

Abstract
Precise control of oligodendrocyte migration and development is crucial for myelination of axons in the central nervous system (CNS), but important questions remain unanswered about the mechanisms controlling these processes. In a zebrafish screen for myelination mutants, we identified a mutation in zinc finger protein 16-like (znf16l). znf16l mutant larvae have reduced myelin basic protein (mbp) expression and reduced CNS myelin. Marker, time-lapse and ultrastructural studies indicated that oligodendrocyte specification, migration and myelination are disrupted in znf16l mutants. Transgenic studies indicated that znf16l acts autonomously in oligodendrocytes. Expression of Zfp488 from mouse rescued mbp expression in znf16l mutants, indicating that these homologs have overlapping functions. Our results defined the function of a new zinc finger protein with specific function in oligodendrocyte specification, migration and myelination in the developing CNS.
KEY WORDS: Myelin, Oligodendrocyte, Zebrafish
Summary: A genetic screen uncovers a role for zinc finger protein 16-like as an important regulator of the oligodendrocyte lineage in zebrafish embryos.
INTRODUCTION
Precise temporal control of cell fate specification and migration is essential to coordinate the development of different cell types. In the nervous system, for example, newly differentiated neurons extend axons over long distances to form connections with their postsynaptic targets (Araújo and Tear, 2003). Oligodendrocytes, the myelinating cells of the central nervous system (CNS), provide another example. During development, oligodendrocyte precursor cells (OPCs) migrate throughout the CNS. After completing their migration, some OPCs differentiate as oligodendrocytes and myelinate their associated axons immediately, whereas other OPCs delay differentiation until much later stages (Young et al., 2013). Formation of new myelin from OPCs in the adult is essential for motor learning, underscoring the importance of the precise temporal control of differentiation of myelinating oligodendrocytes in the healthy CNS (Scholz et al., 2009; McKenzie et al., 2014). In some demyelinated lesions in individuals with multiple sclerosis, OPCs are present but apparently unable to form myelin (Kotter et al., 2006; Boyd et al., 2013), highlighting the need to understand the temporal control of oligodendrocyte maturation in health and disease.
OPCs are derived from a group of Olig2-expressing progenitors in the motoneuron progenitor (pMN) domain in the ventral CNS. Cells of the pMN domain generate motoneurons at early stages, when they express Olig2 and the neurogenic factor Ngn2 (Mizuguchi et al., 2001; Novitch et al., 2001), and they form OPCs at later stages, when they express Olig2 and the gliogenic factor Sox10 (Stolt et al., 2002; Pozniak et al., 2010). This transition between neurogenic fate and gliogenic fate is mediated by Notch receptors and their ligands (Wang et al., 1998; Park and Appel, 2003), organizing the pMN domain into temporal compartments. Diverse signals, including Pdgf and Netrin-1, direct OPC migration away from the midline and towards the axons that they eventually myelinate (de Castro and Bribián, 2005; Tsai et al., 2006; Miyamoto et al., 2008). After contacting their target axons and initiating differentiation, maturing oligodendrocytes express transcription factors, such as Zfp191 and Myrf, that initiate and maintain the expression of the genes essential for myelination (Emery et al., 2009; Howng et al., 2010). Despite much progress in identifying essential regulators of oligodendrocyte development and myelination, many aspects of the developmental program are not well understood. In particular, little is known about the factors that specify OPCs within the pMN domain and initiate their migration.
To define new genes that regulate oligodendrocyte specification and differentiation, we have conducted a genetic screen for mutations that disrupt myelin gene expression in zebrafish. In this study, we report that the zinc finger protein Znf16-like (Znf16l) regulates the onset of OPC migration and differentiation. In late embryonic stages when OPCs normally begin their migration, znf16l mutants had Olig2-expressing progenitors at the CNS midline but lacked OPCs in most regions of the CNS. Time-lapse imaging revealed that although OPCs were present in the mutants at these early stages, their migration was delayed. At later stages, OPC migration recovered in the mutants, but CNS myelin remained significantly reduced. Through transgenic analyses, we determined that Znf16l acts autonomously in the oligodendrocyte lineage. We also discovered that the normal OPC development could be rescued in znf16l mutants by transgenic expression of the mammalian zinc finger protein Zfp488, which was previously shown to cooperate with Notch signaling to specify OPC fate in the chicken (Wang et al., 2006). Lastly, we showed that despite similar myelination defects in both znf16l mutants and previously studied notch3 mutants (Zaucker et al., 2013), the two genes have different roles in regulating development of the oligodendrocyte lineage. These findings indicate that Znf16l is an essential regulator of specification, migration and myelination in the oligodendrocyte lineage.
RESULTS
Znf16-like is essential for normal myelination in the CNS
In a genetic screen for zebrafish mutants with abnormal myelination, we identified st78 as a recessive mutation that reduced expression of myelin basic protein (mbp) mRNA expression in the CNS at 5 days postfertilization (dpf; Fig. 1A). Homozygous st78 mutants exhibited normal mbp expression in the peripheral nervous system (PNS) and normal gross morphology at 5 dpf (Fig. 1A,B). High-resolution meiotic mapping localized the st78 mutation to a 0.9 Mb region of linkage group 7 (LG7; Fig. 1C). Sequence analyses of genomic DNA from st78 mutants revealed a G-to-T transversion in the previously uncharacterized gene XM_694039, which is predicted to encode a zinc finger protein, Zinc finger 16-like (Znf16l, also known as Zinc finger 697-like/Znf697l). This mutation changed a cysteine in one of the six predicted Cys2His2 zinc fingers of Znf16l to a phenylalanine (C428→F; Fig. 1D,E).
Fig. 1.

Mutations in znf16-like reduce mbp mRNA expression in the CNS. (A) At 5 dpf, expression of mbp was specifically reduced in the CNS but not the PNS of st78 homozygote compared with the heterozygous sibling. (B) st78 mutant had normal gross morphology at 5 dpf compared with the wild-type sibling. (C) Genetic mapping linked st78 locus to XM_694039 in linkage group 7. (D) Sequence chromatogram shows the lesion (red box) in the coding sequence of znf16-like (XM_694039). The point mutation in st78 changes a cysteine in a Cys2His2 zinc finger of Znf16l to a phenylalanine. (E) Schematic diagram depicting Znf16-like protein with all its zinc fingers, the location of the transversion in the st78 allele, and the 4 bp deletion resulting in a frameshift in the st97 allele. (F) Nucleotide sequence of znf16l and the TALE nuclease targeted sequence (black) and diagnostic restriction enzyme site (red). Bottom line shows the st97 allele and the corresponding amino acid sequence resulting from the frameshift. (G) At 5 dpf, expression of mbp was reduced in the CNS of st97 homozygote in comparison to the heterozygous sibling. st97 failed to complement st78, and the transheterozygous mutant lacked mbp in the CNS, similar to the homozygous mutants. The genotypes of all larvae shown in A,B,G,H were determined by PCR tests for their respective lesions.
To construct a null mutation in znf16l, we generated a second mutant allele using transcription activator-like effector nucleases (TALEN; Sanjana et al., 2012). We identified an allele, st97, with a deletion of coding nucleotides 133-136 of znf16l (Fig. 1E,F). This 4 bp deletion caused a frameshift in the open reading frame, resulting in a premature stop codon upstream of all six predicted zinc fingers in the Znf16-like protein. Like homozygotes for the st78 point mutation, homozygous st97 mutants lacked mbp expression in the CNS at 5 dpf and exhibited normal gross morphology (Fig. 1G,H). Furthermore, the st97 frameshift mutation failed to complement the st78 point mutation: st78/st97 transheterozygotes also lacked mbp expression at 5 dpf (Fig. 1G). These results confirmed that Znf16l is essential for normal mbp expression in the larval CNS and further suggested that the disruption of one zinc finger module in st78 mutants abolished the function of Znf16-like.
Specific defects in oligodendrocyte lineage in znf16l mutants
In the developing spinal cord, most oligodendrocytes are derived from a ventral region known as the pMN domain, which also gives rise to motoneurons (Rowitch, 2004). At 24 hours postfertilization (hpf), we detected no difference in the expression of pMN markers between znf16l mutants and wild-type siblings (Fig. 2A,B), indicating that the oligodendrocyte defects did not arise from a delay in the formation of the pMN domain. Likewise, motoneuron development appeared similar in the mutants and wild-type siblings, based on the comparable expression patterns of the motoneuron marker islet1 at 52 hpf (Fig. 2C). By contrast, at this same stage (52 hpf), we detected differences between the mutants and wild-type siblings in the expression of sox10 and olig2 in oligodendrocytes (Fig. 2D,E). olig2-expressing cells had migrated widely in the brain and the spinal cord of the wild-type siblings at this stage, whereas olig2 expression in the mutant was restricted to the ventral midline (Fig. 2D). We also examined expression of sox10, which is restricted to cells in the oligodendrocyte lineage in the CNS. At 52 hpf, sox10-expressing oligodendrocyte-lineage cells were evident in the ventral spinal cord of wild-type embryos but not in mutants (Fig. 2E′). These results support the possibility that znf16l functions specifically in the development of cells in the oligodendrocyte lineage.
Fig. 2.

Developmental disruptions in znf16l mutants are specific to the oligodendrocyte lineage. (A,B) znf16l mutants displayed normal expression of nkx2.2 (A) and olig2 (B) at 24 hpf. (C,C′) znf16l mutants also displayed normal expression of the motoneuron marker islet1 at 52 hpf. (D,E) Defects in znf16l mutants were specific to cells in the oligodendrocyte lineage. (D-D‴) olig2 is expressed by oligodendrocyte-lineage cells and a subset of motoneurons. The expression pattern of olig2 in wild-type siblings revealed groups of cells that had migrated dorsally (arrowhead), ventrally (white brackets) and laterally (box). These cell populations are missing or reduced in the mutants. D′ and D″ show regions in black and red boxes, respectively, in D. (E) sox10 expression is restricted to cells of the oligodendrocyte lineage in the CNS. Wild-type siblings displayed a group of cells (brackets in E′) immediately dorsal to the notochord that were missing in the mutants, confirming the disruption of oligodendrocyte-lineage cells in znf16l mutants. The genotypes of all embryos shown were determined by PCR tests for the st78 lesion. Scale bars: 100 μm.
Loss of znf16l function delays the onset of oligodendrocyte migration
To define the role of znf16l in oligodendrocyte lineage development, we examined znf16l mutants bearing the Tg(olig2:GFP) transgene in timecourse and time-lapse studies. This transgene is expressed in OPCs and throughout differentiation and maturation of myelinating oligodendrocyte-lineage cells (Shin et al., 2003; Zannino and Appel, 2009), allowing observation of different stages of the oligodendrocyte lineage. At 60 and 84 hpf, we observed significantly fewer olig2-expressing cells in the forebrain, midbrain and hindbrain of the mutants compared with their wild-type siblings (Fig. 3A,B,F). By 108 hpf, however, the number of olig2-expressing cells in the mutants was comparable to that of the wild-type siblings (Fig. 3C,F).
Fig. 3.

Disruption of OPC migration in znf16l mutants. (A-C) znf16lst78 mutants and wild-type siblings in Tg(olig2:GFP) background. The number of OPCs is greatly reduced in the mutants at 60 hpf (A,A′) and 84 hpf (B,B′), but recovers quickly by 108 hpf (C,C′). (D,E) Dorsal view of time-lapse imaging from 48-52 and 64-72 hpf, with trajectories of 10 individual OPCs as they migrated laterally out of the midline, shown using the MTrackJ cell-tracking tool (n=3 fish per genotype, per time point). Cells were tracked every 15 min in real time (see Movies 1-4 for time-lapse images). Genotypes of all embryos were determined by PCR after imaging. (F) Quantification of the number of GFP-expressing cells in the hindbrain of Tg(olig2:GFP) znf16lst78 mutants and wild-type siblings (n=15 wild type, n=9 mutant). The numbers were significantly different at 60 and 84 hpf, but not at 108 hpf (two-tailed Student's t-test, ****P<0.0001). (G) Quantification of average OPC displacement (μm) per hour of 30 tracked cells from each timepoint. Mutant OPCs traveled farther than wild-type siblings. (H) Quantification of the fraction of time for which each OPC was actively moving. Mutant OPCs spent more time moving compared with wild-type siblings at either stage. Error bars show s.e.m. significance with one-way ANOVA and post hoc comparisons. ***P=0.0003 in G; ****P<0.0001 in H.
Time-lapse studies showed that olig2:GFP-expressing OPCs migrated from the midline of wild-type embryos between 48 and 52 hpf, but few if any OPCs migrated from the midline in znf16l mutants at these stages (Movies 1, 2; Fig. 3D). In the mutants, OPCs were observed migrating from the midline, starting at about 60 hpf (Movies 3, 4; Fig. 3E). After OPCs began migrating in znf16l mutants, they appeared to move over longer distances and over longer periods of time than their wild-type counterparts. To quantify this, we tracked migration of 30 OPCs in znf16l mutants and wild-type siblings (ten cells per fish from three mutants and three wild-type siblings) and measured their displacement over time. In the 64-72 hpf time period, OPCs migrated faster in mutants than in their wild-type siblings (3.75±0.36 versus 2.20±0.18 μm/h; Fig. 3G). Mutant OPCs also spent a larger fraction of the observation period in movement (0.33±0.02 versus 0.20±0.01; Fig. 3H). We also noted that some mutant OPCs migrated in close proximity to the midline, sometimes re-entering the midline instead of migrating away and dispersing throughout the CNS as wild-type OPCs did. Taken together, these observations indicated that the onset of migration of OPCs is delayed in znf16l mutants and that some mutant OPCs displayed aberrant migratory behavior.
Reduction of mature oligodendrocytes in znf16l mutants
To investigate the progression of oligodendrocyte development in znf16l mutants, we examined the expression of claudin k (cldnk), a marker of mature oligodendrocyte-lineage cells in the CNS of the zebrafish larva (Münzel et al., 2012). At 3 dpf, znf16lst78 mutants expressed little or no detectable cldnk in the CNS, in contrast to their wild-type siblings (Fig. 4A). At 5 dpf, the mutant larvae expressed some cldnk in the CNS but at a much lower level than their wild-type siblings (Fig. 4B). To determine whether mutant oligodendrocytes might initiate myelination at later stages, we analyzed the expression of mbp at 9 dpf (Fig. 4C). Despite the strong reduction of mbp expression at 5 dpf (Fig. 1A), znf16lst78 mutants had detectable mbp expression at 9 dpf, although the level of expression was less than in their wild-type siblings. These results indicated that oligodendrocyte maturation is delayed in the mutants, despite the recovery in OPC number that occurred at earlier stages.
Fig. 4.

CNS myelin is reduced in znf16l mutants. (A-C) Expression of mature oligodendrocyte markers claudin k (A,B) and mbp (C) was reduced in znf16lst78 mutants between 3 and 9 dpf. Expression of cldnk was reduced but detectable at 5 dpf (B), and expression of mbp was reduced but detectable at 9 dpf (C). (D,E) Transverse transmission electron microscopy images of ventral spinal cord showed that many wild-type axons were already myelinated in wild type (D) but not mutants (E). (F,G) At 9 dpf, more axons were myelinated in the mutants (G) than at 5 dpf, but the number was still much less than in wild-type siblings at 9 dpf (F). (H) Quantification of the number of myelinated axons in ventral spinal cord at 5 and 9 dpf. Error bars show s.d.; significance was determined with two-tailed Student's t-test. ***P<0.001 in H. (I) Adult znf16l homozygous mutants are viable and fertile, with no gross morphological defects compared with their wild-type siblings for both of the alleles. Genotypes of all fish analyzed were determined by PCR assay.
Myelin is reduced in znf16l mutants
To assess whether the abnormal expression of oligodendrocyte markers reflects any defects in myelin in znf16l mutants, we examined the ultrastructure of the myelinated axons in the ventral spinal cord. In accordance with the marker analyses, the number of myelinated axons in the mutants was significantly lower than in their wild-type siblings at 5 dpf (1.7±0.8 per hemisegment of the ventral spinal cord in mutants, n=6, compared with 18.7±0.9 in wild type, n=6; Fig. 4D,E,H). At 9 dpf, the number of myelinated axons increased in mutants (7.7±0.7, n=3), but the number remained significantly reduced compared with the wild-type siblings at the same stage (27.3±0.3, n=3; Fig. 4F-H). These results indicated that znf16l is required for myelination for at least several days after OPC migration is complete. Although myelin was reduced in znf16l mutants, some myelin did form and some homozygous mutants survived to become fertile adults (Fig. 4I; in the progeny of heterozygous intercrosses: 6.8% homozygous st78 mutants at 90 dpf, n=177; 7.2% homozygous st97 mutants at 90 dpf, n=111).
Znf16l functions autonomously in oligodendrocytes for proper CNS myelination
To determine which cell types in the CNS require Znf16l function for myelination, we performed transgenic rescue experiments using full-length Znf16l expressed under the control of different tissue-specific regulatory elements. Control uninjected mutants were raised and stained alongside to ensure that rescued mbp expression was not a result of the weak expression of mbp seen in older mutant embryos. In transient transgenic assays, we analyzed constructs that expressed Znf16l in neurons (huC, mnx1), oligodendrocytes (cldnk, sox10) and macrophages or microglia (mpeg1; Fig. 5A,B). The cldnk:znf16l and sox10:znf16l transgenes rescued mbp expression in the CNS of mutants at 5 dpf (cldnk: 45% of the mutants, n=77; sox10: 57% of the mutants, n=47; Fig. 5B,C). No rescue was detected with the mnx1:znf16l construct (Fig. 5B). The huc:znf16l construct rescued at a low frequency (17% of the mutants, n=59; Fig. 5B,C), perhaps because of expression of the huC regulatory sequences in some cells of the oligodendrocyte lineage (Fig. S1). The mpeg1:znf16l construct did not rescue mbp expression in the mutants at 5 dpf (Fig. 5B). These results provide evidence that znf16l acts autonomously in OPCs and early oligodendrocytes to promote CNS myelination.
Fig. 5.
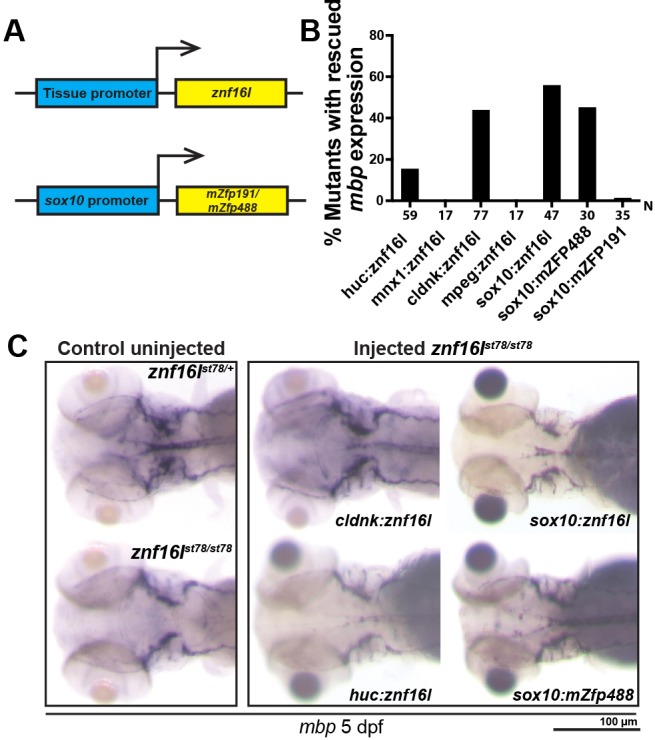
Znf16l functions cell autonomously in the oligodendrocyte lineage. (A) Diagram of the transgenic constructs driving expression of znf16l by tissue-specific gene promoters in neurons (huC), oligodendrocytes (cldnk, sox10) or macrophages (mpeg). Bottom panel shows another transgenic construct strategy to express mammalian zinc finger proteins, mZfp191 and mZfp488, in oligodendrocytes under the control of the sox10 promoter. (B) Plot quantifies the percentage of mbp rescue using different tissue promoters and different zinc finger proteins. (C) Representative images of mbp rescue in cldnk:znf16l-, sox10:znf16l-, huc:znf16l- and sox10:mZfp488-injected znf16l mutants. There is no visible difference in the level of rescues with the different constructs. Genotypes of all fish analyzed were determined by PCR assay.
Mouse Zfp488, but not Zfp191, can rescue znf16l mutants
Sequence analysis did not identify a clear ortholog of Znf16l in mammals. Previous studies have identified several zinc finger proteins that have specific functions in the oligodendrocyte lineage in mammals, including Zfp191, which is required for mature oligodendrocyte-lineage cells to myelinate (Howng et al., 2010), and Zfp488, which can promote oligodendrocyte lineage differentiation and maturation in combination with Notch signaling (Wang et al., 2006). The zebrafish genome contains an ortholog of Zfp191, but no gene clearly orthologous to Zfp488. Thus, we analyzed the previously characterized mouse genes in these functional studies. To test whether either of these zinc finger proteins shares any functional overlap with Znf16l, we performed a transgenic rescue experiment with full-length mZfp191 or mZfp488 under the control of sox10 promoter (Fig. 5A). Zfp488 rescued mbp expression in the CNS of znf16l mutants (47% of the mutants, n=30), whereas expression of Zfp191 had little or no effect (2.9% of the mutants, n=35; Fig. 5B,C). These data suggest that mammalian Zfp488 and zebrafish Znf16l have a shared function in promoting oligodendrocyte-lineage cell migration and differentiation despite the divergent sequences of these zinc finger proteins.
Different requirements for znf16l and notch3 in oligodendrocyte development
Previous analysis showed that notch3 mutants share phenotypic similarities with znf16l mutants (Zaucker et al., 2013). In notch3 mutants, an early reduction in olig2-expressing OPCs underlies a reduction in CNS mbp expression (Zaucker et al., 2013). In notch3 mutants, mbp expression begins to recover by 7 dpf, and some homozygous notch3 mutants survive to adulthood (Zaucker et al., 2013), similar to znf16l mutants. In addition, previous work in the chicken indicates that Zfp488, which can compensate for loss of Znf16l in our in vivo rescue assays, cooperates with Notch signaling, because overexpression of Zfp488 promotes ectopic oligodendrocyte precursor formation only when Notch signaling is activated (Wang et al., 2006).
To investigate the possible relationship of Znf16l and Notch3 in oligodendrocyte development, we examined the expression of early markers in both mutants in parallel. As previously described (Zaucker et al., 2013), notch3st51 and notch3ZM homozygous mutants show marked reductions in expression of deltaD and notch3, whereas heterozygous siblings had increased expression of both genes compared with wild-type siblings (Fig. 6B,C,E,F). By contrast, znf16lst78 mutants expressed both deltaD and notch3 at the same level as their wild-type siblings (Fig. 6A,D), suggesting that Znf16l and Notch3 have distinct functions, despite the similar timecourse of mbp reduction and recovery in the mutants. In addition, we observed that znf16l mutants lacked a ventral population of olig2-expressing progenitors in the hindbrain (Fig. 2D′; Fig. 6G″), whereas notch3 mutants lack a more dorsal population (Fig. 6H″,I″). Furthermore, mutants in these two genes displayed differences in the lateral migration of olig2-expressing OPCs (Fig. 2D‴; Fig. 6G′-I′). These results indicate that Znf16l and Notch3 have different effects on OPCs in the developing embryo.
Fig. 6.

znf16l and notch3 mutants lack distinct populations of OPCs. In situ hybridization of znf16lst78, notch3st51 and notch3ZM embryos at 54 hpf for deltaD (A-C), notch3 (D-F), and olig2 (G-I) mRNA expression. (A-F) znf16l homozygous mutants do not exhibit decreased deltaD (A) or notch3 (D) expression seen in notch3 homozygous mutants (asterisks in B,C,E). (G′-I′) znf16l mutants exhibit a delay in lateral migration of OPCs (red brackets in G′) that is not seen in either notch3 mutant (H′,I′). (G-I,G″-I″) Lateral view of the hindbrain shows that znf16l mutants are mostly lacking the ventral OPC population (white arrowheads), whereas notch3 mutants are mostly lacking the dorsal OPC population (black arrowheads). Genotypes of all fish analyzed were determined by PCR assay. Scale bars: 100 μm.
Severe oligodendrocyte defects in znf16l;notch3 double mutants
To investigate the relationship of znf16l and notch3 in oligodendrocyte development further, we analyzed znf16l;notch3 double mutants. The double mutants had a greater reduction of olig2-expressing OPCs at 52 hpf compared with either single mutant (Fig. 7A-H). The double mutants also lacked laterally migrating OPCs, similar to znf16l mutants (Fig. 7C,D,G,H). By 4 dpf, znf16l and notch3 single mutants significantly recovered the number and distribution of their OPCs (Fig. 7I-K), but znf16l;notch3 double mutants were mostly devoid of OPCs (Fig. 7L). At 5 dpf, notch3 single mutants had reduced mbp expression in the hindbrain and in spinal cord (Fig. 7N), whereas znf16l mutants lacked mbp in both the hindbrain and the spinal cord (Fig. 7O). Like znf16l single mutants, the znf16l;notch3 double mutants also lacked mbp in the CNS at 5 dpf (Fig. 7P). At 9 dpf, the double mutants still had a strong reduction of mbp mRNA in the CNS (Fig. 7T), whereas mbp expression had recovered in both znf16l and notch3 single mutants (Fig. 7R,S).
Fig. 7.

Severe defects in oligodendrocyte development in mutants lacking function of Znf16l and Notch3. In situ hybridization analyses of oligodendrocyte development and myelination in mutants lacking Notch3, Znf16l, or both. (A,E,I,M,Q) Wild-type oligodendrocyte precursors migrating laterally can be detected at 52 hpf (A), and strong expression of mbp can be detected at 5 dpf (M) continuing to 9 dpf (Q). (B,F,J,N,R) notch3 mutants (st51) exhibit reduced lateral migration of OPCs at 52 hpf and gaps in olig2 expression (red arrows in B,F and bracket in B). The number and distribution of OPCs recovers by 4 dpf (J), although mbp expression is still reduced strongly in hindbrain of notch3 mutants (N). (R) By 9 dpf mbp expression recovers, but remains distinguishably reduced compared with wild type. (C,G,K,O,S) znf16l single mutants (st97) show reduction of OPCs at an early stage (bracket in C,G) and a lack of mbp at 5 dpf that recovers by 9 dpf as shown before (O and S). (D,H,L,P,T) Mutants lacking both Znf16l and Notch3 have a more severe phenotype than either single mutant. Numbers of OPCs are strongly reduced and no OPCs migrate laterally similar to the znf16l mutant at 52 hpf (red arrows and bracket in D,H). Numbers of OPCs are still reduced by 4 dpf (L). mbp expression in double mutants is strongly reduced at 5 dpf (P) and very slight expression is observed by 9 dpf (T) compared with either single mutant (R and S). Genotypes of all fish analyzed were determined by PCR assay. Scale bars: 50 μm.
DISCUSSION
Many studies indicate that Hedgehog signaling activity specifies the progenitors in the pMN domain in the ventral neural tube (Richardson et al., 2006) and that these pMN progenitors sequentially form motoneurons at early stages of embryogenesis and oligodendrocyte precursor cells at later stages (Wu et al., 2006). Notch signaling activity is important for this transition (Wang et al., 1998; Genoud et al., 2002; Park and Appel, 2003; Kim et al., 2008; Zaucker et al., 2013), but the other factors that direct pMN progenitors to become migratory OPCs are not well understood. Our study demonstrates that Znf16l is essential for neural progenitors to become migratory OPCs. In znf16l mutants, early patterning of the pMN domain is normal, but oligodendrocyte precursors are specifically delayed in initiating their migration from the midline of the CNS. Moreover, CNS myelin is greatly diminished in znf16l mutants. Despite the severe disruption of OPC migration in mutant embryos, migratory OPCs are evident in the mutant CNS at early larval stages. CNS myelin also partly recovers in larval mutants, but myelin remained significantly reduced in mutants at stages after OPCs have dispersed throughout the CNS. This reduction in myelin indicates that znf16l also regulates the onset of myelination, in addition to early events in OPC specification and migration. Expression of Znf16l in the oligodendrocyte lineage rescues mbp expression in the mutants, indicating that Znf16l acts autonomously in these cells. Our analysis identifies Znf16l as a key autonomous regulator of OPC specification, migration and myelination.
In light of the severe defects in oligodendrocyte development in znf16l mutant embryos and early larvae, it is interesting that some CNS myelin is present at later stages. Likewise, previous work in zebrafish indicates that the notch3 gene is required for OPC specification and myelination in the early larva and that myelin partly recovers at later stages (Zaucker et al., 2013). These studies point to redundancy in the control of oligodendrocyte development in zebrafish. There are at least two levels at which this redundancy might operate. First, it is possible that there are genetically distinct populations of OPCs that are able to compensate for loss of each other. Second, it possible that different members of the Zinc finger and Notch receptor families have overlapping functions, such that homologous genes are able to substitute for each other at some stages.
Previous studies discovered heterogeneity in oligodendrocyte populations in mammals (Tomassy and Fossati, 2014). Spatially, oligodendrocytes arise in distinct regions of the neocortex (Kessaris et al., 2006), cerebellum (Buffo and Rossi, 2013), dorsal spinal cord and ventral spinal cord (Cai et al., 2005). Temporally, multiple waves of oligodendrogenesis occur in the healthy, developing CNS (Kessaris et al., 2006). Ablation studies show that these OPC populations can compensate for the loss of each other, highlighting the redundancy in the lineage despite the apparent heterogeneity in origins of OPCs (Kessaris et al., 2006). Our study is consistent with, but does not demonstrate conclusively, the possibility that such redundancy in the oligodendroglial lineage also exists in the zebrafish. Different domains of olig2 expression are disrupted in znf16l and notch3 single mutants, and the combined loss of both Notch3 and Znf16l in the double mutants resulted in a more severe disruption of oligodendrocyte development and myelination. Thus, it is possible that znf16l and notch3 are required in distinct populations of OPCs that can compensate for loss of each other. A conclusive test of this possibility will require identification of molecular markers that distinguish different populations of OPCs.
There is also evidence for redundancy of individual gene functions in the control of oligodendrocyte development. For example, Notch receptors other than Notch3, notably Notch1, regulate neural progenitor differentiation to promote gliogenesis (Genoud et al., 2002). It is likely that Notch1 activity can partly compensate for the loss of Notch3, contributing to the recovery of CNS myelination in notch3 mutants. Likewise, our in vivo rescue experiments indicate that a mammalian homolog of znf16l, Zfp488, can substitute for znf16l despite their sequence divergence. Although we have not detected an ortholog of Zfp488 in the zebrafish genome, its characteristic structure of two zinc finger motifs flanking a nuclear localization signal is shared with another protein, PR domain containing 8 (Prdm8), that is conserved among zebrafish, mice and humans (Wang et al., 2006; Ross et al., 2012). Prdm8 has been shown to interact with Bhlhb5, a basic helix-loop-helix protein related to Olig2, to form a repressor complex in the context of neurogenesis (Ross et al., 2012). It will be of interest to examine the role of prdm8 and the possibility that Prdm8 and Znf16l have overlapping functions in the control of oligodendrocyte specification, migration and myelination.
Future work is required to identify Znf16l target genes, but genes controlling OPC migration are among the likely candidates. Netrin-1 in the ventral CNS repels migrating OPCs, and in Netrin-1 mutants, OPCs remain near the CNS midline rather than dispersing throughout the CNS as they do in wild type (Tsai et al., 2006). The phenotypic similarity between Netrin-1 and znf16l mutants suggested that receptors for Netrin-1 or other midline repellants might be among the genes activated by Znf16l. In preliminary analyses, however, we observed normal expression of the Netrin receptor genes dcc and unc5a in znf16l mutants (data not shown), suggesting that that Znf16l promotes OPC migration by other mechanisms.
Concluding remarks
Precise control of different steps of OPC development is critical for the development, function and repair of the CNS, but the regulatory mechanisms controlling these processes are not well understood. We have identified Znf16-like as a novel regulator of OPC specification, migration and myelination. Our in vivo rescue assays established that Znf16-like shows functional overlap with its mammalian homolog, Zfp488. Comparison of defects in znf16l and notch3 mutants indicates a degree of redundancy in oligodendrocyte development. Many znf16l mutants survive to adulthood, providing an opportunity to test the role of this gene in response to CNS injury and myelin repair.
MATERIALS AND METHODS
Zebrafish lines and embryos
Experiments involving zebrafish were approved by the Stanford University Institutional Animal Care and Use Committee. Embryos from wild-type (TL, AB/TU and WIK), Tg(olig2:GFP) (Park et al., 2002), znf16lst78, znf16lst97, notch3st51 and notch3ZM (Zaucker et al., 2013) strains were raised at 28.5°C and staged as described by Kimmel et al. (1995). Embryos were treated with 0.003% 1-phenyl-2-thiourea (PTU) in Methylene Blue embryo water to inhibit pigmentation.
N-ethyl-N-nitrosourea mutagenesis, genetic screen and genetic mapping
Founder P0 wild-type males were mutagenized with N-ethyl-N-nitrosourea and subsequently crossed to raise F1 and F2 families for screening as described by Pogoda et al. (2006). An F3 genetic screen was conducted to identify putative mutants with defects in PNS or CNS myelination by whole-mount in situ hybridization at 5 dpf using a riboprobe for myelin basic protein (mbp; Pogoda et al., 2006). st78 mutant embryos were phenotypically sorted from wild-type siblings by a lack of mbp expression in the CNS. Bulk segregant analysis with 480 simple sequence length polymorphisms (SSLPs; Talbot and Schier, 1999) identified markers on LG7 flanking the st78 mutation. High-resolution mapping was conducted using additional SSLPs and single nucleotide polymorphisms linked to the mutation, which were found by sequencing PCR fragments amplified from genomic DNA of mutants and wild-type siblings.
TALEN-mediated genomic deletion
Transcription activator-like effector nucleases (TALEN; Sanjana et al., 2012) were employed to generate the st97 allele. Transcription activator-like effector binding sites were identified in the znf16l genomic sequence using the TALE-NT 2.0 tool (Cermak et al., 2011; Doyle et al., 2013). pCS2+ vectors expressing TALENs targeting the following sequences were cloned: 5′-TCCGAGCTCGAGCCCGAC-3′ and 5′-TCCGTGATCTCGGTCACCGA-3′. Expression vectors were linearized with SmaI and transcribed in vitro using the mMessage mMachine T7 Ultra Kit (Ambion). A mixture containing equal amounts of each mRNA (∼400 pg each) was injected into one-cell stage zebrafish embryos. On the next day, some surviving injected embryos were lysed and genotyped following the protocols described below to measure the efficiency of inducing genomic deletions. The remaining fish were raised to adulthood, and mosaic carriers were identified by assaying for transmission of genomic deletions to their progeny. Sequencing identified st97 as a 4 bp deletion that induces a frameshift in the open reading frame of znf16l.
Genotyping
To genotype the st78 lesion, PCR was conducted (primers: 5′-CAGAGTCCATTCCCTTGTCCACAAT-3′ and 5′-CAGTTTGCACCTTGTCTTCTTG-3′), and the amplification products were cleaved with the restriction enzyme MluCI; the st78 mutation disrupts one of the two restriction sites in the PCR product, so that the mutant allele produces fragments of 170 and 130 bp, whereas the wild-type allele produces fragments of 170, 110 and 20 bp. To genotype the st97 allele, PCR was conducted (primers: 5′-TGGTGCTCTATGGTGTCTGTCT-3′ and 5′-GTGATGTTCCTGCCCAGATG-3′), and the amplification products were cleaved with PvuII; the 4 bp deletion in st97 removes the restriction site in the PCR product, so that the mutant allele results in an uncleaved, full-length PCR product.
Whole-mount RNA in situ hybridization
In situ hybridization on whole zebrafish embryos and larvae from 24 hpf to 9 dpf was performed using standard methods (Thisse and Thisse, 2008). Antisense riboprobes were transcribed from the following cDNAs cloned in the pCRII-Topo vector: mbp (Pogoda et al., 2006), claudin k (Münzel et al., 2012), nkx2.2a (Barth and Wilson, 1995), olig2 (Park et al., 2002), islet1 (Inoue et al., 1994), sox10 (Dutton et al., 2001), deltaD and notch3 (Zaucker et al., 2013). Imaging was performed using a Zeiss Stemi SV 11 Apo stereomicroscope using the 1.6× and 10× objectives and images were captured using the AxioCam Hrc and AxioVision imaging software. For differential interference contrast microscopy, a Zeiss Axio Imager M2 microscope was used using the 20× objective, and images were captured on an AxioCam Mrc with AxioVision imaging software.
Transmission electron microscopy
Transmission electron microscopy was performed as described by Lyons et al. (2009). Stained copper grids were imaged on a JEOL JEM-1400 transmission electron microscope.
Expression constructs
Full-length znf16l coding sequence (XM_694039) of 1587 bp with a Kozak sequence at the 5′ end was amplified from a 3 dpf embryonic cDNA pool (using the primers: 5′-GCCGCCACCATGAGCCGAAAAAGGAA-3′ and 5′-TTACCCAGTTTGCACCTTGTCT-3′) and directionally inserted into the pCR8-GW-TOPO vector (Invitrogen). Tissue-specific expression plasmids were made by LR recombination between this plasmid, pTol2, p3′E-polyA plasmid and the following tissue-specific promoter-containing p5′E plasmids: huC promoter (neurons; Shiau et al., 2013), mpeg1 (macrophages and microglia; Shiau et al., 2013) and claudin k (oligodendrocytes; Münzel et al., 2012). sox10 promoter flanked by Tol2 sequences was previously described by Glenn and Talbot (2013). Briefly, znf16l with adapter sequences were amplified with the following primers: 5′-GTGGCCGCAGAACGAGTGGACCGGCCGCCACCatgagccgaaaaaggaa-3′ and 5′-CATGTCTGGATCATCATCGATTttacccagtttgcaccttgtc-3′ (upper case indicates homology to the sox10 promoter vector and lower case indicates gene-specific sequence) and cloned into the sox10-promoter vector with CloneEZ PCR-Cloning kit (GenScript). mZfp191 and mZfp488 were amplified from mouse cDNA (a gift from Natasha O'Brown, Kingsley Lab, Stanford) using the following primers: mZfp191: 5′-GTGGCCGCAGAACGAGTGGACCGGCCGCCACCatgtctgcacagtcagtggaa-3′ and 5′-CATGTCTGGATCATCATCGATTtcctaatttcttaaaccttcacaacat-3′; and mZfp488: 5′-GTGGCCGCAGAACGAGTGGACCGGCCGCCACCatggctgctggaacatc-3′ and 5′-CATGTCTGGATCATCATCGATTctctgtagccacctgctaactatgt-3′. PCR fragments were cloned into sox10-promoter vector described above with CloneEZ PCR-Cloning kit. Another neuronal expression construct was made by inserting a 3.1 kb fragment containing previously defined regulatory sequences from mnx1/hb9 into the p5′E donor vector (Arkhipova et al., 2012). Transgenes were transiently expressed by co-injecting ∼12-25 pg of Tol2 constructs and ∼50-100 pg of Tol2 transposase mRNA at the one-cell stage. Uninjected embryos were raised in parallel as negative controls.
Time-lapse and fluorescent imaging
Live imaging was performed by anesthetizing embryos in 0.016% (w/v) tricaine and mounting them with the dorsal side up in 1.5% low-melting-point agarose. Static fluorescent images were captured using a Zeiss LSM 5 Pascal confocal microscope with the Axioplan 2 imaging system under the Plan-Neofluar 10× objective (numerical aperture 0.30) and the 488 nm laser line. Time-lapse imaging was performed on a Zeiss Axio Observer microscope coupled to a PerkinElmer UltraView vox spinning-disk confocal microscope. Images were captured using a Hamamatsu camera, using the Volocity Acquisition suite set to record one z-stack every 3 min. Image analyses were performed on the Volocity software and tracking was performed on ImageJ software using the MtrackJ plugin (Meijering et al., 2012). Tracking was done manually by marking the center of each cell every five frames (15 min in real time). As embryos continued to grow througout the imaging period, tracked cells drifted over time as embryos grew larger. Drift was measured individually for every embryo, and cells were actively moving when the displacement was greater than the drift.
Statistical analyses
Statistical analyses were performed using the Prism6 software (GraphPad). Student's t-test was used for all comparisons between mutants and heterozygous siblings. Oligodendrocyte displacement and activity appeared to be normally distributed, and analysis was performed with one-way ANOVA with post hoc comparisons between individual means.
Acknowledgements
We are grateful to M. Barna and A. Villeneuve for sharing equipment. We thank C. E. Shiau for help with the genetic screen and expert advice; A. M. Meireles, K. Shen and D. Lysko for helpful discussions; T. Reyes and C. Hill for maintaining the fish facility; and T. D. Glenn and J. Perrino for help with electron microscopy.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
H.S. identified and mapped the st78 mutation, generated st97 and conducted all experiments on znf16l and notch3 mutants. H.S. and W.S.T. analyzed the data and wrote the manuscript.
Funding
H.S. was supported by an A*STAR fellowship (Singapore). W.S.T. is a Catherine R. Kennedy and Daniel L. Grossman Fellow in Human Biology. This work was supported by the National Institutes of Health [R01NS050223]; and the National Multiple Sclerosis Society [RG-4756-A-3 to W.S.T]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.128215/-/DC1
References
- Araújo S. J. and Tear G. (2003). Axon guidance mechanisms and molecules: lessons from invertebrates. Nat. Rev. Neurosci. 4, 910-922. 10.1038/nrn1243 [DOI] [PubMed] [Google Scholar]
- Arkhipova V., Wendik B., Devos N., Ek O., Peers B. and Meyer D. (2012). Characterization and regulation of the hb9/mnx1 beta-cell progenitor specific enhancer in zebrafish. Dev. Biol. 365, 290-302. 10.1016/j.ydbio.2012.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth K. A. and Wilson S. W. (1995). Expression of zebrafish nk2.2 is influenced by sonic hedgehog/vertebrate hedgehog-1 and demarcates a zone of neuronal differentiation in the embryonic forebrain. Development 121, 1755-1768. [DOI] [PubMed] [Google Scholar]
- Boyd A., Zhang H. and Williams A. (2013). Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol. 125, 841-859. 10.1007/s00401-013-1112-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffo A. and Rossi F. (2013). Origin, lineage and function of cerebellar glia. Prog. Neurobiol. 109, 42-63. 10.1016/j.pneurobio.2013.08.001 [DOI] [PubMed] [Google Scholar]
- Cai J., Qi Y., Hu X., Tan M., Liu Z., Zhang J., Li Q., Sander M. and Qiu M. (2005). Generation of oligodendrocyte precursor cells from mouse dorsal spinal cord independent of Nkx6 regulation and Shh signaling. Neuron 45, 41-53. 10.1016/j.neuron.2004.12.028 [DOI] [PubMed] [Google Scholar]
- Cermak T., Doyle E. L., Christian M., Wang L., Zhang Y., Schmidt C., Baller J. A., Somia N. V., Bogdanove A. J. and Voytas D. F. (2011). Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 39, e82 10.1093/nar/gkr218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Castro F. and Bribián A. (2005). The molecular orchestra of the migration of oligodendrocyte precursors during development. Brain Res. Brain Res. Rev. 49, 227-241. 10.1016/j.brainresrev.2004.12.034 [DOI] [PubMed] [Google Scholar]
- Doyle E. L., Stoddard B. L., Voytas D. F. and Bogdanove A. J. (2013). TAL effectors: highly adaptable phytobacterial virulence factors and readily engineered DNA-targeting proteins. Trends Cell Biol. 23, 390-398. 10.1016/j.tcb.2013.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutton K. A., Pauliny A., Lopes S. S., Elworthy S., Carney T. J., Rauch J., Geisler R., Haffter P. and Kelsh R. N. (2001). Zebrafish colourless encodes sox10 and specifies non-ectomesenchymal neural crest fates. Development 128, 4113-4125. [DOI] [PubMed] [Google Scholar]
- Emery B., Agalliu D., Cahoy J. D., Watkins T. A., Dugas J. C., Mulinyawe S. B., Ibrahim A., Ligon K. L., Rowitch D. H. and Barres B. A. (2009). Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell 138, 172-185. 10.1016/j.cell.2009.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genoud S., Lappe-Siefke C., Goebbels S., Radtke F., Aguet M., Scherer S. S., Suter U., Nave K.-A. and Mantei N. (2002). Notch1 control of oligodendrocyte differentiation in the spinal cord. J. Cell Biol. 158, 709-718. 10.1083/jcb.200202002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenn T. D. and Talbot W. S. (2013). Analysis of Gpr126 function defines distinct mechanisms controlling the initiation and maturation of myelin. Development 140, 3167-3175. 10.1242/dev.093401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howng S. Y. B., Avila R. L., Emery B., Traka M., Lin W., Watkins T., Cook S., Bronson R., Davisson M., Barres B. A. et al. (2010). ZFP191 is required by oligodendrocytes for CNS myelination. Genes Dev. 24, 301-311. 10.1101/gad.1864510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A., Takahashi M., Hatta K., Hotta Y. and Okamoto H. (1994). Developmental regulation of islet-1 mRNA expression during neuronal differentiation in embryonic zebrafish. Dev. Dyn. 199, 1-11. 10.1002/aja.1001990102 [DOI] [PubMed] [Google Scholar]
- Kessaris N., Fogarty M., Iannarelli P., Grist M., Wegner M. and Richardson W. D. (2006). Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci. 9, 173-179. 10.1038/nn1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Shin J., Kim S., Poling J., Park H.-C. and Appel B. (2008). Notch-regulated oligodendrocyte specification from radial glia in the spinal cord of zebrafish embryos. Dev. Dyn. 237, 2081-2089. 10.1002/dvdy.21620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B. and Schilling T. F. (1995). Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253-310. 10.1002/aja.1002030302 [DOI] [PubMed] [Google Scholar]
- Kotter M. R., Li W.-W., Zhao C. and Franklin R. J. M. (2006). Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J. Neurosci. 26, 328-332. 10.1523/JNEUROSCI.2615-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons D. A., Naylor S. G., Scholze A. and Talbot W. S. (2009). Kif1b is essential for mRNA localization in oligodendrocytes and development of myelinated axons. Nat. Genet. 41, 854-858. 10.1038/ng.376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie I. A., Ohayon D., Li H., Paes de Faria J., Emery B., Tohyama K. and Richardson W. D. (2014). Motor skill learning requires active central myelination. Science 346, 318-322. 10.1126/science.1254960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijering E., Dzyubachyk O. and Smal I. (2012). Methods for cell and particle tracking. Methods Enzymol. 504, 183-200. 10.1016/B978-0-12-391857-4.00009-4 [DOI] [PubMed] [Google Scholar]
- Miyamoto Y., Yamauchi J. and Tanoue A. (2008). Cdk5 phosphorylation of WAVE2 regulates oligodendrocyte precursor cell migration through nonreceptor tyrosine kinase Fyn. J. Neurosci. 28, 8326-8337. 10.1523/JNEUROSCI.1482-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi R., Sugimori M., Takebayashi H., Kosako H., Nagao M., Yoshida S., Nabeshima Y.-i., Shimamura K. and Nakafuku M. (2001). Combinatorial roles of olig2 and neurogenin2 in the coordinated induction of pan-neuronal and subtype-specific properties of motoneurons. Neuron 31, 757-771. 10.1016/S0896-6273(01)00413-5 [DOI] [PubMed] [Google Scholar]
- Münzel E. J., Schaefer K., Obirei B., Kremmer E., Burton E. A., Kuscha V., Becker C. G., Brösamle C., Williams A. and Becker T. (2012). Claudin k is specifically expressed in cells that form myelin during development of the nervous system and regeneration of the optic nerve in adult zebrafish. Glia 60, 253-270. 10.1002/glia.21260 [DOI] [PubMed] [Google Scholar]
- Novitch B. G., Chen A. I. and Jessell T. M. (2001). Coordinate regulation of motor neuron subtype identity and pan-neuronal properties by the bHLH repressor Olig2. Neuron 31, 773-789. 10.1016/S0896-6273(01)00407-X [DOI] [PubMed] [Google Scholar]
- Park H. C. and Appel B. (2003). Delta-Notch signaling regulates oligodendrocyte specification. Development 130, 3747-3755. 10.1242/dev.00576 [DOI] [PubMed] [Google Scholar]
- Park H.-C., Mehta A., Richardson J. S. and Appel B. (2002). olig2 is required for zebrafish primary motor neuron and oligodendrocyte development. Dev. Biol. 248, 356-368. 10.1006/dbio.2002.0738 [DOI] [PubMed] [Google Scholar]
- Pogoda H.-M., Sternheim N., Lyons D. A., Diamond B., Hawkins T. A., Woods I. G., Bhatt D. H., Franzini-Armstrong C., Dominguez C., Arana N. et al. (2006). A genetic screen identifies genes essential for development of myelinated axons in zebrafish. Dev. Biol. 298, 118-131. 10.1016/j.ydbio.2006.06.021 [DOI] [PubMed] [Google Scholar]
- Pozniak C. D., Langseth A. J., Dijkgraaf G. J. P., Choe Y., Werb Z. and Pleasure S. J. (2010). Sox10 directs neural stem cells toward the oligodendrocyte lineage by decreasing Suppressor of Fused expression. Proc. Natl. Acad. Sci. USA 107, 21795-21800. 10.1073/pnas.1016485107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson W. D., Kessaris N. and Pringle N. (2006). Oligodendrocyte wars. Nat. Rev. Neurosci. 7, 11-18. 10.1038/nrn1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross S. E., McCord A. E., Jung C., Atan D., Mok S. I., Hemberg M., Kim T.-K., Salogiannis J., Hu L., Cohen S. et al. (2012). Bhlhb5 and Prdm8 form a repressor complex involved in neuronal circuit assembly. Neuron 73, 292-303. 10.1016/j.neuron.2011.09.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowitch D. H. (2004). Glial specification in the vertebrate neural tube. Nat. Rev. Neurosci. 5, 409-419. 10.1038/nrn1389 [DOI] [PubMed] [Google Scholar]
- Sanjana N. E., Cong L., Zhou Y., Cunniff M. M., Feng G. and Zhang F. (2012). A transcription activator-like effector toolbox for genome engineering. Nat. Protoc. 7, 171-192. 10.1038/nprot.2011.431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz J., Klein M. C., Behrens T. E. J. and Johansen-Berg H. (2009). Training induces changes in white-matter architecture. Nat. Neurosci. 12, 1370-1371. 10.1038/nn.2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau C. E., Monk K. R., Joo W. and Talbot W. S. (2013). An anti-inflammatory NOD-like receptor is required for microglia development. Cell Rep. 5, 1342-1352. 10.1016/j.celrep.2013.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J., Park H.-C., Topczewska J. M., Mawdsley D. J. and Appel B. (2003). Neural cell fate analysis in zebrafish using olig2 BAC transgenics. Methods Cell Sci. 25, 7-14. 10.1023/B:MICS.0000006847.09037.3a [DOI] [PubMed] [Google Scholar]
- Stolt C. C., Rehberg S., Ader M., Lommes P., Riethmacher D., Schachner M., Bartsch U. and Wegner M. (2002). Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes Dev. 16, 165-170. 10.1101/gad.215802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot W. S. and Schier A. F. (1999). Positional cloning of mutated zebrafish genes. Methods Cell Biol. 60, 259-286. 10.1016/S0091-679X(08)61905-6 [DOI] [PubMed] [Google Scholar]
- Thisse C. and Thisse B. (2008). High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59-69. 10.1038/nprot.2007.514 [DOI] [PubMed] [Google Scholar]
- Tomassy G. S. and Fossati V. (2014). How big is the myelinating orchestra? Cellular diversity within the oligodendrocyte lineage: facts and hypotheses. Front. Cell Neurosci. 8, 201 10.3389/fncel.2014.00201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai H.-H., Macklin W. B. and Miller R. H. (2006). Netrin-1 is required for the normal development of spinal cord oligodendrocytes. J. Neurosci. 26, 1913-1922. 10.1523/JNEUROSCI.3571-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Sdrulla A. D., diSibio G., Bush G., Nofziger D., Hicks C., Weinmaster G. and Barres B. A. (1998). Notch receptor activation inhibits oligodendrocyte differentiation. Neuron 21, 63-75. 10.1016/S0896-6273(00)80515-2 [DOI] [PubMed] [Google Scholar]
- Wang S.-Z., Dulin J., Wu H., Hurlock E., Lee S.-E., Jansson K. and Lu Q. R. (2006). An oligodendrocyte-specific zinc-finger transcription regulator cooperates with Olig2 to promote oligodendrocyte differentiation. Development 133, 3389-3398. 10.1242/dev.02522 [DOI] [PubMed] [Google Scholar]
- Wu S., Wu Y. and Capecchi M. R. (2006). Motoneurons and oligodendrocytes are sequentially generated from neural stem cells but do not appear to share common lineage-restricted progenitors in vivo. Development 133, 581-590. 10.1242/dev.02236 [DOI] [PubMed] [Google Scholar]
- Young K. M., Psachoulia K., Tripathi R. B., Dunn S.-J., Cossell L., Attwell D., Tohyama K. and Richardson W. D. (2013). Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron 77, 873-885. 10.1016/j.neuron.2013.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannino D. A. and Appel B. (2009). Olig2+ precursors produce abducens motor neurons and oligodendrocytes in the zebrafish hindbrain. J. Neurosci. 29, 2322-2333. 10.1523/JNEUROSCI.3755-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaucker A., Mercurio S., Sternheim N., Talbot W. S. and Marlow F. L. (2013). notch3 is essential for oligodendrocyte development and vascular integrity in zebrafish. Dis. Model. Mech. 6, 1246-1259. 10.1242/dmm.012005 [DOI] [PMC free article] [PubMed] [Google Scholar]