

ABSTRACT
Neuronal excitability is strictly regulated by various mechanisms, including modulation of ion channel activity and trafficking. Stimulation of m1 muscarinic acetylcholine receptor (also known as CHRM1) increases neuronal excitability by suppressing the M-current generated by the Kv7/KCNQ channel family. We found that m1 muscarinic acetylcholine receptor stimulation also triggers surface transport of KCNQ subunits. This receptor-induced surface transport was observed with KCNQ2 as well as KCNQ3 homomeric channels, but not with Kv3.1 channels. Deletion analyses identified that a conserved domain in a proximal region of the N-terminal tail of KCNQ protein is crucial for this surface transport – the translocation domain. Proteins that bind to this domain were identified as α- and β-tubulin and collapsin response mediator protein 2 (CRMP-2; also known as DPYSL2). An inhibitor of casein kinase 2 (CK2) reduced tubulin binding to the translocation domain, whereas an inhibitor of glycogen synthase kinase 3 (GSK3) facilitated CRMP-2 binding to the translocation domain. Consistently, treatment with the GSK3 inhibitor enhanced receptor-induced KCNQ2 surface transport. M-current recordings from neurons showed that treatment with a GSK3 inhibitor shortened the duration of muscarinic suppression and led to over-recovery of the M-current. These results suggest that m1 muscarinic acetylcholine receptor stimulates surface transport of KCNQ channels through a CRMP-2-mediated pathway.
KEY WORDS: Channel trafficking, Muscarinic acetylcholine receptor, KCNQ2, CK2, GSK3, CRMP-2
Summary: Activation of m1 muscarinic acetylcholine receptor is well known to suppress KCNQ channel activities, but it also induces surface transport of KCNQ channels through a CRMP-2 mediated mechanism.
INTRODUCTION
The M-type K+ current, which regulates the firing rate of neurons is generated by channels containing Kv7/KCNQ 2–5 subunits (Delmas and Brown, 2005; Jentsch, 2000). Dysfunction of the M-channel, caused by various mutations in KCNQ2 and KCNQ3 genes, leads to neurological disorders such as epilepsy (Jentsch, 2000) and encephalopathy (Weckhuysen et al., 2012). Therefore, proper surface expression of the M-channel is important in order to maintain physiological neuronal excitability. However, the mechanism regulating M-channel surface density is not well understood.
KCNQ2 homomeric channels are known to localize largely in the endoplasmic reticulum (ER), with inefficient delivery to the plasma membrane (Alaimo et al., 2009; Choveau and Shapiro, 2012; Etxeberria et al., 2008). It has been shown that the rate of KCNQ2 exit from the ER is regulated by KCNQ2-bound calmodulin (Alaimo et al., 2009; Cavaretta et al., 2014; Etxeberria et al., 2008). Alternatively, co-expression of KCNQ3 with the KCNQ2 subunit has been shown to increase the delivery of KCNQ2–KCNQ3 heteromeric channels to the surface, compared to that of KCNQ2 homomeric channels (Schwake et al., 2000). Therefore, the KCNQ3 subunit has been considered as the regulatory subunit for KCNQ channel trafficking. However, recent studies demonstrate that KCNQ3 knockout mice show normal M-currents (Soh et al., 2014; Tzingounis and Nicoll, 2008), suggesting that the role for the KCNQ3 subunit in channel trafficking is more limited than was originally anticipated. In addition, many KCNQ2 channel mutations are known to cause benign familial neonatal seizures that usually cease spontaneously several months after birth (Weckhuysen et al., 2012), which implies that there are feedback mechanisms that adjust neuronal excitability.
Collapsin response mediator protein 2 (CRMP-2; also known as DPYSL2) is a kinesin-binding protein, which promotes the recognition of cargo proteins and is known to regulate neural polarity (Fukata et al., 2002). It has been shown that GSK3β-mediated phosphorylation of CRMP-2, which is trigged by cyclin-dependent kinase 5 (CDK5), inhibits its binding to tubulin dimers, whereas dephosphorylation facilitates CRMP-2–tubulin binding and promotes axonal growth by delivering tubulin dimers to microtubule terminals for elongation (Yoshimura et al., 2005). Recent studies show that CRMP-2 is also important for ion channel trafficking and other neural functions (Brittain et al., 2009; Kawano et al., 2005; Yoshida et al., 1998).
Stimulation of Gq-coupled receptors, including m1 muscarinic acetylcholine receptor (mAChR; also known as CHRM1) suppresses the M-current by activating multiple signaling cascades (Delmas and Brown, 2005; Kosenko et al., 2012). In this study, we demonstrate that stimulation of m1 mAChR also triggers surface transport of KCNQ channels. We identified that casein kinase II (CK2) is required for this mode of surface transport. Using a mapping analysis, we identified that a region of 26 conserved amino acid residues at the proximal N-terminal tail of the KCNQ2 subunit is responsible for this surface transport. A pulldown assay followed by tandem mass spectrometric analyses identified α- and β-tubulin and CRMP-2 as proteins that interact with this domain. Consistent with the involvement of a tubulin–CRMP-2 complex, treatment with a GSK3 inhibitor facilitated surface transport of KCNQ2 channels. Moreover, in superior cervical ganglion (SCG) neurons, treatment with a GSK3 inhibitor facilitated recovery from M-current suppression, which resulted in larger M-currents than under basal conditions. We propose that the identified KCNQ2 surface transport process is an important regulatory mechanism for adjusting neuronal excitability.
RESULTS
Stimulation of m1 mAChR triggers surface transport of KCNQ channels
Total internal reflection fluorescence (TIRF) microscopy selectively illuminates a thin area (<100 nm) proximal to the cover glass and has been extensively used to detect membrane-associated events such as exocytosis (Axelrod, 2008; Steyer and Almers, 2001). During our previous study using TIRF analyses (Kosenko et al., 2012), we observed that m1 mAChR stimulation increased TIRF signals of KCNQ2 homomeric channels that had been tagged with monomeric citrine (mCit), KCNQ2–mCit, which indicated an increase in surface KCNQ2 channels. However, this phenomenon appeared to be counterintuitive because m1 mAChR stimulation has been well characterized to suppress KCNQ2 current. We reasoned that this mAChR-induced KCNQ2 surface transport might be one of the feedback mechanisms for stabilizing neuronal excitability.
The mAChR-induced increase in TIRF signal was further characterized using the same conditions under which we initially observed the phenomenon – expressing KCNQ2–mCit in a Chinese hamster ovary cell line that stably expressed human m1 mAChR, CHO hm1. Transient expression of KCNQ2–mCit in CHO hm1 cells resulted in a level of functional expression equivalent to that of non-tagged KCNQ2, as evaluated by measuring the current density at 0 mV; 39.9±5.5 pA/pF for KCNQ2–mCit (n=13) versus 35.3±4.9 pA/pF for KCNQ2 (n=10). Stimulation of m1 mAChR with 3 µM oxotremorine-M (oxo-M) triggered a gradual increase in TIRF signals, as we had observed previously (Fig. 1A,B; Movie 1). This effect was not due to changes in the fluorescence intensity of mCit per se because epifluorescence, which represents whole-cell fluorescence, did not change (Fig. 1A,B). To further test whether the increased TIRF signal was derived from increased surface KCNQ2 channels at the plasma membrane, we performed surface biotin-labeling of FLAG-tagged KCNQ2 channels. After a 5-min incubation with 3 µM oxo-M, surface proteins were biotinylated (Fig. 1C). Biotinylated proteins were purified using NeutrAvidin beads and KCNQ2-FLAG was detected by immunoblotting with an antibody against FLAG. Biotin labeling efficiency was determined relative to the KCNQ2–FLAG signal from whole-cell lysate. The absence of biotinylation of co-expressed cytosolic GFP confirmed selective biotinylation of cell surface proteins (Fig. 1C). Activation of m1 mAChR increased surface KCNQ2 channels by 63.8±21.2%, n=5 (Fig. 1C). Because our initial TIRF assay with a 4-min duration did not reach a steady state (Fig. 1B), we extended the observation period to 30 min (Fig. 1D). The TIRF signal continued to increase after application of 3 µM oxo-M. This increase was best fit with a single exponential function with a time constant, τ=11.06±0.61 min. When m1 mAChR activation was terminated by 100 µM pirenzepine, the TIRF signal halted further increase, and signals slowly returned to basal levels (Fig. 1D). These results confirm that activation of m1 mAChR induces surface transport of KCNQ2 channel.
Fig. 1.

Stimulation of m1 mAChR triggers surface transport of KCNQ2 channels. (A) Representative cell images in pseudocolor showing that application of 3 µM oxo-M increases the TIRF signal but not the epifluorescent signal (Epi) of KCNQ2–mCit expressed in CHO hm1 cells. (B) Pooled quantification of the experiments shown in A. Application of 3 µM oxo-M, shown as a black box (oxo), increases the TIRF signal but not epifluorescent signal of KCNQ2–mCit. (C) Surface biotinylation of the KCNQ2–FLAG channel in CHO hm1 cells, showing that 3 µM of oxo-M increased the amount of surface KCNQ2 homomeric channels. Co-expressed GFP was not biotinylated, confirming selective surface biotin labeling. A summary histogram displaying biotinylation of KCNQ2 relative to that of untreated KCNQ2 is also shown. *P<0.05. (D) 3 µM oxo-M induced a TIRF response during a 30 min observation period and the response was terminated by the addition of 100 µM pirenzepine at t=10 min. (E) Superecliptic-pHluorin-tagged KCNQ2 was expressed in CHO hm1 cells and rat SCG neurons, showing that stimulation with 3 µM oxo-M triggers surface transport of KCNQ2 in both cell types. Insets show representative pseudocolor epifluorescent images of pHluorin-tagged KCNQ2 signals from an SCG neuron. Error bars show s.e.m. Scale bars: 10 µm.
Because the KCNQ2 channel is a neuronal channel, a key question is whether this surface transport observed in CHO hm1 represents a neuronal phenomenon. For decades, SCG neurons have been a standard experimental system for M-current research (Delmas and Brown, 2005; Hoshi et al., 2005). However, we found that performing the TIRF assay on cultured SCG neurons was very difficult because neurons often grew on top of other cell types and made poor contact with the cover glass, as described previously by another group (Pettinger et al., 2013). Therefore, we constructed a pH-sensitive GFP construct, superecliptic pHluorin, which was inserted within the S1–S2 loop of the KCNQ2 subunit, a region that has also been used for insertion of the hemagglutinin (HA) epitope for surface detection (Chung et al., 2006; Schwake et al., 2003). Expression of pHluorin-KCNQ2 in CHO hm1 cells exhibited voltage-gated currents with a current density of 21.7±3.1 pA/pF (n=15) at 0 mV, which confirmed that pHluorin-KCNQ2 proteins formed functional channels. Superecliptic pHluorin has been used to measure surface transport of membrane proteins because it senses changes between the acidic pH in vesicular lumen and the physiological pH at the extracellular space (Sankaranarayanan et al., 2000). However, pHluorin-based experiments have limitations because it also responds to changes in intracellular pH caused by factors other than exocytosis (Rathje et al., 2013; Wilkinson et al., 2014). Therefore, we first evaluated pHluorin-KCNQ2 responses to oxo-M in CHO hm1 cells by comparing oxo-M responses of TIRF signals from KCNQ2–mCit in CHO hm1 cells (Fig. S1). Application of 3 µM oxo-M increased the fluorescence of pHluorin-KCNQ2 (Fig. 1E) with an almost identical time course to that observed when using TIRF to analyze KCNQ2–mCit, with a high correlation (r=0.97, Fig. S1). Because the mAChR response of KCNQ2–mCit TIRF signals parallels with surface labeling of KCNQ2 protein as mentioned above, the high correlation between TIRF measurements and the pHluorin-KCNQ2 signals suggests that pHluorin-KCNQ2 responds to the exposure of KCNQ2 channels to the extracellular environment.
We then expressed pHluorin-KCNQ2 channels in SCG neurons. pHluorin-KCNQ2 channels showed an increase in fluorescent signal upon oxo-M stimulation at the soma, similar to that obtained from CHO hm1 cells (Fig. 1E); however, responses from the neurites of these neurons were inconsistent and did not reach significance (Fig. 1E, inset). Overall, these results suggest that mAChR-induced surface transport of KCNQ2 channels is mediated by a common mechanism in neurons and CHO cells.
mAChR-induced surface transport of ion channels is specific to the KCNQ channel family and requires CK2
TIRF assays using KCNQ2–mCit in CHO hm1 cells were used to further characterize this mAChR-induced KCNQ2 surface transport. Dose–response relationships indicated that oxo-M stimulates KCNQ2 channel surface transport at a 50% effective concentration (EC50) of 0.15±0.04 µM (Fig. 2A). Next, we examined whether m1 mAChR stimulation can induce translocation of other channels. KCNQ3 is another subtype of the KCNQ subfamily that has been implicated in KCNQ channel trafficking, and Kv3.1a (encoded by a splice variant of KCNC1) is a voltage-gated K+ channel from a different subfamily. TIRF signals from mCit-tagged KCNQ3 increased after stimulation of m1 mAChR to a similar extent as KCNQ2–mCit; however, signals from mCit-tagged Kv3.1a showed only marginal changes (Fig. 2B,C). In addition, co-expression of KCNQ2 and KCNQ3 did not further facilitate mAChR-induced surface transport (Fig. 2C). These results suggest that subunits from the KCNQ family have a common mechanism for surface transport, which is triggered by mAChR activation.
Fig. 2.

mAChR-induced surface transport is specific to KCNQ channels and is mediated by CK2. (A) Dose–response curve of the KCNQ2–mCit response to oxo-M. (B,C) Time course (B) and summary histogram (C) of TIRF signals from KCNQ2–mCit (Q2), KCNQ3–mCit (Q3) and Kv3.1a–mCit showing that mAChR-induced surface transport is specific to the KCNQ channel family but not to Kv3.1 channels. ***P<0.001; NS, not significant. (D) 10 µM of ionomycin decreased the TIRF signal of KCNQ2–mCit and Kv3.1a–mCit. (E,F) Time course (E) and summary histogram (F) showing that application of 50 µg/ml concanavalin A (ConA) induced a sustained increase of KCNQ2–mCit and Kv3.1a–mCit at the cell surface. Con A facilitated the oxo-M-induced increase in the KCNQ2–mCit TIRF signal. ***P<0.001. (G,H) Time course (G) and summary histogram (H) showing that pre-treatment with TBB inhibited the mAChR-induced TIRF signal increase of KCNQ2–mCit. ***P<0.001. (I) Application of 10 μM TBB or 100 nM calyculin A (cal) did not increase the TIRF signal of KCNQ2–mCit. Error bars show s.e.m. and n values are given in brackets on each graph.
Because cytosolic Ca2+ is an important regulator for selective exocytosis (Südhof, 2004), we applied 10 µM of ionomycin to raise the concentration of cytosolic Ca2+. However, ionomycin did not increase the TIRF signals of KCNQ2–mCit or Kv3.1a–mCit (Fig. 2D). We then asked whether mAChR-induced surface transport is a result of reduced endocytosis. To test this, we applied an inhibitor of clathrin-mediated endocytosis, concanavalin A. Application of 50 µg/ml concanavalin A induced a gradual increase of TIRF signals of KCNQ2–mCit (Fig. 2E,F) as well as Kv3.1a–mCit (Fig. 2F), indicating constitutive exocytosis of these channels to the plasma membrane. Subsequent application of oxo-M augmented the increase in the TIRF signal of KCNQ2–mCit (Fig. 2E,F) but had no effect on Kv3.1a–mCit (Fig. 2F). Because inhibition of endocytosis facilitated mAChR-induced surface transport, these results suggest that mAChR stimulation facilitates exocytosis of KCNQ2 channels. Furthermore, these results suggest that mAChR-induced surface transport of KCNQ2 channels is not mediated by non-selective constitutive exocytosis.
To elucidate the signaling pathway that mediates mAChR-induced KCNQ2 surface transport, we tested several kinase inhibitors (Fig. 2G,H). An inhibitor of protein kinase C (PKC), 1 µM bisindolylmaleimide I, as well as a phosphoinositide-3 kinase inhibitor, 10 µM LY294002, had no effect on mAChR-induced KCNQ2 surface transport. However, CK2-selective inhibitors, 10 µM tetrabromobenzotriazole (TBB) and 5 µM tetrabromocinnamic acid (TBCA), suppressed mAChR-induced KCNQ2 surface transport (Fig. 2G,H). Furthermore, application of TBB induced a minor reduction in TIRF signals (Fig. 2I). Because CK2 is a constitutively active kinase (Meggio and Pinna, 2003), we wondered whether a phosphatase inhibitor, 100 nM calyculin A, would mimic receptor stimulation. However, application of calyculin A did not induce KCNQ2 translocation (Fig. 2I). These results suggest that KCNQ2 surface transport is a multistep process that requires other factors in addition to CK2 activation.
ER retention of KCNQ2 channels has been established as an important regulatory mechanism for KCNQ2 channel trafficking (Etxeberria et al., 2008). Therefore, we asked whether mAChR stimulation changes ER retention of KCNQ2 channels. Because non-confluent CHO cells are thin (3–5 µm thickness), we used epifluorescent images for colocalization analysis using Pearson's correlation coefficient. As expected from tetramer formation of KCNQ2 subunits, a positive control using cyan fluorescent protein (CFP)-tagged KCNQ2-CFP and KCNQ2–mCit showed a high correlation coefficient (Fig. 3A). Next, we optimized our conditions to differentiate ER versus post-Golgi vesicles, visualized with live cell markers – monomeric turquoise carrying the ER retention signal (ER–mTur) to label the ER (Goedhart et al., 2012), and monomeric cerulean-tagged VAMP2 (VAMP2–mCer) or monomeric citrine-tagged VAMP2 (VAMP2-mCit) to label post-Golgi vesicles (Jahn and Scheller, 2006). Unfortunately, the Pearson's correlation coefficient from raw cell images of CHO hm1 cells expressing ER-mTur and VAMP2-mCit showed a moderate correlation coefficient derived from homogenous background fluorescence (Fig. 3B, top). Therefore, we applied the median-subtraction method to remove local background because our focus was on small cellular vesicles (Dunn et al., 2011). This process drastically removed background noise and improved this assay. The Pearson's correlation coefficient obtained from median-subtracted images indicated no correlation between ER and post-Golgi vesicular compartments (Fig. 3B,E), but a high correlation between KCNQ2–CFP and KCNQ2–mCit was maintained (Fig. 3A,E).
Fig. 3.

Colocalization analysis of KCNQ2–mCit with ER and post-Golgi markers. (A) Representative original epifluorescent images (top), median-subtracted images (middle) and enlarged images from median-subtracted images (bottom) from cells expressing KCNQ2–CFP (left) or KCNQ2–mCit (middle). Merged (right) images obtained by using a CFP channel (green) and a YFP channel (red) are shown. Scatter plots of fluorescent signals and the Pearson's correlation coefficient from regions of interest (indicated by the white outline) are shown. (B) Negative control for colocalization analyses of ER–mTur and VAMP2–mCit, a post-Golgi vesicular marker, are shown. Pearson's correlation coefficient from original fluorescent images shows moderate correlation owing to background fluorescence. The median-subtracted image shows low background and an improved ability to distinguish the ER- and VAMP2-positive vesicles. (C) Colocalization analyses of ER–mTur and KCNQ2–mCit. (D) Colocalization analyses of VAMP2–mCer and KCNQ2–mCit. (E) Summary histogram of colocalization analyses for the indicated protein pairs shown in panels A–D. The correlation coefficient from each pair was significantly different, at least P<0.01, from that of the others. (F) Changes in the Pearson's correlation coefficient upon application of 3 µM oxo-M between ER–mTur and KCNQ2–mCit and VAMP2–mCer and KCNQ2–mCit. Oxo-M application reduced colocalization between VAMP-2 and KCNQ2. ***P<0.001. (G) Treatment with 10 μM TBB facilitated colocalization and disrupted oxo-M-induced disassembly of KCNQ2–mCit and VAMP2–mCer colocalization. Error bars show s.e.m., n values are given in brackets on the graphs. *P<0.05; **P<0.01; NS, not significant. Scale bars: 10 µm.
Using this procedure, we examined the correlation coefficient between KCNQ2–mCit and ER–mTur or VAMP2–mCer. To our surprise, the correlation coefficient between KCNQ2–mCit and ER–mTur (Fig. 3C,E) was lower than that between KCNQ2–mCit and VAMP2–mCer (Fig. 3C,E), suggesting that KCNQ2 channels reside more in post-Golgi vesicles than in the ER. To examine whether these pools respond to m1 mAChR stimulation, we calculated the Pearson's correlation coefficient before and after applying 3 µM oxo-M. The correlation coefficient between KCNQ2 and ER was not changed by application of oxo-M. In contrast, the correlation coefficient between KCNQ2 and VAMP2 was reduced by oxo-M (Fig. 3F).
Because CK2 inhibitors prevented mAChR-induced KCNQ2 surface transport, we tested whether TBB changed the colocalization of KCNQ2 with these two pools of cytosolic vesicles. Application of 10 μM TBB had no effect on colocalization between KCNQ2 and the ER (Fig. 3G). In contrast, TBB application increased colocalization of KCNQ2 and VAMP2, and prevented reduction of this colocalization upon mAChR stimulation (Fig. 3G). These results suggest that a pool of KCNQ2 channels residing in post-Golgi vesicles is responsible for mAChR-induced KCNQ2 surface transport in a CK2-dependent manner.
A conserved proximal N-terminal region of the KCNQ subunit is crucial for mAChR-induced KCNQ surface transport
Because mAChR activation selectively induces surface transport of KCNQ channels, but not of Kv3.1 channels, we reasoned that the KCNQ protein contains a domain that dictates vesicular trafficking. To identify the responsible translocation domain, we performed deletion analyses on the KCNQ2 subunit using mCit-tagged KCNQ2 deletion mutants and TIRF assays (Fig. 4A). Deletion of the C-terminal tail of the KCNQ2 subunit, which contains the calmodulin-binding site regulating ER retention (Etxeberria et al., 2008), as well as the ankyrin G binding domain (Pan et al., 2006), did not disrupt mAChR-induced translocation. In contrast, an N-terminal deletion in the KCNQ2 protein resulted in attenuated translocation. A transmembrane segment of the KCNQ2 protein that lacked the N- and C-terminal tails, Q2(98–320)–mCit, also showed reduced translocation (Fig. 4A). These results suggest that the N-terminus of KCNQ2 is important for mAChR-induced surface translocation. Because the assumed translocation domain should bind to vesicular trafficking machinery that delivers KCNQ2-containing vesicles to the plasma membrane, we reasoned that cytosolic fragments containing the translocation domain should translocate near to the plasma membrane upon mAChR stimulation. Indeed, cells expressing an mCit-tagged N-terminal tail construct, Q2(1–97)–mCit, showed an increased TIRF signal after stimulation of m1 mAChR (Fig. 4B). Further deletion analysis within the N-terminus showed that a KCNQ2 fragment comprising amino acid residues 70–96, Q2(70–96)–mCit, was able to translocate upon mAChR stimulation (Fig. 4B). Q2(70–96)–mCit before mAChR stimulation showed homogenous subcellular localization (Fig. S2A), suggesting that this domain does not bind to specific cellular compartments, such as the cytoskeleton. The identified translocation domain was highly conserved among KCNQ subtypes but did not contain conserved serine or threonine residues, which indicates that this domain is not a substrate for serine/threonine kinases (Fig. 4C). Interestingly, this translocation domain has been previously shown to regulate targeting and ER retention of KCNQ1 channels (Dahimene et al., 2006; Jespersen et al., 2004).
Fig. 4.

Identification of the regulatory domain involved in mAChR-induced KCNQ2 channel surface transport. (A) Summary of deletion analyses, showing that the N-terminal tail is crucial for the mAChR-induced increase in the TIRF signal. Filled bars show responses that were significantly different (P<0.05) from the oxo-M response of full-length KCNQ2, Q2(FL)–mCit. TM, transmembrane domain. (B) Deletion analysis within the N-terminal tail. Q2(70–96)–mCit was the smallest fragment that showed mAChR-induced translocation. Filled bars indicate significant difference (P<0.05) from the negative control (cells expressing mCit alone). (C) Q2(70–96), designated as the translocation domain, is highly conserved among the indicated KCNQ (‘Q’) subtypes. (D) 10 µM TBB treatment inhibited mAChR-induced translocation of Q2(70–96)–mCit that had been stimulated by 3 µM oxo-M (oxo, black box). (E) Overexpression of His6-tagged Q2(70–96) interfered with mAChR-induced surface transport of full-length KCNQ2–mCit [Q2(FL)–mCit] that had been stimulated by 3 µM oxo-M. (F) Representative voltage-clamp current traces for wild-type KCNQ2 [KCNQ2(wt)] and KCNQ2(Δ70–96). (G) KCNQ2(Δ70–96) exhibited a suppressed response to oxo-M. The response of the control construct, Q2(FL)–mCit, from panel E is also shown for comparison. (H) The Pearson's correlation coefficient between KCNQ2(Δ70–96)–mCit [Q2(Δ70–96)] and ER–mTur (ER) as well as VAMP2–mCer. NS, not significant. Error bars show s.e.m., n values are given in brackets on the graphs.
We evaluated whether mAChR-induced translocation of Q2(70–96)–mCit uses the same mechanism as surface transport of full-length KCNQ2. TBB blocked translocation of Q2(70–96)–mCit, as was observed with KCNQ2-mCit (Fig. 4D). For further confirmation, we examined whether overexpression of the Q2(70–96) fragment disrupted mAChR-induced surface transport of full-length KCNQ2–mCit. To test this, we co-expressed His6-tagged Q2(70–96) and KCNQ2–mCit in CHO hm1 cells and performed a TIRF assay. Co-expression of His6-tagged Q2(70–96) reduced mAChR-induced surface transport of KCNQ2–mCit (Fig. 4E).
If the Q2(70–96) domain is crucial for mAChR-induced surface transport of the KCNQ2 channel, a channel that lacks the translocation domain should not be transported to the plasma membrane. To test this, we generated a KCNQ2(Δ70–96) deletion construct. As expected, expression of KCNQ2(Δ70-96) protein did not produce any current (0.6±0.3 pA/pF, n=6, Fig. 4F). In addition, mAChR-induced surface transport of KCNQ2(Δ70–96)–mCit was suppressed (Fig. 4G). Because mutations of the translocation domain of the KCNQ1 channel cause ER retention (Dahimene et al., 2006), we examined whether KCNQ2(Δ70–96)–mCit showed distinct vesicular distribution. The correlation coefficient indicated that KCNQ2(Δ70–96)–mCit showed lower colocalization with VAMP2-positive vesicles compared to that of wild-type KCNQ2–mCit (Fig. 3F versus Fig. 4H, P<0.0001). However, values of the correlation coefficient with the ER were unaltered between the two forms of the KCNQ2 channel. Moreover, application of oxo-M did not change the correlation coefficient of KCNQ2(Δ70–96)–mCit with VAMP2 (Fig. 4H), supporting the notion of a lack of mAChR-induced surface transport for this mutant channel. We conclude that the Q2(70–96) construct contains the crucial domain that is required for surface transport of KCNQ2 channels.
CRMP-2 and tubulin bind to the translocation domain
To identify adaptor proteins that mediate mAChR-induced KCNQ channel surface transport, we performed pulldown assays using GST fusion proteins containing the translocation domain, GST–Q2(70–96). Because the KCNQ2 channel is a neural protein, we used mouse brain lysate as a protein source for pulldown analyses. Several proteins were selectively pulled down by GST–Q2(70–96), as evaluated by SDS-PAGE and silver staining (Fig. 5A). The most apparent protein was ∼50 kDa followed by a protein with ∼60 kDa. Tandem mass spectrometric analyses were performed for these two prominent bands. Detailed information for the identified proteins can be found in Tables S1 and S2. The highest ranked proteins from the 50-kDa band were α- and β- tubulin, and the highest ranked proteins from the 60 kDa band were collapsin response mediator proteins (CRMPs), including CRMP-2 (Fig. 5A). CRMP-2 is an adaptor protein that is known to bridge kinesin and tubulin heterodimers (Fukata et al., 2002). Immunoblotting confirmed selective binding of α-tubulin and CRMP-2 to GST–Q2(70-96) (Fig. S2B).
Fig. 5.

Identification and characterization of translocation-domain-binding proteins. (A) Representative silver-staining of an SDS-PAGE gel, showing selective purification by GST–Q2(70–96) fusion protein from mouse brain lysate. Pulldown assays using glutathione beads (GSH beads), GST-bound GSH beads (GST) and GST–Q2(70–96)-bound GSH beads [GST–Q2(70–96)] are shown. Arrowheads indicate GST–Q2(70–96)-purified bands used for mass spectrometric analyses. The highest ranked proteins from mass spectrometry analyses of each band are indicated. (B) Pulldown assays showing selective binding of GST–Q2(70–96) to CRMP-2 and α tubulin from untreated SH-SY5Y cell lysates; the binding was modulated by pre-treatment of cells with 5 µM TBB or 5 µM CHIR99021 (CH). *P<0.05, **P<0.01. IB, immunoblot; UT, untreated. (C) Immunoprecipitation of transiently expressed full-length KCNQ2–FLAG in SH-SY5Y cells, showing selective CRMP-2 binding and its modulation through pre-treatment with 5 µM TBB or 5 µM CHIR99021. **P<0.01. Error bars show s.e.m.
CK2 and GSK3 regulate KCNQ2 channel trafficking by changing protein interactions
Because GSK3β is known to negatively regulate CRMP-2 binding to tubulin dimers (Yoshimura et al., 2005), we examined whether GSK3 regulates the interaction of these proteins using a potent and selective inhibitor of GSK3, CHIR99021. Pulldown assays using GST–Q2(70–96) were performed to evaluate binding of the translocation domain to α-tubulin and CRMP-2 from cell lysates that had been prepared from untreated SH-SY5Y cells, or cells treated with either 5 µM TBB or 5 µM CHIR99021 (Fig. 5B). Tubulin binding to the translocation domain was reduced upon treatment with either TBB or CHIR99021, suggesting that phosphorylation of tubulin promotes binding of the translocation domain (Fig. 5B). In contrast, treatment with CHIR99021, but not TBB, facilitated CRMP-2 binding to the translocation domain (Fig. 5B). TBB treatment did not alter the stoichiometry of binding of the translocation domain to CRMP-2 and tubulin, as estimated from the ratio between pulled down tubulin and CRMP-2. In contrast, CHIR99021 treatment increased the CRMP-2 and tubulin stoichiometry (Fig. 5B), which could be attributed to the increased binding of CRMP-2 to tubulin dimer, as reported previously (Yoshimura et al., 2005). Immunoprecipitation using the full-length KCNQ2–FLAG protein replicated the results of pulldown assays with respect to KCNQ2–CRMP2 binding and the effects of TBB and CHIR99021 (Fig. 5C). These results suggest that CK2 and GSK3 regulate CRMP-2 and tubulin binding to KCNQ2 subunits.
Inhibition of GSK3 facilitates mAChR-induced surface transport of KCNQ2 channels, as well as recovery from muscarinic suppression of the M-current in neurons
Our in vitro binding study suggests that GSK3 is involved in mAChR-induced translocation of the KCNQ2 channel. Indeed, TIRF assays showed that CHIR99021 treatment facilitated mAChR-induced KCNQ2–mCit surface transport (Fig. 6A,B). In addition, treatment with TBB disrupted mAChR-induced KCNQ surface transport, even in cells that had been treated with CHIR99021 (Fig. 6B). By contrast, application of CHIR99021 alone did not result in significant changes in the TIRF signals of KCNQ2 channels, which suggests that promoting CRMP-2 binding to KCNQ is not sufficient to mimic mAChR-induced KCNQ surface transport (Fig. 6C).
Fig. 6.
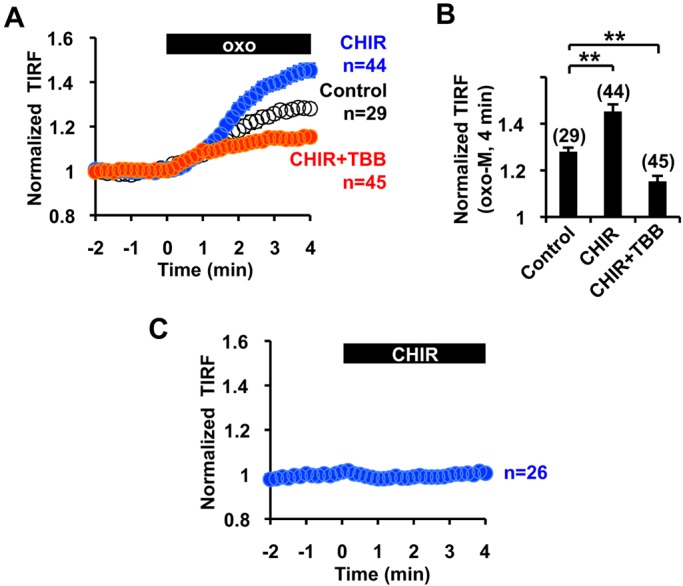
Functional modulation of mAChR-induced KCNQ2–mCit surface transport by CHIR99021 and TBB. (A,B) Time course (A) and summary histogram (B) showing that pretreatment with 5 µM CHIR99021 (CHIR) facilitated mAChR-induced KCNQ2–mCit surface transport and its blockade by 10 µM TBB. **P<0.01. (C) Application of 5 µM CHIR99021 alone did not affect the TIRF signal of KCNQ2–mCit. Error bars show s.e.m. n values are given in brackets on the graphs.
To evaluate the physiological relevance of mAChR-induced KCNQ2 surface transport in neurons, we measured the M-current in SCG neurons and the effect of 5 µM CHIR99021. The standing current at −30 mV, generated by the M-current and leak current, was unaffected by CHIR99021 treatment (∼1 h) [control 11.1±2.0 pA/pF (n=13) versus CHIR99021 treatment 14.6±1.6 pA/pF (n=10), P>0.05, Student's t-test]. We then measured the time course of muscarinic suppression and subsequent recovery of the M-current (Fig. 7A,B). In control SCG neurons, application of 0.1 µM oxo-M for 1 min induced transient M-current suppression, which recovered to the baseline level ∼3 min after wash (Fig. 7B,C). When SCG neurons were pre-treated with CHIR99021, neurons showed equivalent muscarinic suppression of the M-current (t=1 min in Fig. 7B, P>0.05, Student's t-test). However, M-currents recorded from CHIR99021-treated neurons recovered significantly faster to baseline values, ∼1 min after wash (Fig. 7B,C). Moreover, the current amplitudes became larger than those of the control, which is similar to over-recovery of the M-current (Akasu et al., 1993; Pfaffinger, 1988; Villarroel, 1994). These results suggest that CK2 and GSK3 modulate the duration of muscarinic suppression of the M-current and over-recovery of the M-current.
Fig. 7.
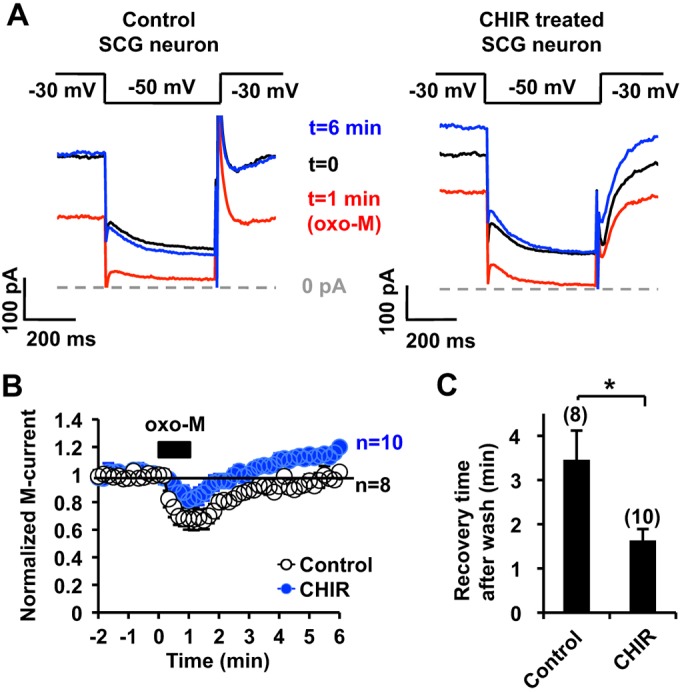
Effect of GSK3 inhibition on muscarinic suppression of the M-current. (A) Representative M-current traces from control (left) and 5 µM CHIR99021 (CHIR)-treated (right) rat SCG neurons showing suppression and recovery after treatment with 0.1 µM oxo-M. The data at the indicated time points are taken from the graph shown in B. (B) Summary time courses of oxo-M-induced M-current suppression from control and CHIR99021-treated SCG neurons, showing that the duration of oxo-M induced M-current suppression is shorter in CHIR99021-treated neurons. The black box indicates the presence of 0.1 µM oxo-M. The horizontal line indicates the baseline level of 1. (C) Quantification of the time required to recover to the control M-current amplitude after washout. *P<0.05. Error bars show s.e.m. n values are given in brackets on the graphs.
DISCUSSION
The present study demonstrates that m1 mAChR activation, which is known to suppress KCNQ channel activity, triggers surface transport of KCNQ channels. This delayed offset action is likely to be one of the underlying feedback mechanisms regulating neuronal excitability.
Several studies on KCNQ trafficking have established that ER retention is an important mechanism for its functional expression (Choveau and Shapiro, 2012; Etxeberria et al., 2008). Our results suggest that m1-mAChR-induced KCNQ surface transport utilizes post-Golgi vesicles as a source of KCNQ channels to be trafficked to the surface. However, we observed a large pool of KCNQ2 channels in cytosolic vesicles after mAChR stimulation, including those channels in the ER. Therefore, we think that ER retention is important for long-term regulation, whereas post-Golgi vesicles serve as a releasable pool for acute regulation of surface KCNQ channels.
We identified that the translocation domain is crucial for KCNQ2 surface transport. This domain overlaps with the domain that has been found to regulate KCNQ1 channel trafficking (Dahimene et al., 2006; Jespersen et al., 2004). Translocation-domain mutant KCNQ1 channels that have been found in Romano-Ward LQT1 syndrome are known to accumulate in the ER (Dahimene et al., 2006). Consistent with these findings, a deletion mutant of the translocation domain in the KCNQ2 channel, KCNQ2(Δ70–96), did not produce any current and showed reduced localization with post-Golgi vesicles, which resulted in a relative accumulation of KCNQ2(Δ70–96) in the ER. These results suggest that the translocation domain is also crucial for steady-state functional expression of the KCNQ2 channel.
We demonstrated that the translocation domain binds to α- and β-tubulin and CRMP-2. Tubulin binding to the translocation domain might indicate that the translocation domain binds to microtubules. However, the translocation-domain-containing fusion protein, Q2(70–96)–mCit, did not localize to the cytoskeleton (Fig. S2A), suggesting that microtubules are not a stable binding substrate for the translocation domain. Furthermore, we think that the α–β tubulin dimer bridges the binding of the channel to CRMP-2 for the following reasons: (1) tubulin was the most prominent protein in the GST–Q2(70–96) pulldown assay, suggesting a primary interaction; (2) inhibition of GSK3, which is known to promote CRMP-2 binding to tubulin dimers, increased CRMP-2 pulled down by GST–Q2(70–96) without changing tubulin binding to GST–Q(70–96).
An involvement of CRMP-2 in KCNQ2 channel trafficking might explain why we could not detect mAChR-induced surface channel transport in neurites of SCG neurons because CRMP-2 is known to localize at the distal axon in developing neurons (Arimura et al., 2004). CRMP-2-mediated channel regulation has also been reported for the N-type voltage-gated Ca2+ channel (Brittain et al., 2009). In this case, it has been demonstrated that CRMP-2 binds directly to the Ca2+ channel at the channel domain I–II intracellular loop and the distal C-terminus. However, these sites do not share amino acid sequence similarity to the translocation domain of KCNQ2 channel.
Our pulldown assays demonstrated that protein interactions between KCNQ2, tubulin and CRMP-2 were regulated by two kinases, CK2 and GSK3 (Fig. 8). Our binding assay suggests that CK2- and GSK3-mediated phosphorylation facilitates tubulin–KCNQ2 binding. Because GSK3 requires a priming phosphorylation, CK2 would serve as the priming kinase for this step. However, an apparent effect of GSK3 inhibition was the increase in the CRMP-2:tubulin ratio without increasing KCNQ2–tubulin binding. This finding suggests that KCNQ2-bound tubulin is not usually occupied by CRMP-2 under basal conditions, which implies that the CRMP-2–tubulin interaction is the key determinant for KCNQ2 surface transport. GSK3β-mediated regulation of the KCNQ channel was originally reported in a genetic study of bipolar affective disorder (Borsotto et al., 2007). In addition, it has been demonstrated that the KCNQ2 protein is a substrate for GSK3β (Borsotto et al., 2007). However, whether GSK3-mediated phosphorylation suppresses (Borsotto et al., 2007) or enhances (Kapfhamer et al., 2010) the M-current is still controversial. Our results showed that inhibition of GSK3 for up to 1 h did not change the M-current amplitude per se but primed surface KCNQ transport that was triggered by m1 mAChR stimulation.
Fig. 8.
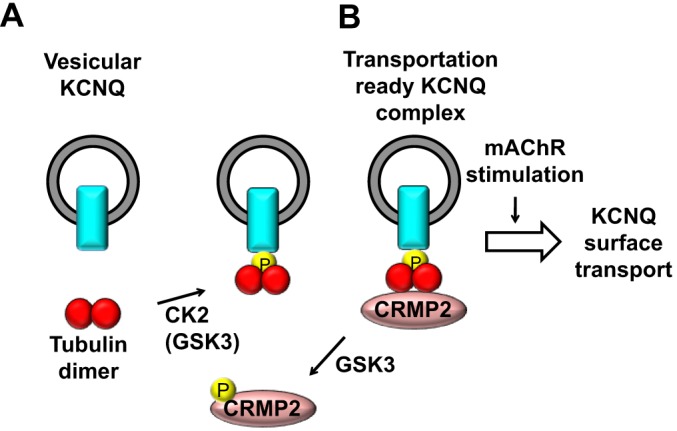
Proposed mechanism for mAChR-induced transport of KCNQ to the surface. (A) Protein kinases CK2 and GSK3 phosphorylate tubulin, facilitating its binding to the KCNQ channel. Because GSK3 requires a priming phosphorylation, CK2 is most likely to be the priming kinase for this step. (B) GSK3-mediated phosphorylation of CRMP-2 negatively regulates the association of CRMP-2 with the KCNQ channel complex through binding to the tubulin dimer. The KCNQ2–tubulin–CRMP-2 complex is required for the functional expression of the KCNQ channel, which is facilitated by stimulation of m1 mAChR.
The promotion of surface KCNQ2 transport upon treatment with the GSK3 inhibitor led to a larger M-current recovery after agonist washout, which is similar to the over-recovery described for the M-current from bull frog sympathetic neurons (Pfaffinger, 1988). It has been shown that arachidonic acid (Villarroel, 1994) and myosin light chain kinase (Akasu et al., 1993) are implicated in over-recovery in frog neurons. However, both proposed mechanisms assume that there are changes in the activities of existing channels at the plasma membrane, which is independent of a GSK-inhibitor-induced mechanism. Considering that over-recovery of the M-current is rarely described in mammalian neurons (Beech et al., 1991; Hoshi et al., 2003; Kosenko et al., 2012), such modulation might have been altered during evolution.
In conclusion, stimulation of m1 mAChR accelerates tubulin–CRMP-2-mediated KCNQ2 trafficking. This tubulin–CRMP-2-mediated KCNQ2 trafficking is also important for steady-state functional expression of KCNQ2 channels. This newly identified form of channel trafficking could function as a feedback mechanism for regulating neuronal excitability.
MATERIALS AND METHODS
Antibodies, reagents and plasmids
Antibodies against the following proteins were used in this study: CRMP-2 (rabbit monoclonal, Abcam), α-tubulin (catalog number T6199, mouse monoclonal, Sigma-Aldrich) and FLAG epitope (M2 mouse monoclonal, Sigma-Aldrich). Protein kinase inhibitors, including bisindolylmaleimide I, LY 294002, TBB and CHIR99021 were purchased from Sigma-Aldrich. Plasmids encoding KCNQ2–mCit and KCNQ2–FLAG have been described previously (Kosenko et al., 2012). Plasmids encoding pmTurquoise2-ER (Goedhart et al., 2012) and mCer-4aa-VAMP2 (Jahn and Scheller, 2006) were obtained through Addgene. Deletion mutations of KCNQ2 were generated by PCR. KCNQ2 deletion mutants and the coding region of VAMP2 were subcloned into the pGFP-N1-derived pmCit-N1 plasmid. For GST-fusion proteins, KCNQ2 fragments were subcloned into pGEX-4T. All PCR-derived fragments were verified by sequencing.
Cell culture and transfection
CHO hm1 cells were cultured in α minimal essential medium supplemented with 5% fetal calf serum (FCS) and 500 µg/ml G418 sulfate. SH-SY5Y cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FCS. CHO hm1 cells were transfected using LT1 transfection reagent (Mirus Bio LLC). SCG neurons were obtained from 14- to 19-day-old rats and cultured as described previously (Kosenko et al., 2012). Introduction of expression plasmids into SCG neurons was achieved through nuclear injection at 2 days in vitro (DIV), and cells were used for experiments at DIV 4.
Live-cell imaging
Live-cell imaging and TIRF imaging was performed, essentially, as described previously (Kosenko et al., 2012). Briefly, one day after transfection, cells were re-plated onto 18-mm round cover glasses. Cells were used for TIRF experiments 2 days after transfection. For recording, medium was replaced with Q2 solution containing 144 mM NaCl, 5 mM KCl, 2 mM CaCl2, 0.5 mM MgCl2, 10 mM glucose and 10 mM HEPES (pH 7.4). Fluorescence emission was acquired using an inverted microscope IX-81 (Olympus Tokyo) and an ImageEM CCD camera, (Hamamatsu Photonics, Hamamatsu, Shizuoka Japan) and was controlled using MetaMorph 7.6.3 (Molecular Devices, Sunnyvale, CA). For the excitation light for TIRF experiments, a 445-nm diode laser (Coherent, Santa Clara, CA), and a 515-nm diode-pumped solid-state laser (Cobolt, Stockholm, Sweden) with an acousto-optic tunable filter were used with a TIRF module (Olympus). For the excitation light for epifluorescence, a xenon arc lamp (Lambda LS, Sutter Instrument, Novato, CA) equipped with excitation filters ET436/20× and ET505/20× (Chroma Technology, Bellows Falls, VT) was used. Emission images were obtained through a dual-view module (Photometrics, Tucson, AZ) with ET535/30m, ET480/40m emission filters and a T505lpxr dichroic mirror (Chroma Technology). For pHluorin experiments, a filter set 41001 (Chroma Technology, Bellows Falls, VT) was used. The exposure time was 100 ms; images were taken every 10 s to measure time-lapse imaging, except for those shown in Fig. 1D, which were taken with 30-s intervals.
Colocalization analysis
In our filter setting, a bleed through from the yellow fluorescent protein (YFP) channel to the CFP channel was minimal (CFP/YFP=0.000032±0.00016). Original and median-subtracted images were used to obtain pixel-to-pixel Pearson's correlation coefficients between CFP and YFP images using the correlation plot function in MetaMorph. To obtain median-subtracted images, median-filtered images (32×32 pixel), which represent background images, were generated from the original images. These background images were subtracted on a pixel-to-pixel basis from corresponding original images to obtain median-subtracted images (Dunn et al., 2011). An area surrounding a single cell was selected as a region of interest to obtain the Pearson's correlation coefficient per cell. All image processing was done using MetaMorph software.
Surface protein biotinylation
Surface protein biotinylation was performed as described previously (Kosenko et al., 2012). GFP- and FLAG-tagged KCNQ2 channels were transiently expressed in CHO hm1 cells. Cells were washed with the Q2 solution described above, then incubated with Q2 solution containing 3 µM oxo-M for 5 min. Cells were then washed twice with ice-cold PBS followed by incubation with sulfo-NHS-LC-biotin (Life Technologies) for 30 min at 4°C. The treated cells were further washed twice with PBS containing 100 mM glycine. Cells were then lysed in HSE buffer containing 1% Triton X-100, 150 mM NaCl, 5 mM EDTA, 5 mM EGTA, 20 mM HEPES (pH 7.4) and protease inhibitors. Biotinylated proteins were purified using NeutrAvidin beads and KCNQ2–FLAG was detected with horseradish peroxidase (HRP)-conjugated FLAG antibody. The ratios of biotinylated KCNQ2 and lysate KCNQ2 immunoblot signals were calculated and normalized to those of control.
Pulldown assay and mass spectrometry analysis
GST fusion constructs were transformed into BL21 (DE3) Escherichia coli for protein expression. Expression of fusion proteins was induced with 0.1 mM isopropyl-β-D-thiogalactopyranoside and incubated for 3 h at 29˚C. Transformed bacteria were pelleted and lysed in HSE buffer, as described above. GST fusion proteins were purified using glutathione–Sepharose beads and washed with HSE buffer. Mouse brain lysate was prepared freshly from adult mouse brains in HSE buffer and a Dounce homogenizer, followed by centrifugation at 18,000 g for 30 min. Mouse brain lysate was then pre-cleared with glutathione beads, followed by centrifugation at 18,000 g for 30 min to remove endogenous GST. GST-bound glutathione beads and GST–Q2(70–96)-bound glutathione beads (GST 10 µg) were incubated with pre-cleared brain extract (1 mg) for 1 h at 4°C, followed by a wash with HSE buffer and analysis by using SDS-PAGE and silver staining or western blotting. Protein bands were cut from the gel for alkylation, which was followed by in-gel trypsin digestion, as described previously (Shevchenko et al., 2007). Tryptic peptides were analyzed with liquid chromatography tandem mass spectroscopy (LC MS/MS) utilizing an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher) coupled on-line with an Easy-nLC 1000 (Thermo Fisher, San Jose, CA) (Kaake et al., 2010). Each MS/MS experiment comprised one mass spectrometry scan in Fourier transform mode (350–1800 m/z, resolution of 60,000 at m/z 400) followed by ten data-dependent MS/MS scans in ion trap mode with normalized collision energy at 29%. Database searching was performed against a concatenated SwissProt database (2014.12.4), comprising a normal and its randomized version using the Batch-Tag software within a developmental version of Protein Prospector (v 5.13.1, University of California San Francisco) (Kaake et al., 2010). Mus musculus was set as the species for searching. The mass tolerances for parent ions and fragment ions were set as ±20 ppm and 0.6 Da, respectively. Trypsin was set as the enzyme with two maximum missed cleavages allowed. Cysteine carbamidomethylation was set as a constant modification. Protein N-terminal acetylation, asparagine deamidation, N-terminal conversion of glutamine to pyroglutamic acid, and methionine oxidation were selected as variable modifications. Proteins were identified with expectation values of ≤0.01 and a minimum of two unique peptides.
For pulldown experiments using SH-SY5Y cells, cells were homogenized in HSE buffer containing 1 mM Na3VO4 and 50 mM NaF.
Electrophysiological measurements
Whole-cell patch-clamp recordings for CHO hm1 cells were performed as described previously (Kosenko et al., 2012). Perforated patch-clamp recordings were performed at room temperature on SCG neurons using an Axopatch 200B patch-clamp amplifier (Molecular Devices, Sunnyvale, CA), as described previously (Hoshi et al., 2005). Briefly, signals were sampled at 2 kHz, filtered at 1 kHz and acquired using pClamp software (version 10.2, Molecular Devices). Cells were perfused with the Q2 solution described above. M-current amplitudes were measured as deactivating currents during 500-ms test pulses to −50 mV from a holding potential of −30 mV.
Statistical analysis
Quantified results are expressed as the mean±s.e.m. The statistical significance of the results was assessed by using ANOVA and Student's t-test using Prism 6 (GraphPad). *P<0.05, **P<0.01, ***P<0.001.
Acknowledgements
We thank Dorus Gadella (University of Amsterdam, The Netherlands) for providing pmTurquoise2-ER, and Robert Zucker (University of California, Berkeley, CA) for providing mCer-4aa-VAMP2.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
N.H. designed the study. L.J., A.K., N.H. performed live-cell imaging and pulldown assays, and analyzed results. N.H. performed the electrophysiological recordings. C.Y. and L.H. designed and performed mass spectrometry analysis and analyzed results. N.H., L.H. and X.L. wrote the paper.
Funding
This work is supported by the National Institutes of Health [grant numbers R01NS067288 to N.H. and R01GM074830 to L.H.]; and grants from the National Natural Science Foundation of China [grant numbers 81473235 and 81020108031 to X.L.]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.175547/-/DC1
References
- Akasu T., Ito M., Nakano T., Schneider C. R., Simmons M. A., Tanaka T., Tokimasa T. and Yoshidat M. (1993). Myosin light chain kinase occurs in bullfrog sympathetic neurons and may modulate voltage-dependent potassium currents. Neuron 11, 1133-1145. 10.1016/0896-6273(93)90226-H [DOI] [PubMed] [Google Scholar]
- Alaimo A., Gomez-Posada J. C., Aivar P., Etxeberria A., Rodriguez-Alfaro J. A., Areso P. and Villarroel A. (2009). Calmodulin activation limits the rate of KCNQ2 K+ channel exit from the endoplasmic reticulum. J. Biol. Chem. 284, 20668-20675. 10.1074/jbc.M109.019539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimura N., Menager C., Fukata Y. and Kaibuchi K. (2004). Role of CRMP-2 in neuronal polarity. J. Neurobiol. 58, 34-47. 10.1002/neu.10269 [DOI] [PubMed] [Google Scholar]
- Axelrod D. (2008). Chapter 7: Total internal reflection fluorescence microscopy. Methods Cell Biol. 89, 169-221. 10.1016/S0091-679X(08)00607-9 [DOI] [PubMed] [Google Scholar]
- Beech D. J., Bernheim L., Mathie A. and Hille B. (1991). Intracellular Ca2+ buffers disrupt muscarinic suppression of Ca2+ current and M current in rat sympathetic neurons. Proc. Natl. Acad. Sci. USA 88, 652-656. 10.1073/pnas.88.2.652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsotto M., Cavarec L., Bouillot M., Romey G., Macciardi F., Delaye A., Nasroune M., Bastucci M., Sambucy J.-L., Luan J.-J. et al. (2007). PP2A-Bgamma subunit and KCNQ2 K+ channels in bipolar disorder. Pharmacogenomics J. 7, 123-132. 10.1038/sj.tpj.6500400 [DOI] [PubMed] [Google Scholar]
- Brittain J. M., Piekarz A. D., Wang Y., Kondo T., Cummins T. R. and Khanna R. (2009). An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J. Biol. Chem. 284, 31375-31390. 10.1074/jbc.M109.009951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaretta J. P., Sherer K. R., Lee K. Y., Kim E. H., Issema R. S. and Chung H. J. (2014). Polarized axonal surface expression of neuronal KCNQ potassium channels is regulated by calmodulin interaction with KCNQ2 subunit. PLoS ONE 9, e103655 10.1371/journal.pone.0103655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choveau F. S. and Shapiro M. S. (2012). Regions of KCNQ K+ channels controlling functional expression. Front. Physiol. 3, 397 10.3389/fphys.2012.00397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H. J., Jan Y. N. and Jan L. Y. (2006). Polarized axonal surface expression of neuronal KCNQ channels is mediated by multiple signals in the KCNQ2 and KCNQ3 C-terminal domains. Proc. Natl. Acad. Sci. USA 103, 8870-8875. 10.1073/pnas.0603376103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahimene S., Alcolea S., Naud P., Jourdon P., Escande D., Brasseur R., Thomas A., Baro I. and Merot J. (2006). The N-terminal juxtamembranous domain of KCNQ1 is critical for channel surface expression: implications in the Romano-Ward LQT1 syndrome. Circ. Res. 99, 1076-1083. 10.1161/01.RES.0000250262.12219.95 [DOI] [PubMed] [Google Scholar]
- Delmas P. and Brown D. A. (2005). Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat. Rev. Neurosci. 6, 850-862. 10.1038/nrn1785 [DOI] [PubMed] [Google Scholar]
- Dunn K. W., Kamocka M. M. and McDonald J. H. (2011). A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 300, C723-C742. 10.1152/ajpcell.00462.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etxeberria A., Aivar P., Rodriguez-Alfaro J. A., Alaimo A., Villace P., Gomez-Posada J. C., Areso P. and Villarroel A. (2008). Calmodulin regulates the trafficking of KCNQ2 potassium channels. FASEB J. 22, 1135-1143. 10.1096/fj.07-9712com [DOI] [PubMed] [Google Scholar]
- Fukata Y., Itoh T. J., Kimura T., Menager C., Nishimura T., Shiromizu T., Watanabe H., Inagaki N., Iwamatsu A., Hotani H. et al. (2002). CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat. Cell Biol. 4, 583-591. 10.1038/ncb825 [DOI] [PubMed] [Google Scholar]
- Goedhart J., von Stetten D., Noirclerc-Savoye M., Lelimousin M., Joosen L., Hink M. A., van Weeren L., Gadella T. W. J. Jr. and Royant A. (2012). Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat. Commun. 3, 751 10.1038/ncomms1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N., Zhang J.-S., Omaki M., Takeuchi T., Yokoyama S., Wanaverbecq N., Langeberg L. K., Yoneda Y., Scott J. D., Brown D. A. et al. (2003). AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat. Neurosci. 6, 564-571. 10.1038/nn1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N., Langeberg L. K. and Scott J. D. (2005). Distinct enzyme combinations in AKAP signalling complexes permit functional diversity. Nat. Cell Biol. 7, 1066-1073. 10.1038/ncb1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R. and Scheller R. H. (2006). SNAREs — engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 7, 631-643. 10.1038/nrm2002 [DOI] [PubMed] [Google Scholar]
- Jentsch T. J. (2000). Neuronal KCNQ potassium channels: physiology and role in disease. Nat. Rev. Neurosci. 1, 21-30. 10.1038/35036198 [DOI] [PubMed] [Google Scholar]
- Jespersen T., Rasmussen H. B., Grunnet M., Jensen H. S., Angelo K., Dupuis D. S., Vogel L. K., Jorgensen N. K., Klaerke D. A. and Olesen S.-P. (2004). Basolateral localisation of KCNQ1 potassium channels in MDCK cells: molecular identification of an N-terminal targeting motif. J. Cell Sci. 117, 4517-4526. 10.1242/jcs.01318 [DOI] [PubMed] [Google Scholar]
- Kaake R. M., Milenković T., Pržulj N., Kaiser P. and Huang L. (2010). Characterization of cell cycle specific protein interaction networks of the yeast 26S proteasome complex by the QTAX strategy. J. Proteome Res. 9, 2016-2029. 10.1021/pr1000175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapfhamer D., Berger K. H., Hopf F. W., Seif T., Kharazia V., Bonci A. and Heberlein U. (2010). Protein Phosphatase 2a and glycogen synthase kinase 3 signaling modulate prepulse inhibition of the acoustic startle response by altering cortical M-Type potassium channel activity. J. Neurosci. 30, 8830-8840. 10.1523/JNEUROSCI.1292-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano Y., Yoshimura T., Tsuboi D., Kawabata S., Kaneko-Kawano T., Shirataki H., Takenawa T. and Kaibuchi K. (2005). CRMP-2 is involved in kinesin-1-dependent transport of the Sra-1/WAVE1 complex and axon formation. Mol. Cell. Biol. 25, 9920-9935. 10.1128/MCB.25.22.9920-9935.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosenko A., Kang S., Smith I. M., Greene D. L., Langeberg L. K., Scott J. D. and Hoshi N. (2012). Coordinated signal integration at the M-type potassium channel upon muscarinic stimulation. EMBO J. 31, 3147-3156. 10.1038/emboj.2012.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meggio F. and Pinna L. A. (2003). One-thousand-and-one substrates of protein kinase CK2? FASEB J. 17, 349-368. 10.1096/fj.02-0473rev [DOI] [PubMed] [Google Scholar]
- Pan Z., Kao T., Horvath Z., Lemos J., Sul J.-Y., Cranstoun S. D., Bennett V., Scherer S. S. and Cooper E. C. (2006). A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J. Neurosci. 26, 2599-2613. 10.1523/JNEUROSCI.4314-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettinger L., Gigout S., Linley J. E. and Gamper N. (2013). Bradykinin controls pool size of sensory neurons expressing functional delta-opioid receptors. J. Neurosci. 33, 10762-10771. 10.1523/JNEUROSCI.0123-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffinger P. (1988). Muscarine and t-LHRH suppress M-current by activating an IAP-insensitive G-protein. J. Neurosci. 8, 3343-3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathje M., Fang H., Bachman J. L., Anggono V., Gether U., Huganir R. L. and Madsen K. L. (2013). AMPA receptor pHluorin-GluA2 reports NMDA receptor-induced intracellular acidification in hippocampal neurons. Proc. Natl. Acad. Sci. USA 110, 14426-14431. 10.1073/pnas.1312982110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan S., De Angelis D., Rothman J. E. and Ryan T. A. (2000). The use of pHluorins for optical measurements of presynaptic activity. Biophys. J. 79, 2199-2208. 10.1016/S0006-3495(00)76468-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwake M., Pusch M., Kharkovets T. and Jentsch T. J. (2000). Surface expression and single channel properties of KCNQ2/KCNQ3, M-type K+ channels involved in epilepsy. J. Biol. Chem. 275, 13343-13348. 10.1074/jbc.275.18.13343 [DOI] [PubMed] [Google Scholar]
- Schwake M., Jentsch T. J. and Friedrich T. (2003). A carboxy-terminal domain determines the subunit specificity of KCNQ K+ channel assembly. EMBO Rep. 4, 76-81. 10.1038/sj.embor.embor715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A., Tomas H., Havliš J., Olsen J. V. and Mann M. (2007). In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856-2860. 10.1038/nprot.2006.468 [DOI] [PubMed] [Google Scholar]
- Soh H., Pant R., LoTurco J. J. and Tzingounis A. V. (2014). Conditional deletions of epilepsy-associated KCNQ2 and KCNQ3 channels from cerebral cortex cause differential effects on neuronal excitability. J. Neurosci. 34, 5311-5321. 10.1523/JNEUROSCI.3919-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyer J. A. and Almers W. (2001). A real-time view of life within 100 nm of the plasma membrane. Nat. Rev. Mol. Cell Biol. 2, 268-275. 10.1038/35067069 [DOI] [PubMed] [Google Scholar]
- Südhof T. C. (2004). The synaptic vesicle cycle. Annu. Rev. Neurosci. 27, 509-547. 10.1146/annurev.neuro.26.041002.131412 [DOI] [PubMed] [Google Scholar]
- Tzingounis A. V. and Nicoll R. A. (2008). Contribution of KCNQ2 and KCNQ3 to the medium and slow afterhyperpolarization currents. Proc. Natl. Acad. Sci. USA 105, 19974-19979. 10.1073/pnas.0810535105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarroel A. (1994). On the role of arachidonic acid in M-current modulation by muscarine in bullfrog sympathetic neurons. J. Neurosci. 14, 7053-7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weckhuysen S., Mandelstam S., Suls A., Audenaert D., Deconinck T., Claes L. R. F., Deprez L., Smets K., Hristova D., Yordanova I. et al. (2012). KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 71, 15-25. 10.1002/ana.22644 [DOI] [PubMed] [Google Scholar]
- Wilkinson K. A., Ashby M. C. and Henley J. M. (2014). Validity of pHluorin-tagged GluA2 as a reporter for AMPA receptor surface expression and endocytosis. Proc. Natl. Acad. Sci. USA 111, E304 10.1073/pnas.1319322111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H., Watanabe A. and Ihara Y. (1998). Collapsin response mediator protein-2 is associated with neurofibrillary tangles in Alzheimer's disease. J. Biol. Chem. 273, 9761-9768. 10.1074/jbc.273.16.9761 [DOI] [PubMed] [Google Scholar]
- Yoshimura T., Kawano Y., Arimura N., Kawabata S., Kikuchi A. and Kaibuchi K. (2005). GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120, 137-149. 10.1016/j.cell.2004.11.012 [DOI] [PubMed] [Google Scholar]