

Abstract
Test batteries to screen chemicals for mutagenic hazard include several endpoints regarded as effective for detecting genotoxic carcinogens. Traditional in vivo methods primarily examine clastogenic endpoints in haematopoietic tissues. Although this approach is effective for identifying systemically distributed clastogens, some mutagens may not induce clastogenic effects; moreover, genotoxic effects may be restricted to the site of contact and/or related tissues. An OECD test guideline for transgenic rodent (TGR) gene mutation assays was released in 2011, and the TGR assays permit assessment of mutagenicity in any tissue. This study assessed the responses of two genotoxicity endpoints following sub-chronic oral exposures of male Muta™Mouse to 9 carcinogenic polycyclic aromatic hydrocarbons (PAHs). Clastogenicity was assessed via induction of micronuclei in peripheral blood, and mutagenicity via induction of lacZ transgene mutations in bone marrow, glandular stomach, small intestine, liver, and lung. Additionally, the presence of bulky PAH-DNA adducts was examined. Five of the 9 PAHs elicited positive results across all endpoints in at least one tissue, and no PAHs were negative or equivocal across all endpoints. All PAHs were positive for lacZ mutations in at least one tissue (sensitivity = 100%), and for 8 PAHs, one or more initial sites of chemical contact (i.e., glandular stomach, liver, small intestine) yielded a greater response than bone marrow. Five PAHs were positive in the micronucleus assay (sensitivity = 56%). Furthermore, all PAHs produced DNA adducts in at least one tissue. The results demonstrate the utility of the TGR assay for mutagenicity assessment, especially for compounds that may not be systemically distributed.
Keywords: Genotoxicity, Mutation, Micronucleus, Polycyclic aromatic hydrocarbon, Muta™Mouse
Highlights
-
•
The Muta™Mouse is a reliable tool for in vivo mutagenicity assessment of PAHs.
-
•
All 9 PAHs induced lacZ transgene mutations in small intestine.
-
•
Only 5 of 9 PAHs induced lacZ mutations and micronuclei in haematopoietic tissue.
-
•
Tissue-specific results are likely related to metabolism, repair, and proliferation.
-
•
For oral exposures, it is important to examine effects at the site-of-contact.
1. Introduction
In 1973 Ames and colleagues published a scientific paper proclaiming “Carcinogens are Mutagens” (Ames et al., 1973). Since then, a flurry of publications has debated the validity of this statement. Currently, the genetic toxicology community acknowledges that 65 to 90% of rodent carcinogens will elicit a positive response in at least one of the standard regulatory assays for genetic toxicity (Hernandez et al., 2009, Kirkland et al., 2005, Waters et al., 2010, Zeiger, 1998).
Genetic toxicology test batteries are generally effective for detecting carcinogenic compounds, and the sensitivity of the three-test battery is quite high (i.e., 85–90%, depending on the assays included) (Kirkland et al., 2005). However, not all chemicals fit this paradigm, and there is increasing recognition of response inconsistency. In vivo “false negatives” are known rodent carcinogens that fail to induce a positive response in an in vivo genotoxicity assay and may be the result of either (i) a lack of target tissue exposure, or (ii) endpoint incompatibility, or potentially both. With respect to the former, the standard test battery often examines haematopoietic tissue; however, the target tissue for mutation or chromosome damage may be a solid organ such as the liver or the gastrointestinal tract. With respect to the latter, in vitro tests may assess the induction of mutations, whereas the in vivo tests often assess clastogenicity. Yet, mutagenic compounds do not necessarily induce chromosome damage, and examining only clastogenicity in vivo may result in a missed positive response induced by a mutagenic carcinogen. By examining mutagenicity both in vitro and in vivo, it may be possible to resolve some in vivo false negatives.
Researchers have attempted to resolve problems related to a lack of target tissue exposure by designing assays that assess genotoxicity in the tissue that is suspected to be a target for induced genetic damage or carcinogenesis. Examples include unscheduled DNA synthesis in liver (OECD, 1997b), the comet assay for DNA strand breaks in various target tissues (Collins, 2004, JaCVAM, 2012, OECD, 2013, Rothfuss et al., 2010), and the liver micronucleus (MN) assay (Suzuki et al., 2004, Suzuki et al., 2005, Suzuki et al., 2009, Takasawa et al., 2010, Takasawa et al., 2013). Several in vivo mammalian gene mutation assays exist, however, most are not well suited to regulatory use because they are labour intensive, require large numbers of animals, and are prohibitively costly (e.g., the in vivo Hprt mutation assay). A novel endogenous gene mutation assay based on the Pig-a gene was more recently developed (Bryce et al., 2008); however, it is also currently restricted to haematopoietic tissue, and will require validation before it can be adopted for routine use.
Tests for gene mutations in TGRs (transgenic rodents) may be capable of resolving the issues leading to false negative results since they can detect induced transgene mutations in vivo in almost any target tissues. An OECD test guideline for the TGR somatic and germ cell gene mutation assay (OECD test guideline #488) was approved on July 28, 2011 (OECD, 2011). The Muta™Mouse TGR assay has been tested with a wide range of known mutagens, and it has accurately returned positive results in several target tissues (for a review, see Lambert et al., 2005 & Lambert et al., 2009). Additionally, it can be combined with other assays that detect cytogenetic damage, such as the peripheral blood MN assay (OECD, 1997a). This is advantageous, as it is difficult to combine the comet assay, which can also provide tissue-specific indications of DNA damage, with other genotoxicity endpoints (e.g., TGR mutations, MN, or Pig-a mutations) as the timing of sample collection for comet cannot be accommodated in the OECD test guidelines or recommended protocols for the other endpoints (OECD, 2013). A previous study published by our group simultaneously examined the frequency of transgene (lacZ) mutations, Pig-a mutations, and MN induced in Muta™Mouse by oral exposure to benzo(a)pyrene (BaP), a prototypical polycyclic aromatic hydrocarbon (PAH) (Lemieux et al., 2011).
PAHs are a ubiquitous group of combustion-derived organic compounds containing at least two fused benzene rings. Several PAHs, particularly the larger five- and six-ring compounds, are mutagens in mammals and other vertebrates, and are classified by the International Agency for Research on Cancer (IARC) as carcinogens (IARC, 1983, IARC, 2010). IARC currently lists one PAH (i.e., BaP) as a known human carcinogen (Group 1), three PAHs as probable human carcinogens (Group 2A), and eleven PAHs as possible human carcinogens (Group 2B) (IARC, 1983, IARC, 2010).
PAHs must be metabolically transformed to become DNA reactive. The major mutagenic pathway of PAH activation is via the production of DNA-reactive dihydrodiol-epoxides via CYP1 isozymes and epoxide hydrolase. The PAH-dihydrodiol-epoxides exert their mutagenic effect by covalently binding to a nucleotide, thereby forming a bulky DNA adduct. If these adducts are mis-repaired they can cause permanent DNA sequence changes (i.e., mutations); mutations in critical genes can lead to uncontrolled cell proliferation and replicative immortality, eventually leading to the establishment of neoplasia (Cairns, 1998, Josephy, 1997).
Cytochrome P450 1a1 (CYP1A1) is the major enzyme responsible for catalysing the initial oxidation of PAHs; however, other P450 isozymes such as CYP1B1 and CYP1A2 are also capable of catalysing oxidative reactions (Shimada et al., 2002, Shimada and Fujii-Kuriyama, 2004, Xue and Warshawsky, 2005). CYP1A2 is a major hepatic P450 isozyme, whereas CYP1A1 and 1B1 are primarily expressed extra-hepatically in several murine tissues including lung and small intestine (Choudhary et al., 2003, Hart et al., 2009, Renaud et al., 2011, Zhang et al., 2003). CYP1A1 is generally present only at low levels unless its expression is induced via aryl hydrocarbon receptor (AhR) agonism, whereas CYP1B1 is constitutively expressed in certain tissues, and a substantial amount of CYP1A2 activity is present in liver. Both of these enzymes can also be upregulated following induction via AhR agonism (Nebert et al., 2004).
A second PAH-activation pathway, known as the radical cation pathway, involves one-electron oxidation that produces PAH radical cations that contribute to the formation of depurinating adducts. A third activation pathway involves the PAH-dihydrodiol intermediate produced by CYP1 metabolism and further metabolism by aldo-keto reductases (AKRs) to produce a catechol that can undergo oxidation to generate o-quinones. The resulting o-quinones can form covalent DNA adducts or undergo redox cycling that generates oxidative DNA damage. This third pathway is known as the o-quinone or AKR pathway (Penning, 2014).
To expedite assessments of PAH-contaminated matrices (e.g., contaminated soil, urban air particulates) regulatory agencies have identified a select number of PAHs that are routinely the focus of concern and control (i.e., the US EPA 16 priority PAHs). The United States Environmental Protection Agency (US EPA) designates seven priority PAHs as B2 carcinogens (i.e., probable human carcinogens) (Keith and Telliard, 1979). The Canadian Environmental Protection Act (CEPA) identifies five PAHs as probable human carcinogens (CEPA, 1994). Few studies have examined the in vivo genotoxic potency of the aforementioned B2 carcinogens; an assessment of their relative potency across a number of tissues would permit an evaluation of existing in vivo endpoints for genotoxicity assessment, and the identification of critical tissues for effective hazard identification and assessment.
The objectives of this study were to evaluate the ability of the Muta™Mouse TGR lacZ mutation assay to correctly return a positive result in various tissues for selected carcinogenic PAHs, and to compare the sensitivity of the TGR response to another regulatory genotoxicity endpoint (i.e., peripheral blood MN assay). Nine genotoxic PAHs were selected for this study (Table 1); they are either listed as IARC Group 1 and 2 carcinogens, are US EPA B2 carcinogens, or were previously found to be genotoxic in Muta™Mouse FE1 cells (IARC, 2010, Keith and Telliard, 1979, Lemieux et al., 2015). BghiP is classified as Group 3 by IARC due to inadequate evidence to evaluate its carcinogenicity in experimental animals; however numerous studies have shown that BghiP can induce the formation of stable adducts (IARC, 2010), and it elicits a positive mutation response in Muta™Mouse FE1 cells. Therefore, it was also included in the current study. The PAHs examined herein are benz(a)anthracene (BaA; IARC Group 2B), dibenz(a,h)anthracene (DBahA; IARC Group 2A), benzo(b)fluoranthene (BbF; IARC Group 2B), chrysene (CHRY; IARC Group 2B), benzo(k)fluoranthene (BkF; IARC Group 2B), indeno(1,2,3-c,d)pyrene (INDENO, IARC Group 2B), and DBalP (IARC Group 2A; also known as dibenzo(def,p)chrysene), and BghiP (IARC Group 3). The previously published results for BaP (IARC Group 1) (Labib et al., 2012, Lemieux et al., 2011), the liver lacZ mutation and DNA adduct data for DBahA (Malik et al., 2013), as well as lung lacZ mutation and DNA adduct data recently published by Labib et al. (2015) (for all compounds except DBalP) are also included in our analyses.
Table 1.
Description of chemicals, doses, animal ages at the commencement of exposures, and data sources for the analyses presented in this work.
| Compound | CAS # | Purity | Dose (mg/kg BW/day) |
Animal age | Data source | ||
|---|---|---|---|---|---|---|---|
| Low | Med | High | |||||
| BaP | 50–32-8 | 99% | 25 | 50 | 75 | 25 weeks | Lemieux et al., 2011, Labib et al., 2012 |
| BaA | 53–55-3 | 99% | 20 | 40 | 80 | 10 weeks | This studya |
| DBahA | 53–70-3 | ≥ 98% | 6.25 | 12.5 | 25 | 10 weeks | Malik et al., 2013 (lacZ and DA in Lv), this studya |
| BbF | 205–99-2 | ≥ 98% | 25 | 50 | 100 | 16 weeks | This studya |
| CHRY | 218–01-9 | ≥ 98% | 20 | 40 | 80 | 14 weeks | This studya |
| BkF | 207–08-9 | ≥ 98% | 25 | 50 | 100 | 11 weeks | This studya |
| INDENO | 193–39-5 | ≥ 98% | 12.5 | 25 | 50 | 13 weeks | This studya |
| BghiP | 191–24-2 | ≥ 98% | 6.25 | 12.5 | 25 | 10 weeks | This studya |
| DBalP | 191–30-0 | ≥ 98% | 0.2 | 0.6 | 2 | 12 weeks | This study |
DA: DNA adducts; Lv: liver.
Mutation and DA data for lung only published in Labib et al. (2015).
2. Materials & methods
2.1. Animal treatment
Adult male Muta™Mouse (strain 40.6) specimens were individually housed in a microVENT ventilated rack (Allentown Inc., Allentown, NJ) on a 12-h light/12-h dark cycle. Animals received standard rodent chow (2014 Teklad Global standard rodent diet) and water ad libitum for the duration of the study. Animals were administered BaP, BaA (Sigma-Aldrich, Oakville, ON, Canada), DBahA, BbF, CHRY, BkF, INDENO, BghiP, and DbalP (Cambridge Isotopes, Tewksbury, MA) dissolved in highly refined olive oil (Sigma-Aldrich), and administered at 0.005 ml/g body weight. Compound purity, Chemical Abstract Service (CAS) numbers, doses and animal ages at the commencement of each study, as well as data source (i.e., from this study or previously published) are listed in Table 1. The doses were administered daily by oral gavage for 28 consecutive days. Doses were selected based on preliminary range-finding experiments for each compound, and the selected doses did not elicit overt signs of toxicity. There were five animals in each dose group and the vehicle control group. As per OECD guideline #474, 2-days following the last dose (i.e., one day prior to necropsy), all animals were bled via the saphenous or facial vein in order to obtain peripheral blood for the MN assay (OECD, 1997a). All animals were euthanized at 3 days post-dosing (OECD, 2011) via cardiac puncture under isoflurane anaesthesia, followed by cervical dislocation and chest cavity opening. The femurs were removed and the bone marrow was flushed out, pelleted via brief centrifugation, and flash frozen in liquid nitrogen. The small intestine and glandular stomach were flushed with PBS and flash-frozen. The liver and lung were removed and flash frozen. All tissues were stored at − 80 °C. Mice were bred, maintained, and treated in accordance with the Canadian Council for Animal Care Guidelines, and the protocols were approved by Health Canada's Animal Care Committee.
2.2. Genomic DNA isolation for lacZ mutation scoring and PAH-DNA adduct analysis
Murine tissues were prepared for total genomic DNA isolation in the following manner:
Bone marrow: Thawed bone marrow was combined with 5-ml ice cold lysis buffer (1 mM Na2EDTA, 100 mM NaCl, 20 mM Tris–HCl, pH 7.4, 1% SDS (w/v)) and incubated overnight at 37 °C with gentle shaking.
Liver: The right lobe of the liver was thawed and homogenized in TMST (50 mM Tris, pH 7.6, 3 mM magnesium acetate, 250 mM sucrose, 0.2% Triton X-100) using a Dounce tissue grinder. The homogenized tissue was then pelleted by centrifugation for 6 min at 800 × g (4 °C) and washed twice in TMST. The pellet was then re-suspended in 5 ml lysis buffer and incubated overnight at 37 °C with gentle shaking.
Small intestine: Tissue from the jejenum was slit open and the mucus layer and intestinal contents were removed by rinsing the tissue in ice-cold buffer (75 mM KCl, 20 mM EDTA) via repeated aspiration (i.e., 3–4 times) in and out of a 1-ml needleless syringe. The epithelial lining was then removed from the supporting tissue by repeatedly (10 times) forcing the tissue in and out of the syringe in fresh buffer. The sample was then centrifuged for 10 min at 2800 rpm (4 °C), re-suspended in 5 ml lysis buffer, and incubated overnight at 37 °C with gentle shaking.
Lung: Lung tissue (right lobe) was minced and placed in a sterile tube containing PBS. The tissue was then de-gassed by applying a vacuum to the top of the tube. The tubes were then centrifuged and re-suspended in 5 ml lysis buffer for an overnight incubation at 37 °C with gentle shaking.
Glandular stomach: Stomach tissue was opened and flushed with PBS, and then scraped using the edge of a scalpel blade. The scraped tissue was transferred to a glass Dounce tissue grinder and homogenized. The homogenized tissue was combined with 5 ml lysis buffer and incubated for 1 h at 37 °C with gentle shaking. RNAse A (0.1 mg/ml) was then added and samples incubated overnight at 37 °C with gentle shaking.
Genomic DNA was isolated from all lysed tissues using a phenol/chloroform extraction procedure described previously (Douglas et al., 1994, Vijg and Douglas, 1996). Isolated DNA was dissolved in 100 μl TE buffer (10 mM Tris pH 7.6, 1 mM EDTA) and stored at 4 °C until use.
2.3. Mutant frequency analysis
The PGal (phenyl-β-D-galactoside) positive selection assay was used for the determination of lacZ mutant frequency in DNA samples from bone marrow, liver, lung, glandular stomach, and small intestine, as previously described (Gossen et al., 1992, Lambert et al., 2005, Vijg and Douglas, 1996). Mutant frequency was calculated as the ratio of mutant plaque forming units (pfu) to total pfu.
2.4. PAH-DNA adduct analysis
The nuclease P1 enrichment version of the 32P-postlabelling assay was used to determine DNA adduct frequency in DNA samples from bone marrow, liver, lung, glandular stomach, and small intestine. The procedure was performed as described in Phillips and Arlt (2014) and Arlt et al. (2008), with modifications as described in Malik et al. (2013).
2.5. Micronucleus assay
MicroFlow® kits (Litron Laboratories, Rochester, NY) were used for enumeration of micronucleated reticulocytes (RET) and normochromatic erythrocytes (NCE). Briefly, approximately 60 μl of peripheral blood was immediately combined with 350 μl of anticoagulant. Blood samples were then fixed by transferring to ice-cold methanol and stored at − 80 °C. After 3–5 days, fixed blood samples were then centrifuged, rinsed, and transferred to a long-term storage solution. Coded specimens were shipped to Litron Laboratories (Rochester, NY) for analysis. Micronuclei were scored in NCEs (i.e., MN-NCEs) and RETs (i.e., MN-RETs) by flow cytometry via a 3-colour labelling method described in (Dertinger et al., 2004, Torous et al., 2001).
2.6. Data analysis
The lacZ and MN dose–response data were analysed in SAS v.9.1 (SAS Institute, Cary, NC) using Poisson regression. The data were fit to the model log(E(Yi)) = log ti + βxi, where E(Yi) is the expected value for the ith observation, β is the vector of regressions coefficients, xi is a vector of covariates for the ith observation, and ti is the offset variable used to account for differences in observation count period (e.g., total pfu). The offset (e.g., natural log of pfu) was given a constant coefficient of 1.0 for each observation, and log-linear relationships between mutant count and test article concentration were specified by a natural log link function.
Genotoxic potencies were calculated for each endpoint in each tissue as the slope of the linear portion of the dose–response functions. BaP MN (in blood), as well as DNA adducts and lacZ data in bone marrow, liver, small intestine and glandular stomach were previously published in Lemieux et al. (2011). Lung data were previously published in Labib et al. (2012). DBahA lacZ mutant frequency data for liver only were previously published in Malik et al. (2013), and lung lacZ and DNA adduct data for BaA, DBahA, BbF, CHRY, BkF, INDENO, and BghiP were recently published in Labib et al. (2015). These data were re-analysed in the manner stated above to permit direct comparison to the results generated specifically for this study.
3. Results
3.1. DNA adducts
For simplicity, the DNA adduct data were only qualitatively examined (i.e., positive or negative) to investigate target tissue exposure to reactive metabolites (Table 2). The adduct data will be analysed and discussed in more detail (i.e., quantitative analyses) in a separate manuscript. Six of the 9 PAHs (i.e., BaP, BaA, DBahA, BbF, BkF, and DBalP) induced significant increases in DNA adduct frequencies in all 5 tissues examined (i.e., glandular stomach, small intestine, liver, lung, bone marrow). CHRY elicited a positive response in all tissues except bone marrow. INDENO only elicited a positive response in liver and lung. BghiP only elicited a positive response in lung. Interestingly, lung was the only tissue to show a significant response for all 9 PAHs.
Table 2.
PAH-DNA adduct result across all 5 tissues for each PAH examined.
| Compound | Glandular stomach | Small intestine | Liver | Lung | Bone marrow |
|---|---|---|---|---|---|
| BaP | + | + | + | + | + |
| BaA | + | + | + | + | + |
| DBahA | + | + | + | + | + |
| BbF | + | + | + | + | + |
| CHRY | + | + | + | + | − |
| BkF | + | + | + | + | + |
| INDENO | − | − | + | + | − |
| BghiP | − | − | − | + | − |
| DBalP | + | + | + | + | + |
3.2. lacZ mutagenicity
The Muta™Mouse TGR assay was able to correctly identify the selected PAHs as in vivo mutagens, and all 9 PAHs elicited positive responses in at least one tissue (Table 3). BaP, BbF, BkF, INDENO, and DBalP elicited significant increases in lacZ mutant frequency in all 5 tissues examined (Table 3; Fig. 1A, D, F, G, I). BaA and DBahA elicited a positive response for 4 out of the 5 tissues, with negative responses for lung and bone marrow, respectively (Table 3; Fig. 1B, C). CHRY and BghiP elicited positive responses in only 2 of the 5 tissues examined, both were negative in glandular stomach, liver, and bone marrow (Table 3; Fig. 1E, H). For all PAHs examined, with the exception of DBalP, the fold-increase in mutant frequency (i.e., over control) for site of contact or related tissues (i.e., small intestine, glandular stomach, liver) was higher than for bone marrow (Fig. 1). The largest fold-increase in mutant frequency for BaP, DBahA, BbF, CHRY, BkF, and INDENO was observed for small intestine (Fig. 1A, C, D, E, F, G). For BaA, only glandular stomach yielded a larger fold-change than small intestine (Fig. 1B), and for BghiP, only lung yielded a larger fold-change than small intestine (Fig. 1H). For DBalP, the largest fold-change over control was obtained for bone marrow (Fig. 1I). For BbF, BkF, and INDENO (Fig. 1D, F, G), bone marrow results showed the smallest fold-change over control, and for DBahA, CHRY, and BghiP, the bone marrow responses were negative (i.e., no significant increase) (Fig. 1C, E, H).
Table 3.
Genotoxic response for lacZ mutagenicity results in glandular stomach (GS), small intestine (SI), liver, (Lv), lung (Lg), and bone marrow (BM), and micronucleus (MN) results in reticulocytes (RETs) and normochromatic erythrocytes (NCEs). Sensitivity for each tissue is also presented.
| Compound |
lacZ |
MN |
|||||
|---|---|---|---|---|---|---|---|
| GS | SI | Lv | Lg | BM | RETs | NCEs | |
| BaP | +++ | +++ | +++ | +++ | +++ | + | ++ |
| BaA | + | + | WP | − | + | − | − |
| DBahA | + | +++ | + | +++ | − | + | + |
| BbF | +++ | +++ | +++ | ++ | ++ | + | + |
| CHRY | − | + | − | WP | − | − | − |
| BkF | + | ++ | + | + | + | + | + |
| INDENO | + | +++ | + | + | + | − | − |
| BghiP | − | + | − | + | − | − | − |
| DBalP | ++ | + | ++ | + | +++ | ++ | + |
| Total Pos | 7 | 9 | 7 | 8 | 6 | 5 | 5 |
| Total Neg | 2 | 0 | 2 | 1 | 3 | 4 | 4 |
| Sensitivity | 78% | 100% | 78% | 89% | 67% | 56% | 56% |
+: Dose–response, 1.5–5-fold above background; ++: Dose–response, 5–10-fold above background; +++: Dose–response, > 10-fold above background; WP (weak positive): dose–response, but below 1.5-fold above background; −: Negative.
Fig. 1.

Muta™Mouse lacZ mutagenicity results for 9 PAHs in bone marrow (BM), liver (Lv), glandular stomach (GS), small intestine (SI), and lung (Lg). Plots show results for (A) benzo(a)pyrene, (B) benz(a)anthracene, (C) dibenz(a,h)anthracene, (D) benzo(b)fluoranthene, (E) chrysene, (F) benzo(k)fluoranthene, (G) indeno(1,2,3-c,d)pyrene, (H) benzo(g,h,i)perylene, and (I) dibenzo(a,l)pyrene. Mutant frequencies ± standard error are displayed for each tissue and dose. Statistical results for the overall dose–response relationship are presented for each tissue. Results of the custom contrasts for each dose vs. control are marked with an *, indicating significance at p < 0.05. Ŧ: dose–response data were truncated prior to X2 analysis. NS indicates not significant.
3.3. Micronucleus (MN) frequency
BaP, DBahA, BbF, BkF, and DBalP elicited statistically significant positive results in the MN assay for both RETs and NCEs (Table 3, Fig. 2A–E). BaA, CHRY, INDENO, and BghiP did not elicit significant positive responses for the MN assay (Table 3). Larger fold-changes over control were consistently seen in RETs, versus NCEs, with the sole exception being BaP (Fig. 2A). This highlights the importance of scoring micronuclei in the immature red blood cell population (i.e., RETs), which captures the effects of recent damage.
Fig. 2.

Micronucleus (MN) frequency results for the 5 PAHs that elicited positive responses in this assay in reticulocytes (RETs) and normochromatic erythrocytes (NCEs). Plots show results for (A) benzo(a)pyrene, (B) dibenz(a,h)anthracene, (C) benzo(b)fluoranthene, (D) benzo(k)fluoranthene, and (E) dibenzo(a,l)pyrene. Percent micronucleated blood cells (i.e., % MN) ± standard error are displayed for each tissue and dose. Statistical results for the overall dose–response relationship are presented for both tissues. Results of the custom contrasts for each dose vs. control are marked with an *, indicating significance at p < 0.05.
CHRY, BaA, and INDENO are all IARC Group 2B carcinogens for which experimentally-induced genetic damage is well documented; however, the results obtained herein indicate an inability of these compounds to induce MN. This is noteworthy and concerning since the results support the contention that the MN assay in peripheral blood, which is routinely employed for regulatory screening, has a limited ability to detect PAH-induced genetic damage.
3.4. Potency comparisons
The potency (i.e., slope of the linear portion of the dose–response function) of each compound was ranked by tissue for each endpoint (i.e., 1 = most potent, 5 = least potent) (Table 4). For the lacZ mutation endpoint, the highest potency (i.e., rank = 1) was observed in small intestine for 6 out of 9 PAHs (i.e., BaP, DBahA, BbF, CHRY, BkF, INDENO). Potencies for small intestine were second highest (i.e., rank = 2) for the remaining 3 compounds (i.e., BaA, BghiP, DBalP). Conversely, 3 compounds failed to elicit a positive response in bone marrow (i.e., DBahA, CHRY and BghiP), and bone marrow responses were least potent (i.e., rank = 5) for an additional 3 compounds (i.e., BbF, BkF, INDENO). Interestingly, DBalP elicited the most potent response (i.e., rank = 1) in bone marrow. Effects on glandular stomach and lung showed intermediate potency for all compounds, with potency ranks ranging from 1 to 5, whereas responses in the liver were the least potent after bone marrow, with potency ranks ranging from 2 to 4. A mean relative potency score was calculated for each tissue, across all compounds, in order to provide a general potency ranking for PAHs in the tissues examined. The most potent response was observed in small intestine (1.3), followed by glandular stomach (2.9), lung (3.0), liver (3.3), and finally bone marrow (3.7).
Table 4.
Relative ranking of lacZ mutagenic potency for each compound in glandular stomach (GS), small intestine (SI), liver, (Lv), lung (Lg), and bone marrow (BM), as well as micronucleus (MN) induction response in reticulocytes (RETs) and normochromatic erythrocytes (NCEs). The last row shows mean rank across all compounds for a given tissue/endpoint.
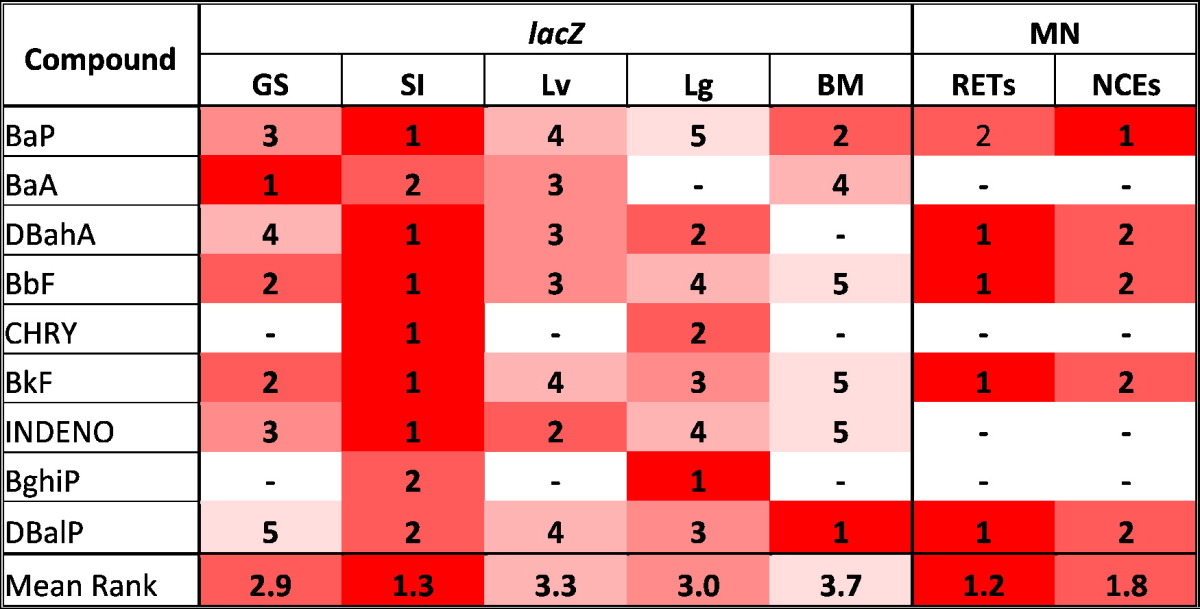
Potencies are ranked 1–5. 1: highest potency for each compound per assay. 5: lowest potency for each compound per assay. –: negative response.
For the MN endpoint, the most potent responses were observed in RETs for all compounds that yielded a significant positive response (i.e., DBahA, BbF, BkF, DBalP), with the sole exception of BaP (Fig. 2). BaP yielded a more potent response in NCEs (Fig. 2), resulting in mean potencies of 1.2 for RETs, and 1.8 for NCEs.
3.5. Cross-tissue comparisons
For the Muta™Mouse lacZ mutation endpoint BaP elicited the largest fold-change increase over control across all 5 tissues, which is not surprising since BaP is known to be a potent mutagen and carcinogen and the only PAH declared by IARC as a Group 1 human carcinogen. Unlike most other PAHs, BaP induced a potent lacZ response in bone marrow. DBalP also elicited a positive lacZ mutagenicity response in all tissues. Again, this is not surprising since DBalP is an extremely potent in vivo mutagen. It is noteworthy that, in contrast to the results for the other tested PAHs, the most potent response for this compound was observed in bone marrow. This suggests that there is substantial systemic distribution of DBalP, which is then metabolically converted in bone marrow to a reactive metabolite (e.g., DBalP-11,12-diol-13,14-epoxide). In comparison with BaP, DBalP elicited a smaller fold-change increase for most tissues; however, due to the extreme toxicity of DBalP (i.e., as observed in preliminary range-finding experiments), it was necessary to select doses that are far lower than those used for BaP (i.e., highest responses for DBalP observed at doses almost 40-fold lower than BaP).
The fact that DBahA elicited a positive response in all tissues except bone marrow is concerning since regulatory assessments often only examine haematopoietic tissue. It seems reasonable to assert that reactive DBahA metabolites do not reach the bone marrow, and therefore are unable to induce mutations in this tissue. However, DBahA did elicit positive responses for the MN endpoint in RETs and NCEs, therefore systemic exposure of reactive metabolites is likely. Indeed, systemic exposure of the reactive metabolite was confirmed by quantification of DBahA-induced bulky adducts in bone marrow at all three doses. The fact that stable, bulky adducts do not appear to have been converted to stable mutations in a rapidly proliferating tissue such as bone marrow may be an indication that there is sufficient capacity for repair of DNA lesions or error-free translesion DNA synthesis. The positive MN response for DBahA may have been induced by secondary metabolites, such as reactive oxygen species, that did not contribute to the formation of mutations in the bone marrow.
CHRY is a relatively weak carcinogen, causing lung and liver cancer in mice following intraperitoneal (i.p.) administration, and lung cancer in rats following intrapulmonary administration (IARC, 2010). IARC recently declared CHRY a Group 2B carcinogen (i.e., possibly carcinogenic to humans), which constitutes an upgrade from the earlier Group 3 declaration. In our study, CHRY only elicited a clear positive response in small intestine and a weak positive response in lung (i.e., significant dose response, but response at high dose below 1.5-fold above control). The lacZ mutation assay results suggest that lung was exposed to reactive CHRY metabolites; indeed CHRY-DNA adducts were detected in lung. In comparison, a lack of lacZ and MN responses in bone marrow and peripheral blood, respectively, suggest that haematopoietic tissue was not exposed. This was confirmed by a lack of CHRY-induced DNA adducts in bone marrow.
BkF and BbF, which are both IARC Group 2B carcinogens, elicited positive responses across all tissues, with the greatest lacZ mutant frequency induction occurring in the small intestine. Both BbF and BkF induced a positive response in the MN assay.
INDENO elicited positive lacZ mutagenicity responses across all tissues (i.e., including bone marrow), but failed to induce positive MN responses in RETs or NCEs, nor did the results reveal measureable adducts in bone marrow. Given the lack of bulky adducts in bone marrow, it is unlikely that INDENO is being metabolized via the diol-epoxide pathway in this tissue. Alternatively, the positive bone marrow response may result from oxidative DNA damage produced via the o-quinone activation pathway.
BaA, which is also classified as an IARC Group 2B carcinogen, induced a significant positive response in the lacZ mutagenicity assay in all tissues except lung. The compound elicited a weak positive response in the liver.
BghiP, which is classified by IARC as Group 3 (i.e., not classifiable due to limited or inadequate data in humans and experimental animals), elicited a positive response in small intestine and lung. Similar to the results obtained for DBahA, the lack of positive response in bone marrow is problematic from a regulatory point of view since assessments are most commonly based on results for haematopoietic tissue. BghiP also failed to elicit a positive MN response in both RETs and NCEs; therefore, it is possible that there is no systemic distribution of activated metabolites. Indeed, DNA adducts were not detected in the bone marrow of BghiP-exposed animals. However, since a positive lacZ response was observed in the lung, activated metabolites are either reaching the lung via pulmonary circulation, or BghiP is being metabolically activated in the lung itself.
3.6. Assay sensitivity
Table 3 summarizes the tissue-specific responses of the PAHs examined herein, and the response patterns therein are briefly outlined below. Importantly, the Muta™Mouse lacZ mutation assay was more sensitive for all tested tissues (i.e., correct identification of a genotoxic carcinogen) in comparison with the MN assay in either RETs or NCEs (Table 3). All of the PAHs investigated in this study elicited significant positive responses in the Muta™Mouse lacZ mutation assay in at least one tissue.
For the lacZ mutagenicity assay, small intestine was the most sensitive tissue, with a positive response for all 9 PAHs tested (100%) (Table 3). Small intestine is a site-of-contact tissue for an oral exposure, which may contribute to its high sensitivity for this assay. Lung, a remote tissue for which responses were consistently lower in comparison to site of contact, was the second most sensitive tissue (i.e., 89%). Lung is the only common tumour site for all 9 PAHs, and therefore is a toxicologically-relevant tissue for an oral exposure. The only compound that did not elicit a positive response in lung was BaA. Seven of the 9 PAHs examined elicited positive responses in both glandular stomach and liver (78%). CHRY and BghiP were both negative in these tissues, and also negative in bone marrow. In the context of this study the glandular stomach constitutes the site of first contact, with compounds interacting with stomach epithelium prior to contact with the small intestine or liver. It is reasonable to assert that the enzymatic activity and/or residence time in the stomach may not be sufficient to achieve adequate conversion of the compounds into DNA-reactive metabolites that elicit bulky adducts and transgene mutations. Bone marrow was the least sensitive tissue for induction of lacZ mutations, with only 6 of 9 compounds eliciting significant positive responses; DBahA, CHRY, and BghiP were all negative. As already noted, lack of responses for known mutagenic carcinogens are potentially a result of ineffective target tissue exposure to the reactive metabolite.
The sensitivity of the MN assay was equal for RETs and NCEs; with both endpoints returning a positive response for 5 out of the 9 PAHs examined (56%).
From a regulatory evaluation point of view it is interesting to consider the results as though the study only examined haematopoietic tissues (i.e., bone marrow, blood), which would be the case if the MN assay alone was employed. The results would have suggested that CHRY and BghiP are not in vivo genotoxicants, despite the fact that both of these compounds induced significant positive responses in other Muta™Mouse tissues. If the study had examined the Muta™Mouse TGR endpoint in bone marrow only, we would have also missed the positive response for DBahA, an IARC Group 2A carcinogen that elicited significant positive responses in all other tissues examined and response induction levels more than 75- and 12-fold above control for small intestine and lung, respectively. Finally, if we had only examined induced increases in MN frequency in peripheral blood (i.e., RETs and NCEs), we would have missed significant positive responses for BaA, INDENO, CHRY, and BghiP. BaA and INDENO elicited positive lacZ mutagenicity responses in several tissues, including bone marrow. The fact that these compounds are unable to elicit reliable positive responses in haematopoietic tissues raises a concern regarding their ability to be detected using standard regulatory assays for genetic toxicity, and highlights the importance of examining site-of-contact tissues and known sites of tumour formation, either alone or in combination with haematopoietic tissues.
4. Discussion
This study employed the Muta™Mouse TGR system to examine the in vivo genetic toxicity of 9 PAHs. The compounds are all mutagenic carcinogens in animal models, and, with the exception of BghiP, are IARC Group 1, 2A, or 2B human carcinogens. It is well-known that the primary mode of action underlying PAH-induced carcinogenicity is genotoxicity, and key events leading to the adverse outcome (i.e., tumours) include the conversion of stable adducts to point mutations or small insertions/deletions (IARC, 2010). Indeed, all 9 PAHs elicited significant positive responses in at least one tissue in the Muta™Mouse lacZ mutation assay, with 5 out of the 9 PAHs eliciting a positive response in all 5 tissues, thereby demonstrating not only their mutagenic potential, but also the ability of this assay to correctly identify genotoxic carcinogens.
Several PAHs have also been shown to induce micronuclei as a result of clastogenicity (Abramsson Zetterberg et al., 2013, Crofton Sleigh et al., 1993, Glatt et al., 1990, He and Baker, 1991, Nishikawa et al., 2005, Schober et al., 2006, Warshawsky et al., 1995, Whong et al., 1994). Interestingly, only 5 of the 9 PAHs examined here elicited significant positive responses in the MN assay, indicating that a complementary assay to detect in vivo mutagenic activity is necessary to prevent “false negatives”.
The MN results in RETs consistently elicited larger fold-changes over control, as well as greater potency values in comparison with the results for NCEs. RETs only make up ~ 10% of circulating red blood cells and their life span is only 1–2 days prior to their maturation into NCEs. Thus, by examining RETs we are able to see evidence of the most recent damage (Clark and Korst, 1969). As they mature, the mutant RETs begin to accumulate as mutant NCEs, which have a life span of 38–46 days in mice (Horky et al., 1978). Therefore, the lower response in NCEs is likely to be due to dilution; in other words, whereas all RETs were recently exposed, only a subset of the sampled NCEs were exposed during the 28-day treatment period. Since affected cells accumulate, a longer sampling time (i.e., > 2 days post-exposure) would have increased the magnitude of the MN frequency response for NCEs (i.e., relative to RETs).
In this study BaP, DBalP, BbF, and BkF were positive across all tissues for both endpoints. A number of positive results for BaP-exposed TGRs have been previously reported for several tissues including small intestine, lung, liver, bone marrow, and spleen in Big Blue® mouse and rat, Muta™Mouse, gpt delta mouse, Dlb-1 congenic mice, and the lacZ plasmid mouse (Boerrigter, 1999, Brooks et al., 1999, Delker et al., 2008, Horibata et al., 2013, Leavitt, 2008, Monroe et al., 1998, Shane et al., 1997, Skopek et al., 1996). BaP has previously been shown to induce positive MN results in several tissues/cell types including skin, peripheral blood erythrocytes, and bone marrow (Abramsson Zetterberg et al., 2013, Lemieux et al., 2011, Nishikawa et al., 2005). DBalP has previously been tested in TGR assays, and a significant increase in lacI mutant frequency was reported in Big Blue® mouse lung following an i.p. administration (Leavitt, 2008), as well as a significant increase in cII mutant frequency in Big Blue® mouse tongue, following a topical application in the oral cavity (Chen et al., 2013). Our group recently published positive DNA adduct and lacZ results in spleen and bone marrow, as well as positive MN results in peripheral blood, following a 3-day oral exposure (Chepelev et al., 2015). The current study is the first study that examined DBalP MN induction and mutagenicity following a sub-chronic oral exposure, and the first report of its mutagenicity in glandular stomach, small intestine, and liver. Interestingly, DBalP has been demonstrated to be much more carcinogenic than BaP in animal models, and perhaps the most carcinogenic of all PAHs tested to date (Cavalieri et al., 1991, Cavalieri et al., 2005, Chen et al., 2013, Higginbotham et al., 1993). Surprisingly, there are no previous reports of TGR or MN results for BbF and BkF.
In our study, DBahA failed to elicit a positive TGR response for bone marrow, yet it was positive in the MN assay. The only previous report of TGR assay results for DBahA, which was published by our group and incorporated into the analyses presented herein, showed a positive response in liver (Malik et al., 2013). Previous publications examining DBahA genetic toxicity reported positive results for micronucleus induction in skin, bone marrow, spleen, and lung (Nishikawa et al., 2005, Whong et al., 1994, Zhong et al., 1995); however the current study appears to be the first to demonstrate a positive MN response in blood.
The results obtained indicate that INDENO, BaA, and BghiP were negative in the MN assay. INDENO elicited a positive response in the TGR assay for all tissues examined, whereas BaA was positive in the TGR assay in all tissues except lung, and BghiP only elicited a positive TGR response in small intestine and lung. There are no previous reports of TGR results for any of these three compounds. In addition, to our knowledge there are no reports in the literature regarding the ability of INDENO or BghiP to induce MN in vitro or in vivo; however, several studies have documented that BaA induces MN in skin, bone marrow, spleen, and lung (Nishikawa et al., 2005, Whong et al., 1994, Zhong et al., 1995).
CHRY elicited positive TGR results for small intestine and lung only, and was negative in the MN assay. Only one study has previously reported positive TGR assay results for CHRY. Following an i.p. exposure of Muta™Mouse, significant elevations in lacZ mutant frequencies were observed in liver, spleen, lung, kidney, bone marrow, and colon (Yamada et al., 2005). Our study did not observe positive results in liver or bone marrow; however, the Yamada et al. (2005) study used an i.p. administration, which would contribute to elevated, direct exposures of organs in the peritoneum (e.g., liver). To our knowledge, the literature only contains a single report of a CHRY-induced increase in MN; He and Baker (1991) documented a significant increase in hairless mouse skin following topical administration (He and Baker, 1991). However, a similar study by Nishikawa (2005) failed to detect an increase in MN in the same species and tissue following topical treatment (Nishikawa et al., 2005).
Small intestine was not only the most sensitive among the tissues examined, but also displayed the largest induction of lacZ mutant frequency (i.e., the most potent response). It has previously been demonstrated that inducible levels of Cyp1a1 gene expression in the small intestine are relatively high, which is not the case for most other tissues (Choudhary et al., 2003, Renaud et al., 2011). In the epithelium of the GI tract, maximum induction levels of CYP1A1 have been shown to be 3–10 times greater than CYP1B1, which in turn, have been shown to be 3–10 times greater than CYP1A2 (Uno et al., 2008). Studies with Cyp1a1/1a2/1b1 single, double, and triple knockout mice have highlighted and clarified the roles of these enzymes in metabolizing BaP and distributing BaP-metabolites following oral exposures (reviewed in Nebert, et al. 2013). Knocking out Cyp1a1 expression results in increased expression of Cyp1b1, concomitant bioactivation of BaP, and adenocarcinoma formation in the proximal small intestine. In contrast, BaP treated Cyp1b1 knockout were healthy (Shi et al., 2010b). The relatively high levels of inducible CYP1A1 appear to be especially important in the detoxification of BaP in the GI tract, whereas CYP1B1 is implicated in the conversion of BaP to DNA-damaging metabolites (Nebert, et al. 2013). Following an oral exposure, compounds such as PAHs are absorbed from the small intestine and transported to the liver, where they undergo oxidative metabolism. This metabolism contributes to clearance of the parent compound, but also in the formation of DNA adducts and mutations in the liver itself. Additionally, as PAH metabolites have been previously demonstrated to undergo enterohepatic circulation, the small intestine would likely be re-exposed to reactive metabolites, and this process would contribute to increased levels of DNA damage that can, in turn, contribute to an increased frequency of mutations (Miller and Ramos, 2001, Ramesh et al., 2004). The combination of in situ metabolism in the small intestine, coupled with re-exposure to reactive metabolites produced in the liver and the high mitotic index of the epithelial lining of the small intestine, likely led to the high levels of transgene mutations.
Although the liver is the major site of PAH metabolism, both CHRY and BghiP failed to induce significant increases in lacZ mutations in this tissue. Liver cells generally have a much lower mitotic index than parenchymal cells in the other examined tissues (Edwards and Klein, 1961, White et al., 2015); therefore, for less potent chemicals, a 28-day exposure regime with a 3-day sampling time may not be sufficient to allow for conversion of DNA damage to lacZ mutations. Both CHRY and BghiP are relatively weak mutagens/carcinogens, with BghiP being classified as IARC Group 3, and CHRY being recently upgraded from Group 3 to 2B. If slowly proliferating tissues such as liver are deemed to be of particular importance, the OECD protocol for TGR assays indicates that a longer sampling time (e.g., 28-days post-exposure) may be more appropriate (OECD, 2011).
The lacZ mutagenic potency values for glandular stomach were penultimate with a sensitivity of 78%. For an oral exposure the stomach is the first site-of-contact tissue, and although the metabolic capacity of stomach epithelium is low in comparison with liver, the magnitude of the response highlights the importance of examining tissues deemed relevant to the exposure route. Only CHRY and BghiP, two of the weakest PAHs examined, failed to elicit a positive response in this tissue.
The fact that lacZ mutagenic activities were observed across all 5 tissues, including remote tissues such as bone marrow and lung, indicates that several PAHs are capable of being metabolized and converted into DNA-reactive substances at remote tissues in situ, or alternatively, that the active metabolites are being systemically distributed. Lung was the second most sensitive tissue for the TGR assay, returning a positive result for all compounds except BaA. Both Cyp1a1 and Cyp1b1 are highly expressed in the mouse lung (Arlt et al., 2015, Choudhary et al., 2003, Renaud et al., 2011); thus, lung has the ability to carry out in situ conversion of PAHs to reactive diol-epoxides that would form bulky adducts. This also appears to be the case for DBahA, CHRY, and BghiP, which failed to elicit positive lacZ results in bone marrow, but elicited positive results for lung. The negative BaA result for lung is troubling since BaA-DNA adducts were detected in this tissue; however, due to the relatively low mitotic index of most lung parenchymal cells, DNA replication may not be sufficient to fix a detectable level of mutations during the exposure and sampling period employed here (White et al., provisionally accepted).
In this study, bone marrow was the least sensitive tissue for the TGR assay, and the lowest potency lacZ responses were also observed in bone marrow, with DBahA, CHRY, and BghiP failing to elicit positive responses. Additionally, CHRY, BaA, INDENO, and BghiP were unable to induce MN in peripheral blood. The negative lacZ mutation and MN responses in haematopoietic tissue from CHRY- and BghiP-exposed animals may be the result of insufficient exposure of the target tissue to reactive metabolites. This is likely due to the lack of sufficient distribution of the parent compound to bone marrow and/or peripheral blood via systemic circulation. As mentioned, following an oral exposure, induction of GI tract Cyp1a1 appears to be especially important for the detoxification of BaP, and this likely holds true for several PAHs. Thus, following an oral exposure to PAHs that are effective AhR agonists, upregulation of Cyp1a1 expression in the GI tract/liver will contribute to enhanced clearance, leaving little parent compound for systemic distribution to bone marrow where reactive metabolites can be generated in situ via catalysis by isozymes such as CYP1B1. This supposition is supported by the aforementioned knock-out studies that showed increased damage at peripheral tissues if Cyp1a1 expression is knocked out either globally or in the GI tract itself; moreover, the effect is reversed by also knocking out Cyp1b1 (Shi et al., 2010a, Uno et al., 2006). However, it should be emphasized that the PAHs examined are not equipotent AhR agonists. In this study, BaP and DBalP induced the most potent lacZ and MN responses in bone marrow/blood, and interestingly, BaP is a relatively poor AhR agonist in comparison to most of the other PAHs examined. Furthermore, it is not clear whether DBalP is an AhR ligand at all (Machala et al., 2001, Ziccardi et al., 2002), and if it is not, one would not expect induction of Cyp1a1 expression in the GI tract, leaving more parent compound available to be metabolized in bone marrow by the constitutively expressed Cyp1b1. This deduction is supported by our previous DBalP study where we observed significant bone marrow cytotoxicity that resulted in a substantial decrease in the percentage of circulating RETs following a 3-day exposure (Chepelev et al., 2015). In contrast to the aforementioned toxicokinetic processes that likely modify the in vivo responses to BaP and DBalP, effective AhR agonism, which has been observed for BkF, DBahA, and INDENO (Ziccardi et al., 2002), and upregulation of Cyp1a1 expression in the GI tract, would likely contribute to weak or negative bone marrow responses. Indeed, this is what was observed.
The results obtained failed to show a detectable increase in DNA adducts in bone marrow from CHRY-, BghiP-, and INDENO-treated animals, which supports the lack of exposure of haematopoietic tissue to reactive metabolites. However, a significant increase in adduct formation in the bone marrow of BaA-treated animals indicates that circulation of this compound did occur, and that the bone marrow was exposed to reactive, DNA-damaging metabolites. This is confirmed by the positive lacZ mutagenicity response in bone marrow. Therefore, the aforementioned negative MN results may result from the inability of the mutagenic metabolites to produce chromosome breaks and MN.
MN are produced during nuclear division when whole chromosome or acentric chromosome or chromatid fragments lag behind at anaphase. This can result from mis-repair (or lack of repair) of double-strand breaks, or simultaneous excision repair of damaged or incorrect bases on opposite strands (Fenech et al., 2011). PAHs do not primarily cause double strand breaks, but rather exert their mutagenic effect via formation of bulky adducts that are repaired via nucleotide excision repair. Although it has been well documented that several PAHs (i.e., BaP, DBahA) are capable of inducing MN (Zhong et al., 1995), it is possible that not all PAHs are capable of causing double strand breaks that contribute to MN formation.
DBahA was one of the 3 compounds that failed to elicit a positive lacZ mutagenicity response in the bone marrow. As already noted, lack of responses for known mutagenic carcinogens are potentially indicative of ineffective target tissue exposure. However, for DBahA, a positive response in bone marrow was obtained for the adduct frequency and MN endpoints, indicating that the haematopoietic tissue was exposed to reactive metabolites. However, the tissue levels of reactive metabolites were likely not sufficient to overwhelm damage repair processes that would inhibit the establishment of mutations. Alternatively, it is possible that the positive MN response for DBahA resulted from a secondary metabolite (e.g., reactive oxygen) that did not contribute to the formation of mutations in the bone marrow. It has previously been shown that an alternate metabolic pathway via dihydrodiol dehydrogenase leads to the auto-oxidation of PAH metabolites to o-quinones, which can undergo redox cycling that generates DNA-damaging ROS and depurinating adducts (IARC, 2014, Miller and Ramos, 2001, Penning et al., 1996, Smithgall et al., 1988).
The haematopoietic tissue results for INDENO are interesting since adducts were not detected in bone marrow, and the MN assay result was negative; however, we did observe a significant increase in lacZ mutations. Given the lack of bulky adducts, it is unlikely that the reactive metabolites are reaching the bone marrow via systemic circulation. A likely alternative cause of the observed lacZ mutations, which would not contribute to the formation of bulky INDENO-DNA adducts, is reactive oxygen species (ROS) generated via the aforementioned o-quinone pathway. Some PAHs, especially those with higher molecular weights (e.g., INDENO), have been found to have greater redox activity that can readily lead to ROS formation and oxidative DNA damage, among other types of cellular injury (Jeng et al., 2011). Oxidative DNA damage cannot be detected using the 32P-postlabelling methodology employed in this study.
Several published studies have previously examined the in vivo genetic toxicity of PAHs; however, the vast majority of these studies assessed responses following i.p. administrations (i.e., no first-pass metabolism), which is generally not considered useful for regulatory decisions that must consider the most likely route of receptor exposure. The most common route of human PAH exposure is oral, followed by inhalation, and dermal. This study is the first to employ an identical oral exposure regime to simultaneously evaluate induction of transgene (lacZ) mutations in various tissues and MN formation in peripheral blood following TGR exposures to 9 PAHs. This type of study is essential to critically evaluate the utility of TGR tools for regulatory genetic toxicity assessment (i.e., assay sensitivity), to compare and contrast TGR results across various tissues (OECD #488), and perhaps more importantly, to benchmark TGR responses against the MN endpoint in peripheral blood (OECD #474).
As noted, the current study concurrently examined induced lacZ mutations in 5 tissues, and the frequency of MN in RETs and NCEs, following sub-chronic oral administration of 9 PAHs. The evaluation of both of these endpoints was conducted in accordance with the protocols for dosing and sample collection recommended in the relevant OECD guidelines (i.e., OECD #474 & 488), and this work demonstrates that the two complementary endpoints can be integrated into a single sub-chronic study, thus providing concurrent assessment of mutation and chromosome damage. Although the study was restricted to a single compound class (i.e., PAHs), the results nonetheless underscore the utility of simultaneous genetic toxicity assessment in multiple tissues across multiple endpoints (i.e., mutation and cytogenetic damage). Additional research is required to determine whether the observed sensitivity of the TGR mutagenicity endpoint is generally applicable, and additionally, whether specificity is equally suitable. Interestingly, detailed reviews of the TGR endpoints have previously demonstrated the ability of the Muta™Mouse lacZ mutagenicity assay to return positive results for a wide range of genotoxic compounds, including carcinogens that failed to elicit positive responses in the in vivo MN assay. For example, Oxazepam (7-Chloro-3-hydroxy-5-phenyl-1,3-dihydro-1,4-benzodiazepin-2-one), a carcinogen that failed to elicit MN in vitro and in vivo, was shown to elicit a positive response in a TGR mutagenicity assay (Lambert et al., 2009). Indeed, we are currently expanding this type of analysis and employing the Muta™Mouse system to examine the in vivo mutagenicity of carcinogens that failed to elicit positive clastogenicity responses in vivo (i.e., false negatives).
Overall, with respect to the tissues examined in this study, the Muta™Mouse lacZ mutagenicity assay was more sensitive (i.e., correct identification of mutagenic carcinogens) than the peripheral blood MN assay; moreover, for the Muta™Mouse endpoint, bone marrow was the least sensitive of the tissues examined. Small intestine, a site-of-contact tissue for oral exposure, was the most sensitive tissue, with the responses therein constituting the largest observed increases in mutant frequency for the PAHs examined. The results of this study call into question the reliability of only examining tissues that are remote from the site of contact (e.g., blood or bone marrow) when assessing the genotoxic hazard of substances for which the primary route of exposure is oral. Moreover, following up an in vitro positive result for mutation with an in vivo clastogenicity assay is not necessarily effective, and may even lead to the generation of in vivo false negatives. This is particularly important for carcinogens (e.g., PAHs) that must be metabolically converted into DNA-reactive metabolites, since the reactive metabolites may not be systemically distributed and/or may only be generated in situ in remote tissues (e.g., bone marrow). As noted, phenomena such as AhR agonism in site of contact and remote tissues can control the nature of the tissue exposure (e.g., type and level of reactive metabolites), and the effects manifested therein that are indicative of sensitivity.
5. Conclusion
It is evident from the results presented that a dynamic interplay between several phenomena govern tissue-specific induction of genetic toxicity endpoints including DNA damage (i.e., adduct frequency), mutagenicity (i.e., lacZ transgene mutations), and clastogenicity (i.e., MN). Specifically, genetic damage in a target tissue appears to be controlled by dynamic processes that regulate (i) absorption, tissue-specific metabolism and systemic distribution of both metabolites and parent compounds, (ii) tissue-specific damage response and DNA repair capacity, and (iii) tissue-specific cellular proliferation.
The results presented clearly demonstrate the utility of the Muta™Mouse TGR assay for reliable in vivo mutagenicity assessment of suspected carcinogens with a mutagenic mode of action; in particular, for substances that require metabolic conversion to DNA-reactive metabolites (i.e., PAHs). Furthermore, the low sensitivity of the peripheral blood MN and the lacZ mutagenicity responses in bone marrow (i.e., limited ability to identify mutagenic carcinogens) highlights the need to examine site-of-contact tissues in addition to haematopoietic tissues, particularly for weak mutagens. Simultaneous enumeration of induced lacZ mutations in several tissues and micronuclei in peripheral blood, comprises complementary components of a highly sensitive system for the detection of mutagenic carcinogens, and we are currently extending the type of analyses presented herein to encompass prioritized environmental matrices contaminated with complex mixtures of combustion-derived PAHs (e.g., contaminated soil, coal-tar amended consumer products).
Conflict of interest
Dr. Arlt reports grants from Health Canada, grants from Cancer Research UK, grants from Wellcome Trust during the conduct of the study. Ms. Long, Ms. Lemieux, and Dr. White have nothing to disclose.
Transparency document
Transparency document.
Acknowledgements
We are grateful to Sabina Halappanavar, Sarah Labib, John Gingerich, Lynda Soper, Caitlin Ritz, Manon Simard, Cristina Aroche, Julie Cox, Jaime Nead, Julie Todd, and Kevin Kittle for intellectual contributions and assistance with the animal handling and sample processing. We are grateful to Jennifer McAllister, Dr. Azam Tayabali, two anonymous reviewers, and a member of the journal editorial board for their careful review of this manuscript and helpful comments.
This project was funded by the Chemicals Management Plan and Health Canada intramural funding, as well as the Natural Sciences and Engineering Research Council of Canada. Volker M. Arlt is supported by Cancer Research UK (Grant C313/A14329), the Wellcome Trust (Grants 101126/Z/13/Z and 101126/B/13/Z) and is a member of the Wellcome Trust funded COMSIG (Causes of Mutational SIGnatures) consortium.
Footnotes
The Transparency document associated with this article can be found, in online version.
References
- Abramsson Zetterberg L., Carlsson R., Sand S. The use of immunomagnetic separation of erythrocytes in the in vivo flow cytometer-based micronucleus assay. Mutat. Res. 2013;752:8–13. doi: 10.1016/j.mrgentox.2012.12.010. [DOI] [PubMed] [Google Scholar]
- Ames B.N., Durston W.E., Yamasaki E., Lee F.D. Carcinogens are mutagens: a simple test system combining liver homogenates for activation and bacteria for detection. Proc. Natl. Acad. Sci. U. S. A. 1973;70:2281–2285. doi: 10.1073/pnas.70.8.2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlt V.M., Krais A.M., Godschalk R.W., Riffo-Vasquez Y., Mrizova I., Roufosse C.A., Corbin C., Shi Q., Frei E., Stiborova M., van Schooten F.J., Phillips D.H., Spina D. Pulmonary inflammation impacts on CYP1A1-mediated respiratory tract DNA damage induced by the carcinogenic air pollutant benzo[a]pyrene. Toxicol. Sci. 2015;146:213–225. doi: 10.1093/toxsci/kfv086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlt V.M., Stiborova M., Henderson C.J., Thiemann M., Frei E., Aimova D., Singh R., Gamboa da Costa G., Schmitz O.J., Farmer P.B., Wolf C.R., Phillips D.H. Metabolic activation of benzo[a]pyrene in vitro by hepatic cytochrome P450 contrasts with detoxification in vivo: experiments with hepatic cytochrome P450 reductase null mice. Carcinogenesis. 2008;29:656–665. doi: 10.1093/carcin/bgn002. [DOI] [PubMed] [Google Scholar]
- Boerrigter M.E.T.I. Treatment of lacZ plasmid-based transgenic mice with benzo[a]pyrene: measurement of DNA adduct levels, mutant frequencies, and mutant spectra. Environ. Mol. Mutagen. 1999;34:140–147. [PubMed] [Google Scholar]
- Brooks R.A., Gooderham N.J., Edwards R.J., Boobis A.R., Winton D.J. The mutagenicity of benzo[a]pyrene in mouse small intestine. Carcinogenesis. 1999;20:109–114. doi: 10.1093/carcin/20.1.109. [DOI] [PubMed] [Google Scholar]
- Bryce S.M., Bemis J.C., Dertinger S.D. In vivo mutation assay based on the endogenous Pig-a locus. Environ. Mol. Mutagen. 2008;49:256–264. doi: 10.1002/em.20379. [DOI] [PubMed] [Google Scholar]
- Cairns J. Mutation and cancer: the antecedents to our studies of adaptive mutation. Genetics. 1998;148:1433–1440. doi: 10.1093/genetics/148.4.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalieri E.L., Higginbotham S., RamaKrishna N.V., Devanesan P.D., Todorovic R., Rogan E.G., Salmasi S. Comparative dose–response tumorigenicity studies of dibenzo[a,l]pyrene versus 7,12-dimethylbenz[a]anthracene, benzo[a]pyrene and two dibenzo[a,l]pyrene dihydrodiols in mouse skin and rat mammary gland. Carcinogenesis. 1991;12:1939–1944. doi: 10.1093/carcin/12.10.1939. [DOI] [PubMed] [Google Scholar]
- Cavalieri E.L., Rogan E.G., Li K.M., Todorovic R., Ariese F., Jankowiak R., Grubor N., Small G.J. Identification and quantification of the depurinating DNA adducts formed in mouse skin treated with dibenzo[a,l]pyrene (DB[a,l]P) or its metabolites and in rat mammary gland treated with DB[a,l]P. Chem. Res. Toxicol. 2005;18:976–983. doi: 10.1021/tx049682k. [DOI] [PubMed] [Google Scholar]
- CEPA . Priority Substances List Assessment Report; 1994. Polycyclic aromatic hydrocarbons. [Google Scholar]
- Chen K., Guttenplan J.B., Zhang S.M., Aliaga C., Cooper T.K., Sun Y.W., DelTondo J., Kosinska W., Sharma A.K., Jiang K., Bruggeman R., Ahn K., Amin S., El-Bayoumy K. Mechanisms of oral carcinogenesis induced by dibenzo[a,l]pyrene: an environmental pollutant and a tobacco smoke constituent. Int. J. Cancer. 2013;133:1300–1309. doi: 10.1002/ijc.28152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chepelev N.L., Long A.S., Williams A., Kuo B., Gagne R., Kennedy D.A., Phillips D.H., Arlt V.M., White P.A., Yauk C.L. Transcriptional profiling of Dibenzo[def,p]chrysene-induced spleen atrophy provides mechanistic insights into its immunotoxicity in MutaMouse. Toxicol. Sci. 2015 doi: 10.1093/toxsci/kfv232. [DOI] [PubMed] [Google Scholar]
- Choudhary D., Jansson I., Schenkman J.B., Sarfarazi M., Stoilov I. Comparative expression profiling of 40 mouse cytochrome P450 genes in embryonic and adult tissues. Arch. Biochem. Biophys. 2003;414:91–100. doi: 10.1016/s0003-9861(03)00174-7. [DOI] [PubMed] [Google Scholar]
- Clark R.H., Korst D.R. Circadian periodicity of bone marrow mitotic activity and reticulocyte counts in rats and mice. Science. 1969;166:236–237. doi: 10.1126/science.166.3902.236. [DOI] [PubMed] [Google Scholar]
- Collins A.R. The comet assay for DNA damage and repair: principles, applications, and limitations. Mol. Biotechnol. 2004;26:249–261. doi: 10.1385/MB:26:3:249. [DOI] [PubMed] [Google Scholar]
- Crofton Sleigh C., Doherty A., Ellard S., Parry E.M., Venitt S. Micronucleus assays using cytochalasin-blocked MCL-5 cells, a proprietary human cell line expressing five human cytochromes P-450 and microsomal epoxide hydrolase. Mutagenesis. 1993;8:363–372. doi: 10.1093/mutage/8.4.363. [DOI] [PubMed] [Google Scholar]
- Delker D.A., Geter D.R., Kleinert K.M., Gollapudi B.B. Frequency and spectrum of lacI mutations in the liver of big blue mice following the administration of genotoxic carcinogens singly and in series. Int. J. Toxicol. 2008;27:35–42. doi: 10.1080/10915810701876620. [DOI] [PubMed] [Google Scholar]
- Dertinger S.D., Camphausen K., Macgregor J.T., Bishop M.E., Torous D.K., Avlasevich S., Cairns S., Tometsko C.R., Menard C., Muanza T., Chen Y., Miller R.K., Cederbrant K., Sandelin K., Ponten I., Bolcsfoldi G. Three-color labeling method for flow cytometric measurement of cytogenetic damage in rodent and human blood. Environ. Mol. Mutagen. 2004;44:427–435. doi: 10.1002/em.20075. [DOI] [PubMed] [Google Scholar]
- Douglas G.R., Gingerich J.D., Gossen J.A., Bartlett S.A. Sequence spectra of spontaneous lacZ gene mutations in transgenic mouse somatic and germline tissues. Mutagenesis. 1994;9:451–458. doi: 10.1093/mutage/9.5.451. [DOI] [PubMed] [Google Scholar]
- Edwards J.L., Klein R.E. Cell renewal in adult mouse tissues. Am. J. Pathol. 1961;38:437–453. [PMC free article] [PubMed] [Google Scholar]
- Fenech M., Kirsch-Volders M., Natarajan A.T., Surralles J., Crott J.W., Parry J., Norppa H., Eastmond D.A., Tucker J.D., Thomas P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis. 2011;26:125–132. doi: 10.1093/mutage/geq052. [DOI] [PubMed] [Google Scholar]
- Glatt H., Gemperlein I., Setiabudi F., Platt K.L., Oesch F. Expression of xenobiotic-metabolizing enzymes in propagatable cell cultures and induction of micronuclei by 13 compounds. Mutagenesis. 1990;5:241–249. doi: 10.1093/mutage/5.3.241. [DOI] [PubMed] [Google Scholar]
- Gossen J.A., Molijn A.C., Douglas G.R., Vijg J. Application of galactose-sensitive e.Coli strains as selective hosts for LacZ− plasmids. Nucleic Acids Res. 1992;20:3254. doi: 10.1093/nar/20.12.3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart S.N., Cui Y., Klaassen C.D., Zhong X.B. Three patterns of cytochrome P450 gene expression during liver maturation in mice. Drug Metab. Dispos. 2009;37:116–121. doi: 10.1124/dmd.108.023812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S., Baker R. Micronuclei in mouse skin cells following in vivo exposure to benzo[a]pyrene, 7,12-dimethylbenz[a]anthracene, chrysene, pyrene and urethane. Environ. Mol. Mutagen. 1991;17:163–168. doi: 10.1002/em.2850170305. [DOI] [PubMed] [Google Scholar]
- Hernandez L.G., van Steeg H., Luijten M., van Benthem J. Mechanisms of non-genotoxic carcinogens and importance of a weight of evidence approach. Mutat. Res. 2009;682:94–109. doi: 10.1016/j.mrrev.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Higginbotham S., RamaKrishna N.V., Johansson S.L., Rogan E.G., Cavalieri E.L. Tumor-initiating activity and carcinogenicity of dibenzo[a,l]pyrene versus 7,12-dimethylbenz[a]anthracene and benzo[a]pyrene at low doses in mouse skin. Carcinogenesis. 1993;14:875–878. doi: 10.1093/carcin/14.5.875. [DOI] [PubMed] [Google Scholar]
- Horibata K., Ukai A., Kimoto T., Suzuki T., Kamoshita N., Masumura K., Nohmi T., Honma M. Evaluation of in vivo genotoxicity induced by N-ethyl-N-nitrosourea, benzo[a]pyrene, and 4-nitroquinoline-1-oxide in the Pig-a and gpt assays. Environ. Mol. Mutagen. 2013;54:747–754. doi: 10.1002/em.21818. [DOI] [PubMed] [Google Scholar]
- Horky J., Vacha J., Znojil V. Comparison of life span of erythrocytes in some inbred strains of mouse using 14C-labelled glycine. Physiol. Bohemoslov. 1978;27:209–217. [PubMed] [Google Scholar]
- IARC Diesel and gasoline engine exhausts and some nitroarenes. IARC Monogr. Eval. Carcinog. Risks Hum. 2014;105 [PMC free article] [PubMed] [Google Scholar]
- IARC Some non-heterocyclic polycyclic aromatic hydrocarbons and some related exposures. IARC Monogr. Eval. Carcinog. Risks Hum. 2010;92 [PMC free article] [PubMed] [Google Scholar]
- IARC Polynuclear aromatic compounds, Part1, chemical, environmental and experimental data. IARC Monogr. Eval. Carcinog. Risks Hum. 1983;32 [PubMed] [Google Scholar]
- JaCVAM, 2012. Report of the JaCVAM initiative international validation study of the in vivo rodent alkaline Comet assay for the detection of genotoxic carcinogens: the 4th (definitive) phase-1st step.
- Jeng H.A., Pan C.H., Diawara N., Chang-Chien G.P., Lin W.Y., Huang C.T., Ho C.K., Wu M.T. Polycyclic aromatic hydrocarbon-induced oxidative stress and lipid peroxidation in relation to immunological alteration. Occup. Environ. Med. 2011;68:653–658. doi: 10.1136/oem.2010.055020. [DOI] [PubMed] [Google Scholar]
- Josephy P.D. Oxford University Press; New York: 1997. Polycyclic aromatic hydrocarbon carcinogenesis, in: anonymous molecular toxicology; p. 315. [Google Scholar]
- Keith L., Telliard W. ES&T special report: priority pollutants: I — a perspective view. Environ. Sci. Technol. 1979;13:416–423. [Google Scholar]
- Kirkland D., Aardema M., Henderson L., Muller L. Evaluation of the ability of a battery of three in vitro genotoxicity tests to discriminate rodent carcinogens and non-carcinogens I. Sensitivity, specificity and relative predictivity. Mutat. Res. 2005;584:1–256. doi: 10.1016/j.mrgentox.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Labib S., Williams A., Guo C.H., Leingartner K., Arlt V.M., Schmeiser H.H., Yauk C.L., White P.A., Halappanavar S. Comparative transcriptomic analyses to scrutinize the assumption that genotoxic PAHs exert effects via a common mode of action. Arch. Toxicol. 2015 doi: 10.1007/s00204-015-1595-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labib S., Yauk C.L., Williams A., Arlt V.M., Phillips D.H., White P.A., Halappanavar S. Subchronic oral exposure to benzo(a)pyrene leads to distinct transcriptomic changes in the lungs that are related to carcinogenesis. Toxicol. Sci. 2012;129:213–224. doi: 10.1093/toxsci/kfs177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert, I.B., Singer, T.M., Boucher, S.E., Douglas, G.R. 2009. Detailed review paper on transgenic rodent mutation assays. OECD Series on Testing and Assessment 103. [DOI] [PubMed]
- Lambert I.B., Singer T.M., Boucher S.E., Douglas G.R. Detailed review of transgenic rodent mutation assays. Mutat. Res. 2005;590:1–280. doi: 10.1016/j.mrrev.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Leavitt S.A. Mutations induced by benzo[a]pyrene and dibenzo[a,l]pyrene in lacI transgenic B6C3F1 mouse lung result from stable DNA adducts. Mutagenesis. 2008;23:445–450. doi: 10.1093/mutage/gen033. [DOI] [PubMed] [Google Scholar]
- Lemieux C.L., Douglas G.R., Gingerich J., Phonethepswath S., Torous D.K., Dertinger S.D., Phillips D.H., Arlt V.M., White P.A. Simultaneous measurement of benzo[a]pyrene-induced Pig-a and lacZ mutations, micronuclei and DNA adducts in Muta Mouse. Environ. Mol. Mutagen. 2011;52:756–765. doi: 10.1002/em.20688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux C.L., Long A.S., Lambert I.B., Lundstedt S., Tysklind M., White P.A. In vitro mammalian mutagenicity of complex polycyclic aromatic hydrocarbon mixtures in contaminated soils. Environ. Sci. Technol. 2015;49:1787–1796. doi: 10.1021/es504465f. [DOI] [PubMed] [Google Scholar]
- Machala M., Vondracek J., Blaha L., Ciganek M., Neca J.V. Aryl hydrocarbon receptor-mediated activity of mutagenic polycyclic aromatic hydrocarbons determined using in vitro reporter gene assay. Mutat. Res. 2001;497:49–62. doi: 10.1016/s1383-5718(01)00240-6. [DOI] [PubMed] [Google Scholar]
- Malik A.I., Rowan-Carroll A., Williams A., Lemieux C.L., Long A.S., Arlt V.M., Phillips D.H., White P.A., Yauk C.L. Hepatic genotoxicity and toxicogenomic responses in MutaMouse males treated with dibenz[a,h]anthracene. Mutagenesis. 2013;28:543–554. doi: 10.1093/mutage/get031. [DOI] [PubMed] [Google Scholar]
- Miller K.P., Ramos K.S. Impact of cellular metabolism on the biological effects of benzo[a]pyrene and related hydrocarbons. Drug Metab. Rev. 2001;33:1–35. doi: 10.1081/dmr-100000138. [DOI] [PubMed] [Google Scholar]
- Monroe J.J., Kort K.L., Miller J.E., Marino D.R., Skopek T.R. A comparative study of in vivo mutation assays: analysis of hprt, lacI, and cII/cI as mutational targets for N-nitroso-N-methylurea and benzo[a]pyrene in Big Blue(TM) mice. Mutat. Res. 1998;421:121–136. doi: 10.1016/s0027-5107(98)00171-7. [DOI] [PubMed] [Google Scholar]
- Nebert D.W., Dalton T.P., Okey A.B., Gonzalez F.J. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J. Biol. Chem. 2004;279:23847–23850. doi: 10.1074/jbc.R400004200. [DOI] [PubMed] [Google Scholar]
- Nebert D.W., Shi Z., Galvez-Peralta M., Uno S., Dragin N. Oral benzo[a]pyrene: understanding pharmacokinetics, detoxication, and consequences--Cyp1 knockout mouse lines as a paradigm. Mol. Pharmacol. 2013;84(3):304–313. doi: 10.1124/mol.113.086637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa T., Nakamura T., Fukushima A., Takagi Y. Further evaluation of the skin micronucleus test: results obtained using 10 polycyclic aromatic hydrocarbons. Mutat. Res. 2005;588:58–63. doi: 10.1016/j.mrgentox.2005.09.004. [DOI] [PubMed] [Google Scholar]
- OECD, 2013. In vivo mammalian alkaline comet assay. Draft OECD Guideline for the Testing of Chemicals.
- OECD, 2011. Transgenic rodent somatic and germ cell gene mutation assays. OECD Guideline for the Testing of Chemicals 488.
- OECD, 1997a. Mammalian erythrocyte micronucleus test. OECD Guideline for the Testing of Chemicals 474.
- OECD, 1997b. Unscheduled DNA synthesis (UDS) test with mammalian liver cells in vivo. OECD Guideline for the Testing of Chemicals 486. [DOI] [PubMed]
- Penning T.M. Human aldo-keto reductases and the metabolic activation of polycyclic aromatic hydrocarbons. Chem. Res. Toxicol. 2014;27:1901–1917. doi: 10.1021/tx500298n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penning T.M., Ohnishi S.T., Ohnishi T., Harvey R.G. Generation of reactive oxygen species during the enzymatic oxidation of polycyclic aromatic hydrocarbon trans-dihydrodiols catalyzed by dihydrodiol dehydrogenase. Chem. Res. Toxicol. 1996;9:84–92. doi: 10.1021/tx950055s. [DOI] [PubMed] [Google Scholar]
- Phillips D.H., Arlt V.M. 32P-postlabeling analysis of DNA adducts. Methods Mol. Biol. 2014;1105:127–138. doi: 10.1007/978-1-62703-739-6_10. [DOI] [PubMed] [Google Scholar]
- Ramesh A., Walker S.A., Hood D.B., Guillen M.D., Schneider K., Weyand E.H. Bioavailability and risk assessment of orally ingested polycyclic aromatic hydrocarbons. Int. J. Toxicol. 2004;23:301–333. doi: 10.1080/10915810490517063. [DOI] [PubMed] [Google Scholar]
- Renaud H.J., Cui J.Y., Khan M., Klaassen C.D. Tissue distribution and gender-divergent expression of 78 cytochrome P450 mRNAs in mice. Toxicol. Sci. 2011;124:261–277. doi: 10.1093/toxsci/kfr240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothfuss A., O'Donovan M., De Boeck M., Brault D., Czich A., Custer L., Hamada S., Plappert-Helbig U., Hayashi M., Howe J., Kraynak A.R., van der Leede B.J., Nakajima M., Priestley C., Thybaud V., Saigo K., Sawant S., Shi J., Storer R., Struwe M., Vock E., Galloway S. Collaborative study on fifteen compounds in the rat-liver Comet assay integrated into 2- and 4-week repeat-dose studies. Mutat. Res. 2010;702:40–69. doi: 10.1016/j.mrgentox.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Schober W., Luch A., Soballa V.J., Raab G., Stegeman J.J., Doehmer J., Jacob J., Seidel A. On the species-specific biotransformation of dibenzo[a,l]pyrene. Chem. Biol. Interact. 2006;161:37–48. doi: 10.1016/j.cbi.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Shane B.S., Lockhart A.M., Winston G.W., Tindall K.R. Mutant frequency of lacI in transgenic mice following benzo[a]pyrene treatment and partial hepatectomy. Mutat. Res. 1997;377:1–11. doi: 10.1016/s0027-5107(97)00004-3. [DOI] [PubMed] [Google Scholar]
- Shi Z., Dragin N., Galvez-Peralta M., Jorge-Nebert L.F., Miller M.L., Wang B., Nebert D.W. Organ-specific roles of CYP1A1 during detoxication of dietary benzo[a]pyrene. Mol. Pharmacol. 2010;78:46–57. doi: 10.1124/mol.110.063438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z., Dragin N., Miller M.L., Stringer K.F., Johansson E., Chen J., Uno S., Gonzalez F.J., Rubio C.A., Nebert D.W. Oral benzo[a]pyrene-induced cancer: two distinct types in different target organs depend on the mouse Cyp1 genotype. Int. J. Cancer. 2010;127:2334–2350. doi: 10.1002/ijc.25222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T., Fujii-Kuriyama Y. Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and 1B1. Cancer Sci. 2004;95:1–6. doi: 10.1111/j.1349-7006.2004.tb03162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T., Inoue K., Suzuki Y., Kawai T., Azuma E., Nakajima T., Shindo M., Kurose K., Sugie A., Yamagishi Y., Fujii-Kuriyama Y., Hashimoto M. Arylhydrocarbon receptor-dependent induction of liver and lung cytochromes P450 1A1, 1A2, and 1B1 by polycyclic aromatic hydrocarbons and polychlorinated biphenyls in genetically engineered C57BL/6J mice. Carcinogenesis. 2002;23:1199–1207. doi: 10.1093/carcin/23.7.1199. [DOI] [PubMed] [Google Scholar]
- Skopek T.R., Kort K.L., Marino D.R., Mittal L.V., Umbenhauer D.R., Laws G.M., Adams S.P. Mutagenic response of the endogenous hprt gene and lacl transgene in benzo[a]pyrene-treated Big Blue™ B6C3F1 mice. Environ. Mol. Mutagen. 1996;28:376–384. doi: 10.1002/(SICI)1098-2280(1996)28:4<376::AID-EM11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Smithgall T.E., Harvey R.G., Penning T.M. Spectroscopic identification of ortho-quinones as the products of polycyclic aromatic trans-dihydrodiol oxidation catalyzed by dihydrodiol dehydrogenase. A potential route of proximate carcinogen metabolism. J. Biol. Chem. 1988;263:1814–1820. [PubMed] [Google Scholar]
- Suzuki H., Ikeda N., Kobayashi K., Terashima Y., Shimada Y., Suzuki T., Hagiwara T., Hatakeyama S., Nagaoka K., Yoshida J., Saito Y., Tanaka J., Hayashi M. Evaluation of liver and peripheral blood micronucleus assays with 9 chemicals using young rats: a study by the Collaborative Study Group for the Micronucleus Test (CSGMT)/Japanese Environmental Mutagen Society (JEMS) — Mammalian Mutagenicity Study Group (MMS) Mutat. Res. 2005;583:133–145. doi: 10.1016/j.mrgentox.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Suzuki H., Shirotori T., Hayashi M. A liver micronucleus assay using young rats exposed to diethylnitrosamine: methodological establishment and evaluation. Cytogenet. Genome Res. 2004;104:299–303. doi: 10.1159/000077506. [DOI] [PubMed] [Google Scholar]
- Suzuki H., Takasawa H., Kobayashi K., Terashima Y., Shimada Y., Ogawa I., Tanaka J., Imamura T., Miyazaki A., Hayashi M. Evaluation of a liver micronucleus assay with 12 chemicals using young rats (II): a study by the Collaborative Study Group for the Micronucleus Test/Japanese Environmental Mutagen Society — Mammalian Mutagenicity Study Group. Mutagenesis. 2009;24:9–16. doi: 10.1093/mutage/gen047. [DOI] [PubMed] [Google Scholar]
- Takasawa H., Suzuki H., Ogawa I., Shimada Y., Kobayashi K., Terashima Y., Matsumoto H., Aruga C., Oshida K., Ohta R., Imamura T., Miyazaki A., Kawabata M., Minowa S., Hayashi M. Evaluation of a liver micronucleus assay in young rats (III): a study using nine hepatotoxicants by the Collaborative Study Group for the Micronucleus Test (CSGMT)/Japanese Environmental Mutagen Society (JEMS) — Mammalian Mutagenicity Study Group (MMS) Mutat. Res. 2010;698:30–37. doi: 10.1016/j.mrgentox.2010.02.009. [DOI] [PubMed] [Google Scholar]
- Takasawa H., Takashima R., Hattori A., Narumi K., Kawasako K., Morita T., Hayashi M., Hamada S. Development of a repeated-dose liver micronucleus assay using adult rats (II): further investigation of 1,2-dimethylhydrazine and 2,6-diaminotoluene. Mutat. Res. 2013;751:12–18. doi: 10.1016/j.mrgentox.2012.10.004. [DOI] [PubMed] [Google Scholar]
- Torous D.K., Hall N.E., Dertinger S.D., Diehl M.S., Illi-Love A.H., Cederbrant K., Sandelin K., Bolcsfoldi G., Ferguson L.R., Pearson A., Majeska J.B., Tarca J.P., Hewish D.R., Doughty L., Fenech M., Weaver J.L., Broud D.D., Gatehouse D.G., Hynes G.M., Kwanyuen P., McLean J., McNamee J.P., Parenteau M., Van Hoof V., Vanparys P., Lenarczyk M., Siennicka J., Litwinska B., Slowikowska M.G., Harbach P.R., Johnson C.W., Zhao S., Aaron C.S., Lynch A.M., Marshall I.C., Rodgers B., Tometsko C.R. Flow cytometric enumeration of micronucleated reticulocytes: high transferability among 14 laboratories. Environ. Mol. Mutagen. 2001;38:59–68. doi: 10.1002/em.1051. [DOI] [PubMed] [Google Scholar]
- Uno S., Dalton T.P., Dragin N., Curran C.P., Derkenne S., Miller M.L., Shertzer H.G., Gonzalez F.J., Nebert D.W. Oral benzo[a]pyrene in Cyp1 knockout mouse lines: CYP1A1 important in detoxication, CYP1B1 metabolism required for immune damage independent of total-body burden and clearance rate. Mol. Pharmacol. 2006;69:1103–1114. doi: 10.1124/mol.105.021501. [DOI] [PubMed] [Google Scholar]
- Uno S., Dragin N., Miller M.L., Dalton T.P., Gonzalez F.J., Nebert D.W. Basal and inducible CYP1 mRNA quantitation and protein localization throughout the mouse gastrointestinal tract. Free Radic. Biol. Med. 2008;44:570–583. doi: 10.1016/j.freeradbiomed.2007.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijg J., Douglas G. Bacteriophage lambda and plasmid lacZ transgenic mice for studying mutations in vivo. In: Pfeifer G., editor. Technologies for Detection of DNA Damage and Mutations. Plenum Press; New York: 1996. pp. 391–410. [Google Scholar]
- Warshawsky D., Livingston G.K., Fonouni-Fard M., LaDow K. Induction of micronuclei and sister chromatid exchanges by polycyclic and N-heterocyclic aromatic hydrocarbons in cultured human lymphocytes. Environ. Mol. Mutagen. 1995;26:109–118. doi: 10.1002/em.2850260204. [DOI] [PubMed] [Google Scholar]
- Waters M.D., Jackson M., Lea I. Characterizing and predicting carcinogenicity and mode of action using conventional and toxicogenomics methods. Mutat. Res. 2010;705:184–200. doi: 10.1016/j.mrrev.2010.04.005. [DOI] [PubMed] [Google Scholar]
- White, P.A., Douglas, G.R., Phillips, D.H., Arlt, V.M., (provisionally accepted). Quantitative relationships between lacZ mutant frequency and DNA adduct frequency in MutaMouse tissues and cultured cells exposed to 3-nitrobenzanthrone. Mutagenesis. [DOI] [PMC free article] [PubMed]
- Whong W.Z., Stewart J.D., Cutler D., Ong T. Induction of in vivo DNA adducts by 4 industrial by-products in the rat-lung-cell system. Mutat. Res. 1994;312:165–172. doi: 10.1016/0165-1161(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Xue W., Warshawsky D. Metabolic activation of polycyclic and heterocyclic aromatic hydrocarbons and DNA damage: a review. Toxicol. Appl. Pharmacol. 2005;206:73–93. doi: 10.1016/j.taap.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Yamada K., Suzuki T., Kohara A., Kato T.A., Hayashi M., Mizutani T., Saeki K. Nitrogen-substitution effect on in vivo mutagenicity of chrysene. Mutat. Res. 2005;586:1–17. doi: 10.1016/j.mrgentox.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Zeiger E. Identification of rodent carcinogens and noncarcinogens using genetic toxicity tests: premises, promises, and performance. Regul. Toxicol. Pharmacol. 1998;28:85–95. doi: 10.1006/rtph.1998.1234. [DOI] [PubMed] [Google Scholar]
- Zhang Q.Y., Dunbar D., Kaminsky L.S. Characterization of mouse small intestinal cytochrome P450 expression. Drug Metab. Dispos. 2003;31:1346–1351. doi: 10.1124/dmd.31.11.1346. [DOI] [PubMed] [Google Scholar]
- Zhong B.Z., Gu Z.W., Stewart J., Ong T. Micronucleus formation induced by three polycyclic aromatic hydrocarbons in rat bone marrow and spleen erythrocytes following intratracheal instillation. Mutat. Res. 1995;326:147–153. doi: 10.1016/0027-5107(94)00164-z. [DOI] [PubMed] [Google Scholar]
- Ziccardi M.H., Gardner I.A., Denison M.S. Application of the luciferase recombinant cell culture bioassay system for the analysis of polycyclic aromatic hydrocarbons. Environ. Toxicol. Chem. 2002;21:2027–2033. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transparency document.