the 3′ and 5′ ends. Both lncRNAs and mRNAs primarily contain the Alu J and S subfamilies that were amplified relatively early in primate evolution. Alu J subfamilies are typically overrepresented in lncRNAs, whereas the Alu S dimer is overrepresented in mRNAs. The sequences of Alu dimers tend to be constrained in both lncRNAs and mRNAs, whereas the left and right monomers are constrained within particular Alu subfamilies and classes of RNA. Collectively, these findings suggest that Alu-containing RNAs are capable of forming stable structures and that some of these Alu domains might have novel biological functions.
Keywords: Alu, RNA, lincRNA, lncRNA, noncoding
INTRODUCTION
Approximately 75% of the human genome is transcribed (Djebali et al. 2012) and many of these transcripts contain repetitive elements. Repetitive elements are located in the 5′ and 3′ UTRs of many mRNAs (Yulug et al. 1995; Lin et al. 2009) and are also a major component of long noncoding RNAs (lncRNAs) (Kelley and Rinn 2012; Kapusta et al. 2013). In particular, the Alu elements are known to perform molecular functions in mRNAs and some lncRNAs. For example, exonized Alu RNAs (defined here as Alu elements contained within exons) interact with proteins that regulate RNA editing, staufen-mediated RNA decay, translation, and transcription (Ricci et al. 2000; Berger and Strub 2010; Gong and Maquat 2011; Yang et al. 2013).
Alu elements are the most abundant mobile element in the human genome and are unique to primates (for review, see Batzer and Deininger 2002; Berger and Strub 2010; Deininger 2011 and summarized below). Within the major Alu subfamilies, the AluJ and AluS subfamilies were primarily amplified 35–55 million years ago, whereas the active AluY subfamily was primarily amplified 5–10 million years ago. As these elements do not contain ORFs, their amplification in the genome is dependent on trans-acting factors encoded by LINE-1 mobile elements. These insertion events can disrupt a coding region or a splice signal and cause disease (Deininger 2011). Alu elements are ∼280 bases long and typically consist of monomeric left and right arms joined by an A-rich linker. The two monomers are related to the 7SL RNA gene and the complete Alu element is often described as a dimeric structure. Through mutation, splice sites can evolve in different parts of an Alu element (Makałowski et al. 1994). Many of these exonized Alu elements are part of alternatively spliced transcripts (Sorek et al. 2002). Left Alu monomers can be expressed as stable small cytoplasmic (scAlu) RNA and right monomers are thought to be less stable than left monomers (Chang et al. 1996; Sarrowa et al. 1997; Li and Schmid 2004).
Although Alu elements have already been studied in lncRNAs (Gong and Maquat 2011), the functions of many lncRNAs are only beginning to emerge (Amaral et al. 2011). lncRNAs account for some of the pervasive low-level transcription in the human genome (Guttman et al. 2009; Djebali et al. 2012) and are arbitrarily defined as noncoding transcripts that are larger than 200 nt. They appear to be transcribed by RNA polymerase II and can be both capped and polyadenylated (Guttman et al. 2009; Khalil et al. 2009). As lncRNAs tend to have fewer exons than protein-coding genes, their spliced transcripts are usually shorter than most mRNAs (Derrien et al. 2012). Individual lncRNAs are only expressed in a small fraction of cell types (Derrien et al. 2012) or subcellular locations (Mercer et al. 2008) and are therefore likely to be involved in controlling very specific tasks. lncRNAs are involved in various biological processes such as embryonic development, differentiation of skin cells, metastasis, and regulation of cell cycle (Rinn and Chang 2012). Some lncRNAs have already been implicated in the pathogenesis of disease (e.g., breast cancer, prostate cancer, cardiac disease) and it is conceivable that they will emerge as another cause of disease. Along these lines, numerous genetic studies have associated hundreds of noncoding regions in the genome with a broad spectrum of diseases (Ward and Kellis 2012), and lncRNA cis-eQTL SNPs have been associated with disease (Kumar et al. 2013).
To date, lncRNAs have been shown to regulate a variety of molecular processes that include gene transcription, mRNA decay, alternative splicing, and translation (for review, see Rinn and Chang 2012). lncRNAs typically regulate transcription by forming complexes with various proteins that interact with regulatory regions of DNA. The two major types of proteins that are known to be involved in lncRNA-mediated transcription are heterogeneous nuclear ribonucleoproteins (hnRNPs) and chromatin-modifying complexes. hnRNPs are a family of RNA-binding proteins that perform a diverse set of molecular functions that include regulation of mRNA stability, mRNA turnover, pre-mRNA processing, mRNA trafficking, alternative splicing, translational regulation, and packaging of nascent transcripts (Han et al. 2010). More recently, hnRNPs have been shown to regulate gene transcription through their interactions with lncRNAs. For example, hnRNP-K interacts with lincRNA-p21 and binds to promoter regions that regulate genes involved in p53-dependent regulation of cell cycle (Huarte et al. 2010). hnRNP A/B and hnRNP A2/B1 repress transcription of immune genes through their interactions with lincRNA-Cox2 (Carpenter et al. 2013).
Chromatin-modifying complexes remodel chromatin structure by altering the chemical structure of histones. Common modifications to histones include methylation of specific lysines and arginines, acetylation, deacetylation, phosphorylation, and ubiquitination. These modifications affect several biological processes that include gene transcription, chromosome condensation, DNA repair, and DNA replication (Kouzarides 2007). For example, the PRC2 complex that methylates lysine-27 of histone 3 interacts with the lncRNAs Xist, HOTAIR, ANRIL, COLDAIR, Gtl2, and Kcnq1ot1. Each PRC2-lncRNA complex typically represses gene expression in the surrounding chromatin (for review, see Rinn and Chang 2012). The functionally related PRC1 exists in multiple forms and it has been suggested that PRC1 relies on noncoding RNA to methylate lysine-27 on histone 3 (Bracken and Helin 2009). Additional chromatin-modifying proteins that interact with lncRNAs include DNMT3B, G9a, LSD1-CoREST, MLL-WDR5, Set1 and Hda1/2/3 (for review, see Rinn and Chang 2012). lncRNAs employ a variety of molecular mechanisms to post-transcriptionally regulate mRNAs. MALAT1 is an lncRNA that interacts with serine/arginine splicing factors and influences their distribution in nuclear speckle domains (nonmembranous compartments in the nucleus that are believed to store and assemble the pre-mRNA splicing machinery) (Tripathi et al. 2010). MALAT1 is believed to change alternative splicing of pre-mRNAs by modulating the levels and phosphorylation states of splicing factors. AS-Uchl1 is a natural antisense lncRNA that is on the complementary strand that encodes ubiquitin carboxy-terminal hydrolase L1 mRNA (Carrieri et al. 2012). The 5′ end of AS-Uchl1 is complementary to exons 1–2 of Uchl1 and its 3′ end contains the SINEB2 repetitive element. Both of these regions are required for AS-Uchl1 to enhance translation of Uchl1 in a rapamycin-dependent manner. The lncRNAs termed 1/2-sbsRNAs contain an Alu element that is believed to hybridize with partially complementary Alu repeats in the 3′ UTRs of target genes to form a staufen1 binding site (SBS) (Gong and Maquat 2011). The SBS recruits staufen1 and UPF1, which trigger staufen1-mediated mRNA decay.
The above-mentioned study (Gong and Maquat 2011) suggests that Alu elements may exist as functional domains within some lncRNAs. However, the evolution and domain structure of Alu elements has not been extensively studied in lncRNAs or compared with mRNAs. As Alu elements are primate-specific, it is conceivable that they perform important functions in primate-specific lncRNAs. In this study, we investigate the distribution and domain structure of exonized Alu elements in lncRNAs and mRNAs. We show that the domain structure, orientation, and distribution of Alu subfamilies vary considerably across the different transcript types. Indeed, the preference for particular Alu domains and their restricted evolution suggest that a subset of exonized Alu elements can indeed be repurposed as functional domains in lncRNAs and mRNAs.
RESULTS
Overview of data
To investigate the evolution of Alu elements in different classes of RNA, we retrieved representative transcripts from version 73 of Ensembl (Flicek et al. 2011), which corresponds to version 18 of GENCODE (Harrow et al. 2012). GENCODE contains the largest manually curated set of lncRNAs. The data set consisted of six major transcript types that included mRNAs (N = 20,165) and five types of lncRNA (N = 24,468) (Table 1). For comparison, the data set also included sequences from different mRNA regions (5′ UTRs, CDs, and 3′ UTRs). As the number of splice variants encoded by a gene varies considerably and can introduce biased sampling, a representative transcript was retrieved for each transcript class encoded by a gene. The representative transcript was defined as the longest transcript in each case.
TABLE 1.
Summary of lncRNA and mRNA data set
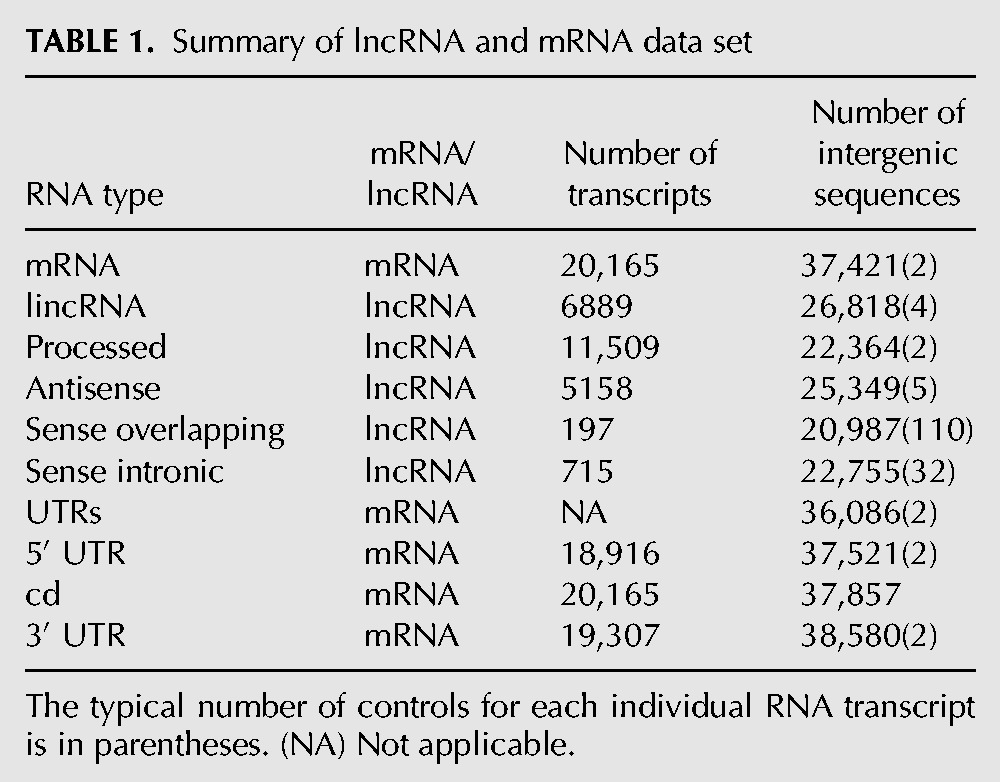
The five types of lncRNA transcripts are lincRNAs (N = 6889), antisense RNAs (N = 5158), sense overlapping RNAs (N = 197), sense intronic RNAs (N = 715), and processed RNAs (N = 11,509) (Derrien et al. 2012). lincRNAs are long intergenic noncoding RNAs that do not intersect with the boundaries of a protein-coding gene (5-kb upstream of the start codon and 30-kb downstream from the stop codon). An antisense RNA gene intersects with any exon of a protein-coding gene or has published evidence for antisense regulation of a protein-coding gene. Sense overlapping RNAs contain a protein-coding gene within one of their introns on the same strand. Sense intronic RNAs are located within the intron of a protein-coding gene on the same strand and do not overlap any exons. All lncRNA that do not belong to any of the above-mentioned types are classified as processed RNA. As detailed in the figure legends, sense intronic RNAs and sense overlapping RNAs were occasionally omitted from figures as there were not enough sequences to perform a robust analysis or the results were simply not informative.
To assess the significance of Alu-related features in different classes of RNA, we retrieved random intergenic sequence controls for each class of RNA (Table 1). A total of N random sequences were retrieved for each individual RNA transcript to ensure that at least 20,000 controls were retrieved for each class of RNA. Each random sequence was “spliced” using the internal intron/exon coordinates of the corresponding RNA transcript that it was size-matched to. Each sequence was sampled from a chromosome with a probability equal to the relative size of the chromosome. The chromosome locations were sampled uniformly at random and the sequences were included in the data set whenever they did not overlap with known transcripts. Because of these stringent criteria, occasionally an intergenic control sequence could not be located for a particular RNA transcript. Therefore, the number of control sequences was not an exact multiple of N and the total number of RNAs in a class. For example, there were 5518 antisense RNAs and we successfully retrieved five control sequences for most of them (25,349/25,790). Finally, as coding regions and UTRs are under different evolutionary constraints, we also retrieved intergenic control sequences that were “spliced” using the exon coordinates of UTRs in the corresponding mRNA transcript.
Depletion of Alu elements in lncRNAs and mRNAs
The percentage of transcripts with Alu elements varied considerably among the different transcript types (18%–41%; Fig. 1A). mRNAs, lincRNAs, processed RNAs, and antisense RNAs had significantly fewer Alu-containing sequences than their corresponding intergenic controls (Fisher's exact test, P = 0 / 5.5 × 10−298, P = 6.9 × 10−57, P = 2.5 × 10−320, P = 1.8 × 10−19). In contrast, sense intronic RNAs and sense overlapping RNAs did not differ significantly from their controls. As expected, longer intergenic controls were more likely to contain Alu elements (Fig. 1B) and confirmed the need for “spliced” intergenic controls that are the same lengths as their corresponding RNA types. We also confirmed that mRNAs, lincRNAs, and processed RNAs had significantly fewer Alu-containing sequences than their corresponding intergenic controls at each chromosome. More than 50% of Alu-containing transcripts had a single Alu element and more than 80% of them had less than three Alu elements. As expected, Alu elements were primarily located in 3′ UTRs of mRNA, and were rarely located in 5′ UTRs and coding regions (Fig. 1A).
FIGURE 1.
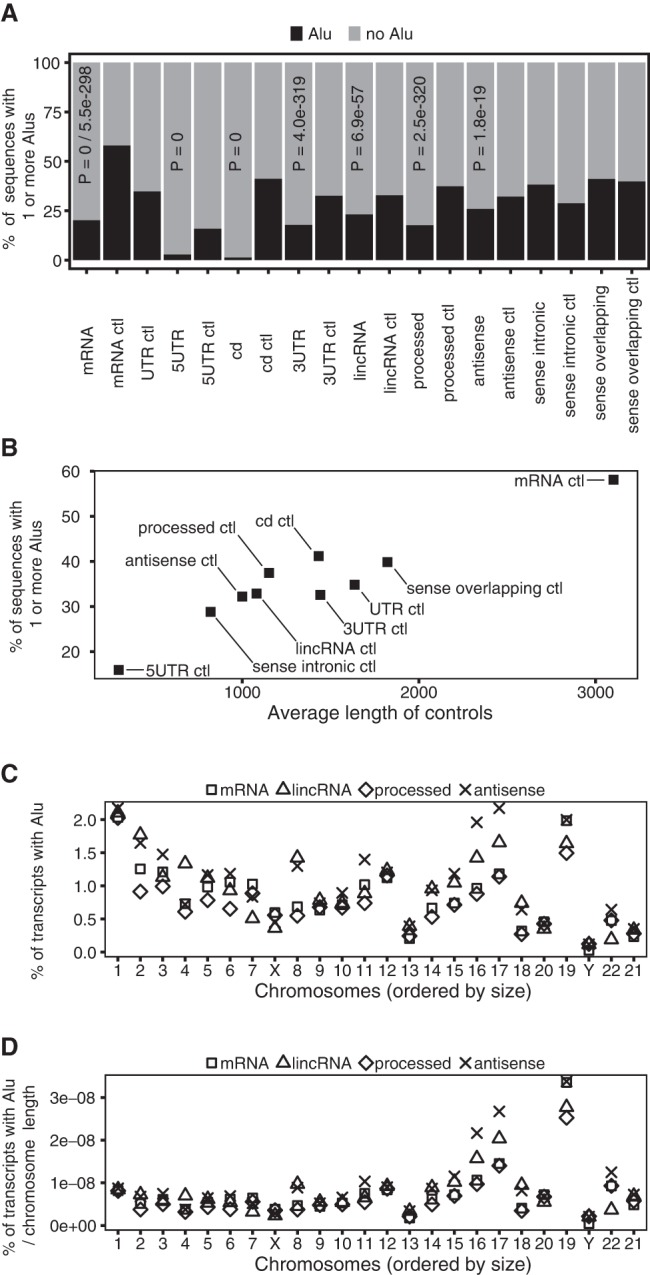
Depletion of Alu elements in mRNAs and lncRNAs. (A) The percentage of each RNA type with one or more Alu elements. The P-values indicate that Alu elements are significantly depleted in the RNA relative to its corresponding control. For mRNAs, the second P-value was generated using the UTR-like control sequences. (B) The percentage of intergenic controls with one or more Alu elements is greater in longer sequences. (C) The percentage of transcripts with an Alu element at each chromosome. The percentages at each chromosome sum up to the percentages in A. Sense intronic RNAs and sense overlapping RNAs were omitted because of the small number of sequences at each chromosome. (D) Percentages from C are divided by the length of each chromosome.
Although we observed a depletion of Alu-containing RNA at each chromosome, the previously reported distribution of Alu elements across chromosomes (Lander et al. 2001) prompted us to examine the percentage of Alu-containing transcripts encoded at each chromosome (Fig. 1C,D). We omitted sense intronic and sense overlapping transcripts as their sample sizes were too small at each chromosome. In general, the percentages across each chromosome were correlated between the different transcript types (Fig. 1C,D). In particular, the percentages for processed RNA and mRNA were highly correlated across chromosomes. A relatively high percentage of the Alu-containing transcripts mapped to the smaller chromosomes (16, 17, and 19). On chromosome 17, there was a high percentage of antisense and lincRNA transcripts with Alu elements. On chromosome 19, every class of transcript had a relatively high percentage of Alu-containing transcripts (Fig. 1C,D).
Alu-containing mRNAs on chromosome 19 are associated with intrachromosomal gene duplication
As chromosome 19 is Alu-rich (Lander et al. 2001) and contains many duplicated genes (Lander et al. 2001; Grimwood et al. 2004), we investigated whether the propensity for chromosome 19 genes to contain Alu elements (Fig. 1C,D) was associated with intrachromosomal gene duplications. Although this hypothesis could not be tested for lncRNAs (current methods cannot reliably determine nearest paralogs for lncRNAs), our analysis of protein-coding genes provided valuable insight.
Using EnsemblCompara gene trees (Vilella et al. 2009), we determined the nearest paralog (see Materials and Methods) for each protein-coding gene and whether the paralog was on the same chromosome or a different chromosome. In general, the majority of protein-coding genes had its nearest paralog on a different chromosome (Fig. 2). However, 42% of Alu-containing mRNA encoded by chromosome 19 had its nearest paralog on the same chromosome. In contrast, only 22% of mRNA encoded by chromosome 19 that lacked an Alu element had its nearest paralog on the same chromosome. The distance between nearest intrachromosomal paralogs on chromosome 19 varied considerably (mean = 2191 kb, SD = 5602 kb). Most pairs of intrachromosomal paralogs did not possess the same Alu subfamilies (Fig. 2C). Therefore, the duplication of genes within chromosome 19 is probably associated with a subsequent exonization of Alu elements in this Alu-rich region.
FIGURE 2.
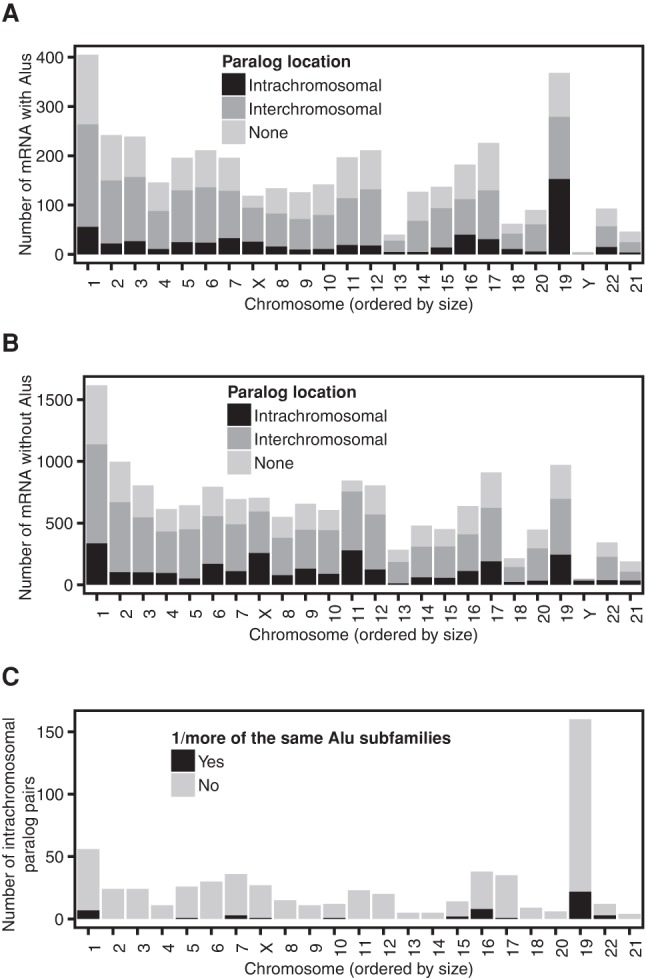
Alu-containing mRNAs on chromosome 19 are associated with intrachromosomal gene duplication. (A) Type of gene duplication associated with mRNAs that contain Alu elements. (B) Type of gene duplication associated with mRNAs that lack Alu elements. (C) Number of intrachromosomal paralog pairs that have one or more of the same Alu subfamilies.
Orientation and position of Alu elements in lncRNAs and mRNAs
As forward-oriented Alu elements can sometimes contain poly(A) signals (Lee et al. 2008; Chen et al. 2009), we examined the orientation and relative position of Alu elements in each class of RNA. lincRNAs, processed RNAs, and antisense RNAs had a significantly greater proportion of reverse-oriented Alu elements than their corresponding intergenic controls (Fisher's exact test, P = 1.0 × 10−17, P = 8.6 × 10−16, P = 5.0 × 10−16, Fig. 3A). Only sense intronic RNAs and mRNAs had similar proportions of forward- and reverse-oriented Alu elements. Among the small number of Alu elements located within 5′ UTRs and coding regions, there was a clear preference for the reverse orientations (P = 6.0 × 10−28 and P = 9.3 × 10−16).
FIGURE 3.
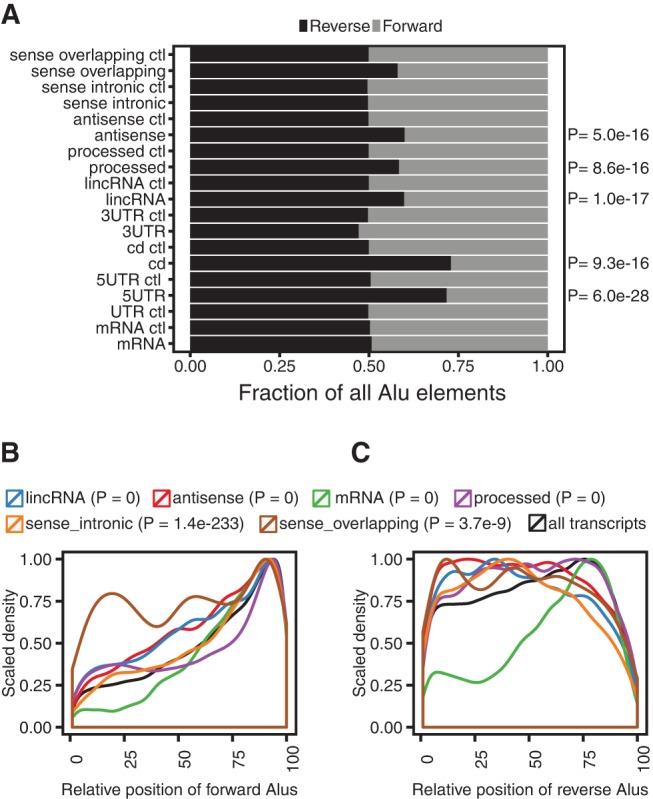
Orientation and position of Alu elements in lncRNAs and mRNAs. (A) Orientation of Alu elements in each RNA type. P-values indicate a significant enrichment of reverse-oriented Alu elements in the class of RNA relative to its corresponding control. (B,C) The relative position of forward-oriented and reverse-oriented Alu elements in mRNA, the five types of lncRNA, and six transcripts combined. There are 100 bins for each RNA molecule. Each bin spans 1% of an RNA molecule.
To determine whether there was a positional bias for reverse-oriented and forward-oriented Alu elements, each RNA sequence was divided into 100 bins of equal size (to account for differences in transcript length) and the number of transcripts with an Alu element at each bin location was counted. For example, a transcript with 700 nt would be assigned 100 bins that each spanned 7 nt. In mRNAs, the majority of forward- and reverse-oriented Alu elements were positioned near the 3′ end (Fig. 3B,C). However, there were proportionally fewer forward-oriented Alu elements in the 5′ end than reverse-oriented Alu elements, and the relative positions of forward-oriented Alu elements had a greater 3′ bias than reverse-oriented Alu elements in mRNA (Mann–Whitney test, P = 0). The distance between Alu elements and coding regions was generally correlated with the length of the UTR that contained the Alu element (data not shown).
In all five types of lncRNA, there was a striking difference in the locations of reverse- and forward-oriented Alu elements (Fig. 3B,C). Reverse-oriented Alu elements were tolerated throughout the sequence, whereas forward-oriented Alu elements were primarily located in the 3′ end. Consistent with this, the locations of forward-oriented Alu elements were significantly greater than reverse-oriented Alu elements in antisense RNAs, lincRNAs, processed RNAs, sense intronic RNAs, and sense overlapping RNAs (Mann–Whitney test, P = 0, P = 0, P = 0, P = 1.4 × 10−233, P = 3.7 × 10−9).
Putative poly(A) signals (AAUAAA) were less common in reverse-oriented Alu elements than in forward-oriented Alu elements in both lncRNAs and mRNAs. However, only a small portion of Alu's in lncRNAs (7%) and mRNAs (10%) contained canonical poly(A) signals, which only partially explains the different locations of reverse- and forward-oriented Alu elements in lncRNA. Consistent with this, only a small percentage of Alu elements contained the reverse complement of the canonical poly(A) signal in mRNAs (8%) and lncRNAs (6%). Collectively, these results are consistent with Alu elements primarily occurring in the 3′ UTRs of mRNA (Yulug et al. 1995) and demonstrate that reverse-oriented Alu elements are tolerated in a variety of positions within lncRNAs.
Exonization of Alu domains
The dimeric (full-length) Alu element, the left Alu monomer, and the right Alu monomer are known to form RNA structural domains, whereas random regions of an Alu element are less likely to be structured (Sinnett et al. 1991). We therefore determined the regions of each Alu element that was exonized and whether it spanned a structured region (see Materials and Methods). There were three categories that corresponded to structured regions (dimeric, left monomer, right monomer), and one category (other) that is likely to consist of unstructured RNAs. Structured Alu elements were enriched in mRNAs, lincRNAs, processed RNAs, antisense, and sense intronic RNAs relative to their corresponding controls (Fig. 4A; Fisher's exact test, P = 2.5 × 10−236 / 4.4 × 10−113, P = 1.2 × 10−41, P = 2.5 × 10−85, P = 6.0 × 10−31, P = 0.0008). Dimeric Alu elements were the predominant Alu domain in mRNAs, whereas all three of the structured domains were quite common in the different classes of lncRNA. Within mRNAs, 3′ UTRs primarily contained dimeric Alu domains, 5′ UTRs contained similar proportions of each domain, and coding regions primarily contained left and right monomers.
FIGURE 4.

Alu domains are enriched in lncRNAs and mRNAs. (A) The fraction of Alu domains (dimeric, left monomer, right monomer) and non-domains (other) in mRNAs and the five types of lncRNA. (B) The exon location of forward- and reverse-oriented Alu domains in mRNAs. (C) The exon location of forward- and reverse-oriented Alu domains in the three major types of lncRNA (lincRNAs, processed RNAs, and antisense RNAs). Sense intronic and sense overlapping RNAs were not included as the distribution of their exon locations were slightly different than in the other classes of lncRNA.
Next, we examined the propensity of these different domains to occur in different exon locations. In mRNAs, both forward- and reverse-oriented dimeric Alu's were primarily located in the terminal exon of mRNAs (Fig. 4B). Although the majority of dimeric Alu's also occurred in the terminal exon of lncRNAs (lincRNAs, processed RNAs, and antisense RNAs), there was also a relatively large proportion of dimeric Alu's in the initial exon of lncRNAs (Fig. 4C). The exon locations were slightly different in sense intronic and sense overlapping RNAs (data not shown). Consistent with Figure 3, reverse-oriented Alu elements were more frequent than forward-oriented Alu elements in initial and internal exons of lncRNAs. In particular, the right monomer of reverse-oriented Alu elements was primarily incorporated into the internal exons of lncRNAs. Collectively, the above results demonstrate that mRNAs and lncRNAs have a clear preference for structured Alu domains that reside within particular exon locations.
lncRNAs and mRNAs primarily contain Alu J and S subfamilies
As many human lncRNAs appears to have evolved during primate evolution (Derrien et al. 2012; Necsulea et al. 2014; Washietl et al. 2014), we investigated the composition of exonized Alu subfamilies that were amplified at different periods. Both lncRNAs and mRNAs were primarily composed of Alu J and Alu S subfamilies that were amplified 35–55 million years ago (Fig. 5; Batzer and Deininger 2002). The dimeric Alu J subfamily was enriched in antisense RNAs, and sense intronic RNAs relative to their respective controls (P = 9.3 × 10−06, P = 0.0398). In the other types of exonized Alu elements (left monomer, right monomer, other), the Alu J subfamily was typically overrepresented (see P-values in Fig. 5). Interestingly, the dimeric Alu S subfamily was only overrepresented in mRNAs (Fisher's exact test, P = 8.8 × 10−13/6.7 × 10−13), with 3′ UTRs having a preference for this subfamily. Collectively, these results indicate that lncRNAs and mRNAs primarily contain Alu elements that were amplified relatively early in primate evolution, providing sufficient time for exon formation.
FIGURE 5.

Alu subfamilies in mRNAs and lncRNAs. The fraction of each Alu subfamily is shown for each Alu domain. P-values indicate significant enrichment of an Alu subfamily (red = Alu J, blue = Alu S) relative to the corresponding control. For mRNAs, the second P-value was generated using the UTR-like control sequences.
Evolutionary constraints in the Alu elements of lncRNAs and mRNAs
To investigate whether the sequences of exonized Alu elements might be under evolutionary constraints, we examined percentage identities between each Alu element and its corresponding consensus sequence in Repbase (Jurka et al. 2005). We used this measure of evolutionary constraint as reliable multiple sequence alignments cannot be generated for homologous lncRNAs (very few lncRNAs sequences are defined in other primates). Alu elements that are similar to consensus sequences are likely to be subject to evolutionary constraints (potential purifying/negative selection). In contrast, Alu elements that are dissimilar to consensus sequences could be subject to either neutral drift or positive selection. Thus, we focused our attention on Alu elements that shared a high percentage identity with consensus sequences relative to their controls.
Within the Alu J subfamily, sequences were constrained in mRNAs, processed RNAs, and antisense RNAs (Fig. 6A–C). The most significant sequence constraints were in the dimeric (Mann–Whitney test, P = 5.3 × 10−46, P = 2.4 × 10−04, P = 2.4 × 10−04) and left (P = 2.3 × 10−05, P = 0.0579, P = 0.0242) domains. Within the Alu S subfamily (Fig. 6D–F), the most significant sequence constraints were in the dimeric domains of mRNAs, lincRNAs, processed RNAs, and antisense RNAs (P = 5.3 × 10−99, P = 0.0046, P = 2.7 × 10−08, P = 0.0034). Within the Alu Y subfamily (data not shown), dimeric sequences were constrained in mRNAs (P = 0.001) and the left monomers were constrained in processed RNAs (P = 0.0014). We did not observe any relationship between the position of Alu elements within different RNA types and their evolutionary constraints. Overall, the results indicate that dimeric Alu elements are significantly constrained in lncRNAs and mRNAs, whereas the left and right monomers are constrained within particular Alu subfamilies and classes of RNA.
FIGURE 6.

Sequence constraints in dimeric Alu elements. Percent identities between Alu sequences and consensus Alu sequences. (A) Dimeric Alu J subfamily in mRNAs and the three major classes of lncRNA. (B, left) Alu J subfamily in mRNAs and the three major classes of lncRNA. (C, right) Alu J subfamily in mRNAs and the three major classes of lncRNA. (D) Dimeric Alu S subfamily in mRNAs and the three major classes of lncRNA. (E, left) Alu S subfamily in mRNAs and the three major classes of lncRNA. (F, right) Alu S subfamily in mRNAs and the three major classes of lncRNA. P-values indicate that the Alu elements in the RNA (colored by RNA type) are under greater sequence constraint than their respective controls.
DISCUSSION
In general, the majority of repetitive elements in the genome are expected to be harmless nonfunctional junk DNA. To a lesser extent, some repetitive elements are expected to be deleterious garbage DNA that has not been removed from the population yet. Functional repetitive elements are expected to be at even lower frequencies. To date, a small number of repetitive elements have emerged as potential regulators of various biological processes (de Souza et al. 2013). They are believed to regulate gene transcription, polyadenylation of mRNA, alternative splicing, RNA editing, and RNA translation (Berger and Strub 2010; Deininger 2011). lncRNAs have been reported to contain Alu and SINEB2 elements that function in staufen-mediated decay (Gong and Maquat 2011) and RNA translation (Carrieri et al. 2012), respectively. Alu elements are of particular interest as they are primate-specific and have the potential to alter the domain structure of lncRNAs and mRNAs. To further assess the potential for Alu elements to be exapted in mRNAs and lncRNAs, we examined and compared the evolution of Alu elements in mRNA and five types of lncRNA (lincRNA, antisense RNA, sense overlapping RNA, sense intronic RNA, processed RNA).
This study revealed several key findings that require careful interpretation: (1) There is a significant depletion of Alu elements in mRNAs and lncRNAs across the genome and most chromosomes. (2) The percentage of lncRNAs and mRNAs with Alu elements tends to be correlated across chromosomes. (3) The frequent duplication/genesis of coding genes within chromosome 19 is often associated with a subsequent exonization of Alu elements in this Alu-rich chromosome. (4) Unlike 3′ UTRs in mRNAs, reverse-oriented Alu elements are overrepresented in lncRNAs and are located in a variety of positions within lncRNAs. (5) mRNAs and lncRNAs have a clear preference for structured Alu domains that reside within particular exon locations. (6) lncRNAs and mRNAs primarily contain Alu subfamilies that were amplified during early primate evolution. (7) Many Alu elements appear to be subject to evolutionary constraints in lncRNAs and mRNAs.
The depletion of Alu elements in lncRNAs and mRNAs suggests that most exonized Alu elements arising in the population are deleterious to RNA function or stability. Although Alu-containing RNAs are commonly encoded by Alu-rich chromosomes (e.g., chromosome 17 and 19), they are depleted relative to the amount of Alu-containing controls detected on these chromosomes. While mRNAs encoded by chromosome 19 appear to exonize Alu elements after gene duplication, the mechanism of lncRNA genesis in Alu-rich regions may be less reliant on gene duplication. Nevertheless, we still suspect that Alu exonization primarily occurs in lncRNAs after their initial genesis. We also note that the extent of Alu depletion may be greater than our results suggest, as the transcripts in our data set may not accurately represent the predominant splice variants in a cell. Also, it is conceivable that the inclusion or exclusion of an Alu element in different splice variants may have a specific regulatory effect that is associated with particular conditions. Despite the general depletion of Alu elements, many mRNAs and lncRNAs appear to tolerate exonized Alu elements, suggesting that they are at least harmless junk and perhaps functional in some instances.
Although most Alu elements are located in the 3′ UTRs of mRNAs and are equally likely to occur in either orientation, lncRNAs contain a significant enrichment of reverse-oriented Alu elements that are not restricted to the 3′ end. In rare instances when Alu elements are located in the 5′ UTR or coding region they are primarily in the reverse orientation. Although reverse-oriented Alu elements have less canonical poly(A) signals than forward-oriented Alu elements, it should be noted that canonical poly(A) signals are relatively rare in all Alu elements and only partially explains this observation.
The preferences for structured RNA domains (corresponding to the left monomer, the right monomer, and a full-length dimeric element) are striking. There is a clear preference for dimeric Alu elements in mRNAs and these are primarily encoded by the Alu S subfamily. In lncRNAs, the three domain structures tend to be similarly enriched, and the Alu J subfamily tends to be overrepresented. The extent that these preferences might relate to required functions (e.g., essential biochemical processes that are protected from deleterious mutations) versus other activities that may not be functionally required (e.g., the propensity for nonessential Alu elements to be spliced) (Makałowski et al. 1994; Sorek et al. 2002) will need to be experimentally determined.
Because of the lack of well-defined RNA sequences in primates, it is difficult to assess whether their exonized Alu elements are under selection. However, by comparing exonized Alu elements to consensus sequences, we were able to determine whether the evolution of these sequences was constrained (a potential indicator of negative/purifying selection). Dimeric Alu S and Alu J elements appear to be under evolutionary constraint in mRNAs and lncRNAs. The left and right Alu arms also appear to be subject to evolutionary constraints within particular Alu subfamilies and classes of RNA. We believe that those Alu RNAs with the greatest sequence constraints are prime candidates for experimental characterization.
Similar to protein domains, it is conceivable that Alu domains have been exapted as modular functional domains in different RNA types (Gong and Maquat 2011; Johnson and Guigo 2014). The extent that Alu domains perform modular functions should become apparent soon as several research groups are investigating the function of Alu-containing RNA.
MATERIALS AND METHODS
lncRNA and mRNA sequences
Using the Ensembl API, human lncRNA and mRNA sequences were retrieved from version 73 of Ensembl (Flicek et al. 2011), which was based on assembly GRCh37 and is identical to version 18 of GENCODE (Harrow et al. 2012).
GENCODE contains the largest manually curated set of lncRNAs and mRNAs. We retrieved sequences that were classified as lincRNA, antisense RNA, sense overlapping RNA, sense intronic RNA, processed RNA, or mRNA within GENCODE/Ensembl (Table 1). As the number of splice variants encoded by a gene varies considerably and can introduce biased sampling, a representative transcript was retrieved for each transcript class encoded by a gene. The representative transcript was defined as the longest transcript in each case. The Ensembl API was used to determine the transcript coordinates for all exons, which were further classified as initial, internal, or terminal. For comparison, 5′ UTR, CD, and 3′ UTR sequences were extracted from mRNAs. There were slightly fewer UTRs than CDs (Table 1), as some mRNAs do not have UTRs.
Random intergenic control sequences
Random intergenic sequence controls were retrieved for each class of RNA (Table 1). A total of N random sequences were retrieved for each individual RNA transcript to ensure that at least 20,000 controls were retrieved for each class of RNA. Each random sequence was “spliced” using the relative intron/exon coordinates of the corresponding RNA transcript that it was size-matched to. Each sequence was sampled from a chromosome with a probability equal to the relative size of the chromosome. The locations within a chromosome were sampled uniformly at random and the sequences were included in the data set whenever they did not overlap with known transcripts. Because of these stringent criteria, occasionally an intergenic control sequence could not be located for a particular RNA transcript. Therefore, the number of control sequences were not an exact multiple of N and the total number of RNAs in a class. For example, there were 5518 antisense RNAs and we successfully retrieved five control sequences for most of them (25,349/25,790). As coding regions and UTRs are under different evolutionary constraints, we also retrieved intergenic control sequences that were “spliced” using the exon coordinates of UTRs in the corresponding mRNA transcript (coding regions were treated as introns). Related to this, we retrieved intergenic controls that had the same lengths and exon structures as 5′ UTRs, coding regions, and 3′ UTRs.
Identification and analysis of Alu elements
We used RepeatMasker (Smit) and Repbase (Jurka et al. 2005) to identify repetitive elements in lncRNA and mRNA transcripts. The NCBI/RMBLAST search engine was used with the following options: -norna -html -source -gff -nolow –species human. We did not specify the –alu option, which only reports Alu elements, as we had a casual interest in other repetitive elements. Key information for each repetitive element was parsed from the RepeatMasker result files (the repeat name, the length of the repeat in the transcript, the orientation of the repeat, the start and end of the repeat relative to the matching consensus sequence, the start and end of the repeat in the transcript, the percent identity shared with matching consensus sequence, Alu subfamilies, etc.).
An exonized Alu element was defined as a complete dimer when it aligned to position 50 (or less) and position 240 (or greater) in the matching consensus sequence. An exonized Alu element was defined as a right arm when its start and end positions aligned to position 120 (or greater) and position 241 (or greater), respectively, in the matching consensus sequence. An exonized Alu element was defined as a left arm when its start and end positions aligned to position 49 (or less) and position 179 (or less), respectively, in the matching consensus sequence. An exonized Alu element was defined as dimeric when its start and end positions aligned to position 49 (or less) and position 241 (or greater), respectively, in the matching consensus sequence. An exonized Alu elements was defined as “other” when it spanned other regions.
Identification of nearest paralogs
Ensembl gene trees (Vilella et al. 2009) were used to identify nearest paralogs for each protein-coding gene. The ancestral nodes that connected each within-species paralog to the gene of interest were retrieved. The ancestral node with the shortest distance to the gene of interest was used to identify the nearest paralog. When the nearest ancestor node was connected to multiple within-species paralogs, the within-species paralog with the shortest distance to the gene of interest was defined as the nearest paralog. The nearest paralog was classified as intrachromosomal when it was located on the same chromosome as the gene of interest and interchromosomal when it was on a different chromosome.
Positional bias of Alu elements
To determine whether there was a bias in the position of reverse and forward-oriented Alu elements within lncRNAs and mRNAs, each RNA molecule was divided into 100 bins of equal size to account for differences in transcript length. The number of lncRNAs and mRNAs that contained an Alu element at each bin location was counted and plotted.
Putative poly(A) signals
All Alu elements were scanned for the presence of the canonical poly(A) signal (AAUAAA), its reverse complement (UUUAUU), as well as 12 other less common signals (Chen et al. 2009).
Statistical analysis and plotting
Statistical analysis was performed using the R package (Ihaka and Gentleman 1996). The nonparametric Mann–Whitney test was used to compare unpaired data. The Fisher's exact test was used to identify overrepresented and underrepresented categories within contingency tables. All P-values were adjusted to control for false discovery in multiple testing (Benjamini and Hochberg 1995). All plots were generated using the R package.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.048280.114.
REFERENCES
- Amaral PP, Clark MB, Gascoigne DK, Dinger ME, Mattick JS. 2011. lncRNAdb: a reference database for long noncoding RNAs. Nucleic Acids Res 39: D146–D151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzer MA, Deininger PL. 2002. Alu repeats and human genomic diversity. Nat Rev Genet 3: 370–379. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Methodol 57: 289–300. [Google Scholar]
- Berger A, Strub K. 2010. Multiple roles of Alu-related noncoding RNAs. In Long non-coding RNAs of progress in molecular and subcellular biology (ed. Ugarkovic D), Vol. 51, pp. 119–146. Springer, Berlin, Heidelberg. [DOI] [PubMed] [Google Scholar]
- Bracken AP, Helin K. 2009. Polycomb group proteins: navigators of lineage pathways led astray in cancer. Nat Rev Cancer 9: 773–784. [DOI] [PubMed] [Google Scholar]
- Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, Byron M, Monks B, Henry-Bezy M, Lawrence JB, et al. 2013. A long noncoding RNA mediates both activation and repression of immune response genes. Science 341: 789–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrieri C, Cimatti L, Biagioli M, Beugnet A, Zucchelli S, Fedele S, Pesce E, Ferrer I, Collavin L, Santoro C, et al. 2012. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 491: 1–6. [DOI] [PubMed] [Google Scholar]
- Chang DY, Hsu K, Maraia RJ. 1996. Monomeric scAlu and nascent dimeric Alu RNAs induced by adenovirus are assembled into SRP9/14-containing RNPs in HeLa cells. Nucleic Acids Res 24: 4165–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Ara T, Gautheret D. 2009. Using Alu elements as polyadenylation sites: a case of retroposon exaptation. Mol Biol Evol 26: 327–334. [DOI] [PubMed] [Google Scholar]
- de Souza FSJ, Franchini LF, Rubinstein M. 2013. Exaptation of transposable elements into novel cis-regulatory elements: is the evidence always strong? Mol Biol Evol 30: 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deininger P. 2011. Alu elements: know the SINEs. Genome Biol 12: 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. 2012. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22: 1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. 2012. Landscape of transcription in human cells. Nature 489: 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, et al. 2011. Ensembl 2012. Nucleic Acids Res 40(Database issue): D84–D90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong C, Maquat LE. 2011. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 470: 284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimwood J, Gordon LA, Olsen A, Terry A, Schmutz J, Lamerdin J, Hellsten U, Goodstein D, Couronne O, Tran-Gyamfi M, et al. 2004. The DNA sequence and biology of human chromosome 19. Nature 428: 529–535. [DOI] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al. 2009. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458: 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SP, Tang YH, Smith R. 2010. Functional diversity of the hnRNPs: past, present and perspectives. Biochem J 430: 379–392. [DOI] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. 2012. GENCODE: the reference human genome annotation for the ENCODE project. Genome Res 22: 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, et al. 2010. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142: 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihaka R, Gentleman R. 1996. R: a language for data analysis and graphics. J Comput Graph Stat 5: 299–314. [Google Scholar]
- Johnson R, Guigo R. 2014. The RIDL hypothesis: transposable elements as functional domains of long noncoding RNAs. RNA 20: 959–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. 2005. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110: 462–467. [DOI] [PubMed] [Google Scholar]
- Kapusta A, Kronenberg Z, Lynch VJ, Zhuo X, Ramsay L, Bourque G, Yandell M, Feschotte C. 2013. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet 9: e1003470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley D, Rinn J. 2012. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol 13: R107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, et al. 2009. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci 106: 11667–11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. 2007. Chromatin modifications and their function. Cell 128: 693–705. [DOI] [PubMed] [Google Scholar]
- Kumar V, Westra HJ, Karjalainen J, Zhernakova DV, Esko T, Hrdlickova B, Almeida R, Zhernakova A, Reinmaa E, Võsa U, et al. 2013. Human disease-associated genetic variation impacts large intergenic non-coding RNA expression. PLoS Genet 9: e1003201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. [DOI] [PubMed] [Google Scholar]
- Lee JY, Ji Z, Tian B. 2008. Phylogenetic analysis of mRNA polyadenylation sites reveals a role of transposable elements in evolution of the 3′-end of genes. Nucleic Acids Res 36: 5581–5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li TH, Schmid CW. 2004. Alu's dimeric consensus sequence destabilizes its transcripts. Gene 324: 191–200. [DOI] [PubMed] [Google Scholar]
- Lin L, Jiang P, Shen S, Sato S, Davidson BL, Xing Y. 2009. Large-scale analysis of exonized mammalian-wide interspersed repeats in primate genomes. Hum Mol Genet 18: 2204–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makałowski W, Mitchell GA, Labuda D. 1994. Alu sequences in the coding regions of mRNA: a source of protein variability. Trends Genet 10: 188–193. [DOI] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS. 2008. Specific expression of long noncoding RNAs in the mouse brain. Proc Natl Acad Sci 105: 716–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Necsulea A, Soumillon M, Warnefors M, Liechti A, Daish T, Zeller U, Baker JC, Grützner F, Kaessmann H. 2014. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 505: 635–640. [DOI] [PubMed] [Google Scholar]
- Ricci EP, Kucukural A, Cenik C, Mercier BC, Singh G, Heyer EE, Ashar-Patel A, Peng L, Moore MJ. 2000. Staufen1 senses overall transcript secondary structure to regulate translation. Nat Struct Mol Biol 21: 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Chang HY. 2012. Genome regulation by long noncoding RNAs. Annu Rev Biochem 81: 145–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrowa J, Chang DY, Maraia RJ. 1997. The decline in human Alu retroposition was accompanied by an asymmetric decrease in SRP9/14 binding to dimeric Alu RNA and increased expression of small cytoplasmic Alu RNA. Mol Cell Biol 17: 1144–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnett D, Richer C, Deragon JM, Labuda D. 1991. Alu RNA secondary structure consists of two independent 7 SL RNA-like folding units. J Biol Chem 266: 8675–8678. [PubMed] [Google Scholar]
- Smit AFA. RepeatMasker. http://www.repeatmasker.org/.
- Sorek R, Ast G, Graur D. 2002. Alu-containing exons are alternatively spliced. Genome Res 12: 1060–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA, et al. 2010. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell 39: 925–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, Birney E. 2009. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res 19: 327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward LD, Kellis M. 2012. Interpreting noncoding genetic variation in complex traits and human disease. Nat Biotechnol 30: 1095–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washietl S, Kellis M, Garber M. 2014. Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mammals. Genome Res 24: 616–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Zhou X, Jin Y. 2013. ADAR-mediated RNA editing in non-coding RNA sequences. Sci China Life Sci 56: 944–952. [DOI] [PubMed] [Google Scholar]
- Yulug IG, Yulug A, Fisher EM. 1995. The frequency and position of Alu repeats in cDNAs, as determined by database searching. Genomics 27: 544–548. [DOI] [PubMed] [Google Scholar]