

Abstract
Spliceosomal Prp38 proteins contain a conserved amino-terminal domain, but only higher eukaryotic orthologs also harbor a carboxy-terminal RS domain, a hallmark of splicing regulatory SR proteins. We show by crystal structure analysis that the amino-terminal domain of human Prp38 is organized around three pairs of antiparallel α-helices and lacks similarities to RNA-binding domains found in canonical SR proteins. Instead, yeast two-hybrid analyses suggest that the amino-terminal domain is a versatile protein–protein interaction hub that possibly binds 12 other spliceosomal proteins, most of which are recruited at the same stage as Prp38. By quantitative, alanine surface-scanning two-hybrid screens and biochemical analyses we delineated four distinct interfaces on the Prp38 amino-terminal domain. In vitro interaction assays using recombinant proteins showed that Prp38 can bind at least two proteins simultaneously via two different interfaces. Addition of excess Prp38 amino-terminal domain to in vitro splicing assays, but not of an interaction-deficient mutant, stalled splicing at a precatalytic stage. Our results show that human Prp38 is an unusual SR protein, whose amino-terminal domain is a multi-interface protein–protein interaction platform that might organize the relative positioning of other proteins during splicing.
Keywords: pre-mRNA processing factor 38, pre-mRNA splicing, protein–protein interactions, spliceosomal B complex, spliceosome, yeast two-hybrid analysis
INTRODUCTION
Splicing entails the removal of noncoding intervening sequences (introns) from precursor messenger RNAs (pre-mRNAs) and the ligation of neighboring coding regions (exons), and it constitutes an essential step in the maturation of most eukaryotic primary protein-coding transcripts. A splicing event encompasses two consecutive transesterification reactions, steps 1 and 2, which in the vast majority of cases in humans are carried out by the major spliceosome. A hallmark of spliceosomes is their stepwise assembly from five small nuclear ribonucleoprotein particles (snRNPs U1, U2, U4, U5, and U6 in the major spliceosome) and many non-snRNP splicing factors, which occurs de novo for each splicing event (Wahl et al. 2009; Will and Lührmann 2011). In the consensus view of constitutive splicing, a splicing process is initiated by formation of an A complex, in which U1 snRNP is bound at a 5′-splice site (SS) and U2 snRNP at a branch point sequence (BPS) of an intron. Subsequently, the B complex is formed by incorporation of the U4, U5, and U6 snRNPs as a preformed U4/U6•U5 tri-snRNP. Neither the spliceosomal snRNPs nor the B complex contain a functional active site for the splicing transesterification reactions. Instead, the B complex must undergo major compositional and conformational rearrangements to first yield the Bact and, after additional rearrangements, the B* complex, which carries out the first step of splicing. Further remodeling after step 1 gives rise to the C complex, which catalyzes the second transesterification reaction, after which the mature mRNA is released as a mRNP and the remaining subunits of the spliceosome are recycled for further rounds of splicing.
Spliceosome catalytic activation involves a number of processes that depend on the activities of spliceosome-associated RNA helicases: that is, (i) removal of U1 snRNP from the 5′-SS, which requires the Prp28 protein (Chen et al. 2001); (ii) unwinding of U4 and U6 snRNAs, which are extensively base paired in the U4/U6•U5 tri-snRNP and in the B complex, via the Brr2 protein (Laggerbauer et al. 1998; Raghunathan and Guthrie 1998); and (iii) remodeling of the U2 snRNP-associated heteromeric splicing factors 3a and 3b around the BPS, which is facilitated by the Prp2 protein (Warkocki et al. 2009; Lardelli et al. 2010). Additional proteins have been functionally linked to spliceosome catalytic activation, including the Prp38 protein in yeast. Saccharomyces cerevisiae cells bearing temperature-sensitive (ts) prp38 alleles exhibit severe splicing defects at repressive temperatures, with splicing being stalled before the first transesterification reaction (Blanton et al. 1992; Xie et al. 1998).
Prp38 is a universally conserved splicing factor and is also essential in Drosophila melanogaster (Andersen and Tapon 2008). Although S. cerevisiae (sc) Prp38 is a stable subunit of the U4/U6•U5 tri-snRNP, the human (hs) counterpart is recruited to the spliceosome independently of the tri-snRNP at the B-complex stage, together with a group of eight other non-snRNP proteins (hsSnu23, hsMFAP1, hsSmu1, hsRED, hsFBP21, hsUBL5, hsNPW38, and hsNPW38BP) (Agafonov et al. 2011). hsPrp38 and the latter group of non-snRNP proteins are released again during the next step of spliceosome maturation, the formation of the activated Bact complex (Agafonov et al. 2011). Thus, we refer to hsPrp38 and the other proteins recruited and released at the same stages as “B-specific” factors.
Although an amino-terminal domain (NTD) of unknown structure and function is found in all Prp38 orthologs, only higher eukaryotic Prp38 proteins additionally contain a carboxy-terminal arginine–serine (RS) repeat-containing region (Fig. 1A–C). RS domains are a signature of serine/arginine-rich splicing regulatory proteins (SR proteins) (Long and Caceres 2009). The RS domains of SR proteins can be serine-phosphorylated by SR protein kinases (SRPKs) and can interact with other SR proteins and pre-mRNAs in a phosphorylation-dependent manner (Long and Caceres 2009). Consistent with the carboxy-terminal region of hsPrp38 constituting an authentic RS domain, it has been found to bind other SR proteins and SRPKs in a yeast two-hybrid (Y2H) assay (Hegele et al. 2012). In canonical SR proteins, the RS domain is preceded or followed by one or several RNA recognition motif (RRM) and/or Zinc-finger (ZnF) domains, which often bind exonic splicing enhancer (ESE) sequences on pre-mRNAs (Long and Caceres 2009). More rarely, SR proteins contain other RNA-binding domains, such as RNA helicase or PWI domains, in addition to or instead of RRMs or ZnFs (Long and Caceres 2009). There are only very few cases of SR proteins with RNA-unrelated additional domains, such as a cyclophilin domain in the case of CypRS (Stegmann et al. 2009) or a kinase domain in ClkSty-1 (Long and Caceres 2009). The Prp38 NTD is unrelated to known RS domain–associated domains in other SR proteins. Presently, it is unclear which precise functions are exerted by the universal Prp38 NTD and which additional, higher eukaryote–specific functions are maintained through the RS domain.
FIGURE 1.

Domain organization and conservation of Prp38 orthologs. (A) Domain organization of Saccharomyces cerevisiae and Homo sapiens Prp38 proteins. Numbers indicate domain borders. (B) Multiple sequence alignment of Prp38 orthologs. Numbers above the alignment refer to the human protein. Secondary structure elements based on the crystal structures of hsPrp38NTD and hsPrp38NTD+ are indicated by red cylinders (α-helices) and yellow arrows (β-strands). Residues, at which substitutions selectively interfere with binding to specific hsPrp38-interacting proteins, according to clusters IV–VII (see Fig. 4) are pointed out by purple triangles. Blue “RS” symbols above the alignment indicate RS dipeptides found in the carboxy-terminal region of hsPrp38. (C) Number of RS repeats in Prp38 orthologs of different species. A higher number of RS repeats correlates with a higher complexity of the host.
Here, we elucidated the crystal structure of the hsPrp38 NTD (hsPrp38NTD) and showed by Y2H analysis that it binds a large number of other spliceosomal proteins, including, but not limited to, previously characterized Prp38 interactors. We also delineated four distinct binding sites for several interacting proteins on the hsPrp38 NTD and found that at least two Y2H interaction partners can bind the NTD simultaneously in vitro. When added in excess to splicing reactions, the isolated NTD stalls the spliceosome at a precatalytic B-complex stage. Our results show that hsPrp38 is an unusual SR protein, in which the RS domain is coupled to a multi-interface protein–protein interaction (PPI) domain.
RESULTS
Crystal structure analysis suggests that hPrp38NTD is a protein–protein interaction domain
Humans bear two scPrp38-like proteins, produced from separate genes, hsPrp38A (UniProt ID: Q8NAV1; Gene ID: 84950) and hsPrp38B (UniProt ID: Q5VTL8; Gene ID: 55119). The two proteins show 38.5% sequence identity for 112 aligned residues, but hsPrp38B (546 residues) is significantly longer than hsPrp38A (312 residues) and bears a number of insertions and a carboxy-terminal appendix (not shown). Both proteins are ubiquitously produced in human tissues (http://www.proteinatlas.org) but so far only hsPrp38A has been found in spliceosomal complexes (Wahl et al. 2009; Agafonov et al. 2011).
hsPrp38 contains an NTD (residues 1–179) of unknown fold and function, which is universally conserved in Prp38 orthologs, followed by a stretch rich in acidic amino acids (acidic linker, AL; residues 180–202) and a carboxy-terminal region that harbors a series of RS dipeptide repeats (residues 203–312), resembling the RS domains of SR proteins (Fig. 1A,B). Although it is lacking from scPrp38, an equivalent of the RS domain is also found in other higher eukaryotic Prp38 orthologs, and the number of RS repeats in this region increases from simple multicellular organisms (three RS repeats in Caenorhabditis elegans) via more complex invertebrates (seven RS repeats in Drosophila melanogaster) to mammalian Prp38 proteins (16 RS repeats in hsPrp38; Fig. 1B,C).
The conserved Prp38 NTD does not bear obvious sequence similarity to known RNA-binding domains as found in canonical SR proteins. To start investigating its possible functions, we determined a crystal structure of hsPrp38NTD (residues 1–179) at 1.28-Å resolution via single-wavelength anomalous dispersion (SAD), using selenomethionine-derivatized protein (Table 1). Crystals of hsPrp38NTD contained a single molecule per asymmetric unit, all residues of which were well defined in the electron density. The core of the hsPrp38NTD structure is formed by three pairs of antiparallel α-helices (α4/α5, α6/α7, and α8/α9), which are connected and surrounded by long loops, β-hairpins, and four additional helices (α1, α2, and α3 on one end and α10 on the other; Fig. 2A). We also determined a structure of hsPrp38NTD+ (residues 1–205), containing NTD and AL, at 1.9 Å resolution by molecular replacement, using the hsPrp38NTD structure as a search model (Table 1). Again, the first 179 residues were clearly visible in the electron density, whereas residues 180–205, including the AL, could not be traced, presumably because of intrinsic disorder. The structure of hsPrp38NTD+ is very similar to that of hsPrp38NTD (root-mean-square deviation [rmsd] of 0.54 Å for 165 structurally equivalent Cα atoms) with the exception of the last 14 ordered residues, 166–179. In hsPrp38NTD+ this region forms a loop that folds back onto the tandem helical repeats α6/α7 and α8/α9 instead of forming a short α-helix (α10) as seen in hsPrp38NTD (Fig. 2A,B).
TABLE 1.
Crystallographic data
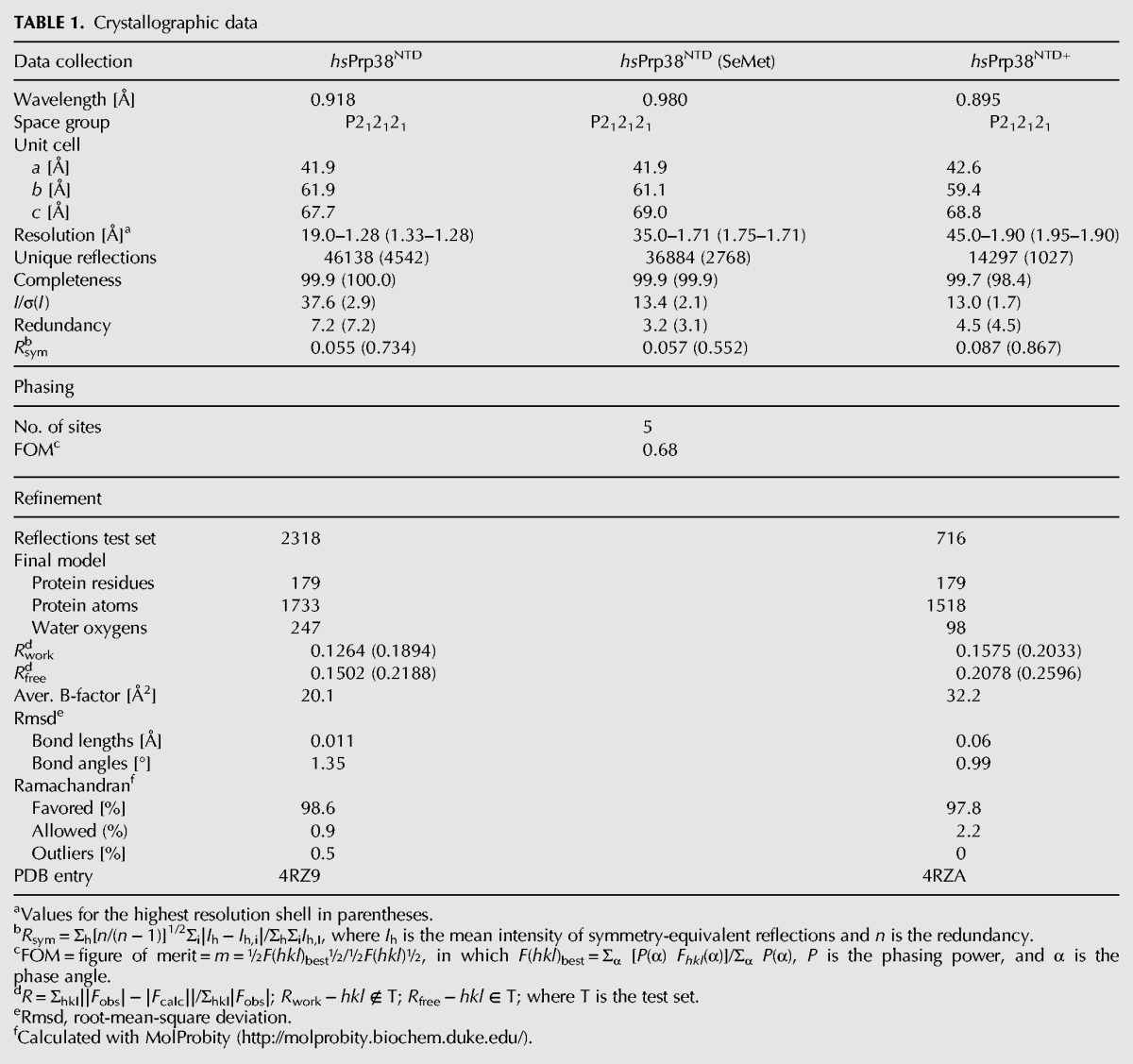
FIGURE 2.
Crystal structures of hsPrp38 amino-terminal domains. Cartoon representation of (A) the crystal structure of hsPrp38NTD (residues 1–179) and (B) the crystal structure of hsPrp38NTD+ (residues 1–205). α-Helices—red colors; β-strands—yellow; loops—gray. Three α-helical repeats forming the cores of the structures (α4/α5, α6/α7, and α8/α9) are shown in different shades of red.
As expected from sequence comparisons, the structure of hsPrp38NTD lacks similarity to known RNA-binding domains. Instead, a similarity search with the Dali server (Holm and Sander 1998) revealed that the central helical repeats of hsPrp38NTD structurally resemble HEAT repeats as found, for example, in the phosphatase 2A subunit PR65 (root-mean-square deviation of 2.9 Å for 105 structurally equivalent Cα atoms to PDB ID 1B3U; Groves et al. 1999). As HEAT repeat regions comprise versatile PPI platforms (Palidwor et al. 2009), we next investigated the possibility that the NTD of Prp38 proteins functions as a PPI element.
The spliceosomal hsPrp38NTD interactome
A recent Y2H analysis identified PPIs among approximately 240 human spliceosomal proteins (Hegele et al. 2012). This screen included the AL and carboxy-terminal RS domain of hsPrp38, whereas the universally conserved NTD was not represented in this study. Therefore, to test if hsPrp38NTD acts as a protein-binding platform in the spliceosome, we performed Y2H-based interaction screens with hsPrp38NTD or full-length hsPrp38 (hsPrp38FL) as bait and the above prey collection of human spliceosomal proteins represented by more than 440 clones. Bait and prey constructs were tested in a pairwise manner with double (hsPrp38FL) or fourfold (hsPrp38NTD) redundancy. Prey constructs that yielded colonies in both independent tests (in the case of hsPrp38FL) or in at least three independent experiments (in the case of hsPrp38NTD) were considered potential binding partners (Fig. 3A, top and middle). We screened in parallel specific domain constructs of other spliceosomal proteins, which yielded very different interaction candidates (Fig. 3A, bottom), demonstrating the specificity of the Y2H results.
FIGURE 3.
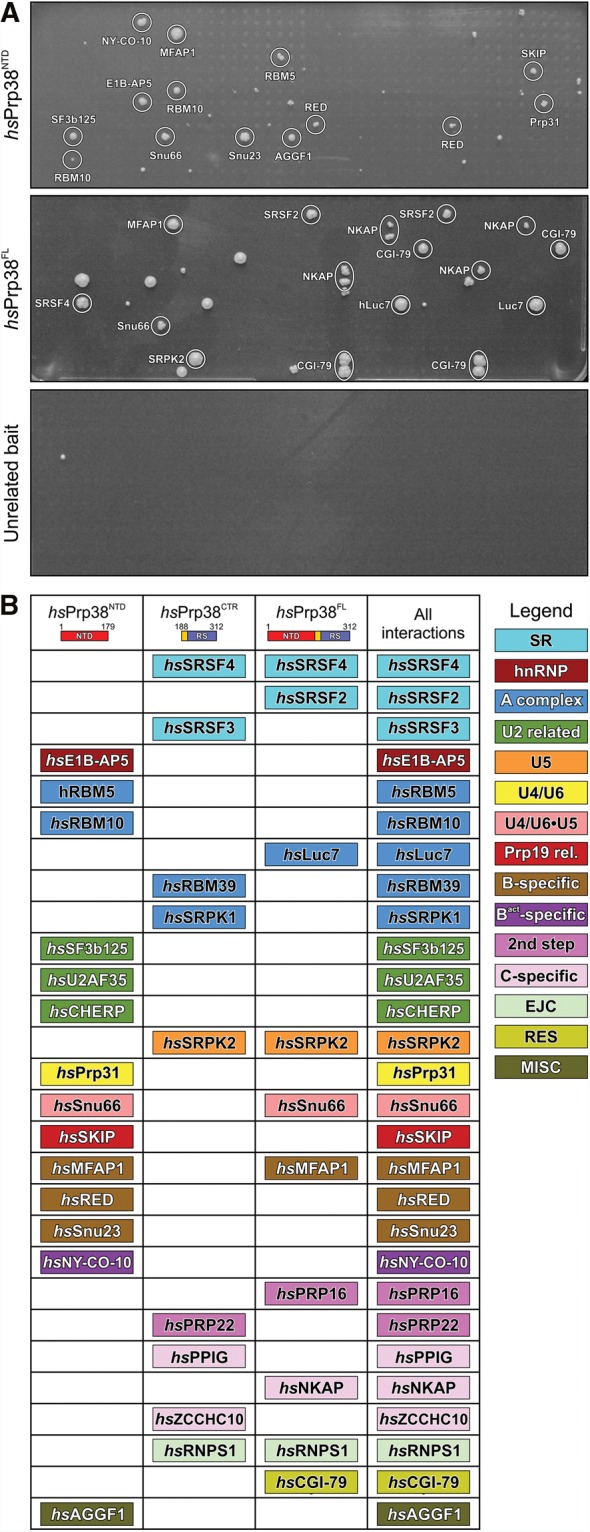
hsPrp38 binding partners identified by yeast two-hybrid analyses. (A) Representative Y2H screens of hsPrp38NTD (top), hsPrp38FL (middle), and an unrelated bait as negative control (bottom). Y2H hits were considered putative interactions if they appeared in at least 75% of the repetitions. (B) Interaction partners of hsPrp38NTD (this study), a carboxy-terminal region (CTR) of hsPrp38 (comprising the AL and RS domain; residues 188–312; Hegele et al. 2012) and hsPrp38FL (this study) are listed and color-coded according to their affiliation with spliceosomal subcomplexes or groups of proteins (legend on the right).
Based on these criteria, 12 putative, direct interaction partners of hsPrp38NTD were identified (Fig. 3A,B). One major group of Y2H interactors consists of other B-specific proteins (i.e., hsSnu23, hsMFAP1, and hsRED). hsPrp38NTD also showed interactions with the tri-snRNP proteins, hsPrp31 and hsSnu66, which are also recruited at the B-complex stage, the Prp19-related protein hsSKIP, which starts to get recruited at B-complex stage but is fully present only at the following Bact stage, as well as with hsNY-CO-10/hsCwc27, which first enters at the Bact stage (Agafonov et al. 2011). In addition, there were hsPrp38NTD Y2H interactions with several splicing factors that are recruited early during spliceosome assembly (Agafonov et al. 2011) (i.e., hsRBM5, hsRBM10, hsSF3b125, hsE1B-AP5, and hsAGGF1). hsRBM5, hsRBM10, and hsSF3b125 bind directly to the spliceosomal discard and disassembly factor hsPrp43 (which is at least in part recruited during A complex formation; Agafonov et al. 2011), to the spliceosomal activation factor hsPrp19, and to the alternative splicing factor hsSR140, respectively (Hegele et al. 2012). hsE1B-AP5 interacts with heterogeneous nuclear (hn) RNP proteins and, like hsAGGF1, with the U1 snRNP component hsU1C (Hegele et al. 2012). Notably, hsPrp38NTD did not interact with any of the canonical SR proteins, SRPKs, or any proteins, besides hsSKIP and hsNY-CO-10/hsCwc27, that are recruited after the B-complex stage (Agafonov et al. 2011).
As expected, hsPrp38FL interacted with proteins previously identified as interactors of its AL/RS domain (hshRNPS1, hsSRSF4, and hsSRPK2; Hegele et al. 2012), and with a subset of the hsPrp38NTD binding partners (hsMFAP1 and hsSnu66). It showed additional interactions not observed with hsPrp38NTD or the AL/RS domain (hsSRSF2, hsLuc7, hsPrp16, hsNKAP, and hsCGI-79; Fig. 3A,B). These observations could indicate that the presence of the AL/RS domain can influence protein binding at the NTD and vice versa, as well as that some hsPrp38 interactors might recognize epitopes on both the NTD and the RS domain. However, differences in the interaction profiles might also reflect differences in the Y2H constructs (such as steric constraints due to the proximity of the fused DNA-binding domain) and/or differences in the expression levels for the various constructs. Therefore, we presently consider all proteins that show up as Y2H interactors of any of the tested hsPrp38 regions as candidate binding partners. Together, our Y2H results suggest that the NTD of hsPrp38 serves as a PPI hub in the course of assembly of the spliceosomal B complex.
Systematic mutational analysis identifies four distinct protein interaction interfaces on hsPrp38NTD
Several of the hsPrp38NTD/hsPrp38FL interaction partners identified here are simultaneously present in a spliceosomal complex, such as the B-specific proteins (Agafonov et al. 2011). Thus the question arises: Do these proteins bind to the same or overlapping sites on the hsPrp38 surface, or do certain groups of interaction partners recognize distinct binding sites enabling simultaneous binding of two or more proteins. Based on hydrophobicity, charge distribution, or conservation patterns, no obvious protein interaction interfaces were directly apparent from the hsPrp38NTD or hsPrp38NTD+ structures. Therefore, we systematically substituted surface-exposed amino acids, individually or in neighboring pairs, which could potentially contribute to protein binding, with alanines, and we tested interactions of the resulting 56 hsPrp38NTD variants with the previously identified interactors of wild-type (wt) hsPrp38NTD in a Y2H array. In this approach, all candidates were tested in triplicates and in parallel against each alanine mutant version of hsPrp38NTD. In principle, specific Y2H pairs might result in cell toxicity and thus give rise to weaker growth. However, during selection for the diploid strains after mating, both baits and preys are constitutively expressed. In this step, we obtained uniform growth across the entire array, demonstrating a lack of toxicity for any bait/prey combination. Because our Y2H system is tuned toward very low protein production levels to increase specificity, typically no toxic effects are observed (Worseck et al. 2012). The setup, therefore, enabled us to reliably quantify the growth as a measure of relative interaction signal (pixel intensities of grayscale bitmap images).
We normalized the growth signals within each array to the hsPrp38NTD–hsMFAP1 interaction, as this interaction gave rise to the most robust and strongest growth signal among all hsPrp38NTD interactions and was invariant in all but one mutant pairing. Overall, highly correlated values were obtained for interacting pairs, suggesting that growth is not affected by possible differences in the expression levels of bait and prey constructs. Mean values of triplicate experiments for hsPrp38NTD variants were then hierarchically clustered revealing patterns of decreased or increased interaction with the 12 previously identified wt hsPrp38NTD interaction partners (Fig. 4A,B).
FIGURE 4.

Binding site mapping. (A) Fifty-six single or double alanine variants of hsPrp38NTD are clustered according to the degree of similarity in their effects on the interactions with the 12 identified hsPrp38NTD binding partners (clusters I–VII on the right). As a measure of relative binding strength, the growth signal of Y2H hits was quantified, normalized to the growth signal of the hsPrp38NTD–hsMFAP1 interaction and presented in a heat map by a white-to-red color gradient. White boxes—no growth; red boxes—strong growth. The wt binding profile is boxed in cluster I. (B) Growth of representative Y2H colonies of the 12 hsPrp38NTD binding partners with respective hsPrp38NTD variants of all seven clusters on selective agar (I–VII). Red or green boxes indicate reduction or increase of the growth signal relative to wt hsPrp38NTD, respectively. V↑—subgroup of cluster V with increased interaction to hsSnu66; V↓—subgroup of cluster V with reduced interaction to hsSnu66. (C) Cartoon representation of hsPrp38NTD with clusters IV–VII mapped to the protein surface. Residues mutated in clusters IV–VII that negatively affect binding of an interaction partner are colored in red; the residue that upon mutation positively affects binding of hsSnu66 is colored in green.
More than half of the mutant constructs (31) gave rise to interaction patterns very similar to wt hsPrp38NTD (cluster I), suggesting that no critical protein–protein contact residues were affected by the corresponding residue exchanges. All other variants led to reduced Y2H interactions or, in a single case, to an enhanced Y2H interaction. Based on pattern similarity in the cluster analysis, we grouped these variants into six distinct sets (clusters II–VII; Fig. 4A,B). hsPrp38NTD mutations in clusters II and III abolished all interactions except with hsMFAP1 or with hsMFAP1 and hsE1B-AP5, respectively. Residues exchanged by the mutations of clusters II and III are scattered all over the surface of hsPrp38NTD, and thus do not point to a confined interaction region on wt hsPrp38NTD. Rather it suggests that this set of amino acid changes affects protein expression, folding, or stability. In contrast, clusters IV–VII represent hsPrp38NTD variants, for which only a selected subset of Y2H interactions was changed, whereas Y2H interactions with all other hsPrp38NTD Y2H interactors were hardly affected. The exchanged residues in clusters IV–VII show distinct local distributions on the hsPrp38NTD surface (Fig. 4C). The large set of mutants that show differential Y2H interactions provide further evidence for the specificity of our assays.
In cluster IV, mutations K23A/I24A and R28A are located at the amino terminus and center of helix α2, respectively, and interfered specifically with hsE1B-AP5 binding, whereas all other hsPrp38NTD interaction partners showed wt-like growth signals. Thus, hsE1B-AP5 might directly interact with the helix α2 region of hsPrp38NTD (Fig. 4C). In cluster V, three double mutations selectively altered hsSnu66 binding. Of these, V48A/D49A and M52A/E53A are located in hsPrp38NTD helix α4, where V48 is engaged in hydrophobic interactions with F90 of helix α6 and V99 of helix α7, whereas M52 interacts with V99 and engages in a hydrogen bond with D49 of helix α4. The corresponding mutations might thus not only change the local physico-chemical surface properties, but the absence of the intramolecular contacts involving the affected residues could additionally influence positioning of helix α4. The E45A/L46A mutations at the amino terminus of helix α4 increased hsSnu66 binding relative to wt hsPrp38NTD. Although E45 is a surface-exposed residue, L46 binds to L42 located in the loop preceding helix α4, which could again change the position of this secondary structure element. These results suggest that helix α4 and the immediate surroundings on hsPrp38NTD represent a binding site for hsSnu66 (Fig. 4C). Construct M52A/E53A, in addition to hsSnu66, also decreased Y2H interactions with hsRBM5, hsRBM10, and hsSF3b125, suggesting that these proteins take advantage of an interaction region on hsPrp38NTD, which at least partly overlaps with the hsSnu66-interaction site (Fig. 4C). Residues affected by the E83A and E83A/K84A exchanges (cluster VI) are positioned in a rather flat surface area formed by the loop connecting helix α5 to helix α6. The corresponding mutations abolished Y2H interactions of hsPrp38NTD with hsAGGF1 and hsSnu23, suggesting that hsAGGF1 and hsSnu23 share an interaction site in the corresponding region (Fig. 4C). Finally, residues affected by the D145A/E146A exchanges of cluster VII are located at the amino-terminal tip of helix α9 and the residue exchanges selectively decreased the hsMFAP1 and hsSF3b125 interactions (Fig. 4C).
hsRED, hsPrp31, hsSKIP, and hsNY-CO-10/hsCwc27 interaction sites could not be localized unambiguously with the set of hsPrp38NTD mutant constructs used. Prey constructs representing these four proteins already showed weaker growth with the wt hsPrp38NTD bait than the other identified interactors, making it more difficult to reliably detect effects of single hsPrp38NTD point mutations on these interactions. In addition, these proteins may involve more extensive contact surfaces than those tested in the individual constructs or may interact at sites not well covered in our alanine scan.
Taken together, our growth signal–based, quantitative Y2H analysis provided amino acid–level information to distinguish four distinct interaction interfaces distributed across various surfaces of the compactly folded hsPrp38NTD: Relative to the hsSnu66-binding area in the center of one face of hsPrp38NTD (which is possibly also used by hsRBM5, hsRBM10, and hsSF3b125), a hsSnu23/hsAGGF1-binding site lies at the top on the same face, whereas a hsE1B-AP5 interaction site and a hsMFAP1/hsSF3b125-binding site are located on the right and left side, respectively, of the opposite face of hsPrp38NTD (Fig. 4C). Although proteins mapping to the same or overlapping interfaces on hsPrp38NTD (such as hsSnu66/hsRBM5/hsRBM10/hsSF3b125, hsSnu23/hsAGGF1, and hsMFAP1/hsSF3b125) might thus bind mutually exclusively to hsPrp38, proteins that take advantage of spatially separated contact regions on hsPrp38NTD have the potential to bind concomitantly to the protein.
Concomitant binding of several Y2H interactors to Prp38NTD
To validate some of the Y2H interactors and their putative binding sites on hsPrp38NTD and to confirm that two proteins with spatially separated, putative binding sites can interact concomitantly with Prp38NTD, we performed biochemical binding assays. We recombinantly produced and purified hsMFAP1 (cluster VII) and hsSnu23 (cluster VI) and analyzed complex formation of these proteins with hsPrp38NTD by analytical size exclusion chromatography (SEC). Instead of full-length hsMFAP1, we used a long, soluble fragment of the protein (residues 30–344, identified by amino-terminal sequencing and mass spectrometric fingerprinting), which resulted from endogenous limited proteolysis upon recombinant production of full-length hsMFAP1 in Escherichia coli. Consistent with the strong and reproducible interaction of hsPrp38NTD and hsMFAP1 in the Y2H screens, hsPrp38NTD and hsMFAP130-344 coeluted from the SEC column at a smaller volume compared with the individual proteins (Fig. 5A, panels I, III, IV). In contrast, hsPrp38NTD variants bearing the D145A/E146A double exchanges, which led to abrogation of the hsMFAP1 Y2H interaction, or only the D145A single exchange did not coelute with hsMFAP130-344 (Fig. 5A, panels II, V, VI). These results suggest that our Y2H analyses indeed represent direct, binary hsPrp38NTD interactions and that our systematic mutational analyses point to direct contact sites on hsPrp38NTD with the respective interactor.
FIGURE 5.

Analysis of Prp38-MFAP1, Prp38-Snu23, and Prp38-MFAP1-Snu23 complexes. (A) SEC elution profiles and corresponding SDS-PAGE gels of wt hsPrp38NTD or cluster VII mutation (D145A/E146A, D145A) with a large soluble fragment of hsMFAP1 (residues 30–344). Fractions with a gray background are analyzed on the SDS gels. (B) Binary (panels I–V) and ternary (panel VI) SEC interaction analysis of wt ctPrp38NTD+ (residues 1–220) with ctMFAP1 and ctSnu23. ctMFAP1Frag—fragment obtained by endogenous limited proteolysis during production and purification of ctMFAP1FL. Fractions with a darker gray background were analyzed on the SDS gels.
In contrast to hsMFAP130-344, hsSnu23 did not stably interact with hsPrp38NTD (or hsPrp38NTD+) alone or with hsPrp38NTD (or hsPrp38NTD+) prebound to hsMFAP130-344 in SEC. Under the nonequilibrium SEC conditions, only kinetically and thermodynamically relatively stable complexes are expected to persist. We reasoned that the corresponding interactions might be more stable at ambient temperatures for proteins originating from a thermotolerant species. We therefore recombinantly produced Prp38NTD+, MFAP1, and Snu23 from the thermotolerant fungus, Chaetomium thermophilum (ct), and repeated the SEC analysis. Our ctMFAP1 preparation contained both the full-length protein ctMFAP1FL and a long fragment ctMFAP1Frag, which appeared during recombinant production and was copurified. Indeed, ctPrp38NTD+ (residues 1–220 of ctPrp38) stably interacted with ctMFAP1FL/Frag, as well as with ctSnu23, in SEC (Fig. 5B, panels I–V; note that isolated ctMFAP1FL/Frag and their complexes with ctPrp38NTD+ eluted in the same fractions, possibly because of an unusual shape of ctMFAP1FL/Frag, but that binding is clearly revealed by shifting of ctPrp38NTD+ into the ctMFAP1FL/Frag elution fractions). To test whether both ctMFAP1FL/Frag and ctSnu23 bind to ctPrp38NTD+ simultaneously, we mixed all three proteins and performed SEC runs. The three proteins (i.e., ternary complexes containing ctPrp38NTD+, ctSnu23, and either ctMFAP1FL or ctMFAP1Frag) coeluted at a smaller volume than any of the binary combinations (Fig. 5B, panel VI). Although formation of the ternary complex may be further supported by direct ctMFAP1–ctSnu23 interactions, the above results together suggest that Prp38 can indeed utilize different interfaces to bind multiple proteins at the same time and thus behaves as a true multi-interface protein interaction platform.
An excess of hsPrp38NTD+ stalls splicing after B-complex formation
As only the NTD and AL are universally conserved among Prp38 orthologs, we next tested whether this region alone is sufficient to mediate spliceosome association of hsPrp38 and to support splicing. To this end, we added increasing amounts of recombinantly produced hsPrp38NTD+ (residues 1–205) to splicing-competent HeLa nuclear extract and monitored the effects on splicing of MINX pre-mRNA and on spliceosome assembly. We observed increasing inhibition of splicing at increasing concentrations of hsPrp38NTD+ compared with the control reaction lacking hsPrp38NTD+ (Fig. 6A, cf. lanes 6–9 to lane 5), with complete inhibition of both steps of splicing at 36 µM hsPrp38NTD+ or above. Monitoring splicing complex formation on native gels indicated that spliceosome assembly was stalled after B-complex formation and does not proceed to activation and catalysis (Fig. 6B, cf. lanes 7–10 to lane 6), consistent with a role for full-length hsPrp38 in the subsequent activation step. In contrast, when we used hsPrp38NTD+,D145A (bearing a residue exchange that led to abrogation of its interaction with hsMFAP130-344; Fig. 5A, panel VI) instead of wt hsPrp38NTD+ in the assay, only very mild inhibition of splicing was observed (Fig. 6A, cf. lanes 10–13 to lane 5) and spliceosome assembly readily proceeded to the C-complex stage (Fig. 6B, compare lanes 11–14 to lane 6), suggesting that interaction of hsPrp38NTD+ with MFAP1 is important for its splicing inhibition activity. These results indicate that hsPrp38NTD+ is recruited to spliceosomes at the B-complex stage and interferes with spliceosome catalytic activation by replacing the endogenous protein or by competing with the endogenous protein for some of its functional interactions. Alternatively, hsPrp38NTD+ might sequester other splicing factors in an inactive complex, possibly through hsMFAP1 association, before interaction with the spliceosome.
FIGURE 6.

Dominant-negative effects of hsPrp38NTD+ on splicing. (A) In vitro splicing of MINX pre-mRNA monitored on a denaturing gel. Educts, intermediates, and products of the reactions are identified on the left. Lanes 1–5—time course of splicing (time points indicated above the gel) in the absence of hsPrp38NTD+. Lanes 6–13—effects of adding increasing amounts of hsPrp38NTD+ (lanes 6–9) or hsPrp38NTD+,D145A (lanes 10–13; indicated above the gel) to the reaction. (B) Native gel monitoring the kinetics of spliceosome assembly (time points above the gel) in the absence (lanes 1–6) or presence of increasing amounts of hsPrp38NTD+ (lanes 7–10) or hsPrp38NTD+,D145A (lanes 11–14; indicated above the gel). Emerging complexes are identified on the left; H—hnRNP complexes; A, B, C—spliceosomal A, B, and C complexes.
DISCUSSION
In contrast to the snRNPs and their stably associated factors, comparatively little is presently known about the functions of non-snRNP proteins in the spliceosome. In humans, a group of nine non-snRNP, B-specific proteins joins the spliceosome during B-complex formation and leaves again during the B-to-Bact transition (Agafonov et al. 2011). Here, we have structurally and functionally dissected the B-specific protein, hsPrp38. hsPrp38 exhibits a domain organization reminiscent of SR proteins, with a universally conserved NTD that is connected to a higher eukaryote–specific, carboxy-terminal RS domain via a short AL. We find that the NTD mediates a multitude of intraspliceosomal PPIs, several of which might have to be realized concomitantly to drive spliceosomes into catalytic activation.
hsPrp38 is an unconventional SR protein
Although canonical SR proteins contain an RS domain with one or more RNA-binding domains (Long and Caceres 2009), our crystal structure analyses show that the RS domain of hsPrp38 is associated with an α-helical repeat-containing NTD, reminiscent of PPI domains. Indeed, using a comprehensive Y2H approach, we find that 12 spliceosomal proteins show Y2H interactions with the hsPrp38 NTD. Our screen not only uncovered proteins that have previously been reported to bind Prp38 orthologs in other organisms (i.e., MFAP1 [Andersen and Tapon 2008] or Snu23 [Krogan et al. 2006; Pandit et al. 2009]), but also revealed many additional putative hsPrp38NTD binding partners. In principle, some of the Y2H interactions may be indirect and mediated by endogenous components of the yeast splicing apparatus, as yeast and mammalian splicing factors are generally well conserved. However, our observation of Y2H interactions between hsPrp38NTD and several human factors that are not present in yeast (hsMFAP1, hsRED, hsE1B-AP5, hsAGGF1, hsRBM5, hsRBM10) argues against this possibility. As two tested Y2H interactions (with MFAP1 and Snu23) prevailed in SEC analyses, the detected Y2H interactions likely correspond to direct, binary interactions in the spliceosome. By using proteins from two different organisms, our results also suggest that these hsPrp38NTD interactions are evolutionarily conserved. The observed differences in Prp38NTD-Snu23 stability between human and C. thermophilum could indicate species-specific variations in binding affinities, at least under the buffer/temperature conditions used in the present experiments.
One strength of the Y2H system is to detect weak and transient interactions (Vinayagam et al. 2009; Worseck et al. 2012), which may contribute significantly to the compositional and conformational dynamics during spliceosome assembly. Therefore, rather than dismissing interactions that cannot be recapitulated in more stringent biochemical or biophysical assays, the different interaction assays should be considered complementary (Stelzl 2014). In any case, our results define hsPrp38 as an unusual SR protein, in which a carboxy-terminal RS domain is associated with an amino-terminal PPI domain.
hsPrp38 is a major PPI hub in the spliceosome
High-throughput approaches, such as affinity purification coupled to mass spectrometry and Y2H assays, have been used to map PPIs in multiprotein complexes (Hegele et al. 2012), specific cellular processes (Weimann et al. 2013), or even entire proteomes (Woodsmith and Stelzl 2014). The deduced interaction networks show that the majority of the involved proteins interact with one or a few partners, whereas a limited set of hub proteins exhibit many interactions (Barabási and Oltvai 2004). Hub proteins can be further classified as “date” hubs, which contact their various interaction partners one at a time, and “party” hubs, which interact with many or all of their partners simultaneously, giving rise to multisubunit complexes (Han et al. 2004; Gursoy et al. 2008). Although date hubs typically exhibit a single interface or overlapping interfaces for the sequential interaction with their partners, party hubs are multi-interface proteins with distinct interaction surfaces for their interactors (Kim et al. 2006). In addition to 11 non-self PPIs of the AL and carboxy-terminal RS domain of hsPrp38 identified in a previous Y2H study (Hegele et al. 2012), we detected 17 additional, nonredundant Y2H interactions using hsPrp38NTD and hsPrp38FL. Thus, hsPrp38 might interact with a total of 28 other spliceosomal proteins, representing one of the major PPI hubs of the human spliceosome along with hshnRNPK (24 PPIs), hsRBM4 (24 PPIs), hsCHERP (23 PPIs), hsRBM10 (23 PPIs), and hsSF3b145 (22 PPIs) (Hegele et al. 2012).
In the majority of known spliceosomal PPIs, the precise interaction interfaces remain unknown. Determining whether the interfaces on a given multi-interaction protein are overlapping or spatially segregated and whether such a protein is capable of binding multiple partners at the same time is important for our understanding of the dynamics of spliceosome assembly. To address this question for hsPrp38NTD, we used our structural knowledge and results from our initial Y2H screen to set up alanine surface-scanning two-hybrid arrays, which we evaluated via growth and clustering analyses. This approach allowed us to suggest four binding interfaces for eight of the 12 identified hsPrp38NTD interaction partners. Our assignment of interfaces was validated by the observations that (i) a selected interaction (hsPrp38NTD-hsMFAP1) persisted between the recombinant proteins in SEC, (ii) this interaction was abrogated in an hsPrp38NTD variant that also failed to show the original Y2H interaction, and (iii) two proteins with suggested separated interfaces on Prp38NTD (MFAP1 and Snu23) can indeed interact with Prp38NTD at the same time when using proteins from an organism in which both individual interactions are stable in SEC.
Based on our results, we suggest that hsPrp38 combines aspects of a date and a party hub. Although several groups of interacting proteins apparently have overlapping binding sites on hsPrp38 (such as hsSnu23 and hsAGGF1, hsMFAP1 and hsSF3b125, or hsSnu66, hsRBM5, hsRBM10, and hsSF3b125), others (such as combinations of individual factors represented in clusters IV–VII) take advantage of different interaction surfaces and may bind concomitantly to hsPrp38.
Possible roles of hsPrp38NTD in spliceosome assembly
The hsPrp38NTD interactors we detected belong to (i) the group of B-specific proteins (hsMFAP1, hsSnu23, hsRED), (ii) proteins of the U4/U6•U5 tri-snRNP (hsSnu66, hsPrp31), (iii) proteins joining first during the B-to-Bact transition (hsSKIP, hsNY-CO-10/hsCwc27), and (iv) proteins belonging to the groups of hnRNPs (hsE1B-AP5), A complex proteins (hsRBM5, hsRBM10), U2 related proteins (hsSF3b125, hsU2AF35, hsCHERP), or miscellaneous factors associated with the spliceosome (hsAGGF1). The first group of interactors (other B-specific proteins) suggests that hsPrp38NTD serves to organize at least some of the B-specific proteins as a multimeric complex. This organization could take effect already outside of the spliceosome, allowing the participating proteins to be recruited en bloc. Alternatively, the interactions could emerge only in the context of the spliceosome, with hsPrp38NTD serving to spatially arrange its B-specific interactors.
The U4/U6•U5 tri-snRNP also joins the spliceosome during B-complex formation and proteomics analyses showed that its recruitment precedes incorporation of the B-specific proteins (Schneider et al. 2010). Therefore, hsSnu66 and hsPrp31 most likely provide additional binding sites for efficient or stable incorporation of hsPrp38, and perhaps other B-specific proteins, at the B-complex stage. Notably, because the mapped binding sites for hsSnu66 (tri-snRNP), hsMFAP1, and hsSnu23 (B-specific) are nonoverlapping on the surface of hsPrp38NTD, hsPrp38 could realize its putative functions as an organizer of a complex of B-specific proteins and as a contact to components of the U4/U6•U5 tri-snRNP at the same time.
We also found Y2H interactions of hsPrp38NTD with factors that are not fully present before the Bact complex stage (Agafonov et al. 2011)—that is, the Prp19-related protein hsSKIP and the peptidyl-prolyl cis–trans isomerase (PPIase) domain-containing protein hsNY-Co-10/hsCwc27 (Ulrich and Wahl, 2014). These findings thus suggest that hsPrp38, via its NTD, could help to integrate Bact-specific proteins into the spliceosome at a transition stage just before hsPrp38 and other B-specific proteins leave the spliceosome.
Finally, our Y2H interactions also point to potential contacts between hsPrp38NTD and factors that join the spliceosome during early stages of assembly (hsE1B-AP5, hsRBM5, hsRBM10, hsSF3b125, hsU2AF35, hsCHERP) (Agafonov et al. 2011). These interactions might help to loosely associate hsPrp38, and possibly other B-specific proteins, to early spliceosomal complexes or to recruit these proteins efficiently during B-complex formation. Notably, binding sites of hsRBM5, hsRBM10, and hsSF3b125 seem to overlap with the binding site of the tri-snRNP protein hsSnu66 on hsPrp38NTD. Because of the putative, ensuing binding competition, we assume that the early hsPrp38 partners could initially recruit hsPrp38 (and perhaps other B-specific proteins) and hand it over to hsSnu66 after the U4/U6•U5 tri-snRNP has been integrated.
MATERIALS AND METHODS
Plasmids
Open reading frames (ORFs) encoding full-length hsPrp38, hsMFAP1, and hsSnu23 were amplified from a human cDNA library and cloned into pETM11 plasmids by EMP cloning (Ulrich et al. 2012). pETM11 plasmids containing synthetic ORFs encoding full-length C. thermophilum proteins were obtained from Thermo Fisher Scientific. The pETM11 vector guides production of amino-terminally His6-tagged, TEV-cleavable fusion proteins. Truncations and point mutations were introduced by inverse PCR as described (Ulrich et al. 2012) or by QuikChange site-directed mutagenesis (Stratagene). All expression constructs were verified by sequencing of the promoter and insert regions.
Protein production and purification
Proteins were produced in E. coli Rosetta 2 (DE3) cells either by induction with IPTG (1 mM) in LB medium or by auto-induction in ZY medium (Studier 2005) at 18°C–23°C and for 24–40 h. Selenomethionine-containing hsPrp38NTD was produced in M9 medium (2 mM MgSO4, 0.4% glucose, 0.5 mg riboflavin, 0.5 mg niacinamide, 0.5 mg pyridoxine monohydrate, 0.5 mg thiamine, 50 mg EDTA, 8 mg FeCl3, 0.5 mg ZnCl2, 0.1 mg CuCl2, 0.1 mg CoCl2, 0.1 mg H3BO3, 16 mg MnCl2, traces of NiSO4 and molybdic acid). One liter M9 cultures were grown at 37°C. At OD600 0.5, amino acids (1 g lysine, 1 g threonine, 1 g phenylalanine, 0.5 g leucine, 0.5 g isoleucine, 0.5 g valine, 0.5 g selenomethionine) were added. After an additional 15 min, cultures were shifted to 18°C and induced with 1 mM IPTG. Cells were harvested 48 h post induction.
The following steps were performed at 4°C. Cells were resuspended in solubilization buffer (50 mM sodium phosphate, pH 8.0, 500 mM NaCl, 30 mM imidazole, 5 mM β-mercaptoethanol) and disrupted using a Sonopuls Ultrasonic Homogenizer HD 3100 (Bandelin). The soluble fractions were incubated with Ni2+-NTA resin (GE Healthcare) for 1 h and washed three times with 10 column volumes of solubilization buffer and then bound protein was eluted with 250 mM imidazole, pH 8.0, 150–500 mM NaCl, and 5 mM β-mercaptoethanol. For removal of the His6-tag, TEV protease (1:30) was added and samples were dialyzed overnight against 10 mM Tris–HCl, pH 8.0, 150–350 mM NaCl, 30 mM imidazole, and 5 mM β-mercaptoethanol. Instead of TEV protease treatment, selenomethionine-containing hsPrp38NTD was digested for 40 h at 4°C by chymotrypsin (1:2000). Cleaved proteins were again passed over Ni2+-NTA resin and the flow-through collected. The hsMFAP1 sample was additionally loaded on a 5 mL HiTrap Q ion-exchange column (GE Healthcare) and eluted via a linear 150–400 mM NaCl gradient. All proteins were concentrated and further purified via Superdex 75 or Superdex 200 size exclusion chromatography (GE Healthcare) in SEC buffer (10 mM Tris–HCl, pH 8.0, 300–350 mM NaCl, 0.1 mM EDTA, and 1 mM DTT). Fractions containing the target proteins were pooled, concentrated, shock-frozen in liquid nitrogen, and stored at −80°C.
Crystallization
All crystals were obtained by sitting drop vapor diffusion with drops containing 1 µL protein and 1 µL reservoir solution at 18°C. Native hsPrp38NTD crystals were obtained at a concentration of 12.8 mg/mL with a reservoir solution containing 0.1 M MES, pH 6.5, 15% (v/v) PEG 400 after 1 d. Selenomethionine-containing hsPrp38NTD crystals were obtained at 4°C with a protein concentration of 6.8 mg/mL with a reservoir solution containing 0.1 M MES, pH 6.0, 16%–18% (w/v) PEG 4000, 200 mM MgCl2 after 1 d. hsPrp38NTD+ crystals were obtained at a protein concentration of 12.0 mg/mL with a reservoir solution containing 0.1 M MES, pH 5.0, 16% PEG 3350, 0.2 M (NH4)2SO4, 5% (v/v) glycerol.
Diffraction data collection and structure determination
For data collection, crystals were flash-cooled in liquid nitrogen after transfer into cryo-protecting solution (crystallization buffer supplemented with 20% (v/v) propylene glycol or 20% (v/v) glycerol). Diffraction data were collected at beamline 14.2 of the BESSY II storage ring (Berlin, Germany) (Mueller et al. 2012) and processed with HKL2000 (Otwinowski and Minor 1997) (hsPrp38NTD) or XDS (Kabsch 2010) (selenomethionine hsPrp38NTD and hsPrp38NTD+). Initial phases for hsPrp38NTD were obtained by the SAD strategy using data collected at the selenium peak wavelength. Five selenium sites were located and used for phasing with the ShelX package (Sheldrick 2008). The experimentally phased map was of high quality and allowed manual building of the structure using COOT (Emsley and Cowtan 2004). The structure of hsPrp38NTD+ was solved by molecular replacement using MOLREP (Vagin and Teplyakov 2010) from the CCP4 package (Collaborative Computational Project, Number 4 1994) and the structure coordinates of the refined hsPrp38NTD model. After placement of the NTD portion, clear negative difference electron density allowed remodeling of the residues 164–179 of hsPrp38NTD+. Structural models were completed through alternating rounds of automated refinement using REFMAC5 (Murshudov et al. 2011) or phenix.refine (Afonine et al. 2012) and manual model building using COOT (Emsley and Cowtan 2004). Structure coordinates and diffraction data have been deposited with the Protein Data Bank (http://www.pdb.org) under accession codes 4RZ9 (hsPrp38NTD) and 4RZA (hsPrp38NTD+) and will be released upon publication.
Yeast two-hybrid assays
PPI matrix Y2H screening was performed as described previously (Hegele et al. 2012; Worseck et al. 2012). Briefly, PRP38 baits, amino-terminally fused to lexA DNA-binding domains in pBTM116-D9 vectors, were mated in duplicate in 384 array format with prey strains representing approximately 240 spliceosomal proteins by approximately 440 clones (Hegele et al. 2012). Interacting bait–prey pairs were identified by growth on selective agar plates (Leu-Trp-Ura-His). Only bait–prey pairs that showed growth in at least 75% of independent experiments were considered for further evaluation.
Cluster analysis
Interaction mating with variant hsPrp38NTD constructs was performed in 96-well MTP format in triplicates. After mating of the bait–prey strains, diploid yeast cultures were first uniformly grown for 2 d on nonselective Leu-Trp agar plates and then transferred to selective Leu-Trp-Ura-His agar plates. Yeast growth after 6 d on selective agar was quantified from grayscale TIFF images by measuring the pixel intensity across the growth area using the AIDA software (raytest). Triplicate values normalized to growth of the corresponding hsPrp38NTD-hsMFAP1 pair were averaged. Because the D145A/E146A variant of hsPrp38NTD showed strongly reduced growth with hsMFAP1, normalization was performed for this mutant version using the average size values of unaffected interaction partners. The data matrix of relative yeast growth (rows: hsPrp38NTD mutant constructs; columns: interaction partner proteins) was subjected to hierarchical clustering using Euclidean distance.
Analytical gel filtration chromatography
Proteins (50 µM), alone or as equimolar mixtures, were incubated in SEC buffer for 30 min at 4°C. Fifty microliter samples were analyzed on a Superdex 200 increase 3.2/300 size exclusion column (GE Healthcare) using an ÄKTAmicro system (GE Healthcare) at 4°C. Elution profiles were inspected by SDS-PAGE.
In vitro splicing assays
Pre-mRNA splicing was performed with 32P-labeled, m7GpppG-capped MINX pre-mRNA in HeLa nuclear extract and analyzed by denaturing PAGE, essentially as described previously (Bessonov et al. 2010), except that the splicing reactions contained 10 nM pre-mRNA. Spliceosomal complexes were analyzed by agarose gel electrophoresis as previously described (Bessonov et al. 2010), and bands were visualized by autoradiography.
ACKNOWLEDGMENTS
We thank Gabi Heyne (Max Planck Institute for Biophysical Chemistry, Department of Cellular Biochemistry) for support in splicing assays, and Karine F. Santos and Claudia Alings (Freie Universität Berlin, Laboratory of Structural Biochemistry) for helpful discussions and for advice in crystallization. We acknowledge access to beamline BL14.2 of the BESSY II storage ring (Berlin, Germany) via the Joint Berlin MX-Laboratory sponsored by the Helmholtz Zentrum Berlin für Materialien und Energie, the Freie Universität Berlin, the Humboldt-Unversität zu Berlin, the Max-Delbrück Centrum and the Leibniz-Institut für Molekulare Pharmakologie. This work was supported by the Max-Planck Society and the Deutsche Forschungsgemeinschaft (grants WA1126/7-1 to M.C.W. and LU294/15-1 to R.L.).
Authors' contributions: All authors designed experiments, analyzed the data, and interpreted the results. T.S., A.K.C.U., L.A., C.L.W., N.B., M.S., and G.W. performed experiments. T.S., A.K.C.U., C.L.W., R.L., U.S., and M.C.W. wrote the manuscript. R.L, U.S., and M.C.W. supervised the project.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.054296.115.
REFERENCES
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. 2012. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68: 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agafonov DE, Deckert J, Wolf E, Odenwälder P, Bessonov S, Will CL, Urlaub H, Lührmann R. 2011. Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol Cell Biol 31: 2667–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen DS, Tapon N. 2008. Drosophila MFAP1 is required for pre-mRNA processing and G2/M progression. J Biol Chem 283: 31256–31267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabási AL, Oltvai ZN. 2004. Network biology: understanding the cell's functional organization. Nat Rev Genet 5: 101–113. [DOI] [PubMed] [Google Scholar]
- Bessonov S, Anokhina M, Krasauskas A, Golas MM, Sander B, Will CL, Urlaub H, Stark H, Lührmann R. 2010. Characterization of purified human Bact spliceosomal complexes reveals compositional and morphological changes during spliceosome activation and first step catalysis. RNA 16: 2384–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanton S, Srinivasan A, Rymond BC. 1992. PRP38 encodes a yeast protein required for pre-mRNA splicing and maintenance of stable U6 small nuclear RNA levels. Mol Cell Biol 12: 3939–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JY, Stands L, Staley JP, Jackups RR Jr, Latus LJ, Chang T-H. 2001. Specific alterations of U1-C protein or U1 small nuclear RNA can eliminate the requirement of Prp28p, an essential DEAD box splicing factor. Mol Cell 7: 227–232. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50: 760–763. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- Groves MR, Hanlon N, Turowski P, Hemmings BA, Barford D. 1999. The structure of the protein phosphatase 2A PR65/A subunit reveals the conformation of its 15 tandemly repeated HEAT motifs. Cell 96: 99–110. [DOI] [PubMed] [Google Scholar]
- Gursoy A, Keskin O, Nussinov R. 2008. Topological properties of protein interaction networks from a structural perspective. Biochem Soc Trans 36: 1398–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J-DJ, Bertin N, Hao T, Goldberg DS, Berriz GF, Zhang LV, Dupuy D, Walhout AJM, Cusick ME, Roth FP, et al. 2004. Evidence for dynamically organized modularity in the yeast protein–protein interaction network. Nat Cell Biol 430: 88–93. [DOI] [PubMed] [Google Scholar]
- Hegele A, Kamburov A, Grossmann A, Sourlis C, Wowro S, Weimann M, Will CL, Pena V, Lührmann R, Stelzl U. 2012. Dynamic protein–protein interaction wiring of the human spliceosome. Mol Cell 45: 567–580. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. 1998. Dictionary of recurrent domains in protein structures. Proteins 33: 88–96. [DOI] [PubMed] [Google Scholar]
- Kabsch W. 2010. XDS. Acta Crystallogr D Biol Crystallogr 66: 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PM, Lu LJ, Xia Y, Gerstein MB. 2006. Relating three-dimensional structures to protein networks provides evolutionary insights. Science 314: 1938–1941. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, et al. 2006. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440: 637–643. [DOI] [PubMed] [Google Scholar]
- Laggerbauer B, Achsel T, Lührmann R. 1998. The human U5-200kD DEXH-box protein unwinds U4/U6 RNA duplices in vitro. Proc Natl Acad Sci 95: 4188–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardelli RM, Thompson JX, Yates JR, Stevens SW. 2010. Release of SF3 from the intron branchpoint activates the first step of pre-mRNA splicing. RNA 16: 516–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JC, Caceres JF. 2009. The SR protein family of splicing factors: master regulators of gene expression. Biochem J 417: 15–27. [DOI] [PubMed] [Google Scholar]
- Mueller U, Darowski N, Fuchs MR, Förster R, Hellmig M, Paithankar KS, Pühringer S, Steffien M, Zocher G, Weiss MS. 2012. Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J Synchrotron Radiat 19: 442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. 2011. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67: 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Palidwor GA, Shcherbinin S, Huska MR, Rasko T, Stelzl U, Arumughan A, Foulle R, Porras P, Sanchez-Pulido L, Wanker EE, et al. 2009. Detection of α-rod protein repeats using a neural network and application to huntingtin. PLoS Comput Biol 5: e1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit S, Paul S, Zhang L, Chen M, Durbin N, Harrison SMW, Rymond BC. 2009. Spp382p interacts with multiple yeast splicing factors, including possible regulators of Prp43 DExD/H-Box protein function. Genetics 183: 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunathan PL, Guthrie C. 1998. RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2. Curr Biol 8: 847–855. [DOI] [PubMed] [Google Scholar]
- Schneider M, Will CL, Anokhina M, Tazi J, Urlaub H, Lührmann R. 2010. Exon definition complexes contain the tri-snRNP and can be directly converted into B-like precatalytic splicing complexes. Mol Cell 38: 223–235. [DOI] [PubMed] [Google Scholar]
- Sheldrick GM. 2008. A short history of SHELX. Acta Crystallogr A Found Crystallogr 64: 112–122. [DOI] [PubMed] [Google Scholar]
- Stegmann CM, Seeliger D, Sheldrick GM, de Groot BL, Wahl MC. 2009. The thermodynamic influence of trapped water molecules on a protein-ligand interaction. Angew Chem Int Ed Engl 48: 5207–5210. [DOI] [PubMed] [Google Scholar]
- Stelzl U. 2014. E. coli network upgrade. Nat Biotechnol 32: 241–243. [DOI] [PubMed] [Google Scholar]
- Studier FW. 2005. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41: 207–234. [DOI] [PubMed] [Google Scholar]
- Ulrich A, Wahl MC. 2014. Structure and evolution of the spliceosomal peptidyl-prolyl cis-trans isomerase Cwc27. Acta Crystallogr D Biol Crystallogr 70: 3110–3123. [DOI] [PubMed] [Google Scholar]
- Ulrich A, Andersen KR, Schwartz TU. 2012. Exponential megapriming PCR (EMP) cloning—seamless DNA insertion into any target plasmid without sequence constraints. PLoS One 7: e53360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. 2010. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr 66: 22–25. [DOI] [PubMed] [Google Scholar]
- Vinayagam A, Stelzl U, Wanker EE. 2009. Repeated two-hybrid screening detects transient protein–protein interactions. Theor Chem Acc 125: 613–619. [Google Scholar]
- Wahl MC, Will CL, Lührmann R. 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718. [DOI] [PubMed] [Google Scholar]
- Warkocki Z, Odenwälder P, Schmitzová J, Platzmann F, Stark H, Urlaub H, Ficner R, Fabrizio P, Lührmann R. 2009. Reconstitution of both steps of Saccharomyces cerevisiae splicing with purified spliceosomal components. Nat Struct Mol Biol 16: 1237–1243. [DOI] [PubMed] [Google Scholar]
- Weimann M, Grossmann A, Woodsmith J, Özkan Z, Birth P, Meierhofer D, Benlasfer N, Valovka T, Timmermann B, Wanker EE, et al. 2013. A Y2H-seq approach defines the human protein methyltransferase interactome. Nat Methods 10: 339–342. [DOI] [PubMed] [Google Scholar]
- Will CL, Lührmann R. 2011. Spliceosome structure and function. Cold Spring Harb Perspect Biol 3: a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodsmith J, Stelzl U. 2014. Studying post-translational modifications with protein interaction networks. Curr Opin Struct Biol 24: 34–44. [DOI] [PubMed] [Google Scholar]
- Worseck JM, Grossmann A, Weimann M, Hegele A, Stelzl U. 2012. A stringent yeast two-hybrid matrix screening approach for protein–protein interaction discovery. Methods Mol Biol 812: 63–87. [DOI] [PubMed] [Google Scholar]
- Xie J, Beickman K, Otte E, Rymond BC. 1998. Progression through the spliceosome cycle requires Prp38p function for U4/U6 snRNA dissociation. EMBO J 17: 2938–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]