

Abstract
Cardiovascular disease is the leading cause of morbidity and mortality in the Western world owing to the limited regenerative capacity of the mammalian cardiovascular system. In lieu of new muscle synthesis, the human heart replaces necrotic tissue with deposition of a noncontractile scar. By contrast, the adult zebrafish is endowed with a remarkable regenerative capacity, capable of de novo cardiomyocyte (CM) creation and scar tissue removal when challenged with an acute injury. In these studies, we examined the contributions of the dynamically regulated microRNA miR-101a during adult zebrafish heart regeneration. We demonstrate that miR-101a expression is rapidly depleted within 3 days post-amputation (dpa) but is highly upregulated by 7-14 dpa, before returning to uninjured levels at the completion of the regenerative process. Employing heat-inducible transgenic strains and antisense oligonucleotides, we demonstrate that decreases in miR-101a levels at the onset of cardiac injury enhanced CM proliferation. Interestingly, prolonged suppression of miR-101a activity stimulates new muscle synthesis but with defects in scar tissue clearance. Upregulation of miR-101a expression between 7 and 14 dpa is essential to stimulate removal of the scar. Through a series of studies, we identified the proto-oncogene fosab (cfos) as a potent miR-101a target gene, stimulator of CM proliferation, and inhibitor of scar tissue removal. Importantly, combinatorial depletion of fosab and miR-101a activity rescued defects in scar tissue clearance mediated by miR-101a inhibition alone. In summation, our studies indicate that the precise temporal modulation of the miR-101a/fosab genetic axis is crucial for coordinating CM proliferation and scar tissue removal during zebrafish heart regeneration.
KEY WORDS: Heart regeneration, MicroRNA-101a, Cardiomyocyte proliferation, Fosab, Scarring, Zebrafish
Summary: The bi-phasic modulation of microRNA-101a, which targets the oncogene fosab (cfos), regulates cardiomyocyte proliferation and scar tissue clearance in the injured adult zebrafish heart.
INTRODUCTION
Heart disease is the leading cause of death worldwide owing in part to the inability to replenish lost cardiomyocytes (CMs) (Odden et al., 2011). Although recent studies have shown that mammals renew CMs throughout their life, there is little de novo synthesis of heart muscle following acute injury such as myocardial infarction (Bergmann et al., 2009; Haubner et al., 2012; Naqvi et al., 2014; Porrello et al., 2011b). Instead, the adult mammalian heart heals with formation of noncontractile collagen-laden scar tissue, leaving the heart with decreased contractility and impaired cardiac output. Adult zebrafish, however, robustly and faithfully regenerate new heart muscle, remove collagen scars and restore lost heart function when challenged with surgical resection of the ventricular apex, genetic ablation of ∼60% of CMs or cryoinjury (Chablais et al., 2011; Gonzalez-Rosa et al., 2011; Poss et al., 2002; Schnabel et al., 2011; Wang et al., 2011). Regeneration of fully functional zebrafish heart muscle and the reconstruction of a contiguous myocardial wall occur through proliferation of spared CMs within the injury border (Jopling et al., 2010; Kikuchi et al., 2010). However, when CM proliferation is disabled via mutation of the gene mps1 (ttk – Zebrafish Information Network) or transgenic and pharmacological blockade of developmental pathways, regeneration is blocked resulting in the formation of a permanent fibrotic scar (Chablais and Jazwinska, 2012; Fang et al., 2013; Huang et al., 2013; Kikuchi et al., 2011; Lepilina et al., 2006; Poss et al., 2002; Zhao et al., 2014). Thus, injury-stimulated CM proliferation and scar tissue clearance are crucial cellular processes for natural heart regeneration.
Proliferation of resident CMs and the removal of scar tissue are accompanied by modulation of developmental genetic programs, suggesting potential regulatory roles by microRNAs (miRNAs). miRNAs are short, noncoding RNAs that control gene expression post-transcriptionally through complementary base-pairing with the 3′-untranslated region (UTR) of target mRNAs thereby inhibiting protein translation (Ha and Kim, 2014). Given the capacity to control hundreds of target genes, miRNAs have been extensively studied as central gene expression regulators in development, disease and cancer (Bernstein et al., 2003; Giraldez et al., 2005; Hatfield et al., 2005; Kloosterman and Plasterk, 2006; Kohlhapp et al., 2015; Lee et al., 1993; Reinhart et al., 2000; Zhao et al., 2005). More recently, with respect to tissue regeneration, we and others have demonstrated that a small subset of differentially regulated miRNAs are required for cellular proliferation of dedifferentiated CMs in adult zebrafish and mouse hearts (Aguirre et al., 2014; Porrello et al., 2013; Yin et al., 2012). It remains vital to define the contributions of other conserved miRNAs in the context of both injury-induced CM proliferation and scar tissue removal.
miR-101a is among a suite of highly conserved miRNAs with documented roles in cellular proliferation in the context of cancer, degenerative diseases and, more recently, in myocardial infarction injury to the adult mammalian heart (Carvalho et al., 2012; Pan et al., 2012; Varambally et al., 2008; Wang et al., 2010). Injury-induced decreases in miR-101a expression stimulated cardiac fibrosis through enhanced fibroblast proliferation but showed no effects on CM proliferation (Pan et al., 2012). However, studies addressing the potential roles for miR-101a in a highly regenerative biological system such as the adult zebrafish have not been explored. In this heart regeneration study, we identified miR-101a from microarray profiling studies as the most highly repressed miRNA at 6 h post-ventricular amputation (hpa).
Here, we employ inducible transgenic tools and antisense locked-nucleic acid (LNA) oligonucleotides to modulate miR-101a activity in vivo in order to elucidate its contributions during adult heart regeneration. We demonstrate that miR-101a regulates both CM proliferation and scar tissue removal by temporal control of the proto-oncogene fosab (cfos). Injury induced-depletion of miR-101a expression levels stimulated CM proliferation. However, prompt upregulation of miR-101a between 7 and 14 dpa is essential to attenuate the scarring process. Intriguingly, sustained suppression of miR-101a for the duration of the regenerative period culminated in new muscle formation but was accompanied with defects in scar tissue clearance. This study documents the dual roles of the miR-101a/fosab circuit in coordinating scar tissue removal, CM proliferation and new cardiac muscle synthesis. To the best of our knowledge, this is the first demonstration that dynamic expression of a single genetic factor pilots two fundamental cellular heart regenerative programs.
RESULTS
miRNAs are modulated at the onset of adult heart injury
To identify potential miRNAs that play a crucial role in the early phases of regeneration, we performed microarray profiling studies, comparing relative RNA expression levels between uninjured and 6 h post-amputation (hpa) ventricles, in biological and technical triplicate. These studies revealed a subset of 17 miRNAs that are either dynamically upregulated or downregulated by at least 1.5-fold with a Student's t-test P-value <0.05 (Fig. 1A). Of this subset, nine miRNAs were highly downregulated and eight miRNAs were upregulated with injury, including the previously studied dre-miR-133a (Yin et al., 2008; Yin et al., 2012). To validate the expression changes revealed by the microarray hybridization studies, we performed real-time quantitative PCR (qPCR) using cDNA synthesized from the same collection of total RNA. Consistent with the microarray studies, qPCR studies of the mature miRNA species showed that miR-101a, miR-19b, miR-29a, miR-214, miR-222, miR-203b and miR-738 display similar changes in expression levels at 6 hpa compared with uninjured ventricles (Fig. 1B). Within the subset of 17 differentially expressed miRNAs, miR-101a was the most robustly depleted miRNA, raising the possibility that it might normally function as a genetic brake to regeneration, and hence was selected for further functional studies.
Fig. 1.

miRNAs are dynamically regulated in response to heart resection injury. Total RNA was extracted from uninjured and 6 h post-amputation (hpa) ventricles. cDNAs were synthesized from small RNAs and hybridized onto microarray chips in triplicate, to detect changes in miRNA expression. (A) Heat-map of the microarray data shows dynamic upregulation and downregulation of miRNAs in response to cardiac injury (green indicates lower expression; red indicates increased expression). (B) Real-time qRT-PCR validation of a subset of the most highly differentially expressed miRNAs from uninjured and 6 hpa wild-type hearts. miRNA levels were normalized to U6 and represented as fold-change relative to expression levels in uninjured hearts. (C) Time course of miR-101a expression levels during heart regeneration. qPCR studies reveal that relative to uninjured hearts (red dashed line) miR-101a expression (blue line) is at its lowest levels at 3 days post-amputation (dpa), increases to 1.5-fold at 14 dpa and is restored to pre-injury levels by 30 dpa. *P<0.05, **P<0.001 (Student's t-test); error bars represent s.e.m.
To understand better the potential roles for miR-101a during the regenerative response, we investigated its temporal expression profile from the onset of ventricular resection until 30 days post-amputation (dpa) (Fig. 1C). Our qPCR studies show that miR-101a expression is profoundly diminished in response to cardiac injury, as early as 6 h post-amputation (hpa), and steadily declines to its lowest levels at 3 dpa. This drop in miR-101a expression correlates with phases of CM dedifferentiation and proliferation (Jopling et al., 2010; Kikuchi et al., 2010; Poss et al., 2002). Interestingly, miR-101a expression levels significantly increase 1.5-fold by 7-14 dpa before returning to uninjured levels at 30 dpa (Fig. 1C). This boost in miR-101a expression levels approximately midway through the regenerative response coincides with active scar tissue removal and continued CM proliferation during zebrafish heart regeneration (Chablais et al., 2011; Gonzalez-Rosa et al., 2011). Thus, ventricular resection triggers a rapid and dynamic bi-phasic modulation in miR-101a expression during natural adult zebrafish heart regeneration.
Depletion of miR-101a promotes CM proliferation
To examine the contributions of miR-101a function in adult tissues, we established inducible transgenic strains under the regulation of the heat-inducible hsp70 promoter to modulate miR-101a activity in vivo. Tg(hs:miR-101a-sp) drives expression of the EGFP cDNA followed by a triplet of perfect binding sites for miR-101a. Upon heat-treatment, the EGFP-miR-101a mRNA binds to and sequesters mature miR-101a RNA. This miR-101a:EGFP-miR-101a association results in ∼80% reduction in miR-101a levels in Tg(hs:miR-101a-sp) hearts, compared with uninjured control hearts (Fig. 2). To elevate miR-101a expression levels experimentally, we employed the hsp70 promoter to drive a 350-nt miR-101a genomic fragment and established a zebrafish heritable strain termed Tg(hs:miR-101a-pre). A single heat-treatment of Tg(hs:miR-101a-pre) stimulates a 1.2- to 1.5-fold increase in mature miR-101a levels that persists for ∼24 h after the completion of heat-treatment (Fig. S2). Importantly, long-term daily heat treatment did not have a significant effect on animal survival (Fig. S2C).
Fig. 2.

Depletion of miR-101a expression promotes cardiomyocyte proliferation in the injured apex. (A-D) Control or Tg(hs:miR-101a-sp) animals were injured and subjected to daily heat treatment. Hearts were collected at 3 dpa for histology, cryosectioned at 10 µm and stained with antibodies to detect Mef2 (green) and Pcna (red) to identify proliferating cardiomyocytes (CMs) (arrowheads) at the injured apex (A,C) and lateral wall (remote zone) (B,D). Insets in A,C are higher magnifications of the areas within dashed boxes. (E) CM proliferation indices were determined by representing Mef+Pcna+ cells as a percentage of total Mef2+ cells. Depletion of miR-101a expression doubled CM proliferation indices at 3 dpa in the injured apex but not in the remote zone. (F) qPCR studies show ∼80% reduction of miR-101a expression levels in Tg(hs:miR-101a-sp) ventricles (blue line) compared with the control group (red dashed line) under conditions of heat treatment. n=5-7; *P<0.05 (Student's t-test); error bars represent s.e.m. HT, heat treatment.
To assess the functional consequences of miR-101a on heart regeneration, we first examined CM proliferation indices under conditions of experimental manipulation of miR-101a at various stages of heart regeneration. Wild-type control, transgenic Tg(hs:miR-101a-sp) and Tg(hs:miR-101a-pre) hearts were injured and animals were subjected to daily heat treatment at 38°C for 1 h. Using antibodies directed against Mef2, a nuclear indicator of CM, and Pcna, a marker of cellular proliferation, we quantified the number of double-labeled cells as a percentage of all CMs within a defined region of each heart (Fig. 2A,C). In Tg(hs:miR-101a-sp) strains, CM proliferation indices were elevated from 3% to 6.5% at 3 dpa, representing a 217% enhancement in comparison with heat-treated control hearts (Fig. 2E). This elevated rate of CM proliferation in Tg(hs:miR-101a-sp) hearts returned to indices comparable to heat-treated control hearts by 14 dpa (Fig. S1). By contrast, overexpression of miR-101a with heat-activation of our Tg(hs:miR-101a-pre) strain inhibited heart regeneration, reducing CM proliferation by 47%, from 4.1% to 2.2% at 3 dpa, relative to control hearts (Fig. S3A). Suppressed regeneration was not a result of increased programmed cell death. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) analysis of control, Tg(hs:miR-101a-sp) and Tg(hs:miR-101a-pre) hearts following heat treatment did not reveal significant differences in apoptotic cells (Fig. S4).
Although our inducible transgenic miR-101a strains were designed to modulate miR-101a function throughout the entire heart, we noted that changes in CM proliferation were regionalized to the injured apex of the ventricle. Analysis of the lateral wall (remote zone) did not reveal a significant difference between heat-treated control and Tg(hs:miR-101a-sp) hearts (Fig. 2B,D,E). Furthermore, this miR-101a-mediated increase in CM proliferation indices was not observed in the absence of heat treatment or injury (Fig. 2E). Collectively, we demonstrate, with inducible transgenic strategies, that injury-induced reduction of miR-101a activity promotes CM proliferation in the regenerating zebrafish heart.
Sustained depletion of miR-101a leads to defects in scar tissue removal
Given that CM proliferation indices were enhanced with greater miR-101a depletion, we reasoned that long-term, sustained reduction of miR-101a activity might also influence other regenerative processes, including scar tissue removal. Therefore, we injured wild-type and Tg(hs:miR-101a-sp) hearts and subjected them to daily heat treatment at 38°C to maintain suppression of miR-101a levels in Tg(hs:miR-101a-sp) animals up to 30 dpa. This heat-treatment protocol was sufficient to suppress miR-101a expression and did not induce lethality in either control or Tg(hs:miR-101a-sp) animals (Fig. S2C). Hearts were collected, cryosectioned and stained with Acid Fuchsin Orange G (AFOG) dyes to assess collagen and fibrin deposition as indicators of scar tissue. In heat-treated control animals, scar tissue was prominent at 7 dpa but was visibly reduced by 14 dpa. By 30 dpa, the majority of the scar tissue was cleared and new heart muscle had penetrated the injured apex. Unexpectedly, Tg(hs:miR-101a-sp) hearts revealed greater fibrin and collagen staining in 14 and 30 dpa samples, compared with control hearts (Fig. 3). Interestingly, 30 dpa Tg(hs:miR-101a-sp) hearts that exhibited greater scar tissue also showed new heart muscle formation in the injured apex, consistent with the observation of increased CM proliferation indices noted at earlier time points (compare Fig. 3E and 3F). Heat-activation of the Tg(hs:miR-101a-pre) miR-101a overexpression strain, however, did not hasten scar tissue removal (Fig. S3).
Fig. 3.

Sustained suppression of miR-101a leads to increased scar tissue. (A-F) Wild-type and Tg(hs:miR-101a-sp) hearts were resectioned, heat treated daily and collected at 7, 14 and 30 dpa for histology. Hearts were sectioned at 10 µm and stained with Acid Fucshin Orange G (AFOG) to detect muscle (brown), collagen (blue) and fibrin (red). Arrowheads in F mark newly regenerated muscle; dashed lines indicate approximate resection injury plane. The penetrance of each phenotype is indicated (number showing the illustrated phenotype/total number of samples). (G) Scarring indices were established by quantifying the percentage of collagen and fibrin within the total injury area at the indicated time points using CellProfiler. Prolonged depletion of miR-101a led to greater scar tissue at 14 and 30 dpa compared with control. (H) Injury size was comparable between the groups as shown by quantification of total injury area. n=5-14; *P<0.001 (Student's t-test); error bars represent s.e.m.
To quantify scarring differences between control and Tg(hs:miR-101a-sp) hearts, we determined scarring indices by representing total collagen and fibrin deposition as a percentage of the total injury area at 7, 14 and 30 dpa (Fig. 3G). In control animals, scarring indices decreased from 47% at 7 dpa to ∼4% at 30 dpa. The 7-14 dpa interval of regeneration showed the largest decrease in collagen and fibrin deposition, a 36% drop in the scarring index (47% to 11%). In Tg(hs:miR-101a-sp) hearts, however, scar tissue indices showed only a minimal decrease from 41% at 7 dpa, 38% at 14 dpa and 28% at 30 dpa. These Tg(hs:miR-101a-sp) scarring indices at 14 and 30 dpa were significantly different from the heat-treated control (P<0.001) (Fig. 3G). Quantification of injury area for each group revealed no significant differences, indicating that defects in scar tissue removal mediated by loss of miR-101a activity were not due to a larger injury size (Fig. 3H). To determine whether miR-101a regulated scar collagen composition, we performed Sirius Red staining and imaged under polarized light microscope in order to differentiate between collagen types III and I. Qualitative analysis indicates that the scar tissue in Tg(hs:miR-101a-sp) hearts is composed primarily of thick collagens (collagen type I), similar to control hearts (Fig. S5). Thus, sustained suppression of miR-101a leads to impaired scar tissue removal without altering collagen composition in the context of enhanced CM proliferation.
Our Tg(hs:miR-101a-sp) strain revealed an unexpected dynamic role for miR-101a in controlling CM proliferation and scar tissue removal. To rule out any potential interactions from long-term heat treatment and miR-101a suppression, we employed antisense locked nucleic acid (LNA) technology as an alternative strategy to specifically knockdown miR-101a activity. Previous studies with LNA reagents directed against the entire mature miRNA sequence have been shown to bind the target sequence with high affinity and specificity (Jepsen et al., 2004; Kumar et al., 1998; Vester and Wengel, 2004). A single nucleotide change is sufficient to abolish complementary binding with the target sequence (Kumar et al., 1998). Based on previous work in the adult mouse (Elmen et al., 2008), we optimized an intraperitoneal (IP) microinjection paradigm that includes daily IP injections of anti-miR-101a oligonucleotides at 10 µg/g for three consecutive days (Fig. S6A). Ventricular resection was performed on day 2.
Treatment with anti-miR-101a oligonucleotides reduced miR-101a expression by ∼55% compared with either vehicle or scrambled LNA-miR-101a administration, as revealed by real-time qPCR studies (Fig. S6B). Importantly, anti-miR-101a mediated suppression of miR-101a activity elevated CM proliferation from 3.8% in vehicle treatment to 7.9% in anti-miR-101a treated hearts, a 208% stimulatory effect, comparable to effects observed in Tg(hs:miR-101a-sp) hearts (P<0.001) (Fig. S6B). A comparison between vehicle and scrambled LNA-miR-101a did not reveal significant differences in CM proliferation indices (Fig. S6C). For studies beyond 7 dpa, we performed additional weekly anti-miR-101a injections as needed. Similar to our observations with Tg(hs:miR-101a-sp) strains, sustained suppression of miR-101a with anti-miR-101a culminated in defects in scar tissue clearance. Vehicle treatment demonstrated a reduction of scar tissue from 20% at 14 dpa to 4.8% by 30 dpa, a decrease of 15.2% (P<0.001). By contrast, there were no significant differences in scarring indices in anti-miR-101a-treated hearts at 14 and 30 dpa (Fig. S6D). Scarring indices at 14 dpa and 30 dpa in anti-miR-101a-treated hearts, however, was significantly different compared with vehicle-treated hearts (P<0.001). In summary, we show with two independent strategies, heat-inducible Tg(hs:miR-101a-sp) and LNA anti-miR, that miR-101a regulates both CM proliferation and scar tissue removal.
Restoration of miR-101a expression by 7-14 dpa is crucial for scar tissue removal
Heart injury triggers a rapid drop in miR-101a expression. However, by 14 dpa, miR-101a expression levels are 1.5-fold greater compared with uninjured ventricles (Fig. 1C). Prolonged experimental suppression of miR-101a activity for 30 dpa inhibited scar tissue resolution (Fig. 3). Given these observations, we hypothesized that the timing of increased miR-101a expression is crucial for scar tissue removal. To identify this critical window, wild-type control and Tg(hs:miR-101a-sp) hearts were resectioned, subjected to defined intervals of heat-treatment and collected for AFOG analysis at 30 dpa (Fig. 4). Quantification of scar tissue in Tg(hs:miR-101a-sp) hearts, showed that neither loss of miR-101a expression between 0 and 7 dpa, when CM proliferation is enhanced, nor between 14 and 30 dpa, revealed significant differences in scarring indices by 30 dpa compared with control hearts (Fig. 4J). Loss of miR-101a activity between 7 and 30 dpa, however, showed greater scarring indices in Tg(hs:miR-101a-sp) hearts (18.5%) compared with control (2.4%) (Fig. 4J). These results indicate three important findings. (1) Enhancing CM proliferation with experimental suppression of miR-101a expression from the onset of injury to 7 dpa did not impact overall scar tissue removal by 30 dpa. Therefore, the consequences of reducing miR-101a expression on CM proliferation and scar tissue removal are likely to be independent of each other. (2) Suppression of miR-101a levels after 14 dpa, a time when miR-101a levels have already increased and scar tissue removal has been initiated, did not interfere with scar clearance. (3) An increase in miR-101a expression levels between 7 and 14 dpa, however, is crucial for scar tissue removal.
Fig. 4.

Upregulation of miR-101a between 7 and 14 dpa is essential for scar tissue removal. (A-I) Control and Tg(hs:miR-101a-sp) animals were subjected to ventricular resection and heat treated at defined intervals. Hearts were collected at 30 dpa and processed for AFOG staining to identify scar tissue. (A-C) Heat-treated induced depletion of miR-101a from 0 to 7 dpa did not alter scar tissue removal between control and Tg(hs:miR-101a-sp) hearts. (D-F) Suppression of miR-101a expression between 7 and 30 dpa resulted in increased scarring in Tg(hs:miR-101a-sp) hearts, compared with controls. (G-I) miR-101a suppression from 14 to 30 dpa, however, led to normal scar tissue clearance in control and Tg(hs:miR-101a-sp) hearts. Dashed lines indicate approximate resection injury plane. (J) Quantification of scarring indices demonstrates that increased miR-101a expression at 7-14 dpa is required for scar tissue clearance. ct, control. n=4-8; *P<0.01 (Student's t-test); NS, not significant; error bars represent s.e.m.
Sustained miR-101a depletion promotes new muscle synthesis while retaining scar tissue
These studies show that depletion of miR-101a activity revealed a unique dual role in cardiac regeneration by stimulating CM proliferation and inhibiting scar tissue removal. AFOG studies suggest that Tg(hs:miR-101a-sp) hearts regenerate new muscle despite maintaining greater scar tissue (Fig. 3). To confirm this observation, we used antibodies directed against Tropomyosin, a muscle-specific marker expressed in differentiated cardiac sarcomeres.
Consistent with normal heart regeneration, wild-type control hearts showed minimal scar tissue and strong Tropomyosin staining within the injured apex, occupying 36% of the total injury site, at the conclusion of a 30-day heat-treatment protocol. In comparison, Tg(hs:miR-101a-sp) hearts displayed ∼28% Tropomyosin in the resected apex but with far greater scar tissue [42% in Tg(hs:miR-101a-sp) and 5.5% in control] (Fig. 5). Both control and Tg(hs:miR-101a-sp) hearts showed formation of a contiguous myocardium wall. These findings indicate that increased fibrosis is observed even in the context of myocardial wall regeneration. In comparison, we performed similar studies on Tg(hs:miR-133a1-pre) hearts, a strain that induces overexpression of miR-133a in CMs in response to heat treatment (Yin et al., 2012). As previously indicated, sustained overexpression of miR-133a resulted in increased scarring at 30 dpa (Fig. 5C,G) (Yin et al., 2012). However, unlike Tg(hs:miR-101a-sp) hearts, Tg(hs:miR-133a1-pre) hearts showed minimal new muscle formation, as indicated by a significant reduction of Tropomyosin expression to 10.5% within the entire injury site of Tg(hs:miR-133a1-pre) hearts, or approximately one-third of the amount observed in heat-treated control hearts (Fig. 5F,H). Quantification of the total injury area showed no significant differences among the three groups (Fig. 5I). In summation, these results show that depletion of miR-101a at the onset of heart injury promotes CM proliferation, thereby stimulating new myocardium regeneration, whereas prolonged suppression of miR-101a impairs scar tissue clearance. Our data reveal two distinct roles for miR-101a during the process of heart regeneration.
Fig. 5.

Long-term miR-101a depletion results in muscle regeneration and defects in scar tissue clearance. (A-F) Control, Tg(hs:miR-101a-sp) and Tg(hs:miR-133a1-pre) hearts were resectioned, heat treated daily for 30 dpa and extracted for histology. Hearts were cryosectioned at 10 µm and stained with either AFOG (A-C) or antibodies directed against Tropomyosin (D-F). Compared with heat-treated control animals, Tg(hs:miR-101a-sp) hearts reveal more scar tissue and less new muscle regeneration in the wounded apex. Tg(hs:miR-133a1-pre) hearts, which overexpress miR-133a1, show defects in both new muscle regeneration and scar tissue removal. Brackets represent approximate injury area; arrowheads in F mark gaps in myocardium and the lack of muscle regeneration. (G-I) Quantification of scarring indices, Tropomyosin expression and injury area were performed with CellProfiler. n=6-7; *P<0.05 (Student's t-test); error bars represent s.e.m.
fosab expression is regulated by miR-101a during heart regeneration
miRNAs regulate gene expression by binding to target mRNA through complementary base pairing in the 3′-UTR and inhibiting protein translation (Kim, 2005). In previous studies of cardiac miRNAs, it was demonstrated that expression of target genes is often inversely correlated with miRNA levels (Hodgkinson et al., 2015; Liu et al., 2008; van Rooij et al., 2006, 2007). Therefore, we used real-time qPCR to profile expression of candidate heart regeneration markers and previously documented miR-101a target genes (Fig. S7, Table S3). Although a subset of known miR-101a target genes was upregulated at 3 dpa, coincident with the downregulation of miR-101a, only FBJ murine osteosarcoma viral oncogene homolog Ab (fosab), the zebrafish ortholog of mammalian Fos, was also downregulated at 14 dpa, a time point of miR-101a upregulation during the regeneration process (Fig. 6A). The combination of an inverse expression relationship with miR-101a, a predicted binding site for miR-101a in the 3′-UTR and previous studies in mammalian cardiac fibrosis suggested fosab as a candidate miR-101a target gene (Pan et al., 2012). Confirming this hypothesis, qPCR studies demonstrate that both anti-miR-101a and Tg(hs:miR-101a-sp)-mediated knockdown of miR-101a activity induced increased levels of fosab (Fig. S8), and Tg(hs:miR-101a-pre)-directed overexpression of miR-101a reduced fosab mRNA levels (Fig. S3B). Confirming the inverse relationship between miR-101a and fosab expression, Fosab protein expression was elevated in miR-101a-suppressed Tg(hs:miR-101a-sp) hearts at 3 and 14 dpa compared with control hearts (Fig. 6B).
Fig. 6.

miR-101a controls fosab activity through interactions at the 3′-UTR. (A) qRT-PCR studies show dynamic fosab expression during a time course of normal heart regeneration. (B) Western blots show increased Fosab protein expression in Tg(hs:miR-101a-sp) ventricles at 3 and 14 dpa relative to controls. Graph shows quantification of western blots using ImageJ software and normalized to β-actin. (C-E′) RNA in situ hybridization studies to detect miR-101a (blue) and fosab (red) in uninjured and 3 dpa regenerating hearts. miR-101a expression is restricted to CMs in uninjured hearts, whereas fosab expression is enriched in CMs at the border zone and non-muscle cells of the wound environment at 3 dpa. (F-H′) Antibody staining to detect Fosab protein expression confirms RNA in situ hybridization expression patterns. E′ and H′ are magnified images of boxed areas in E and H; arrowheads mark miR-101a (E′) and Fosab (H′) expression in CMs; brackets in H′ indicate Fosab in the microenvironment. (I,J) mRNA EGFP-fosab-3′-UTR sensor assays reveal ∼30% decrease in fosab expression when the sensor is co-injected with miR-101a RNA duplex, relative to EGFP-fosab-3′-UTR sensor injection alone. This miR-101a regulation of fosab is abolished in response to a three-nucleotide mutation of the predicted miR-101a binding site (fosabm sensor). n=6-8 embryos per treatment; *P<0.05, **P<0.001 (Student's t-test); NS, not significant; error bars represent s.e.m.
To reveal miR-101a and fosab spatial expression patterns, we performed double RNA in situ hybridization studies with RNAscope technology (www.acdbio.com). These studies showed that miR-101a expression was localized to CMs in uninjured hearts but expression was strongly reduced by 3 dpa (compare blue signal in Fig. 6D and 6E). We detected only a limited number of miR-101a-expressing cells at 3 dpa (Fig. 6E′). Conversely, fosab expression was absent in uninjured heart samples but was strongly upregulated in CMs at the injury border zone and in non-muscle cells within the wound environment by 3 dpa (Fig. 6E,E′, red signal). Immunofluorescence studies to determine Fosab spatial expression pattern confirmed these findings (Fig. 6H).
To demonstrate that miR-101a directly regulates fosab activity in vivo, we performed EGFP-fosab-3′-UTR sensor binding studies in the zebrafish embryo as previously described (Giraldez et al., 2005; Yin et al., 2012). mRNAs corresponding to EGFP-fosab-3′-UTR and mcherry-SV40 were transcribed in vitro and injected in the presence or absence of miR-101a RNA duplex into one-cell-stage embryos (Fig. 6G). EGFP and RFP fluorescence was quantified 48 h post-fertilization. Microinjections with EGFP-fosab-3′-UTR sensor alone produced strong EGFP expression, corresponding to fosab expression, that was repressed by 31% when embryos were co-injected with a miR-101a RNA duplex (P<0.05). This interaction was abolished, however, when three point mutations were introduced in the fosab-3′-UTR sequence predicted to bind with miR-101a (Fig. 6H; Fig. S6B,C). Collectively, these expression and binding studies identify fosab as a direct miR-101a target gene during adult zebrafish heart regeneration.
Restoring fosab activity rescues cardiac phenotypes mediated by experimental depletion of miR-101a expression
To determine whether the effects on CM proliferation and scarring seen with loss of miR-101a are due to the upregulation of fosab, we designed an antisense oligonucleotide to the ATG translation initiation site of fosab mRNA in order to attenuate its activity. This vivo morpholino (MO) is covalently linked to an octa-guanidine dendrimer delivery moiety to enhance cellular uptake into adult tissues. Daily IP microinjection of this fosab vivo MO into adult zebrafish (12.5 µg/g) following heart resection (Fig. 7A,B) was sufficient to lower Fosab protein levels in anti-miR-101a-treated hearts compared with vehicle treatment at 3 and 14 dpa (Fig. 7C). Whereas anti-miR-101a treatment increased Fosab levels by 3.5-fold compared with vehicle treatment at 3 dpa, co-administration with fosab MO reduced Fosab levels to 0.24 arbitrary units (AU), similar to control expression of 0.18 AU (Fig. 7C). Similarly, co-treatment with anti-miR-101a and fosab MO at 14 dpa reduced Fosab levels by 3.9-fold compared with anti-miR-101a treatment alone (Fig. 7C).
Fig. 7.

Knockdown of fosab activity rescues defects mediated by miR-101a depletion. (A,B) Experimental design for fosab vivo MO and combinatorial injections with anti-miR-101a antisense oligonucleotides. (C) Wild-type animals were injured and treated with vehicle, anti-miR-101a oligonucleotides or anti-miR-101a and fosab vivo MO for three days. Hearts were extracted and western blot hybridizations were performed to detect changes to Fosab protein. Fosab levels increase with anti-miR-101a treatment, but expression was restored to near control levels when anti-miR-101a and fosab vivo MO were co-injected. (D) Combinatorial knockdown of fosab and miR-101a activity is sufficient to rescue elevated CM proliferation indices mediated by miR-101a suppression alone. Treatment of wild-type animals with fosab vivo MO alone significantly reduced CM proliferation indices at 3 dpa compared with vehicle treatment. (E) Quantification of scar tissue was assessed at 30 dpa after vehicle, anti-miR-101a, or anti-miR-101a and fosab-vivo-MO treatment. Suppression of fosab expression between 7 and 30 dpa is sufficient to significantly decrease the scarring effect seen with suppression of miR-101a between 7 and 30 dpa. n=6-8; *P<0.05, **P<0.001 (Student's t-test); NS, not significant; error bars represent s.e.m.
Having determined a protocol to attenuate Fosab activity, we asked whether reduction of Fosab activity would perturb the effects on CM proliferation observed at 3 dpa in response to depletion of miR-101a. We injected anti-miR101a with and without fosab MO and assessed CM proliferation (Fig. 7A). Our results show that the reduction of fosab expression to wild-type levels in anti-miR-101a, fosab MO-treated wild-type animals restored CM proliferation indices to similar levels to those observed in vehicle-treated hearts. Administration of fosab MO alone suppressed CM proliferation indices by 55% relative to vehicle treatment (4.5% to 2.5%) (Fig. 7D). These results show that fosab is a potent regulator of CM proliferation and identify the early downregulation of miR-101a in response to injury as a likely mechanism of action for the enhancement of heart regeneration.
To address the long-term consequences of fosab MO treatment on cardiac scarring, we injured wild-type animals and introduced anti-miR-101a with and without fosab vivo MO beginning at 7 dpa (Fig. 7B). This experimental protocol was sufficient to maintain a similar level of Fosab repression, as noted by 3 dpa, and did not induce animal mortality (data not shown). AFOG staining of hearts collected from vehicle, anti-miR-101a and anti-miR-101a, fosab MO treatments revealed that sustained depletion of miR-101a from 7 to 30 dpa led to increased scarring at 30 dpa. Knockdown of fosab by the addition of fosab MO, however, improved scar tissue clearance, reducing scarring indices from 19% in anti-miR-101a-injected hearts to 6% in co-injected samples. This scarring index was not statistically significantly different relative to the 2.6% scarring index observed in vehicle treatment (Fig. 7E). The 13% reduction in scar tissue in anti-miR-101a, fosab MO hearts, relative to anti-miR-101a treated hearts, demonstrates that fosab is an integral miR-101a target gene for scar tissue removal. Collectively, these studies reveal that tight temporal control of the miR-101a/fosab genetic circuit is crucial for heart regeneration.
DISCUSSION
The zebrafish exhibits a remarkable capacity to regenerate missing and damaged heart muscle and fully restore lost cardiac function throughout its life (Kikuchi, 2014). This injury-stimulated de novo creation of heart muscle is executed through a mechanism of dedifferentiation and proliferation of spared CMs and is restricted to the site of injury (Jopling et al., 2010; Kikuchi et al., 2010). Yet, despite the importance for reactivation of quiescent CMs in the context of disease and acute injury, the regulatory circuits that underscore this biology remain underexplored. Although developmental growth factor signaling pathways involving Wingless/Wnts, fibroblast growth factors (FGFs), retinoic acid (RA), platelet-derived growth factor (PDGF) and Notch (Chablais and Jazwinska, 2012; Gupta et al., 2013; Huang et al., 2013; Jopling et al., 2012; Kikuchi et al., 2011; Kim et al., 2010; Lepilina et al., 2006; Zhao et al., 2014) have been the subject of intense studies, only recently have miRNAs emerged as candidate factors for reactivation of quiescent CMs (Aguirre et al., 2014; Yin et al., 2012). Previous studies in adult mice showed that increased expression of hsa-miR-590-3p and hsa-miR-199a-3p in response to myocardial infarction prompted CMs to re-enter the cell cycle and proliferate (Eulalio et al., 2012). Conversely, miRNAs also have important roles as genetic brakes to cellular proliferation. Profiling studies in the neonatal mouse demonstrated that exit from a regeneration competency window following birth is accompanied by upregulation of numerous miRNAs, including the let-7 and miR-15 family (Porrello et al., 2011a, 2013).
Here, we defined the function of miR-101a, a miRNA with a bi-phasic expression pattern in response to ventricular resection in adult zebrafish. Employing inducible transgenic strains and LNA antisense oligonucleotides to perturb miR-101a activity, we demonstrate that miR-101a controls both CM proliferation and scar tissue removal. Our dataset suggests a model whereby injury triggers downregulation of miR-101a expression to near undetectable levels by 3 dpa. This depletion is essential for stimulation of CM proliferation, in part, through the depression of the miR-101a target gene fosab. Whereas the early decrease in miR-101a stimulates CM proliferative activity, its subsequent upregulation by 7-14 dpa is required for removal of scar tissue (Fig. 8). When miR-101a levels are experimentally maintained at low levels, heart regeneration is completed with new muscle synthesis but with intact scar tissue. This defect in scar removal is partially ameliorated by reducing fosab activity. The genetic circuitry of miR-101a and fosab has been implicated in various types of cancers. In a recent study by Liu et al. (2014), a regulatory feedback loop between miR-101a and AP-1, in which fosab is a key factor of AP-1, was discovered in the context of hepatocellular carcinoma (Liu et al., 2014). We have shown, for the first time, the crucial contributions of fosab under the regulation of miR-101a in promoting CM proliferation and scar tissue removal during adult zebrafish heart regeneration.
Fig. 8.
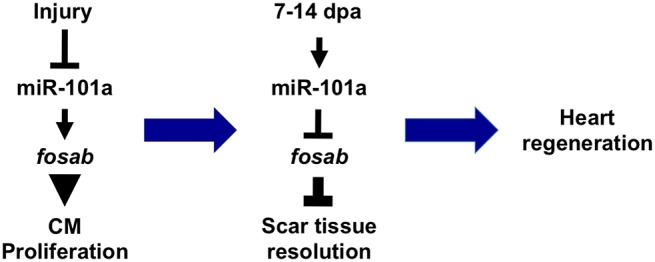
Temporal modulation of miR-101a/fosab genetic circuit controls heart regeneration. In response to injury, miR-101a levels are reduced, enabling fosab expression to increase. This enhanced fosab expression promotes CM proliferation. However, between 7 and 14 dpa, miR-101a levels are significantly elevated, leading to repression of fosab activity. This decline in Fosab protein attenuates scar tissue deposition. miR-101a regulation of fosab during these defined regeneration intervals is crucial for normal heart regeneration.
In our previous work, we showed that miR-133a1, a miRNA expressed in CMs, functions as a cell-autonomous inhibitor of cell proliferation. Whereas injury triggered the decline of miR-133a1 expression, transgenic overexpression of miR-133a1 shifted the injury response towards fibrosis (Yin et al., 2012). Similarly, miR-101a is rapidly depleted at the onset on injury. In fact, within 6 hpa, we noted a 40% drop in expression (Fig. 1C). However, unlike miR-133a1, miR-101a is highly upregulated between 7 and 14 dpa. This dynamic fluctuation in expression prior to the completion of the regenerative process suggested that miR-101a has multiple roles in the healing response. Indeed, our studies revealed that this elevated miR-101a expression by 7-14 dpa is key to halting the scarring response. In the absence of this increase in miR-101a, the heart retained significantly larger scars by 30 dpa, a time when control animals displayed minimal fibrosis (Fig. 4). Interestingly, this defect in scar tissue removal proceeded with new muscle formation (Fig. 5). This suggests that by altering the timing of miR-101a activity, we were able to uncouple CM proliferation from scar tissue removal. This dual role for miR-101a is, therefore, a function of its dynamic expression change, a feature that is absent from injured mouse hearts.
miRNA effects on biology are consequences of both tissue specificity and target gene regulation. Although our studies showed that expression of numerous candidate target genes are inversely expressed with miR-101a at 3 dpa, only fosab exhibited dynamic complementary expression pattern at both 3 and 14 dpa. Through a series of studies, we show that miR-101a binds to and regulates fosab expression and combinatorial reduction of fosab and miR-101a was sufficient to rescue both the proliferative and scarring response (Figs 6, 7). Our in situ hybridization studies revealed that miR-101a expression was enriched in CMs in uninjured hearts but expression was repressed by 3 dpa (Fig. 6D,E). Concomitant with decreases in miR-101a expression, fosab mRNA and protein were elevated in CMs at the injury border as well as non-muscle cells within the wound environment, suggesting at least a cell-autonomous mechanism regulating CM proliferation (Fig. 6E,G). The impact of miR-101a regulation on fosab during CM proliferation is a new finding that appears to be unique to a highly regenerative system like the adult zebrafish. These stimulatory effects on CMs were not observed in miR-101a studies in the injured mouse heart (Pan et al., 2012). However, our studies do not exclude the possibility that fosab activity in the injury microenvironment could exert non-cell-autonomous effects on CM proliferation.
The importance of the injury niche has become increasingly clear in several contexts. Studies of tissue repair and regeneration in both the heart and skeletal muscle have demonstrated that non-muscle cells and immune cells play pivotal roles in inducing proliferative activity. For instance, numerous studies have shown a pro-regenerative capacity of the epicardium and the endocardium in response to injury (Kikuchi et al., 2011; Wang et al., 2015). Moreover, it is becoming increasing clear that cytokine release from inflammatory cells is a crucial modulator of regeneration. Modulation of macrophages attenuates regeneration in salamander limb, zebrafish appendage and neonatal heart regeneration (Aurora et al., 2014; Godwin et al., 2013; Petrie et al., 2014). Previous work on miR-101a in the mammalian heart suggests that fosab is enriched in cardiac fibroblasts and not CMs (Pan et al., 2012). It is intriguing to speculate that differences in the repair response between the zebrafish and mouse heart might be due to crosstalk between non-muscle cells and CMs and/or specific induction of key regulatory factors in spared CMs at the injury border.
The natural decline in miR-101a expression in response to mammalian heart injury promotes fibroblast proliferation and scarring without significantly affecting CM proliferation. Mammalian miR-101a levels do not display the natural sharp increase seen in our zebrafish heart studies (Fig. 1C). Interestingly, experimental elevation of miR-101a in the mouse heart strongly reduced scar tissue deposition in the injured heart (Pan et al., 2012). This finding is in agreement with our results demonstrating that upregulation of miR-101a by 7-14 dpa is instrumental to initiate scar tissue removal. Future studies focused on decoding the transcriptional circuits that promote miR-101a upregulation within this window will provide valuable insight into how removal of fibrosis is managed in a highly regenerative system, and will undoubtedly reveal potential ways of attenuating the cardiac fibrotic response.
CM proliferation and scar tissue removal are the yin and yang of heart muscle repair and regeneration. Understanding how these fundamental repair processes are controlled at the molecular level is essential for therapeutic interventions to restore lost heart function. In this study, we demonstrated, for the first time, that the miR-101a/fosab circuit is a key linchpin in the process of heart regeneration.
MATERIALS AND METHODS
Zebrafish husbandry and heart injury
Zebrafish were maintained at 27°C on a 14 h light/10 h dark cycle in accordance with Institutional Animal Care and Use Committee protocols at the MDI Biological Laboratory. Adult 3- to 6-month-old zebrafish of the Ekkwill (EK) strain were used for all experiments. Transgenic strains were age matched with wild-type siblings. Apical ventricular resection surgeries were performed as previously described (Poss et al., 2002).
Gene expression studies
Total RNA was extracted from ventricles using Tri-Reagent (Sigma) in accordance to the manufacturer's protocol. For real-time qPCR studies, cDNA was synthesized with either the qScript microRNA cDNA Synthesis Kit (Quantabio) or the ProtoScript II First Strand cDNA Synthesis Kit (NEB). Primer sequences for cDNAs are shown in Table S1. qRT-PCR was performed using Brilliant III SYBR Green (Agilent) in a Roche Light cycler 480. Normalization and data analysis were performed as previously described (Yin et al., 2008). For triplicate microarray hybridizations, at least six ventricles per replicate were used. Small RNA labeling and hybridizations were performed and analyzed by LC Sciences to detect expression levels of miRNAs, as previously described (Yin et al., 2012). Microarray data are available at GEO under accession number GSE74494.
Histological methods
Zebrafish hearts were extracted and fixed in 4% paraformaldehyde, embedded in TBS tissue freezing medium (Fisher) and sectioned at 10 µm with a Leica CM1860 cryostat. Immunofluorescence, in situ hybridization and Acid Fuchsin Orange G (AFOG) staining were performed as previously described (Poss et al., 2002; Yin et al., 2012). We used the following primary antibodies: mouse anti-Fosab (Santa Cruz Biotechnology #SC-166940; 1:100), rabbit anti-Mef2 (Santa Cruz Biotechnology #SC-313; 1:75) and mouse anti-PCNA (Sigma #P8825; 1:400). Secondary antibodies used were as follows: Alexa Fluor 488 goat anti-mouse IgG (H+L) (Invitrogen #A11034; 1:200) for Fosab, Alexa Fluor 488 goat anti-rabbit IgG (H+L) for anti-Mef2 and Alexa Fluor594 goat anti-mouse IgG (H+L) for anti-PCNA (Invitrogen #A11032; 1:200). CM proliferation indices were defined as the total number of Mef2+Pcna+ cells represented as a percentage of the total Mef2+ population. Briefly, Mef2+Pcna+ and Mef2+ cells were manually counted within a defined region of 230 pixels (y-axis). Three sections containing the largest injury area were quantified for each heart and included at least five hearts per group.
Quantification of scar tissue was performed using CellProfiler software (www.cellProfiler.org) to calculate total fibrin and collagen over total injured area analyzed. To identify the injury site at 14 and 30 dpa, we examined natural autofluorescence in myocardial tissue to demarcate the amputation plane. Although uninjured muscle has strong EGFP autofluorescence, expression in regenerating muscle appears weaker. The intersection between high and low fluorescence was used as the approximate amputation planes.
To detect the spatial distribution of miR-101a and fosab, we employed RNAscope in situ hybridization technology (Advanced Cell Diagnostics). Hearts were fixed with paraformaldehyde for 1 h at room temperature and processed as previously described (Gemberling et al., 2015). Advanced Cell Diagnostics designed probes for pri-miR-101a (green) and fosab (red). Images were captured at 20× using an Olympus BX53 microscope and Retiga 2000DC camera.
Intraperitoneal microinjections
An antisense locked nucleic acid (LNA) oligonucleotide (Exiqon) (5′-TTATCACAGTACTGT-3′) directed against the mature miR-101a sequence and a scrambled control (Exiqon) were injected into anesthetized animals at 10 μg/g body weight for three consecutive days. Ventricular resection was performed on day 2 of injections. For experiments longer than 1 week, anti-101 LNA oligonucleotides were injected weekly after ventricular resection. Vivo morpholino (5′-GCGTTAAGGCTGGTAAACATCATCC-3′) directed against fosab was injected daily at 12.5 μg/g body weight for the duration of the experiment (Gene Tools).
Immunoblotting
Zebrafish ventricles were harvested in HNTG buffer (20 mM Hepes pH 7.5, 150 mM NaCl, 0.1% Triton X-100, 10% glycerol) and tissues were homogenized using a glass douncer homogenizer. Protein quantification was determined using BCA assay (Thermo Scientific). Samples were reduced in 2× Laemmli's sample buffer and run on an 8% SDS-PAGE gel and transferred onto PVDF membrane. Primary antibodies used were as follows: anti-Fosab (Santa Cruz Biotechnology #SC-166940; 1:100) and β-actin (Cell Signaling #4967; 1:1000). The secondary antibody used was goat anti-mouse-HRP, developed using Super Signal West Dura (Thermo Scientific). Quantification of expression was performed with ImageJ and pixel intensity was represented in arbitrary units.
EGFP sensor studies
To create the EGFP-fosab-3′-UTR cassette for sensor studies, we PCR amplified the 3′-UTR of fosab mRNA and directionally cloned the amplicon downstream of the EGFP cDNA with XbaI (forward primer) and XhoI (reverse primer) restriction sites into the pCS2+ vector (Table S2). For the mutated fosab-3′-UTR, we synthesized a GeneBlock for the complete sequence and cloned it in the same manner (Integrated DNA Technology). In vitro transcription of EGFP-fosab-3′-UTR and mcherry mRNA were performed with HiScribe High Yield RNA Synthesis Kit (NEB). Microinjection and quantification parameters were performed as previously described (Yin et al., 2012).
Construction of and heat-activation of transgenic strains
To construct the Tg(hs:miR-101a-sp) strain, a cassette containing EGFP, triplicate perfect miR-101 binding sites and SV40 polyadenylation sequence was directionally cloned (XhoI/XbaI) behind the hsp70 promoter fragment. To engineer the Tg(hs:miR-101a-pre) strains, a ∼300-bp genomic fragment flanking the miR-101 mature sequence was amplified with PCR and subcloned under the control of the hsp70 promoter fragment. Adult wild-type EK, Tg(hs:miR-101a-sp) and Tg(hs:miR-101a-pre) animals were subjected to a single daily heat treatment for up to 30 days post-amputation. Water temperature was elevated from 26°C to 38°C for a duration of 1 h using experimental conditions previously described (Yin et al., 2012).
Statistical analysis
All statistics were performed using Student's t-test with Welch's correction. P<0.05 was deemed statistically significant.
Acknowledgements
We thank MaryLynn FitzSimons for providing critical comments on the manuscript, Heather Carlisle for establishing the Tg(hs:miR-101a-pre) strain, Ann-Marie Allen for excellent zebrafish care and Dr Kenneth Poss for suggesting the use of RNAscope technology for RNA in situ hybridization studies.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
M.B. and V.P.Y. designed the experiments and analyzed the data. V.P.Y. and A.S. performed the microarray experiments and established the Tg(hs:miR-101a-sp) strain. M.B. conducted all functional studies on miR-101a and fosab and, together with A.S., characterized the Tg(hs:miR-101a-sp) and Tg(hs:miR-101a-pre) strains. M.B. and V.P.Y. wrote the manuscript.
Funding
Research reported in this publication was supported by Institutional Development Awards (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health [P20-GM104318 and P20-GM103423 to V.P.Y.]; an American Heart Association Scientist Development grant [11SDG7210045 to V.P.Y.] and a Department of Defense award [W81XWH-11-1-0425 to V.P.Y.]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.126649/-/DC1
References
- Aguirre A., Montserrat N., Zacchigna S., Nivet E., Hishida T., Krause M. N., Kurian L., Ocampo A., Vazquez-Ferrer E., Rodriguez-Esteban C. et al. (2014). In vivo activation of a conserved microRNA program induces mammalian heart regeneration. Cell Stem Cell 15, 589-604. 10.1016/j.stem.2014.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurora A. B., Porrello E. R., Tan W., Mahmoud A. I., Hill J. A., Bassel-Duby R., Sadek H. A. and Olson E. N. (2014). Macrophages are required for neonatal heart regeneration. J. Clin. Invest. 124, 1382-1392. 10.1172/JCI72181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann O., Bhardwaj R. D., Bernard S., Zdunek S., Barnabe-Heider F., Walsh S., Zupicich J., Alkass K., Buchholz B. A., Druid H. et al. (2009). Evidence for cardiomyocyte renewal in humans. Science 324, 98-102. 10.1126/science.1164680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein E., Kim S. Y., Carmell M. A., Murchison E. P., Alcorn H., Li M. Z., Mills A. A., Elledge S. J., Anderson K. V. and Hannon G. J. (2003). Dicer is essential for mouse development. Nat. Genet. 35, 215-217. 10.1038/ng1253 [DOI] [PubMed] [Google Scholar]
- Carvalho J., van Grieken N. C., Pereira P. M., Sousa S., Tijssen M., Buffart T. E., Diosdado B., Grabsch H., Santos M. A. S., Meijer G. et al. (2012). Lack of microRNA-101 causes E-cadherin functional deregulation through EZH2 up-regulation in intestinal gastric cancer. J. Pathol. 228, 31-44. 10.1002/path.4032 [DOI] [PubMed] [Google Scholar]
- Chablais F. and Jazwinska A. (2012). The regenerative capacity of the zebrafish heart is dependent on TGFbeta signaling. Development 139, 1921-1930. 10.1242/dev.078543 [DOI] [PubMed] [Google Scholar]
- Chablais F., Veit J., Rainer G. and Jaźwińska A. (2011). The zebrafish heart regenerates after cryoinjury-induced myocardial infarction. BMC Dev. Biol. 11, 21 10.1186/1471-213X-11-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmén J., Lindow M., Schütz S., Lawrence M., Petri A., Obad S., Lindholm M., Hedtjärn M., Hansen H. F., Berger U. et al. (2008). LNA-mediated microRNA silencing in non-human primates. Nature 452, 896-899. 10.1038/nature06783 [DOI] [PubMed] [Google Scholar]
- Eulalio A., Mano M., Dal Ferro M., Zentilin L., Sinagra G., Zacchigna S. and Giacca M. (2012). Functional screening identifies miRNAs inducing cardiac regeneration. Nature 492, 376-381. 10.1038/nature11739 [DOI] [PubMed] [Google Scholar]
- Fang Y., Gupta V., Karra R., Holdway J. E., Kikuchi K. and Poss K. D. (2013). Translational profiling of cardiomyocytes identifies an early Jak1/Stat3 injury response required for zebrafish heart regeneration. Proc. Natl. Acad. Sci. USA 110, 13416-13421. 10.1073/pnas.1309810110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemberling M., Karra R., Dickson A. L. and Poss K. D. (2015). Nrg1 is an injury-induced cardiomyocyte mitogen for the endogenous heart regeneration program in zebrafish. eLife 4, e05871 10.7554/eLife.05871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez A. J., Cinalli R. M., Glasner M. E., Enright A. J., Thomson J. M., Baskerville S., Hammond S. M., Bartel D. P. and Schier A. F. (2005). MicroRNAs regulate brain morphogenesis in zebrafish. Science 308, 833-838. 10.1126/science.1109020 [DOI] [PubMed] [Google Scholar]
- Godwin J. W., Pinto A. R. and Rosenthal N. A. (2013). Macrophages are required for adult salamander limb regeneration. Proc. Natl. Acad. Sci. USA 110, 9415-9420. 10.1073/pnas.1300290110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Rosa J. M., Martin V., Peralta M., Torres M. and Mercader N. (2011). Extensive scar formation and regression during heart regeneration after cryoinjury in zebrafish. Development 138, 1663-1674. 10.1242/dev.060897 [DOI] [PubMed] [Google Scholar]
- Gupta V., Gemberling M., Karra R., Rosenfeld G. E., Evans T. and Poss K. D. (2013). An injury-responsive gata4 program shapes the zebrafish cardiac ventricle. Curr. Biol. 23, 1221-1227. 10.1016/j.cub.2013.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha M. and Kim V. N. (2014). Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 15, 509-524. 10.1038/nrm3838 [DOI] [PubMed] [Google Scholar]
- Hatfield S. D., Shcherbata H. R., Fischer K. A., Nakahara K., Carthew R. W. and Ruohola-Baker H. (2005). Stem cell division is regulated by the microRNA pathway. Nature 435, 974-978. 10.1038/nature03816 [DOI] [PubMed] [Google Scholar]
- Haubner B. J., Adamowicz-Brice M., Khadayate S., Tiefenthaler V., Metzler B., Aitman T. and Penninger J. M. (2012). Complete cardiac regeneration in a mouse model of myocardial infarction. Aging 4, 966-977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkinson C. P., Kang M. H., Dal-Pra S., Mirotsou M. and Dzau V. J. (2015). MicroRNAs and cardiac regeneration. Circ. Res. 116, 1700-1711. 10.1161/CIRCRESAHA.116.304377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Harrison M. R., Osorio A., Kim J., Baugh A., Duan C., Sucov H. M. and Lien C.-L. (2013). Igf signaling is required for cardiomyocyte proliferation during zebrafish heart development and regeneration. PLoS ONE 8, e67266 10.1371/journal.pone.0067266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepsen J. S., Sørensen M. D. and Wengel J. (2004). Locked nucleic acid: a potent nucleic acid analog in therapeutics and biotechnology. Oligonucleotides 14, 130-146. 10.1089/1545457041526317 [DOI] [PubMed] [Google Scholar]
- Jopling C., Sleep E., Raya M., Martí M., Raya A. and Izpisua Belmonte J. C. (2010). Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 464, 606-609. 10.1038/nature08899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling C., Suñè G., Morera C. and Izpisua Belmonte J. C. (2012). p38alpha MAPK regulates myocardial regeneration in zebrafish. Cell Cycle 11, 1195-1201. 10.4161/cc.11.6.19637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K. (2014). Advances in understanding the mechanism of zebrafish heart regeneration. Stem Cell Res. 13, 542-555. 10.1016/j.scr.2014.07.003 [DOI] [PubMed] [Google Scholar]
- Kikuchi K., Holdway J. E., Werdich A. A., Anderson R. M., Fang Y., Egnaczyk G. F., Evans T., MacRae C. A., Stainier D. Y. R. and Poss K. D. (2010). Primary contribution to zebrafish heart regeneration by gata4+ cardiomyocytes. Nature 464, 601-605. 10.1038/nature08804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K., Holdway J. E., Major R. J., Blum N., Dahn R. D., Begemann G. and Poss K. D. (2011). Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev. Cell 20, 397-404. 10.1016/j.devcel.2011.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim V. N. (2005). Small RNAs: classification, biogenesis, and function. Mol. Cells 19, 1-15. [PubMed] [Google Scholar]
- Kim J., Wu Q., Zhang Y., Wiens K. M., Huang Y., Rubin N., Shimada H., Handin R. I., Chao M. Y., Tuan T.-L. et al. (2010). PDGF signaling is required for epicardial function and blood vessel formation in regenerating zebrafish hearts. Proc. Natl. Acad. Sci. USA 107, 17206-17210. 10.1073/pnas.0915016107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman W. P. and Plasterk R. H. A. (2006). The diverse functions of microRNAs in animal development and disease. Dev. Cell 11, 441-450. 10.1016/j.devcel.2006.09.009 [DOI] [PubMed] [Google Scholar]
- Kohlhapp F. J., Mitra A. K., Lengyel E. and Peter M. E. (2015). MicroRNAs as mediators and communicators between cancer cells and the tumor microenvironment. Oncogene, doi:10.1038/onc.2015.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R., Singh S. K., Koshkin A. A., Rajwanshi V. K., Meldgaard M. and Wengel J. (1998). The first analogues of LNA (locked nucleic acids): phosphorothioate-LNA and 2′-thio-LNA. Bioorg. Med. Chem. Lett. 8, 2219-2222. 10.1016/S0960-894X(98)00366-7 [DOI] [PubMed] [Google Scholar]
- Lee R. C., Feinbaum R. L. and Ambros V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843-854. 10.1016/0092-8674(93)90529-Y [DOI] [PubMed] [Google Scholar]
- Lepilina A., Coon A. N., Kikuchi K., Holdway J. E., Roberts R. W., Burns C. G. and Poss K. D. (2006). A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 127, 607-619. 10.1016/j.cell.2006.08.052 [DOI] [PubMed] [Google Scholar]
- Liu N., Bezprozvannaya S., Williams A. H., Qi X., Richardson J. A., Bassel-Duby R. and Olson E. N. (2008). microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 22, 3242-3254. 10.1101/gad.1738708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.-J., Lin X.-J., Yang X.-J., Zhou L., He S., Zhuang S.-M. and Yang J. (2014). A novel AP-1/miR-101 regulatory feedback loop and its implication in the migration and invasion of hepatoma cells. Nucleic Acids Res. 42, 12041-12051. 10.1093/nar/gku872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naqvi N., Li M., Calvert J. W., Tejada T., Lambert J. P., Wu J., Kesteven S. H., Holman S. R., Matsuda T., Lovelock J. D. et al. (2014). A proliferative burst during preadolescence establishes the final cardiomyocyte number. Cell 157, 795-807. 10.1016/j.cell.2014.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odden M. C., Coxson P. G., Moran A., Lightwood J. M., Goldman L. and Bibbins-Domingo K. (2011). The impact of the aging population on coronary heart disease in the U.S. Am. J. Med. 124, 827-833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z., Sun X., Shan H., Wang N., Wang J., Ren J., Feng S., Xie L., Lu C., Yuan Y. et al. (2012). MicroRNA-101 inhibited postinfarct cardiac fibrosis and improved left ventricular compliance via the FBJ osteosarcoma oncogene/transforming growth factor-beta1 pathway. Circulation 126, 840-850. 10.1161/CIRCULATIONAHA.112.094524 [DOI] [PubMed] [Google Scholar]
- Petrie T. A., Strand N. S., Tsung-Yang C., Rabinowitz J. S. and Moon R. T. (2014). Macrophages modulate adult zebrafish tail fin regeneration. Development 141, 2581-2591. 10.1242/dev.098459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrello E. R., Johnson B. A., Aurora A. B., Simpson E., Nam Y.-J., Matkovich S. J., Dorn G. W. II, van Rooij E. and Olson E. N. (2011a). MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ. Res. 109, 670-679. 10.1161/CIRCRESAHA.111.248880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrello E. R., Mahmoud A. I., Simpson E., Hill J. A., Richardson J. A., Olson E. N. and Sadek H. A. (2011b). Transient regenerative potential of the neonatal mouse heart. Science 331, 1078-1080. 10.1126/science.1200708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrello E. R., Mahmoud A. I., Simpson E., Johnson B. A., Grinsfelder D., Canseco D., Mammen P. P., Rothermel B. A., Olson E. N. and Sadek H. A. (2013). Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc. Natl. Acad. Sci. USA 110, 187-192. 10.1073/pnas.1208863110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss K. D., Wilson L. G. and Keating M. T. (2002). Heart regeneration in zebrafish. Science 298, 2188-2190. 10.1126/science.1077857 [DOI] [PubMed] [Google Scholar]
- Reinhart B. J., Slack F. J., Basson M., Pasquinelli A. E., Bettinger J. C., Rougvie A. E., Horvitz H. R. and Ruvkun G. (2000). The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403, 901-906. 10.1038/35002607 [DOI] [PubMed] [Google Scholar]
- Schnabel K., Wu C.-C., Kurth T. and Weidinger G. (2011). Regeneration of cryoinjury induced necrotic heart lesions in zebrafish is associated with epicardial activation and cardiomyocyte proliferation. PLoS ONE 6, e18503 10.1371/journal.pone.0018503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E., Sutherland L. B., Liu N., Williams A. H., McAnally J., Gerard R. D., Richardson J. A. and Olson E. N. (2006). A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. USA 103, 18255-18260. 10.1073/pnas.0608791103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E., Sutherland L. B., Qi X., Richardson J. A., Hill J. and Olson E. N. (2007). Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 316, 575-579. 10.1126/science.1139089 [DOI] [PubMed] [Google Scholar]
- Varambally S., Cao Q., Mani R.-S., Shankar S., Wang X., Ateeq B., Laxman B., Cao X., Jing X., Ramnarayanan K. et al. (2008). Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 322, 1695-1699. 10.1126/science.1165395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vester B. and Wengel J. (2004). LNA (locked nucleic acid): high-affinity targeting of complementary RNA and DNA. Biochemistry 43, 13233-13241. 10.1021/bi0485732 [DOI] [PubMed] [Google Scholar]
- Wang H.-J., Ruan H.-J., He X.-J., Ma Y.-Y., Jiang X.-T., Xia Y.-J., Ye Z.-Y. and Tao H.-Q. (2010). MicroRNA-101 is down-regulated in gastric cancer and involved in cell migration and invasion. Eur. J. Cancer 46, 2295-2303. 10.1016/j.ejca.2010.05.012 [DOI] [PubMed] [Google Scholar]
- Wang J., Panakova D., Kikuchi K., Holdway J. E., Gemberling M., Burris J. S., Singh S. P., Dickson A. L., Lin Y.-F., Sabeh M. K. et al. (2011). The regenerative capacity of zebrafish reverses cardiac failure caused by genetic cardiomyocyte depletion. Development 138, 3421-3430. 10.1242/dev.068601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Cao J., Dickson A. L. and Poss K. D. (2015). Epicardial regeneration is guided by cardiac outflow tract and Hedgehog signalling. Nature 522, 226-230. 10.1038/nature14325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin V. P., Thomson J. M., Thummel R., Hyde D. R., Hammond S. M., Poss K. D. (2008). Fgf-dependent depletion of microRNA-133 promotes appendage regeneration in zebrafish. Genes Dev. 22, 728-733. 10.1101/gad.1641808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin V. P., Lepilina A., Smith A. and Poss K. D. (2012). Regulation of zebrafish heart regeneration by miR-133. Dev. Biol. 365, 319-327. 10.1016/j.ydbio.2012.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Samal E. and Srivastava D. (2005). Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 436, 214-220. 10.1038/nature03817 [DOI] [PubMed] [Google Scholar]
- Zhao L., Borikova A. L., Ben-Yair R., Guner-Ataman B., MacRae C. A., Lee R. T., Burns C. G. and Burns C. E. (2014). Notch signaling regulates cardiomyocyte proliferation during zebrafish heart regeneration. Proc. Natl. Acad. Sci. USA 111, 1403-1408. 10.1073/pnas.1311705111 [DOI] [PMC free article] [PubMed] [Google Scholar]