

ABSTRACT
Failure to localize membrane proteins to the primary cilium causes a group of diseases collectively named ciliopathies. Polycystin-1 (PC1, also known as PKD1) is a large ciliary membrane protein defective in autosomal dominant polycystic kidney disease (ADPKD). Here, we developed a large set of PC1 expression constructs and identified multiple sequences, including a coiled-coil motif in the C-terminal tail of PC1, regulating full-length PC1 trafficking to the primary cilium. Ciliary trafficking of wild-type and mutant PC1 depends on the dose of polycystin-2 (PC2, also known as PKD2), and the formation of a PC1–PC2 complex. Modulation of the ciliary trafficking module mediated by the VxP ciliary-targeting sequence and Arf4 and Asap1 does not affect the ciliary localization of full-length PC1. PC1 also promotes PC2 ciliary trafficking. PC2 mutations truncating its C-terminal tail but not those changing the VxP sequence to AxA or impairing the pore of the channel, leading to a dead channel, affect PC1 ciliary trafficking. Cleavage at the GPCR proteolytic site (GPS) of PC1 is not required for PC1 trafficking to cilia. We propose a mutually dependent model for the ciliary trafficking of PC1 and PC2, and that PC1 ciliary trafficking is regulated by multiple cis-acting elements. As all pathogenic PC1 mutations tested here are defective in ciliary trafficking, ciliary trafficking might serve as a functional read-out for ADPKD.
KEY WORDS: Cilium, Polycystin-1, Polycystin-2, Trafficking
Summary: Ciliary trafficking of polycystin-1 is regulated by multiple motifs at its C-terminus and by polycystin-2 but not by cleavage at its G-protein-coupled receptor proteolytic site.
INTRODUCTION
The primary cilium is an evolutionarily conserved microtubule structure that emanates from the cell surface of most mammalian cells. Mounting evidence has indicated that the primary cilium houses a high density of signaling molecules and functions as a center to orchestrate a diverse number of signaling pathways important for development or tissue homeostasis (Dorn et al., 2012; Ezratty et al., 2011; Goetz and Anderson, 2010; Lancaster et al., 2011; Oh and Katsanis, 2012). The primary cilium represents the ‘cell antenna’, and is sensitive to changes in extracellular cues (Barr and Sternberg, 1999; Nauli et al., 2003; Praetorius and Spring, 2005; Singla and Reiter, 2006; Zhang et al., 2004). The paramount importance of cilium is evident, as defects in cilium structure or function cause a variety of diseases, collectively referred to as ciliopathies (Waters and Beales, 2011).
The autosomal dominant polycystic kidney disease (ADPKD) (Zhou, 2009) is such a ciliopathy that manifests as cyst formation in kidney, as well as in liver and pancreas, in humans and mouse models (Lu et al., 1997, 2001). The most common form of this disease is caused by mutations in PKD1 (The International Polycystic Kidney Disease Consortium, 1995; Hughes et al., 1995), whereas the rest results from mutations in PKD2 (Mochizuki et al., 1996). PKD1 encodes polycystin-1 (PC1), a large (over 4300 residues) integral membrane protein with 11 transmembrane domains, a large extracellular domain with multiple predicted motifs, and a small ∼200-amino-acid C-terminal tail (CTT) inside the cell (The International Polycystic Kidney Disease Consortium, 1995; Hughes et al., 1995). PC1 is reported to undergo a notch-like cleavage at its G-protein-coupled receptor (GPCR) proteolytic site (GPS), which is in the extracellular domain, through autoproteolysis (Qian et al., 2002), releasing a small C-terminal fragment (Chauvet et al., 2004; Woodward et al., 2010), presumably through the actions of γ-secretase (Merrick et al., 2012). PC1 is expressed in a wide range of tissues (Geng et al., 1997; Peters et al., 1999), and is localized to apical and basolateral plasma membranes including adherens junctions, desmosomes and the primary cilium (Geng et al., 1996; Nauli et al., 2003; Roitbak et al., 2004; Scheffers et al., 2000; Yoder et al., 2002). PKD2 encodes polycystin-2 (PC2), a member of the transient receptor potential (TRP) family of non-selective cation channels (TRPP2). PC2 colocalizes with PC1 on the primary cilium where they transduce the extracellular fluid flow shear stress into a Ca2+ signal (Nauli et al., 2003; Xu et al., 2003; Yoder et al., 2002). In addition, PC2 is considered as an endoplasmic reticulum (ER)-resident protein where it might also mediate Ca2+ release from ER (Koulen et al., 2002).
To date, proteomic studies have revealed the presence of hundreds of polypeptides inside the cilium (Gherman et al., 2006; Ishikawa et al., 2012). The mechanism by which these proteins are targeted to cilia, however, remains poorly understood. A ciliary-targeting sequence (CTS) recognizable by specific machineries appears to be important for protein delivery to the ciliary compartment. The CTSs of several proteins have been identified and the VxP and Ax(S/A)xQ are two well recognized CTSs found in rhodopsin and several GPCRs. Recognition of the VxP motif by Arf4-based trafficking module (Mazelova et al., 2009) or Ax(S/A)xQ by the BBSome complex (Jin et al., 2010) is a crucial step for the ciliary trafficking of rhodopsin and somatostatin receptor 3 (SSTR3), respectively. Another example of these CTSs has been found in fibrocystin (also known as polyductin, hereafter called FPC), the product of the human autosomal recessive polycystic kidney disease gene (PKHD1). The CTS of FPC contains residues that can be modified by palmitoylation, which appears to be a process important for targeting this protein to lipid rafts and subsequently to the ciliary compartment in a Rab8-dependent manner (Follit et al., 2010). For the majority of the ciliary proteins, the CTSs or the machineries responsible for their ciliary targeting remain to be identified. Previous studies have suggested that PC1 and PC2 could utilize the VxP and Arf4 ciliary trafficking module to target to the primary cilium independently (Geng et al., 2006; Ward et al., 2011). However, given that PC1 and PC2 interact with each other through their C-terminal and N-terminal sequences (Chapin et al., 2010; Qian et al., 1997), it has also been proposed that co-assembly of PC1 and PC2 is a prerequisite for PC2 to reach the cell membrane (Hanaoka et al., 2000; Nauli et al., 2003), and that formation of this complex also enhances PC1 ciliary localization (Chapin et al., 2010).
In this report, we performed a systemic analysis of cis and trans regulators of PC1 ciliary trafficking. We identified crucial sequences and motifs, including a coiled-coil motif in the C-terminal fragment of PC1, that contribute to PC1 ciliary trafficking. Unexpectedly, we found that the VxP motif, previously identified as a CTS in a mini-PC1 construct, is completely dispensable for the ciliary trafficking of full-length PC1. However, we identified a fragment in the PC1 CTT containing CTS activity. PC2 plays a dominant role in facilitating wild-type and mutant PC1 trafficking to the cilia, and interaction with PC1 also promotes localization of PC2 to the primary cilia. Increased expression of PC2 might override the requirement for the PC1 CTT in PC1 ciliary trafficking. We also show that PC1 GPS cleavage is not required for its ciliary trafficking. Taken together, this study suggests that full-length PC1 ciliary trafficking is dependent on the amount of PC2, and that substantial ciliary trafficking of PC1 takes place when PC1 and PC2 form a protein complex.
RESULTS
Multiple sequences in the C-terminus of PC1 contribute to ciliary targeting of full-length PC1
We have recently shown that the CTT of PC1 (hereafter PC1-CTT) is essential for its ciliary localization (Su et al., 2014). To identify the ciliary-targeting sequences and motifs within PC1-CTT, we performed a deletion analysis within this region. PC1-CTT contains several motifs, including a proposed VxP motif, a coiled-coil motif, a G-protein-binding site, and several putative protein phosphorylation and dephosphorylation sites (Fig. S1A).
The VxP motif has previously been proposed to function as a CTS for PC1 because of experiments using a mini chimeric PC1 construct (Ward et al., 2011). To determine whether this motif is responsible for the inability of recombinant full-length PC1 lacking the C-terminal tail to traffic to the cilium, we mutated the VxP sequence to AxA and tested the ciliary trafficking ability of this mutant construct by transiently expressing it in IMCD-3 cells (Fig. 1A). Surprisingly, immunofluorescence showed that YFP–PC1 carrying the AxA mutation trafficked to cilium normally (88% for V4288A/P4290A versus 83% for YFP–PC1), suggesting that this motif is dispensable for the ciliary trafficking of the full-length protein. To our surprise, we found that the C-terminal stretch of 54 residues was dispensable for PC1 trafficking to cilia.
Fig. 1.

The C-terminus of PC1 is required for efficient ciliary targeting of full-length PC1. (A) Ciliary localization of full-length (FL) YFP–PC1 and various truncation, deletion and point mutant variants in IMCD-3 cells. Respective constructs were individually expressed in IMCD-3 cells and ciliary expression of these constructs was evaluated by staining with antibody against GFP (green) and cilia were marked with acetylated α-tubulin (acT, red). Representative images and quantifications of each construct to function are shown. At least 50 ciliated, PC1 transfected cells were counted for the presence of YFP–PC1 signal on cilia under each condition. Error bars represent the s.e.m. between microscope fields from at least three independent experiments. LRR, leucine-rich repeat; REJ, receptor for egg jelly. TM, transmembrane domains. (B) There is no apparent difference in subcellular localization between YFP–PC1 with or without the CTT at the basal body. PC1 was detected with an antibody against GFP (green in merged images), and γ-tubulin (red in merged images) was used as a basal body marker. (C) Apical surface trafficking of YFP–PC1 and YFP–PC1ΔCTT. Live-cell staining using a polyclonal rabbit anti-GFP antibody (green in merged images, *PC1) was first used to detect cell surface PC1. After fixation and permeabilization, a mouse monoclonal antibody against GFP (red in merged images) was used to detect all recombinant PC1, including the intracellular pool. Scale bars: 5 µm.
However, deletion of the last 154 residues of PC1-CTT (YFP–PC1ΔC154) completely abolished the ciliary trafficking of YFP–PC1 (Fig. 1A), indicating that the sequence between the last 54 and 154 amino acids is required for this process. Sequence analyses revealed that this fragment contains a coiled-coil motif and several potential glycogen synthase kinase 3 (GSK3) phosphorylation sites. The role of the coiled-coil motif in the ciliary trafficking of PC1 was tested first using a deletion mutant (YFP–PC1ΔCC), which localized to the cilium at a reduced efficiency (37%), compared to the wild-type construct (Fig. 1A). Study of two additional point mutants in the coiled-coil region (Q4215P and L4229A/L4233A) further pinpointed a role of the coiled-coil motif in the ciliary trafficking of PC1. The Q4215P mutation corresponds to a human PKD1 mutation (Q4224P) that is likely pathogenic (Badenas et al., 1999), whereas L4229A/L4233A represents a double point mutation that abolishes the formation of the coiled-coil structure as predicted using the COILS program. Both mutants are functionally impaired to the same degree as the YFP–PC1ΔCC mutant (Fig. 1A).
Despite of the apparent function of the coiled-coil motif in the ciliary trafficking of PC1, deletion or mutation of the coiled-coil motif did not account for the complete loss of YFP–PC1ΔC154 trafficking to the cilia. This suggests that the sequence upstream of the coiled-coil motif contributes to the ciliary trafficking of PC1. To map the responsible sequences or elements in this region, we made five additional deletion constructs (YFP–PC1ΔC85, YFP–PC1ΔC111, YFP–PC1ΔC121, YFP–PC1ΔC132 and YFP–PC1ΔC143) and compared their function with that of YFP–PC1ΔC154. We found that these deletions led to a gradual reduction in PC1 ciliary trafficking when more amino acids were removed from PC1 (Fig. 1A). This region is highly conserved among species and contains multiple phosphorylation sites.
Because deletion of the last 154 amino acids completely abolished PC1 trafficking to cilia, we went on to test whether the CTS(s) lies within the C-terminal 154-residue fragment and whether the N-terminal 44 amino acids of the CTT are dispensable. Interestingly, deletion of this 44-residue region also severely impaired PC1 trafficking to cilia (ΔN44, Fig. 1A). Sequence inspection of the first 44 amino acids of the CTT suggests that it contains two potential tyrosine-based sorting signals (YHAL and YEMV) responsible for the interaction with the mu subunit of any of the adaptor protein (AP) complexes that might direct protein traffic within the endosomal and the secretory pathways (Dell'Angelica et al., 1999; Ohno et al., 1995). In addition, YEMV is also a consensus sequence for phosphorylation by c-Src and binding of proteins containing SH2 domains (Wilson, 2004). Mutation of each (data not shown) or both of the tyrosine-containing motifs to alanine residues slightly affected the ciliary trafficking of PC1.
The 44-amino-acid region also includes a part of the G-protein-binding site (Parnell et al., 1998) and a leucine residue at amino acid position 4132. Single amino acid deletion of the corresponding leucine residue in human PC1 has previously been shown to be pathogenic in ADPKD patients (Afzal et al., 1999). Interestingly, deletion of this leucine (YFP–PC1ΔL) greatly reduced ciliary trafficking of YFP–PC1 (Fig. 1A).
Western blot analyses showed that six of the mutant PC1 constructs, including YFP–PC1ΔCTT, that totally failed to traffic to the cilium were expressed at a similar level as the full-length wild-type protein, excluding reduced expression levels as a probable cause of reduced ciliary trafficking of these constructs (Fig. S1B). Although YFP–PC1ΔCTT failed to traffic to the primary cilia, it reached the apical plasma membrane as efficiently as YFP–PC1 (Fig. 1B,C), suggesting that trafficking of PC1 lacking the CTT through the ER and the Golgi is not affected. We did not observe apparent accumulation of YFP–PC1ΔCTT at the basal bodies.
The PC1-CTT contains more than one CTS
An N-terminal fragment containing two coiled-coil motifs has been shown to be necessary and sufficient to target Nphp3 to the primary cilia, functioning as a CTS (Nakata et al., 2012). We next tested whether the coiled-coil motif in the PC1-CTT or the PC1-CTT itself is sufficient to target foreign proteins to the cilia. We made a set of chimeric proteins by fusing various mouse PC1-CTT fragments to CD16.7 (Fig. 2A). CD16.7 is a well-characterized chimeric protein often used in chimeric studies aimed at identifying targeting domains and has been used previously to identify the VxP motif in human PC1-CTT as a CTS (Ward et al., 2011). Similar to the findings from the human PC1-CTT, we found that CD16.7-fused mouse PC1-CTT, and CD16.7 fused with the last 112 or last seven amino acids of PC1-CTT (CD16.7–112 and CD16.7–VxP, respectively) trafficked to cilium, whereas their VxP mutants (CD16.7–CTT-AxA and CD16.7–112-AxA) failed (Fig. 2B,C), which indicates that the VxP motif within mouse PC1-CTT also functions as a CTS. To test whether the coiled-coil motif is needed for CD16.7–112 targeting to the cilia, we made two additional constructs, CD16.7–112ΔCC and CD16.7–112ΔC54 (Fig. 2A). Although CD16.7–112ΔCC trafficked to the cilium efficiently, CD16.7–112ΔC54 did not (Fig. 2B,C), suggesting that the coiled-coil motif is neither required nor sufficient for CD16.7–112 targeting to the cilia. Given that the ciliary-targeting activity appears to be located within the fragment between constructs ΔC154 and ΔC54 of full-length PC1 (Fig. 1, 90% efficiency in ΔC54 and 0% in ΔC154), we therefore generated another CD16.7 chimera (CD16.7–ΔN44ΔC54) and tested its ciliary trafficking activity (Fig. 2A). Consistent with what we observed in the full-length PC1 constructs, this short fragment (ΔN44ΔC54) trafficked to cilium efficiently (Fig. 2B,C), suggesting that PC1-CTT contains a CTS, in addition to the known VxP motif, that is sufficient to drive CD16.7 to cilia.
Fig. 2.

The PC1 CTT contains CTS activity but the coiled-coil motif is insufficient to function as a CTS. (A) Schematic representation of CD16.7 and different PC1 C-terminal fragment fusions generated in this study. (B) Expression of indicated constructs in A in the primary cilia of IMCD-3 cells. Cells were stained by antibodies against CD16 (green) and acetylated α-tubulin (Ac-α-tub, red). Scale bar: 5 µm. (C) Quantification of percentage of CD16-positive cilia in cells. Green columns represent VxP motif-containing constructs and red columns represent constructs without the VxP motif. At least 50 ciliated, transfected cells were counted for the presence of CD16 signal on cilia under each condition. Error bars represent the s.e.m. between microscope fields from at least three independent experiments.
To confirm that PC1-CTT is able to serve as a CTS in another fusion protein system, we made a construct using vesicular stomatitis virus G protein (VSVG). VSVG is a membrane protein but is excluded from the primary cilium of IMCD-3 cells (Hu et al., 2010). We generated a VSVG–C112 chimeric construct by fusing the last 112 residues of PC1-CTT to VSVG. We also fused VSVG with the 193 residues of FPC-CTT, which contains a known CTS (Follit et al., 2010) as a positive control. Although VSVG alone failed to localize to the cilia, the positive control VSVG–FCTS as well as VSVG–C112 trafficked to cilium as expected (Fig. S2B,C), thus confirming the presence of CTS activity in PC1-CTT found using the CD16.7 chimeric constructs.
The ciliary targeting of YFP-PC1 is independent of Arf4 and Asap1
Small GTPases such as Arf and Arl are crucial players in ciliogenesis. They are required for the photoreceptor rhodopsin to traffic to the primary cilia, and have been proposed to represent a general mechanism for ciliary targeting of membrane proteins (Hsiao et al., 2012). PC1 has been reported to bind to Arf4 and Asap1, an Arf GTPase-activating protein, and short hairpin RNA (shRNA)-mediated depletion of either protein affects the ciliary trafficking of the CD16.7–PC1 chimeric construct (Ward et al., 2011). Because deletion of the VxP motif from YFP–PC1 did not affect its ciliary trafficking, we sought to investigate whether these small GTPases are required for full-length PC1 to traffic to cilia. For this purpose, we utilized lentivirus-mediated shRNA technology and depleted Arf4 and Asap1 in IMCD-3 cells. Quantitative real-time PCR (qRT-PCR) analyses indicated that we had achieved over 99% knockdown for Arf4 and ∼75% knockdown for Asap1 (Fig. 3A). There were no apparent defects in ciliogenesis (Fig. 3B) or in ciliary targeting of YFP–PC1 in these knockdown cells, when compared to control cells (Fig. 3C,D).
Fig. 3.
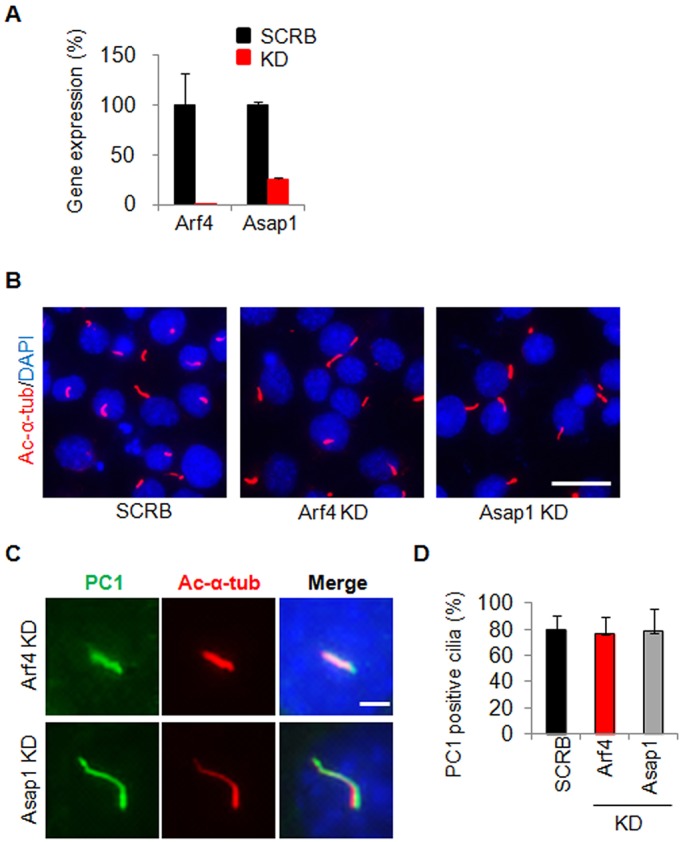
Effect of depletion of Arf4 and Asap1 on the ciliary targeting of PC1. (A) qRT-PCR analysis of Arf4 and Asap1 depletion by lentiviral shRNA in IMCD-3 cells. The relative knockdown (KD) efficiency was over 99% for Arf4 and ∼75% for Asap1. SCRB, scrambled shRNA control. (B) Depletion of Arf4 and Asap1 in IMCD-3 did not affect ciliogenesis of these cells as evaluated by DAPI and acetylated α-tubulin (ac-α-tub, red) staining. Scale bar: 20 µm. (C) Depletion of Arf4 and Asap1 in IMCD-3 did not affect PC1 trafficking to cilia. PC1 was visualized with an antibody against GFP. Representative images taken for the same exposure time in each cell line are shown. Scale bar: 5 µm. (D) Quantification of the percentage of PC1-positive cilia in cells depleted of Arf4 and Asap1. At least 50 ciliated, PC1 transfected cells were counted for the presence of YFP-PC1 signal on cilia under each condition from at least three independent experiments.
PC1 and PC2 mutually promote the ciliary localization of each other
The coiled-coil motif in PC1-CTT is known to mediate the interaction between PC1 and PC2 (Qian et al., 1997; Tsiokas et al., 1997). Recombinant PC2 has been reported to promote ciliary delivery of recombinant PC1 in LLC-PK1 cells, which appears to require the channel activity of PC2 (Chapin et al., 2010). To determine whether endogenous PC2 modulates recombinant PC1 ciliary trafficking in IMCD-3 cells, we depleted PC2 by lentiviral shRNA (Fig. 4A). Notably, YFP–PC1 still trafficked to the primary cilium in this cell line (Fig. 4B), although the intensity of ciliary PC1 signal was reduced to ∼30%, compared to that in the scrambled control cells (Fig. 4C). To determine whether a trace level of PC2 present in PC2 knockdown cells might promote PC1 ciliary trafficking, we transfected full-length PC1 into PC2-knockout (KO) cells. Although YFP–PC1 trafficked to the cilium in up to 49% of wild-type cells (Fig. 4D,E), neither YFP–PC1 nor its mutants was able to localize to the cilium in PC2-KO (Fig. S3) cells. To verify these data, we performed rescue experiments by reintroducing PC2 into PC2 KO cells. Notably, as little as 5 ng of PC2 efficiently promoted PC1 ciliary trafficking (∼43%) in PC2-KO cells. An increase in the dose of PC2 (50 ng and 500 ng) further enhanced ciliary PC1 localization to 67% and 71%, respectively (Fig. 4F,G).
Fig. 4.

PC2 promotes the ciliary trafficking of PC1 in a dose- and CTT-dependent manner but does not affect its channel activity. (A) Lentiviral shRNA depletion of PC2 in IMCD-3 cells. Western blot analysis showed substantial depletion of PC2 protein in two PC2-knockdown IMCD-3 cell lines (KD1 and KD2) using a rabbit anti-PC2 antibody (96525). GAPDH was used as a loading control. Clone KD1 was chosen for latter experiments. SC, scrambled shRNA control. (B) The ciliary trafficking of PC1 in scrambled control (SCRB) and PC2-knockdown (KD) cells. Acetylated α-tubulin (ac-α-tub, red) marks cilia. PC1 was visualized with an antibody against GFP. A line was drawn across the cilia. Scale bar: 5 µm. (C) The relative intensity of PC1 signal along the line was quantified as described in the Materials and Methods and shown in a bar graph. The intensity of YFP–PC1 in cilia (green) is reduced in PC2-knockdown cells, compared to the scrambled control cells. Results are mean±s.d.; n=3. (D) YFP–PC1 failed to localize to the cilia in PC2-KO cells in contrast to in PC2 wild-type cells. Scale bar: 5 µm. (E) The percentage of cilia with YFP–PC1 in PC2-KO and control cells [n=300 in control cells (from two independent experiments); n=450 in KO cells (from five independent experiments)]. (F,G) The failure of PC1 trafficking to the cilia in PC2-KO cells could be rescued by exogenous PC2 in a dose-dependent manner. PC2-KO cells were co-transfected with YFP–PC1 and wild-type PC2 with different doses of plasmid DNA as indicated. YFP–PC1-positive cilia were quantified for all transfections shown in F. The data from multiple experiments are expressed as mean±s.e.m. (H) The C-terminus of PC2 is essential for promoting PC1 targeting to cilia. HA-tagged PC2-703X or its parental plasmid PC2–HA, Myc-tagged PC2-D511V, or its parental plasmid PC2–Myc, were co-transfected with YFP–PC1 at a molecular ratio 1:1 into IMCD-3 cells. Cells were stained with antibodies against GFP (for PC1, green), HA or Myc (for PC2, red). A line was drawn across the cilia. Scale bar: 5 µm. (I) Line graph indicating that the relative intensity of PC1 in cilia (green) was increased in cells co-transfected with full-length PC2 (wild-type or D511V mutant) but not with the PC2-703X mutant. PC2 (HA or Myc, red) was found on the cilia when PC1 was co-expressed. Representative images from at least three independent experiments are shown.
We next examined the properties of PC2 required for the ciliary localization of PC1 using a PC2 C-terminal cytoplasmic tail deletion mutant (PC2-703X) and a channel-dead mutant (PC2-D511V). Although PC2-703X failed to promote the ciliary trafficking of YFP–PC1, as reported in LLC-PK1 cells, PC2-D511V worked as efficiently as the wild-type PC2 (Fig. 4H,I), suggesting that the PC2 C-terminus-mediated interaction, rather than PC2 channel activity, facilitates PC1 ciliary trafficking in IMCD-3 cells. Transfection of the 288 residues of the PC2-CTT alone, however, did not affect the ciliary trafficking of PC1 (Fig. S4A).
Remarkably, in cases when PC1 and PC2 co-expression promoted PC1 targeting to the primary cilia, there was a concurrent increase of recombinant PC2 in the cilium (Fig. 4H,I), indicating that PC1 and PC2 mutually promote each other to enter cilia. We found PC2-703X trafficked to cilium more efficiently than recombinant full-length PC2 in cells transfected with PC2 alone (Fig. S4B).
Complex formation between PC1 and PC2 is crucial for PC2 promoting wild-type and mutant PC1 to the primary cilium
We found that YFP–PC1ΔCTT failed to reach the cilium in IMCD-3 cells that express endogenous PC2. Interestingly, YFP–PC1ΔCTT was found on the cilium in ∼41% of cells when it was co-transfected with an equal molar amount of PC2 DNA (Fig. 5A,B). In addition, the intensity of the ciliary YFP–PC1ΔCTT signal was almost comparable to that of YFP–PC1. These data suggest that the requirement for the PC1-CTT in PC1 ciliary trafficking could be partially bypassed by increased PC2 expression. We also tested the effect of PC2 on two other PC1 mutants, R3269C and M3083R. PC1-R3269C corresponds to a human hypomorphic mutation (PC1-R3277C) that gives rise to polycystic kidney disease (Hopp et al., 2012) whereas PC1-M3083R represents an ENU mouse mutant that has a severe cystic phenotype (Herron et al., 2002). Both mutants were defective in trafficking to cilium when transfected alone into IMCD-3 cells (Fig. 5B), but were also partially rescued by co-expression with PC2, although the rescue was not as efficient as for the YFP–PC1ΔCTT construct.
Fig. 5.

The promoting effect of PC2 on the ciliary trafficking of PC1 is dependent on its interaction with PC1 but not its CTS. (A) Ciliary localization of wild-type PC1 (WT), YFP–PC1ΔCTT, YFP–PC1-R3269C and YFP–PC1-M3083R in the presence of recombinant PC2 in IMCD-3 cells. PC1 was stained with an antibody against GFP (green) and PC2 was stained with an antibody against Myc (red). Representative images of each construct to function are shown. Scale bar: 10 µm. (B) The percentage of ciliary PC1 in the absence and presence of recombinant PC2 was scored. At least 50 ciliated, YFP–PC1-transfected cells from multiple experiments were counted for the presence of PC1 signal on cilia under each condition. (C) PC2 interaction with YFP–PC1, YFP–PC1ΔCTT, YFP–PC1-R3269C and YFP–PC1-M3083R by co-immunoprecipitation (IP). (D) Percentage of PC2 that was co-immunoprecipitated (IPed) with PC1 or its mutants. The experiment was carried out at least two times; the blot shown in Fig. 5C was used for quantification. (E) The CTS mutant PC2-AxA promotes the ciliary trafficking of both YFP–PC1 and YFP–PC1ΔCTT. YFP–PC1 and YFP–PC1ΔCTT together with PC2-AxA–Myc was transfected into IMCD-3 cells and immunofluorescence staining was performed. PC1 was detected with an antibody against GFP and PC2 was detected with an antibody against Myc. The promoting effect of PC2-AxA–Myc on YFP–PC1 and YFP–PC1ΔCTT is comparable with the wild-type PC2–Myc as described in A. Scale bar: 10 μm. (F) The CTS mutant PC2-AxA is able to rescue the ciliary trafficking defect of wild-type YFP–PC1 in PC2-KO cells. YFP–PC1 was co-transfected with or without PC2-AxA into PC2-KO cells. PC1 was detected with an anti-GFP antibody and the cilium was labeled with anti-acetylated-α-tubulin antibody (Ac-α-tub). Representative images from at least three independent experiments are shown.
A plausible explanation of the observed PC2 effect on the ciliary trafficking of PC1 might be due to the fact that PC2 and PC1 form a complex which can traffic to the cilium much more efficiently than PC1 does alone. To test this hypothesis, we co-transfected various PC1 constructs with PC2–Myc into 293T-Rex cells and performed a co-immunoprecipitation assay. As shown in Fig. 5C,D, YFP–PC1 co-immunoprecipitated PC2–Myc most strongly and the amount of co-immunoprecipitated PC2 was reduced greatly in PC1 CTT deletion mutant. The co-immunoprecipitation efficiency decreased even further for the two PC1 mutants YFP–PC1-R3269C and YFP–PC1-M3083R. It is noteworthy that the ability of PC2 to interact with different PC1 constructs correlated well with its ability to promote their ciliary trafficking. Although, for example, both YFP–PC1ΔCTT and YFP–PC1-M3083R failed to reach the cilium completely when transfected alone, both the number of cilia with mutant PC1 expression and the intensity of their ciliary signals varied substantially when co-transfected with PC2 (Fig. 5A,B), in line with their different abilities to interact with PC2 (Fig. 5C,D).
We then tested whether trafficking of YFP–PC1ΔCTT and YFP–PC1-M3083R to the cilium was promoted by PC2 using its CTS when in a complex. Interestingly, however, a CTS mutant PC2 (PC2-AxA) was also capable of mediating YFP–PC1 trafficking to the cilium in both IMCD-3 cells and PC2-KO cells in which YFP–PC1 alone does not traffic to cilia. Moreover, PC2-AxA was able to promote YFP–PC1ΔCTT ciliary trafficking, similar to the wild-type PC2 construct (Fig. 5E,F). The fact that neither PC1 lacking its CTT nor CTS-deficient PC2 was able to traffic to cilium when expressed alone but that both reached cilium when co-expressed is intriguing. These data suggest that PC1 ciliary trafficking is not dependent on the CTS of PC2, and that PC1 and PC2 might mediate the trafficking of each other in earlier steps of the secretory pathway for efficient ciliary targeting.
Notably, the promoting effect of PC2 on the ciliary trafficking of YFP–PC1ΔCTT occurred in IMCD-3 cells where endogenous PC1 exists. To rule out that the ciliary trafficking of YFP–PC1ΔCTT in the presence of PC2 was not due to its homophilic interaction with endogenous PC1, we performed this experiment in PC1-deficient mouse embryonic kidney (MEK) cells (Nauli et al., 2006). We found that neither YFP–PC1ΔCTT nor PC2–Myc trafficked to cilia in these cells when transfected alone, but both PC1ΔCTT and PC2 were found on cilia after co-transfection (Fig. S4C). These data strongly advocate that PC2, not endogenous PC1, promotes the ciliary localization of YFP–PC1ΔCTT.
PC1 cleavage at the GPS site is not required for its ciliary trafficking
PC2 was found to enhance cell surface expression and GPS cleavage of PC1 in HEK293 cells (Chapin et al., 2010). To determine whether PC2 promotes ciliary trafficking of PC1 through facilitating GPS cleavage in IMCD-3 cells, we first tested whether GPS cleavage was required for the ciliary trafficking of PC1. For this purpose, we made several GPS cleavage mutants by mutating the leucine and threonine residues at the conserved GPS cleavage site (HL↓T) (Wei et al., 2007) to histidine, aspartic acid and valine residues, generating YFP–PC1-L3040H, YFP–PC1-T3041D and YFP–PC1-T3041V, respectively. We found that both of the YFP–PC1-L3040H and YFP–PC1-T3041D mutants were resistant to cleavage (Fig. 6A) appearing as a single full-length band on 5% SDS-PAGE gels, in contrast to wild-type control YFP–PC1 which migrated as two larger than 250-kDa bands, full-length PC1 and the N-terminal PC1 fragment that is cleaved at the GPS site.
Fig. 6.

GPS cleavage of PC1 is not required for its ciliary targeting. (A) Western blot showing that both YFP–PC1-L3040H and YFP–PC1-T3041D are GPS cleavage mutants. Wild-type (WT), L3040H and T3041D YFP–PC1 were transfected into HEK293T-Rex cells overnight. The expression of YFP–PC1 was induced for 24 h by adding 1 µg/ml tetracyclin. Non-induced cells were used as a control. YFP–PC1 was detected using an antibody against GFP. The upper arrow denotes the band corresponding to the full-length (FL) non-cleaved PC1 and the lower arrow denotes the band corresponding to the GPS cleaved N-terminal fragment (NTF). (B) The ciliary trafficking of WT, L3040H and T3041D YFP–PC1 in IMCD-3 cells in the absence or presence of PC2. Cells were stained by antibodies against GFP (green) and acetylated α-tubulin (ac-α-tub, red). Representative images from at least three independent experiments are shown. Scale bar: 5 µm.
Next, we examined the ciliary trafficking ability of these mutants in the presence or absence of recombinant PC2. YFP–PC1-L3040H failed to traffic to the cilium when expressed alone, but was able to localize to the cilium when co-transfected with PC2, although not as strongly as YFP–PC1 (Fig. 6B). Interestingly, the GPS cleavage mutant YFP–PC1-T3041D trafficked to cilium like the wild-type YFP–PC1 construct, similar to another GPS cleavage mutant PC1-T3041V (data not shown). PC1-T3041V gives rise to cyst formation in proximal tubes in a mouse knock-in model (Yu et al., 2007). These data suggest that the failure of YFP–PC1-L3040H to reach the cilium is mutation specific and that the ciliary trafficking of PC1 does not require the cleavage at its GPS site per se. Consequently, the PC2 effect on PC1 ciliary trafficking in IMCD-3 cells cannot be attributed to its ability in promoting GPS cleavage of PC1.
DISCUSSION
We have recently shown that the ciliary trafficking of PC1 is regulated by specific BBS proteins and requires the presence of an intact PC1-CTT (Su et al., 2014). Here, we identified that multiple sequences and motifs in PC1-CTT contribute to the ciliary trafficking of PC1. PC1 and PC2 mutually promote the trafficking of each other to the primary cilium, and our results show that PC1 ciliary targeting is highly dependent on the dose of PC2 but not the VxP motifs in PC1 or PC2. GPS cleavage is not required for PC1 ciliary trafficking.
The most important sequences and motifs contributing to PC1 ciliary trafficking in the C-terminal tail we have identified are the coiled-coil motif that is the key for protein–protein interaction, the leucine residue adjacent to the G-protein-binding domain, and the highly conserved sequences containing multiple phosphorylation sites located N-terminal to the coiled-coil motif. The coiled-coil motif in PC1 has been shown to interact with many proteins including PC2 (Qian et al., 1997), Jade1 (Foy et al., 2012) and Pacsin2 (Yao et al., 2014). The sequences N-terminal to coiled-coil motif include serine-rich sequences that might be phosphorylated by various protein kinases.
This work revealed a fundamental difference in studying protein trafficking using a full-length construct versus a chimeric mini-construct. Although a seven-residue VxP-containing motif was sufficient to drive foreign proteins such as CD16.7 fusion proteins to cilia, the VxP motif was completely dispensable for the ciliary trafficking of full-length PC1. This could be due to the fact that the full-length PC1 containing 11 transmembrane domains might interact with multiple proteins that recognize multiple structural determinants for ciliary trafficking, rendering some sequences or motifs dispensable. Our data also suggest that a full-length protein might use a pathway different from its small chimeric form.
Studies on the trafficking of rhodopsin to the primary cilium have suggested that a trafficking module based on the small GTPase Arf4 recognizes the VxP motif, and is involved in the selection and packaging of the cargo destined for delivery to the cilium (Mazelova et al., 2009). Arf4 belongs to the ADP-ribosylation factor family of proteins that include six closely related proteins, Arf1–Arf6. The class II Arf4 is 90% identical to the other class II member Arf5, and 80% identical to the class I Arfs, Arf1, Arf2 and Arf3. Our data suggest that the ciliary trafficking of PC1 is Arf4 independent. We also tested the effect of Arf4 depletion on the ciliary trafficking of PC2 using the PC2-703X construct because PC2 has been shown to interact with Arf4 in an in vitro GST pulldown assay (Ward et al., 2011). However, depletion of Arf4 did not affect PC2 trafficking to the primary cilium (Fig. S2D,E). This is similar to the ciliary trafficking of FPC in which knockdown of Arf4 causes a delay in delivery of the newly synthesized protein from the Golgi to the cilium but does not affect the steady-state level of FPC in the cilium (Follit et al., 2014). Given the high similarity of Arf4 to other family members and the fact that Arf1, Arf3 and Arf5 are upregulated in Arf4-depleted cells (Reiling et al., 2013), it is possible that other Arf family members might participate in ciliary trafficking of PC1 in the absence of Arf4.
We found that PC1 is able to traffic to the primary cilium of IMCD-3 cells depleted of PC2 at a reduced efficiency. In PC2-KO cells, neither the wild-type nor 15 full-length mutant PC1 constructs trafficked to cilium (Fig. S3). Re-expression of PC2 (as little as 5 ng per transfection) in PC2-KO cells robustly promoted PC1 ciliary trafficking, suggesting that the impact of PC2 on PC1 ciliary trafficking is highly dose dependent. This promotion, however, did not rely on PC2 channel activity or the VxP CTS in PC2 but required the presence of the PC2 CTT. This differs from the findings in LLC-PK1 cells, where expression of channel-dead mutant affected the ciliary trafficking of PC1 (Chapin et al., 2010). We also observed that full-length PC2 (wild-type, the channel-dead mutant PC2-D511V or the CTS mutant PC2AxA) was rarely detectable on the primary cilium unless co-expressed with PC1, consistent with previous reports by us (Nauli et al., 2003) and others (Xu et al., 2007) showing that endogenous PC2 is frequently absent from cilia of collecting duct-derived epithelial cells deficient of PC1, although PC2 is able to traffic to cilia independently of PC1 in some instances (Geng et al., 2006). In PC2-KO cells, neither wild-type nor PC2-AxA localizes to cilia except when co-expressed with PC1. Taken together, these observations suggest a mutually dependent model for PC1 and PC2 ciliary trafficking.
One of the most surprising findings is that the CTS-mutant PC2 and various mutant PC1 proteins lacking the PC2-interacting domain (the coiled-coil) remain able to promote the trafficking of each other to the cilium (Fig. 5E,F; and data not shown). PC2 was co-immunoprecipitated by various mutant PC1 proteins lacking the coiled-coil motif (Fig. 5C,D). These findings provide further evidence supporting the observation (Chapin et al., 2010) that PC1 and PC2 interact through a domain other than the coiled-coil motif in PC1-CTT. We conclude that PC1 traffics to the primary cilium in a complex with PC2 and the ciliary trafficking of this complex is far more efficient than that of either protein alone. The formation of a complex between PC1 and PC2 might mutually facilitate their maturation and budding from the ER and/or Golgi complex as proposed for PC2 (Cai et al., 1999) and PC1 (Gainullin et al., 2015). The transmembrane domains are likely required, as expression of a free PC2 CTT is not sufficient to facilitate PC1 trafficking to cilia (Fig. S4A). The trans-acting factors used by the PC1–PC2 complex for ciliary trafficking after leaving the Golgi remain to be elucidated, although both the BBSome (Su et al., 2014) and exocyst complex (Zuo et al., 2011) are likely involved.
The cleavage at the GPS site of PC1 has been reported to correlate with its surface expression level, and PC2 has been reported to promote this cleavage in 293 cells. However, cleavage at the GPS site does not seem to be necessary for ciliary trafficking of PC1 in IMCD-3 cells. Although one of the PC1 GPS cleavage mutants used in our study (YFP–PC1-L3040H) failed to reach cilia, other GPS cleavage mutants (YFP–PC1-T3041D or YFP–PC1-T3041V) trafficked to cilium with only minimum perturbation. In addition, PC2 promoted the ciliary trafficking of both YFP–PC1-L3040H and YFP–PC1-T3041D although the effect of PC2 on the former was much weaker. These data suggest that the GPS cleavage per se is not a prerequisite for PC1 to traffic to the cilium. The L3040H mutation might have affected more than just the cleavage of PC1 at its GPS site, compared to the YFP–PC1-T3041D or YFP–PC1-T3041V mutants. Other PC1 ciliary trafficking mutants described in this study, including YFP–PC1-ΔCTT, YFP–PC1-R3269C and YFP–PC1M3083R, displayed normal GPS cleavage as indicated by the presence of the N-terminal fragment in addition to the uncleaved full-length protein.
A transient expression system can allow a high level expression of recombinant proteins, which might affect the protein sorting machinery. However, recombinant PC1 expression in kidney epithelial cells is generally low (Su et al., 2014), probably due to its large size. Moreover, only those transfected cells with lower levels of PC1 expression were used for our analysis. Combined with co-expression of various PC1 and PC2 mutants in both wild-type and Pkd1- and Pkd2-knockout cells, this expression system served as a robust system for rapid evaluation of a large set of variants and provided a large set of data.
The primary cilium is clearly an important functional site for PC1 and PC2 (Fedeles et al., 2011; Nauli et al., 2003; Zhou, 2009). A recent report (Ma et al., 2013) further substantiates the importance of primary cilia in PKD pathogenesis. In the current study, we found that five out of five single amino acid substitution or deletion mutations that cause PKD in mouse or human were defective in ciliary trafficking. Although ADPKD is a cellularly recessive disease, polycystins are expressed and found in cyst-lining epithelial cells in most ADPKD patients. Given that a large number of ADPKD patients have single amino acid substitution mutations in PKD1 (http://pkdb.mayo.edu/), the fraction of trafficking mutations might be substantial. As proper protein localization is crucial for protein function, ineffective ciliary trafficking likely represents a pathogenic mechanism for ADPKD. Pharmacological correction of polycystin trafficking might lead to a novel personalized therapy of ADPKD. Understanding the mechanism(s) by which PC1 traffics to the cilium might also be instructive to studies of other signaling molecules and structural components of the primary cilia.
MATERIALS AND METHODS
Cell culture and transfection
Mouse inner medullary collecting duct cells (IMCD-3) and mouse embryonic kidney (MEK) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with F12 (50:50) supplemented with 10% fetal bovine serum (FBS). HEK293T-Rex and 293 EBNA cells are cultured in DMEM complemented with 10% FBS. For analysis of ciliary trafficking of PC1 and/or PC2 constructs, cells were transiently transfected at 50–60% confluence with various constructs in PEI (polyethylenimine, Fisher) and MEM medium (Mediatech) and incubated at room temperature for 30 min. After overnight culture, cells were serum starved for 40–48 h in DMEM/F12 containing 0.5% FBS for immunostaining.
Lentiviral knockdown of Arf4 and Asap1
For lentiviral knockdown experiments, plasmids targeting five sites against each gene were purchased from Sigma. Lentiviral particles were assembled in 293 EBNA cells following the manufacturer's instructions. IMCD-3 cells were infected with viruses overnight followed by selection with puromycin at a concentration of 2 µg/ml. Knockdown efficiency in stable cells was tested by qRT-PCR (primer sequences are available upon request). The shRNA target sequences from Sigma found to be effective in this study are Arf4 (TRCN0000311433) and Asap1 (TRCN0000311400).
Plasmids and antibodies
The YFP–PC1-encoding plasmid was generated by inserting YFP immediately downstream of the signal peptide of PC1 as reported previously (Su et al., 2014). All subsequent deletions or mutations derived from YFP–PC1 were performed using PCR-based mutagenesis. The Myc-tagged PC2-CTT expression plasmid was constructed by PCR amplification of the last 288 residues of the C-terminal fragment from mouse cDNA and cloned into the pcDNA3.1-mycHis+ (Invitrogen) plasmid using the BamHI and XhoI sites. The HA-tagged human PC2 and the channel dead PC2-D511V were gifts from Dr Stefan Somlo at Yale University, New Haven, CT. The Myc-tagged mouse PC2 and PC2-703X were gifts from Dr Ralph Witzgall at University of Regensburg, Germany. The CTS-mutated PC2-AxA–Myc was derived from the parental PC2–Myc plasmid by PCR mutagenesis. All CD16.7 and VSVG PC1 chimeric mini-constructs were amplified by recombinant PCR and then cloned into the pcDNA3.1 plasmid. All plasmids constructed in this study were confirmed by DNA sequencing analysis.
For immunofluorescence, the following primary antibodies were used: anti-GFP rabbit polyclonal (Abcam; 1:20,000), anti-GFP mouse monoclonal (Covance; 1:1000) (these antibodies also recognize YFP), anti-HA rat monoclonal (Roche; 1:1000), anti-Myc rat monoclonal (Abcam; 1:500), anti-γ-tubulin mouse monoclonal (Sigma; 1:2000), anti-acetylated α-tubulin mouse monoclonal (Sigma; 1:50,000), anti-acetylated α-tubulin rabbit polyclonal (Cell Signaling; 1:1200), and anti-CD16 mouse monoclonal (Santa Cruz Biotechnology; 1:200). Secondary antibodies (all in 1:500 dilution) used were: goat anti-rabbit-IgG, goat anti-mouse-IgG and goat anti-rat-IgG antibodies conjugated to Alexa Fluor 594, 488 or 405 dyes (Invitrogen).
For western blotting, the following primary antibodies were used: anti-Myc mouse 9e10 monoclonal (Invitrogen; 1:1000); anti-PC2 rabbit polyclonal (96525, generated by our group; 1:1000) (Nauli et al., 2003); anti-PC1 mouse 7e12 monoclonal (Santa Cruz Biotechnology; 1:2000), anti-SNAP rabbit polyclonal (NEB; 1:1000). Horseradish-peroxidase-linked anti-rabbit-IgG and anti-mouse-IgG antibodies (Cell Signaling; 1:5000) were used as secondary antibodies.
Western blotting and co-immunoprecipitation
Western blotting was performed as reported previously (Wang et al., 2014). Briefly, proteins were electrophoresed on SDS-PAGE gels and transferred onto Hybond ECL nitrocellulose membranes. Blots were blocked with 5% nonfat dry milk in PBS, incubated with the primary antibody for 1 h at room temperature or overnight in a cold room and washed three times with PBS. The blots were finally incubated with a horseradish-peroxidase-linked secondary antibody for an hour at room temperature, washed thoroughly and detected with the ECL western blot analysis system.
HEK293T-Rex/PC2–Myc stable cells transfected with desired YFP–PC1 constructs were induced with 1 µg/µl tetracyclin overnight, homogenized with M-PER (Pierce) containing protease inhibitors (Roche) and 1 mM DTT. For co-immunoprecipitation, 2–4 mg of the cell lysate was incubated with 2–4 µl anti-GFP antibody with gentle rocking overnight at 4°C. Protein-A–Sepharose beads (Invitrogen) were added and incubated for another 2 h at 4°C. The washed beads were solubilized in an equal volume of 2× SDS sample buffer and proteins were eluted by boiling for 5 min and analyzed by western blotting. YFP–PC1 was detected by antibody 7e12 and PC2–Myc was detected with an antibody against Myc (9e10). Parental cells were used as a control.
Immunostaining
Cells were fixed with 4% paraformaldehyde (PFA) for 10 min then permeabilized with 0.5% Triton X-100 in PBS for 5 min. For staining with the anti-γ-tubulin antibody, cells were treated with 100% cold methanol at −20°C for an additional 10 min after PFA fixation. After three washes, cells were blocked with 5% BSA for an hour then stained with the desired primary antibodies for another hour at room temperature. After thorough washing, secondary antibodies with appropriate labeling were added for an hour of incubation. After three washes, slides were mounted with ProLong® Gold antifade reagent with or without DAPI (Invitrogen).
For live staining of YFP–PC1 on the cell surface, cells were first rinsed twice with PBS and incubated with anti-GFP antibody on ice. After 1 h, cells were washed, fixed with 4% paraformaldehyde and regular staining was continued according to the protocol above.
Image acquisition and quantification
Fluorescence images were acquired with a Nikon-1000 epifluorescence microscope (Nikon, Tokyo, Japan). All images in the same set of experiments were taken with identical settings and contrast adjusted equally in Photoshop. For each experiment, the number of cells with ciliary expression of recombinant proteins (YFP–PC1 and its variants, PC2 and its variants, or chimeric proteins) was counted in at least ten randomly selected microscopic fields, and then divided by the number of transfected and ciliated cells. In some cases, single line scans were drawn through the middle of randomly selected cilia in the PC1 or PC2 channel using ImageJ software (National Institutes of Health, Bethesda, MD). The sum of the pixel intensities of immunofluorescence along the line over a threshold background was scored, using ImageJ, as the relative intensity of PC1 in cilia. The bar graph of the quantification is an average of multiple random selected cells.
Acknowledgements
We would like to thank the members of the Zhou laboratory and the Harvard Center for Polycystic Kidney Disease Research for scientific discussions and support. We would like to thank Drs Stefan Somlo and Ralph Witzgall for valuable reagents.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
X.S. and J.Z. conceived and designed the project and wrote the manuscript, J.Z. directed the project. X.S., G.Y., W.J., C.L, M.W., and A.T. performed experiments. X.S., M.W., G.Y., W.J., C.L., and J.Z. analyzed data. M.W., C.L. reviewed and contributed to the writing of the manuscript. C.L, W.E. and G.Y. contributed equally to this manuscript.
Funding
This work was supported by grants from the National Institutes of Health [grant numbers RO1DK099532, R37DK51050 and RO1DK53357, RO1DK53357S1 and RO1DK40703 to J.Z.]; and a post-doctoral fellowship from National Kidney Foundation (NKF) to W.E.-J. C.L. was supported by funding from National Nature Science Foundation of the P.R. China [grant number 2012AA02A512]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.160556/-/DC1
References
- Afzal A. R., Hand M., Ternes-Pereira E., Saggar-Malik A., Taylor R. and Jeffery S. (1999). Novel mutations in the 3 region of the polycystic kidney disease 1 (PKD1) gene. Hum. Genet. 105, 648-653. 10.1007/s004399900177 [DOI] [PubMed] [Google Scholar]
- Badenas C., Torra R., San Millan J. L., Lucero L., Mila M., Estivill X. and Darnell A. (1999). Mutational analysis within the 3′ region of the PKD1 gene. Kidney Int. 55, 1225-1233. 10.1046/j.1523-1755.1999.00368.x [DOI] [PubMed] [Google Scholar]
- Barr M. M. and Sternberg P. W. (1999). A polycystic kidney-disease gene homologue required for male mating behaviour in C. elegans. Nature 401, 386-389. 10.1038/43913 [DOI] [PubMed] [Google Scholar]
- Cai Y., Maeda Y., Cedzich A., Torres V. E., Wu G., Hayashi T., Mochizuki T., Park J. H., Witzgall R. and Somlo S. (1999). Identification and characterization of polycystin-2, the PKD2 gene product. J. Biol. Chem. 274, 28557-28565. 10.1074/jbc.274.40.28557 [DOI] [PubMed] [Google Scholar]
- Chapin H. C., Rajendran V. and Caplan M. J. (2010). Polycystin-1 surface localization is stimulated by polycystin-2 and cleavage at the G protein-coupled receptor proteolytic site. Mol. Biol. Cell 21, 4338-4348. 10.1091/mbc.E10-05-0407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvet V., Tian X., Husson H., Grimm D. H., Wang T., Hiesberger T., Igarashi P., Bennett A. M., Ibraghimov-Beskrovnaya O., Somlo S. et al. (2004). Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus. J. Clin. Invest. 114, 1433-1443. 10.1172/JCI21753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Angelica E. C., Mullins C. and Bonifacino J. S. (1999). AP-4, a novel protein complex related to clathrin adaptors. J. Biol. Chem. 274, 7278-7285. 10.1074/jbc.274.11.7278 [DOI] [PubMed] [Google Scholar]
- Dorn K. V., Hughes C. E. and Rohatgi R. (2012). A Smoothened-Evc2 complex transduces the Hedgehog signal at primary cilia. Dev. Cell 23, 823-835. 10.1016/j.devcel.2012.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezratty E. J., Stokes N., Chai S., Shah A. S., Williams S. E. and Fuchs E. (2011). A role for the primary cilium in Notch signaling and epidermal differentiation during skin development. Cell 145, 1129-1141. 10.1016/j.cell.2011.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedeles S. V., Tian X., Gallagher A.-R., Mitobe M., Nishio S., Lee S. H., Cai Y., Geng L., Crews C. M. and Somlo S. (2011). A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat. Genet. 43, 639-647. 10.1038/ng.860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follit J. A., Li L., Vucica Y. and Pazour G. J. (2010). The cytoplasmic tail of fibrocystin contains a ciliary targeting sequence. J. Cell Biol. 188, 21-28. 10.1083/jcb.200910096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follit J. A., San Agustin J. T., Jonassen J. A., Huang T., Rivera-Perez J. A., Tremblay K. D. and Pazour G. J. (2014). Arf4 is required for Mammalian development but dispensable for ciliary assembly. PLoS Genet. 10, e1004170 10.1371/journal.pgen.1004170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foy R. L., Chitalia V. C., Panchenko M. V., Zeng L., Lopez D., Lee J. W., Rana S. V., Boletta A., Qian F., Tsiokas L. et al. (2012). Polycystin-1 regulates the stability and ubiquitination of transcription factor Jade-1. Hum. Mol. Genet. 21, 5456-5471. 10.1093/hmg/dds391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainullin V. G., Hopp K., Ward C. J., Hommerding C. J. and Harris P. C. (2015). Polycystin-1 maturation requires polycystin-2 in a dose-dependent manner. J. Clin. Invest. 125, 607-620. 10.1172/JCI76972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng L., Segal Y., Peissel B., Deng N., Pei Y., Carone F., Rennke H. G., Glücksmann-Kuis A. M., Schneider M. C., Ericsson M. et al. (1996). Identification and localization of polycystin, the PKD1 gene product. J. Clin. Invest. 98, 2674-2682. 10.1172/JCI119090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng L., Segal Y., Pavlova A., Barros E. J., Lohning C., Lu W., Nigam S. K., Frischauf A. M., Reeders S. T. and Zhou J. (1997). Distribution and developmentally regulated expression of murine polycystin. Am. J. Physiol. 272, F451-F459. [DOI] [PubMed] [Google Scholar]
- Geng L., Okuhara D., Yu Z., Tian X., Cai Y., Shibazaki S. and Somlo S. (2006). Polycystin-2 traffics to cilia independently of polycystin-1 by using an N-terminal RVxP motif. J. Cell Sci. 119, 1383-1395. 10.1242/jcs.02818 [DOI] [PubMed] [Google Scholar]
- Gherman A., Davis E. E. and Katsanis N. (2006). The ciliary proteome database: an integrated community resource for the genetic and functional dissection of cilia. Nat. Genet. 38, 961-962. 10.1038/ng0906-961 [DOI] [PubMed] [Google Scholar]
- Goetz S. C. and Anderson K. V. (2010). The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet. 11, 331-344. 10.1038/nrg2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanaoka K., Qian F., Boletta A., Bhunia A. K., Piontek K., Tsiokas L., Sukhatme V. P., Guggino W. B. and Germino G. G. (2000). Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 408, 990-994. 10.1038/35050128 [DOI] [PubMed] [Google Scholar]
- Herron B. J., Lu W., Rao C., Liu S., Peters H., Bronson R. T., Justice M. J., McDonald J. D. and Beier D. R. (2002). Efficient generation and mapping of recessive developmental mutations using ENU mutagenesis. Nat. Genet. 30, 185-189. 10.1038/ng812 [DOI] [PubMed] [Google Scholar]
- Hopp K., Ward C. J., Hommerding C. J., Nasr S. H., Tuan H.-F., Gainullin V. G., Rossetti S., Torres V. E. and Harris P. C. (2012). Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J. Clin. Invest. 122, 4257-4273. 10.1172/JCI64313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao Y.-C., Tuz K. and Ferland R. (2012). Trafficking in and to the primary cilium. Cilia 1, 4 10.1186/2046-2530-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q., Milenkovic L., Jin H., Scott M. P., Nachury M. V., Spiliotis E. T. and Nelson W. J. (2010). A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science 329, 436-439. 10.1126/science.1191054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes J., Ward C. J., Peral B., Aspinwall R., Clark K., San Millan J. L., Gamble V. and Harris P. C. (1995). The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat. Genet. 10, 151-160. 10.1038/ng0695-151 [DOI] [PubMed] [Google Scholar]
- Ishikawa H., Thompson J., Yates J. R. III and Marshall W. F. (2012). Proteomic analysis of mammalian primary cilia. Curr. Biol. 22, 414-419. 10.1016/j.cub.2012.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H., White S. R., Shida T., Schulz S., Aguiar M., Gygi S. P., Bazan J. F. and Nachury M. V. (2010). The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 141, 1208-1219. 10.1016/j.cell.2010.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koulen P., Cai Y., Geng L., Maeda Y., Nishimura S., Witzgall R., Ehrlich B. E. and Somlo S. (2002). Polycystin-2 is an intracellular calcium release channel. Nat. Cell Biol. 4, 191-197. 10.1038/ncb754 [DOI] [PubMed] [Google Scholar]
- Lancaster M. A., Schroth J. and Gleeson J. G. (2011). Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nat. Cell Biol. 13, 700-707. 10.1038/ncb2259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W., Peissel B., Babakhanlou H., Pavlova A., Geng L., Fan X., Larson C., Brent G. and Zhou J. (1997). Perinatal lethality with kidney and pancreas defects in mice with a targetted Pkd1 mutation. Nat. Genet. 17, 179-181. 10.1038/ng1097-179 [DOI] [PubMed] [Google Scholar]
- Lu W., Shen X., Pavlova A., Lakkis M., Ward C. J., Pritchard L., Harris P. C., Genest D. R., Perez-Atayde A. R. and Zhou J. (2001). Comparison of Pkd1-targeted mutants reveals that loss of polycystin-1 causes cystogenesis and bone defects. Hum. Mol. Genet. 10, 2385-2396. 10.1093/hmg/10.21.2385 [DOI] [PubMed] [Google Scholar]
- Ma M., Tian X., Igarashi P., Pazour G. J. and Somlo S. (2013). Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat. Genet. 45, 1004-1012. 10.1038/ng.2715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazelova J., Astuto-Gribble L., Inoue H., Tam B. M., Schonteich E., Prekeris R., Moritz O. L., Randazzo P. A. and Deretic D. (2009). Ciliary targeting motif VxPx directs assembly of a trafficking module through Arf4. EMBO J. 28, 183-192. 10.1038/emboj.2008.267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick D., Chapin H., Baggs J. E., Yu Z., Somlo S., Sun Z., Hogenesch J. B. and Caplan M. J. (2012). The gamma-secretase cleavage product of polycystin-1 regulates TCF and CHOP-mediated transcriptional activation through a p300-dependent mechanism. Dev. Cell 22, 197-210. 10.1016/j.devcel.2011.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki T., Wu G., Hayashi T., Xenophontos S. L., Veldhuisen B., Saris J. J., Reynolds D. M., Cai Y., Gabow P. A., Pierides A. et al. (1996). PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 272, 1339-1342. 10.1126/science.272.5266.1339 [DOI] [PubMed] [Google Scholar]
- Nakata K., Shiba D., Kobayashi D. and Yokoyama T. (2012). Targeting of Nphp3 to the primary cilia is controlled by an N-terminal myristoylation site and coiled-coil domains. Cytoskeleton 69, 221-234. 10.1002/cm.21014 [DOI] [PubMed] [Google Scholar]
- Nauli S. M., Alenghat F. J., Luo Y., Williams E., Vassilev P., Li X., Elia A. E. H., Lu W., Brown E. M., Quinn S. J. et al. (2003). Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 33, 129-137. 10.1038/ng1076 [DOI] [PubMed] [Google Scholar]
- Nauli S. M., Rossetti S., Kolb R. J., Alenghat F. J., Consugar M. B., Harris P. C., Ingber D. E., Loghman-Adham M. and Zhou J. (2006). Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J. Am. Soc. Nephrol. 17, 1015-1025. 10.1681/ASN.2005080830 [DOI] [PubMed] [Google Scholar]
- Oh E. C. and Katsanis N. (2012). Cilia in vertebrate development and disease. Development 139, 443-448. 10.1242/dev.050054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno H., Stewart J., Fournier M. C., Bosshart H., Rhee I., Miyatake S., Saito T., Gallusser A., Kirchhausen T. and Bonifacino J. S. (1995). Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science 269, 1872-1875. 10.1126/science.7569928 [DOI] [PubMed] [Google Scholar]
- Parnell S. C., Magenheimer B. S., Maser R. L., Rankin C. A., Smine A., Okamoto T. and Calvet J. P. (1998). The polycystic kidney disease-1 protein, polycystin-1, binds and activates heterotrimeric G-proteins in vitro. Biochem. Biophys. Res. Commun. 251, 625-631. 10.1006/bbrc.1998.9514 [DOI] [PubMed] [Google Scholar]
- Peters D. J., van de Wal A., Spruit L., Saris J. J., Breuning M. H., Bruijn J. A. and de Heer E. (1999). Cellular localization and tissue distribution of polycystin-1. J. Pathol. 188, 439-446. [DOI] [PubMed] [Google Scholar]
- Praetorius H. A. and Spring K. R. (2005). A physiological view of the primary cilium. Annu. Rev. Physiol. 67, 515-529. 10.1146/annurev.physiol.67.040403.101353 [DOI] [PubMed] [Google Scholar]
- Qian F., Germino F. J., Cai Y., Zhang X., Somlo S. and Germino G. G. (1997). PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat. Genet. 16, 179-183. 10.1038/ng0697-179 [DOI] [PubMed] [Google Scholar]
- Qian F., Boletta A., Bhunia A. K., Xu H., Liu L., Ahrabi A. K., Watnick T. J., Zhou F. and Germino G. G. (2002). Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc. Natl. Acad. Sci. USA 99, 16981-16986. 10.1073/pnas.252484899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiling J. H., Olive A. J., Sanyal S., Carette J. E., Brummelkamp T. R., Ploegh H. L., Starnbach M. N. and Sabatini D. M. (2013). A CREB3-ARF4 signalling pathway mediates the response to Golgi stress and susceptibility to pathogens. Nat. Cell Biol. 15, 1473-1485. 10.1038/ncb2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roitbak T., Ward C. J., Harris P. C., Bacallao R., Ness S. A. and Wandinger-Ness A. (2004). A polycystin-1 multiprotein complex is disrupted in polycystic kidney disease cells. Mol. Biol. Cell 15, 1334-1346. 10.1091/mbc.E03-05-0296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffers M. S., van der Bent P., Prins F., Spruit L., Breuning M. H., Litvinov S. V., de Heer E. and Peters D. J. M. (2000). Polycystin-1, the product of the polycystic kidney disease 1 gene, co-localizes with desmosomes in MDCK cells. Hum. Mol. Genet. 9, 2743-2750. 10.1093/hmg/9.18.2743 [DOI] [PubMed] [Google Scholar]
- Singla V. and Reiter J. F. (2006). The primary cilium as the cell's antenna: signaling at a sensory organelle. Science 313, 629-633. 10.1126/science.1124534 [DOI] [PubMed] [Google Scholar]
- Su X., Driscoll K., Yao G., Raed A., Wu M., Beales P. L. and Zhou J. (2014). Bardet-Biedl syndrome proteins 1 and 3 regulate the ciliary trafficking of polycystic kidney disease 1 protein. Hum. Mol. Genet. 23, 5441-5451. 10.1093/hmg/ddu267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The international Polycystic Kidney Disease Consortium (1995). Polycystic kidney disease: the complete structure of the PKD1 gene and its protein. Cell 81, 289-298. 10.1016/0092-8674(95)90339-9 [DOI] [PubMed] [Google Scholar]
- Tsiokas L., Kim E., Arnould T., Sukhatme V. P. and Walz G. (1997). Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proc. Natl. Acad. Sci. USA 94, 6965-6970. 10.1073/pnas.94.13.6965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Wu M., Yao G., Zhang J. and Zhou J. (2014). The Cytoplasmic Tail of FPC Antagonizes the Full-Length Protein in the Regulation of mTOR Pathway. PloS One. 9, e95630 10.1371/journal.pone.0095630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward H. H., Brown-Glaberman U., Wang J., Morita Y., Alper S. L., Bedrick E. J., Gattone V. H. II, Deretic D. and Wandinger-Ness A. (2011). A conserved signal and GTPase complex are required for the ciliary transport of polycystin-1. Mol. Biol. Cell 22, 3289-3305. 10.1091/mbc.E11-01-0082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters A. M. and Beales P. L. (2011). Ciliopathies: an expanding disease spectrum. Pediatr. Nephrol. 26, 1039-1056. 10.1007/s00467-010-1731-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W., Hackmann K., Xu H., Germino G. and Qian F. (2007). Characterization of cis-autoproteolysis of polycystin-1, the product of human polycystic kidney disease 1 gene. J. Biol. Chem. 282, 21729-21737. 10.1074/jbc.M703218200 [DOI] [PubMed] [Google Scholar]
- Wilson P. D. (2004). Polycystic kidney disease. N. Engl. J. Med. 350, 151-164. 10.1056/NEJMra022161 [DOI] [PubMed] [Google Scholar]
- Woodward O. M., Li Y., Yu S., Greenwell P., Wodarczyk C., Boletta A., Guggino W. B. and Qian F. (2010). Identification of a polycystin-1 cleavage product, P100, that regulates store operated Ca2+ entry through interactions with STIM1. PLoS ONE 5, e12305 10.1371/journal.pone.0012305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G. M., Gonzalez-Perrett S., Essafi M., Timpanaro G. A., Montalbetti N., Arnaout M. A. and Cantiello H. F. (2003). Polycystin-1 activates and stabilizes the polycystin-2 channel. J. Biol. Chem. 278, 1457-1462. 10.1074/jbc.M209996200 [DOI] [PubMed] [Google Scholar]
- Xu C., Rossetti S., Jiang L., Harris P. C., Brown-Glaberman U., Wandinger-Ness A., Bacallao R. and Alper S. L. (2007). Human ADPKD primary cyst epithelial cells with a novel, single codon deletion in the PKD1 gene exhibit defective ciliary polycystin localization and loss of flow-induced Ca2+ signaling. Am. J. Physiol. Renal Physiol. 292, F930-F945. 10.1152/ajprenal.00285.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao G., Su X., Nguyen V., Roberts K., Li X., Takakura A., Plomann M. and Zhou J. (2014). Polycystin-1 regulates actin cytoskeleton organization and directional cell migration through a novel PC1-Pacsin 2-N-Wasp complex. Hum. Mol. Genet. 23, 2769-2779. 10.1093/hmg/ddt672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder B. K., Hou X. and Guay-Woodford L. M. (2002). The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol. 13, 2508-2516. 10.1097/01.ASN.0000029587.47950.25 [DOI] [PubMed] [Google Scholar]
- Yu S., Hackmann K., Gao J., He X., Piontek K., Garcia-Gonzalez M. A., Menezes L. F., Xu H., Germino G. G., Zuo J. et al. (2007). Essential role of cleavage of Polycystin-1 at G protein-coupled receptor proteolytic site for kidney tubular structure. Proc. Natl. Acad. Sci. USA 104, 18688-18693. 10.1073/pnas.0708217104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Taulman P. D. and Yoder B. K. (2004). Cystic kidney diseases: all roads lead to the cilium. Physiology 19, 225-230. 10.1152/physiol.00003.2004 [DOI] [PubMed] [Google Scholar]
- Zhou J. (2009). Polycystins and primary cilia: primers for cell cycle progression. Annu. Rev. Physiol. 71, 83-113. 10.1146/annurev.physiol.70.113006.100621 [DOI] [PubMed] [Google Scholar]
- Zuo X., Fogelgren B. and Lipschutz J. H. (2011). The small GTPase Cdc42 is necessary for primary ciliogenesis in renal tubular epithelial cells. J. Biol. Chem. 286, 22469-22477. 10.1074/jbc.M111.238469 [DOI] [PMC free article] [PubMed] [Google Scholar]