

Abstract
Flow cytometry, a valuable technique that employs the principles of light scattering, light excitation, and emission of fluorochrome molecules, can be used to assess the cell cycle position of individual cells based on DNA content. After the permeabilization of cells, the DNA can be stained with a fluorescent dye. Cells which have a 2N amount of DNA can be distinguished from cells with a 4N amount of DNA, making flow cytometry a very useful tool for the analysis of cell cycle checkpoints following DNA damage. A critical feature of the cellular response to DNA damage is the ability to pause and repair the damage so that consequential mutations are not passed along to daughter generations of cells. If cells arrest prior to DNA replication, they will contain a 2N amount of DNA, whereas arrest after replication but before mitosis will result in a 4N amount of DNA. Using this technique, the role that p53 plays in cell cycle checkpoints following DNA damage can be evaluated based on changes in the profile of the G1, S, and G2/M phases of the cell cycle.
Keywords: Flow cytometry, Fluorescence, Dual parameter, Propidium iodide, BrdU, Phosho-histone H3, p53, DNA damage, DNA damage checkpoints, G1 arrest, G2/M arrest, Apoptosis
1. Introduction
Genomic stability is a critical requirement for cell survival and the prevention of tumorigenesis. In order to ensure that mutations that result from DNA damage are not passed on to daughter generations, the cell must pause and repair the damage. The cellular response pathway is a network that involves sensors of damage that ultimately transmit signals to mediator proteins that regulate the transcription of effector proteins that play an important role in arresting the cell cycle. In the cell cycle, transitions (G1/S, intra S, G2/M) that lead from DNA replication to mitosis are monitored for successful completion. In the event of DNA damage, genotoxic stress, or ribonucleotide depletion, cell cycle checkpoints prevent progression to the next phase of the cell cycle until the damage is repaired, the stress is removed, or nutrients are replenished. Other pathways may be activated that result in programmed cell death if the damage is irreparable (1). When there are defects in the cell cycle checkpoints, gene mutations, chromosome damage, and aneuploidy can result and ultimately, cell transformation can be a consequence of such defects (2).
p53, a transcription factor (3, 4) and tumor suppressor protein (5), can regulate the expression of proteins that play critical roles in growth arrest and apoptosis (programmed cell death) (6). p53 plays a critical role both in the G1/S checkpoint, in which cells arrest prior to DNA replication and have a 2N content of DNA, and in the G2/M checkpoint, in which arrest occurs before mitosis and cells have a 4N content of DNA. The activation of p53 following DNA damage results in the expression of many proteins which are important in cell cycle arrest, repair, and apoptosis (7).
The cyclin-dependent kinase inhibitor, p21, accomplishes cell cycle arrest by inhibiting cyclin–cdk complexes that phosphorylate cell cycle proteins that mediate the passage from G1 to S (8–10). As a result of inhibition, the retinoblastoma protein (pRB) remains hypophosphorylated, E2F remains bound to pRB, and arrest occurs at the G1/S boundary. Proliferating cell nuclear antigen (PCNA), a protein that plays a role in both DNA replication and repair, is a component of the cyclin–cdk complex. p21 binds to and inhibits PCNA from mediating elongation during replication thereby preventing replication in cells that have already entered S phase (11).
Although the G1/S checkpoint is recognized as being entirely p53 dependent, the G2/M checkpoint can be accomplished as a result of multiple pathways. In the presence of DNA damage, p53-dependent and -independent pathways converge to inhibit the activities of cyclin B and Cdc2, proteins that play a role in promoting mitosis (12, 13). Activation of the ATM/CHK2/cdc25 or ATR/CHK1/cdc25 pathways (14) results in the inactivation of phosphatases in the cdc25C family by downregulation and cytoplasmic sequestration. Additionally, p53, itself, is phosphorylated by the kinases in these pathways and in turn becomes stabilized and active. p53 contributes to the maintenance of the G2/M checkpoint by transcriptional repression of both cdc25C and cyclin B (15), upregulation of p21 that can inhibit cyclin B–cdk1 complexes (16), 14-3-3 sigma proteins that target cdc25C proteins to the cytoplasm (17), and GADD45, a protein that can inhibit cyclin B–Cdc2 complexes (18).
Cells in which p53 is deleted or mutated lose the G1 checkpoint and no longer arrest at the G1/S transition. Although they maintain a G2 arrest, this arrest can decay over time thus allowing cells to enter mitosis with unrepaired DNA damage and mutations that increase the risk of progression to malignancy. People who have Li–Fraumeni syndrome, a cancer prone condition in which one allele of the p53 gene is mutated, are susceptible to sarcomas, leukemias, brain and adrenal tumors. In these tumors the remaining allele of p53 is often deleted (loss of heterozygosity) highlighting the importance of the role of p53 in genomic stability (19).
Flow cytometry is a valuable technique used for the analysis of cell cycle checkpoints after DNA damage. Using the principles of light scattering, light excitation, and light emission, fluorescent compounds can be incorporated into the DNA of cells and emit a signal that is detectable and proportional to the number of fluorochrome molecules. A unique feature of flow cytometry is that fluorescence is measured on a per cell basis and not as a bulk volume as in spectrophotometry. In the following method, propidium iodide, a fluorescent compound, is used to intercalate into DNA of cells that have been treated with the DNA damaging agent, doxorubicin. This property can be used to evaluate the DNA content of cells because cells will have a 2N content of DNA prior to cell replication, followed by a 4N amount of DNA after replication.
Briefly, a sample containing stained cells in suspension is taken up by the flow cytometer and hydrodynamically focused through a small nozzle so that single cells pass before a laser, one cell at a time. An argon laser is commonly used in flow cytometry because the 488 nm light that it emits can excite more than one fluorochrome. Therefore multiple fluorochromes can be incorporated simultaneously, provided that their emission wavelengths are far enough apart for detection. In the first protocol, a single parameter assay is performed in which only propidium iodide is incorporated and detected (see Fig. 1). Propidium iodide and a fluorescein isothiocyanate (FITC) conjugated antibody with emissions of 575 nm and 520 nm, respectively, are used to create a dual parameter assay in the second and third protocol. In the second protocol, bromodeoxyuridine (BrdU), a synthetic analog of thymidine, is incorporated into replicating DNA and the antibody that detects BrdU is conjugated to the fluorescent compound (see Fig. 2). In the third protocol, an antibody to phosphorylated histone H3, a marker for mitotic cells, is used which is bound by a secondary antibody that is conjugated to the FITC molecule (see Fig. 3). The light that is emitted is sent to different detectors via band filters and converted to electrical pulses that ultimately are amplified and processed so that the data can be plotted on a graph as digital events. Histograms, which are dot plots of each event, can be constructed to illustrate both parameters. In the second protocol, actively replicating cells can be distinguished because of the BrdU uptake. Cells in G1 (2N content of DNA) and G2/M (4N content of DNA) can be easily discerned because all the cells have incorporated the PI stain. When using the PI stain, however, one cannot distinguish between G2 and mitotic cells since both contain a 4N amount of DNA. The third technique broadens the cell cycle information because mitotic cells are specifically detected using an antibody to phosphorylated histone H3. Once cells exit mitosis, H3 becomes dephosphorylated and is no longer recognized by the antibody.
Fig. 1.
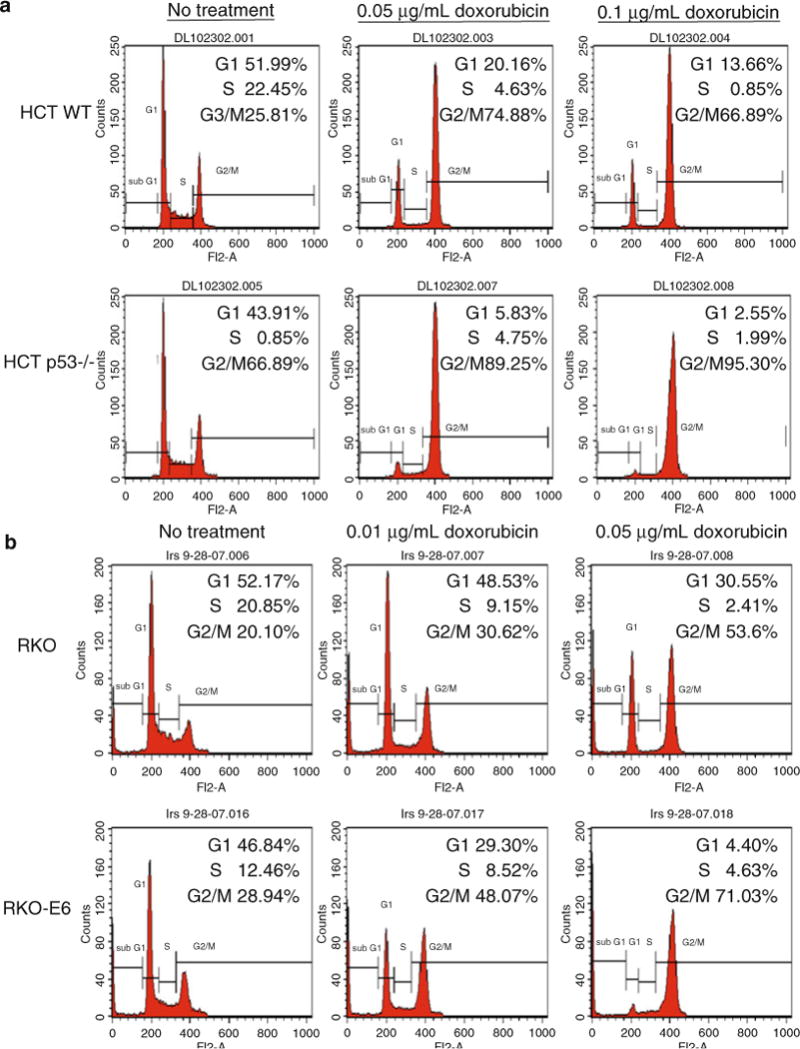
Wild-type p53 cells maintain a G1 and G2/M arrest after treatment with doxorubicin, whereas p53−/− cells primarily arrest in G2/M. (a) HCT 116 WT and p53−/− colorectal carcinoma cells were treated with 0.05 and 0.1 μg/ml doxorubicin for 24 h before harvesting for flow analysis. (b) RKO (wild-type p53) and RKO E6 (p53 is targeted for degradation by HPV E6) colorectal carcinoma cells were treated with 0.01 and 0.05 μg/ml doxorubicin for 24 h before harvesting for flow analysis. The analysis cell profiles are shown for untreated and treated cells. The percentage of cells in G1, S, and G2/M are indicated in the upper right hand corner of each histogram.
Fig. 2.
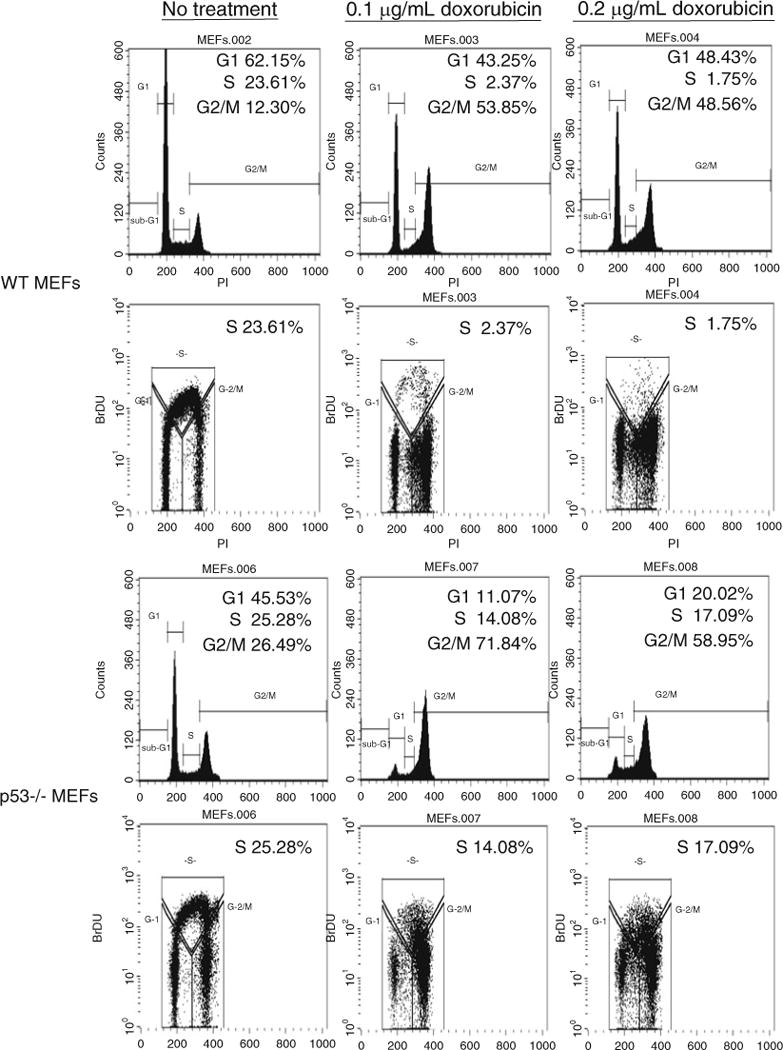
Treatment with doxorubicin in WT p53 mouse embryonic fibroblasts (MEFs) results in a significant reduction of S phase cells. Wild-type and p53−/− MEFs were treated with 0.1 and 0.2 μg/ml doxorubicin for 24 h prior to being pulsed with 10 μM BrdU for 4 h. Following antibody treatment and propidium iodide staining the cells were analyzed by dual parameter flow cytometry. Both single and dual parameter histograms are shown for each condition. The percentage of cells in G1, S, and G2/M are indicated in the upper right hand corner of each histogram.
Fig. 3.
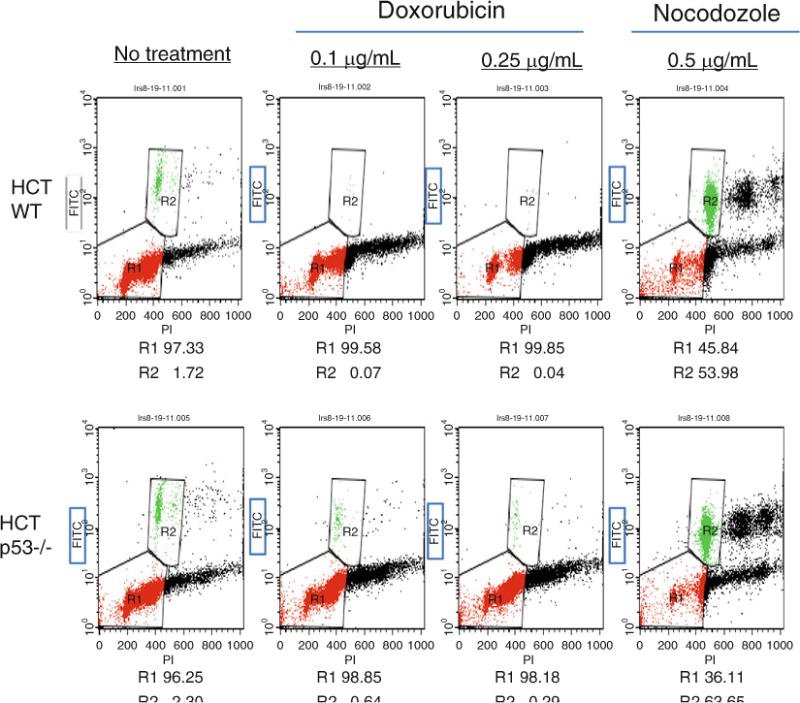
Phosphorylation of histone H3 is diminished after DNA damage with doxorubicin in WT p53 cells. HCT 116 p53 WT and −/− cells were treated with 0.1 and 0.25 μg/ml doxorubicin or 0.5 μg/ml nocodozole for 24 h and stained for Ser10-phosphorylated histone H3 and DNA content (propidium iodide). The dual parameter flow analysis is shown. Phosphorylated histone H3 positive cells are boxed and denoted as R2, whereas cells in G1 and G2 are grouped in R1.
Although the focus of this chapter is the use of flow cytometry to determine cell cycle position, it should be noted that other methods that include western analysis and immunofluorescence may be used to detect the expression of specific proteins that play roles in DNA damage and cell cycle arrest and apoptosis.
2. Materials
2.1. Cell Culture, Lysis, DNA Damaging Agent, and Mitotic Tubule Inhibitor
Dulbecco’s modified Eagle’s medium (DMEM with High Glucose) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals) that has been heat inactivated at 56°C for 30 min.
1× Dulbecco’s phosphate buffered saline without calcium and magnesium (DPBS).
0.05% Trypsin–ethylenediamine tetraacetic acid (EDTA).
Doxorubicin (Sigma-Aldrich, St. Louis, MO) is dissolved in dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St. Louis, MO) to yield a concentration of 1 mg/ml. A sterile solution is prepared by passage through a 0.2 μm filter and aliquots are stored at −20°C. A subsequent dilution of the stock in sterile H2O, 0.1 mg/ml, may be prepared and also stored at −20°C. This can be added directly to tissue culture cells at the appropriate concentration.
Nocodozole (Sigma-Aldrich, St. Louis, MO) is dissolved in distilled water to yield a 500 μg/ml stock that is passed through a 0.2 μm filter and stored at −20°C.
2.2. Propidium Iodide Staining for FACS Analysis
Dulbecco’s phosphate buffered saline without calcium and magnesium (1× DPBS).
Dulbecco’s modified Eagle’s medium (DMEM with High Glucose) is supplemented with 10% FBS (Atlanta Biologicals) that has been heat inactivated at 56°C for 30 min.
0.05% Trypsin–EDTA.
70% Ethanol (ETOH) is prepared from reagent grade, 190 proof (95%) ethanol and diluted with distilled water.
Propidium iodide (PI) (Sigma-Aldrich, St. Louis MO). A 100×stock (2 mg/ml in PBS) is prepared and stored at 4°C in a foil-covered tube for protection from the light.
Bovine pancreatic ribonuclease A (RNase A) (Sigma, St. Louis, MO) is stored at −20°C and for each use an appropriate amount is added fresh to PI/PBS solution.
Polystyrene round-bottom 12 × 75 mm (5 ml) Falcon tubes (BD Falcon, Franklin Lakes, NJ).
2.3. (+)-5-Bromo-2-Deoxyuridine (BrdU) Incorporation
BrdU (Sigma-Aldrich, Fairlawn, NJ) is prepared at a concentration of 10 mM in distilled H2O and stored at −20°C.
1× DPBS.
1% Bovine serum albumin (BSA)/DPBS.
70% ETOH is prepared from reagent grade, 190 proof (95%) ethanol and diluted with distilled H2O.
FITC-conjugated Anti-BrdU antibody (Becton Dickenson).
PI (Sigma Chemical Co.).
2 N HCl with 0.5% Triton-X-100 (v/v).
0.1 M Sodium tetraborate (Na2B4O7·10 H2O), pH 8.5.
0.5% Tween-20/1% BSA/DPBS.
12 × 75 Polystyrene Falcon tubes (BD Falcon, Franklin Lakes, NJ).
2.4. Phosphorylated Histone H3 Assay
0.5% Formaldehyde in 1 × DPBS.
90% Methanol is prepared with distilled H2O.
Anti-phosphorylated ser 10 histone 3 antibody (Cell Signaling) is diluted in DPBS.
RNase A (Sigma, St. Louis, MO) is stored at −20°C and for each use an appropriate amount is added fresh to antibody/PBS solution.
FITC-conjugated secondary antibody (Jackson Laboratories) is appropriately diluted in DPBS.
BSA.
3. Methods
3.1. Treatment with DNA Damaging Agent
3.1.1. Day 0
In preparation for treatment with specific DNA damaging agents, cell lines containing wild type, mutant, or no p53 are trypsinized and plated at an appropriate number into 60 mm or 100 mm tissue culture dishes to yield dishes that will be subconfluent the following day. It is best to start by seeding a sufficient number of dishes in order to do a dose–response curve for your drug.
3.1.2. Day 1
Doxorubicin (0.1 mg/ml) is thawed and added directly to cell cultures at a broad range of concentrations (0.01–0.5 μg/ml) to establish a dose that is suitable. The cell cultures are maintained for an additional 24 h at 37°C before harvesting (see Note 1).
3.2. Staining with Propidium Iodide and Preparation for FACS Analysis
3.2.1. Day 0 and Day 1
Follow the above given protocol.
3.2.2. Day 2
The treated and untreated cells are harvested by trypsinization, making sure to retain the medium from each dish. This prevents the loss of dying cells that lifted off of the dish as a result of the drug treatment. This medium can be used to neutralize the trypsin.
The cell suspensions are pelleted by centrifugation at 1,000 rpm for 5 min at 4°C.
The cell pellets are washed by resuspension in 1× DPBS and spun again at 1,000 rpm for 5 min at 4°C.
The supernatant is carefully removed and the pellet is gently dislodged. 1 ml of 70% ETOH is added dropwise to the cells while vortexing in order to disrupt the clumping of the cells (see Notes 2 and 3).
Once the cells are fixed they can be stored at −20°C for a minimum of 12 h or up to 2 weeks prior to PI staining.
3.2.3. Day 3
The fixed cells are centrifuged at 1,000 rpm for 5 min at 4°C.
The cells are rehydrated with DPBS and spun again at 1,000 rpm for 5 min at 4°C.
The pellet is resuspended in 20 μg/ml PI in PBS containing 1 mg/ml of RNase A and the cells are transferred to polystrene 12 × 75 mm Falcon tubes. Generally, 1–2 ml of the PI stain is adequate for 106 cells. The PI is prepared from a 100× stock (2 mg/ml) but the RNase is added fresh each time. The tubes are placed in the dark at room temperature for 2 h prior to FACS analysis. They may also be stored overnight at 4°C in the dark.
A BD BioSciences flow cytometer (FACSCalibur) is used to analyze samples for cell cycle position. Acquisition and analysis plots of 10,000 cells are generated using CellQuest software. The FL2 laser is employed for detection of the propidium iodide staining of the DNA.
3.3. Dual Parameter, BrdU Incorporation Assay, and Propidium Iodide Staining
3.3.1. Day 0
An appropriate number of cells are seeded for optimal growth in 100 mm tissue culture dishes.
3.3.2. Day 1
The cells are treated with doxorubicin. The stock (0.1 mg/ml) is thawed and added directly to cell cultures. A dose–response curve for each cell line should be performed in order to attain a dose that produces a p53-dependent response. This can be done as a single parameter experiment using PI before setting up a BrdU incorporation assay.
3.3.3. Day 2
BrdU is added directly to the culture medium to attain a final concentration of 10 μM. Washing the cells prior to addition of BrdU is not recommended because it might slow the growth of the cells and the incorporation of the analog. Plan on including a dish of cells that will not be pulsed with BrdU (see Note 4).
Incubate cells at 37°C for 45–60 min, but note that this time will vary depending on the doubling time of the cells.
The BrdU pulsed cells are trypsinized, making sure to save the medium so as not to lose cells that are no longer adhered to the tissue culture dish and spun for 5 min at 1,000 rpm.
The number of cells per sample is adjusted to approximately 106 by resuspension of the cell pellet in 1× PBS and spun again for 5 min at 1,000 rpm.
The pellet is retained and resuspended in 1 ml 70% ETOH. It is important to prevent clumping of cells by gently vortexing the pellet while adding ETOH.
The cells can be stored overnight at −20°C before proceeding. This step can be shortened but usually a minimum of 12 h in fixative is standard before the denaturation step.
The cells are centrifuged at 1,000 rpm for 5 min. 1 ml of 2 N HCL/0.5%Triton X-1000 (v/v) is slowly added to the pellet, a few drops at a time while maintaining a vortex. This will disrupt clumps of cells that may result from addition of the HCL/Triton X mixture. This treatment will denature the DNA and create single-stranded molecules.
Incubate at room temperature for 30 min.
The cell suspension is spun at 1,000 rpm for 5 min. The pelleted cells are resuspended in 1 ml of 0.1 M Na2B4O7·10H2O pH 8.5, a sodium tetraborate solution that neutralizes the acid.
You can pause at this point and store the BrdU-labeled cells at −20°C. If you choose to do this, the cells are centrifuged at 1,000 rpm for 5 min and the pellet is resuspended in 70% ETOH. Otherwise you can proceed directly from steps 9 to 11.
The cells are spun down at 1,000 rpm for 5 min and the pellet is resuspended in 500 μl 0.5% Tween-20/1% BSA/PBS containing an appropriate concentration of anti-BrdU-FITC secondary antibody (Becton Dickinson).
The cells are maintained at room temperature (RT) and protected from the light for 30 min.
The cells are centrifuged at 1,000 rpm for 5 min, followed by resuspension of the pellet in 1 ml of PBS containing 10 μg/ml propidium iodide. The labeled cells are transferred into 12 × 75 mm polystyrene Falcon tubes and stored at RT in the dark for 2 h.
The samples are analyzed for cell cycle position using a BD BioSciences flow cytometer (FACSCalibur). Although the excitation, 488 nm, is the same for both stains, the FL1 and FL3 detection lasers can discriminate between the emission of the green fluorescent FITC stain and the red propidium iodide staining of the DNA. CellQuest software is used to generate the acquisition and analysis plots of 10,000 cells.
3.4. Phosphorylated Histone H3 Assay
3.4.1. Day 0
An appropriate number of cells are seeded for optimal growth in 100 mm tissue culture dishes.
3.4.2. Day 1
The cells are treated with doxorubicin. The stock (0.1 mg/ml) is thawed and added directly to cell cultures. A dose–response curve for each cell line should be performed previously in order to attain a dose that produces a p53-dependent response. To aid in analysis, a dish of cells should also be treated with nocodozole at a final concentration of 500 ng/ml (see Note 5). This agent is an inhibitor of microtubules and will result in a population of cells that are arrested in M phase. The cell cultures are maintained for an additional 24 h at 37°C.
3.4.3. Day 2
The cells are trypsinized, making sure to save the medium that may contain cells that are no longer adhered to the tissue culture dish, and centrifuged for 5 min at 1,000 rpm.
The pellets are resuspended in PBS and spun down for 5 min at 1,000 rpm.
The pelleted cells are fixed in 1 ml of 0.5% formaldehyde for 10 min at 37°C.
The fixed cells are spun out at 1,000 rpm for 5 min and washed in 1 ml PBS to dilute out the fixative.
The cells are spun again at 1,000 rpm for 5 min and permeabilized using 1 ml of 90% methanol.
The cells are incubated on ice for 30 min or can be stored at −20°C for up to 10 days.
The cells are centrifuged and resuspended in PBS.
Once again the cells are centrifuged at 1,000 rpm for 5 min followed by resuspension in the primary antibody, anti-phosphorylated Ser 10 histone 3. The antibody is diluted according to the manufacturer’s suggestion in PBS that contains 100 μg/ml RNase A.
The cells are incubated for 1 h with occasional shaking at RT.
The cells are pelleted and subsequently washed twice with 1% BSA/PBS.
After centrifugation the cells are taken up in the secondary, anti-FITC-conjugated antibody that is diluted accordingly in PBS.
The cells are incubated for 1 h at RT in the dark.
The samples are centrifuged for 5 min at 1,000 rpm and resuspended 1% BSA/PBS.
Once again the cells are centrifuged and resuspended in PBS containing 20 μg/ml propidium iodide and transferred into polystyrene 12 × 75 mm Falcon tubes.
The stained cells are incubated in the dark at RT for 30 min.
A FACS brand flow cytometer can be used for the analysis of the labeled samples. FL1 and FL3 detectors are selected to distinguish between the emission of PI and FITC. CellQuest software is used to generate the acquisition and analysis histograms of labeled cells.
Acknowledgments
The author is appreciative to Melissa Mattia-Sansorbrino and Emir Senturk for their contributions to the figures presented in this chapter. The authors are supported by grants from the National Cancer Institute (P01 CA080058, R01 CA125741, and R01 CA086001).
Footnotes
The cellular response to a particular DNA damaging drug will vary for individual cell lines. Therefore it is important to empirically derive a concentration that will produce a biologic effect that is p53 dependent. It is important to establish a concentration curve of each drug for each cell line. Moreover it is a good idea to use parallel dishes to harvest for protein. An SDS-PAGE gel can be run, blotted, and probed for p53 and its target genes. One can then determine at which dose p53 is being induced and at what levels the response remains p53 dependent.
Cells are impermeable to PI. Therefore for this assay, the cells must be fixed so that the PI stain can penetrate the cells and intercalate into the DNA. Although ETOH is used, paraformaldehyde can also be used and might be preferable, when you want to prevent transfected proteins, like green fluorescent protein (GFP) from leaking out of the cell.
Some cell lines tend to be very sticky and are prone to clumping. Use of 12 × 75 mm Falcon polystyrene tubes with strainer caps will filter out clumps of cells from the final cell suspension.
A sample should be included to which BrdU has not been added. This control will be helpful when gating cells in the analysis portion of this assay.
Nocodozole-treated cells will serve as a positive control for the mitotic population that contains phosphorylated histone H3. This control will aid in proper gating in the analysis.
References
- 1.Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- 2.Paulovich AG, Toczyski DP, Hartwell LH. When checkpoints fail. Cell. 1997;88:315–321. doi: 10.1016/s0092-8674(00)81870-x. [DOI] [PubMed] [Google Scholar]
- 3.Kern SE, Kinzler KW, Bruskin A, Jarosz D, Friedman P, Prives C, Vogelstein B. Identification of p53 as a sequence-specific DNA-binding protein. Science. 1991;252:1708–1711. doi: 10.1126/science.2047879. [DOI] [PubMed] [Google Scholar]
- 4.Pietenpol JA, Tokino T, Thiagalingam S, el-Deiry WS, Kinzler KW, Vogelstein B. Sequence-specific transcriptional activation is essential for growth suppression by p53. Proc Natl Acad Sci USA. 1994;91:1998–2002. doi: 10.1073/pnas.91.6.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989;57:1083–1093. doi: 10.1016/0092-8674(89)90045-7. [DOI] [PubMed] [Google Scholar]
- 6.Burns TF, El-Deiry WS. The p53 pathway and apoptosis. J Cell Physiol. 1999;181:231–239. doi: 10.1002/(SICI)1097-4652(199911)181:2<231::AID-JCP5>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 7.Bargonetti J, Manfredi JJ. Multiple roles of the tumor suppressor p53. Curr Opin Oncol. 2002;14:86–91. doi: 10.1097/00001622-200201000-00015. [DOI] [PubMed] [Google Scholar]
- 8.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 9.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 10.Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55:5187–5190. [PubMed] [Google Scholar]
- 11.Waga S, Hannon GJ, Beach D, Stillman B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature. 1994;369:574–578. doi: 10.1038/369574a0. [DOI] [PubMed] [Google Scholar]
- 12.Agarwal ML, Agarwal A, Taylor WR, Stark GR. p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc Natl Acad Sci USA. 1995;92:8493–8497. doi: 10.1073/pnas.92.18.8493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flatt PM, Tang LJ, Scatena CD, Szak ST, Pietenpol JA. p53 regulation of G(2) checkpoint is retinoblastoma protein dependent. Mol Cell Biol. 2000;20:4210–4223. doi: 10.1128/mcb.20.12.4210-4223.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998;282:1893–1897. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- 15.Taylor WR, DePrimo SE, Agarwal A, Agarwal ML, Schonthal AH, Katula KS, Stark GR. Mechanisms of G2 arrest in response to overexpression of p53. Mol Biol Cell. 1999;10:3607–3622. doi: 10.1091/mbc.10.11.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 17.Chan TA, Hermeking H, Lengauer C, Kinzler KW, Vogelstein B. 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature. 1999;401:616–620. doi: 10.1038/44188. [DOI] [PubMed] [Google Scholar]
- 18.Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ., Jr A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 19.Malkin D. p53 and the Li-Fraumeni syndrome. Cancer Genet Cytogenet. 1993;66:83–92. doi: 10.1016/0165-4608(93)90233-c. [DOI] [PubMed] [Google Scholar]