

Abstract
Transcription factors influence gene expression through their ability to bind DNA at specific regulatory elements. Specific DNA-protein interactions can be isolated through the chromatin immunoprecipitation (ChIP) procedure, in which DNA fragments bound by the protein of interest are recovered. ChIP is followed by high-throughput DNA sequencing (Seq) to determine the genomic provenance of ChIP DNA fragments and their relative abundance in the sample. This chapter describes a ChIP-Seq strategy adapted for budding yeast to enable the genome-wide characterization of binding sites of transcription factors (TFs) and other DNA-binding proteins in an efficient and cost-effective way.
Yeast strains with epitope-tagged TFs are most commonly used for ChIP-Seq, along with their matching untagged control strains. The initial step of ChIP involves the cross-linking of DNA and proteins. Next, yeast cells are lysed and sonicated to shear chromatin into smaller fragments. An antibody against an epitope-tagged TF is used to pull down chromatin complexes containing DNA and the TF of interest. DNA is then purified and proteins degraded. Specific barcoded adapters for multiplex DNA sequencing are ligated to ChIP DNA. Short DNA sequence reads (28–36 base pairs) are parsed according to the barcode and aligned against the yeast reference genome, thus generating a nucleotide-resolution map of transcription factor-binding sites and their occupancy.
Keywords: ChIP-Seq, ChIP, Yeast, Chromatin, Transcription factor, Binding site, Genomics, Multiplex
1 Introduction
Gene expression is often regulated by the binding of transcription factors (TFs) to specific DNA sequences within intergenic regions termed transcription factor-binding sites. The genome of the budding yeast Saccharomyces cerevisiae contains about 6,000 predicted ORFs, of which 200–300 encode TFs [1]. Transcription factors bind preferentially to regions containing a consensus motif, enabling computational prediction of putative binding sites. However, these predictions must be validated experimentally [2], as many regions with perfect consensus motifs can remain unbound while those displaying imperfect motifs can show high level of protein binding [3]. The method of choice for validation of TF binding to DNA, chromatin immunoprecipitation (ChIP), was first developed to characterize RNA polymerase II binding in bacteria [4]. Briefly, DNA-protein complexes are covalently cross-linked by formaldehyde. Cross-linked yeast cells are lysed, and the lysates are then sonicated to shear chromatin fragments into smaller pieces, amenable to subsequent immunoprecipitation (IP) [5]. Antibodies raised against the TF of interest, or against a specific epitope (if the TF is epitope tagged) are used to recover DNA-protein complexes containing the TF of interest. DNA is purified from proteins by reversing the cross-links using heat, followed by proteinase K protein degradation of the proteins. Enrichment for regions bound by a particular TF can be determined by PCR quantification, comparing a yeast strain with a TF-epitope fusion to its isogenic control strain, either to a ChIP in the untagged parental strain or to a mock IP if using a TF-specific antibody. PCR detection is not suitable to discovery of novel binding regions given the low throughput and need for specific primers for amplification. With the development of DNA microarrays, it became possible to query the entire genome for sites bound by a particular TF, using a ChIP approach coupled to hybridization of the recovered DNA to microarrays [6]. This technology, called ChIP-chip, has been successful to identify globally transcription factor-binding profiles [7]. Recently, massively parallel, high-throughput sequencing technologies such as Roche’s 454, Illumina’s Genome Analyzer and HiSeq, LifeTechnologies’ SOLiD and IonTorrent, Helicos’ HeliScope, Pacific Biosciences’ PacBio RS, and Complete Genomics’ DNA nanoball sequencing have revolutionized large-scale genomics projects by generating millions of short DNA sequence reads in a few days, at single-nucleotide resolution. ChIP followed by high-throughput sequencing (ChIP-Seq) has emerged as a powerful method to discover and characterize functional elements of any genome, and was first developed for mammalian applications [8, 9]. The reduced background, decreasing cost of sequencing, lack of cross-hybridization, increased sensitivity, single-nucleotide resolution, and high dynamic range are among the advantages that helped to establish ChIP-Seq over ChIP-chip as the current gold standard in gene regulation studies [10]. The multinational consortiums, ENCODE in humans [11], and modENCODE in worms and flies [12], have taken advantage of novel sequencing technologies to characterize the entire repertoire of functional genomic elements. While the initial transition from ChIP-chip to ChIP-Seq quickly gained momentum in higher eukaryotes, ChIP-Seq studies in organisms with smaller genome remained rare, given high cost per sample and excessive generation of sequence reads compared to the number required to map binding sites at high confidence [13]. Our group developed a multiplex ChIP-Seq strategy to process multiple samples simultaneously and in a cost-effective way, which was used to characterize the distribution of several DNA-binding proteins, including RNA polymerase II, the centromeric histone H3-variant Cse4, and the TF Ste12 [13]. ChIP-Seq experiments in yeast have been valuable in determining the effect of chromatin structures during ChIP that affect several organisms [14, 15]. Novel binding sites were found for key transcription factors involved in various important cellular functions, including ribosome biogenesis [16, 17], transcriptional silencing [18], noncoding RNA regulation [19], ubiquitination [20], stress response [21], metabolism [22], and DNA replication [23] among others. In particular, the sensitivity of yeast ChIP-Seq can be exploited to characterize on a wide spectrum the variation among individuals, at the level of transcription factor binding [24]. ChIP-Seq studies in other fungi, such as Saccharomyces bayanus [18], Cryptococcus neoformans [25], and Neurospora crassa [26], are also feasible given that high-quality genome assemblies exist. In the future, given the rapid development in sequencing technologies, more ChIP-Seq samples could be analyzed in parallel using increased multiplexing [27], all at a lower cost. Challenges still remain for data storage and, very importantly, for the development of simple and fast, yet efficient and precise, computational tools and algorithms that can be used daily by bench scientists [28].
This chapter details a strategy to perform ChIP-Seq in yeast (Fig. 1). It first describes chromatin immunoprecipitation in yeast to prepare ChIP DNA from yeast strains with epitope-tagged TFs. Next, we present the protocol to generate high-quality Illumina DNA sequencing libraries for subsequent high-throughput sequencing on Illumina’s Genome Analyzer IIx, with a special focus on a multiplexing strategy. Briefly, purified ChIP DNA is ligated to barcoded DNA adapters and then PCR-amplified for a few cycles. Multiplex sequencing libraries are mixed in equimolar ratio. This mixture is added to a single Illumina flowcell lane using a cluster station. During this step, single-DNA molecules are amplified to form a cluster containing a 1,000 identical clones. Clusters are then submitted to sequencing-by-synthesis, during which fluorescently labeled nucleotides with a reversible terminator are incorporated at each sequencing cycle. Finally, many aspects of ChIP-Seq data analysis are covered, including alignment of short sequence reads, peak calling for identification of TF-binding sites, data visualization, and some downstream analyses.
Fig. 1.
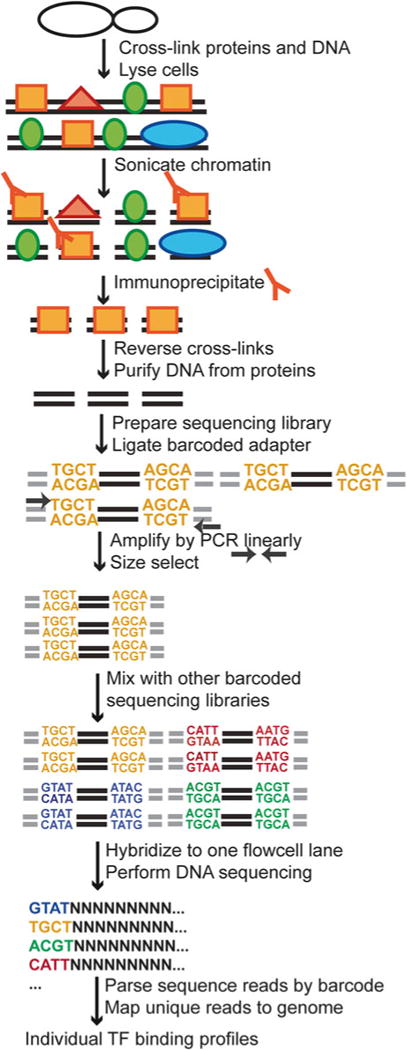
Overview of the ChIP-Seq procedure in budding yeast, focusing on a multiplex high-throughput DNA sequencing approach on the Illumina platform
2 Materials
Yeast cells grown in the appropriate liquid medium, in a 2 L Erlenmeyer flask.
37 % formaldehyde.
2.5 M glycine (sterile).
Nuclease-free water.
1 L PES 0.2 μm filter unit.
0.5 mm zirconium/silica beads.
Syringe needle.
Lysis/IP buffer: 50 mM Hepes/KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1 % Triton X-100, 9. 0.1 % sodium deoxycholate.
100 mM PMSF (phenylmethanesulfonylfluoride).
Roche Complete protease inhibitor cocktail tablet (Roche).
FastPrep-24 machine (MP Biomedicals).
Branson Digital 450 Sonifier (Branson).
Sigma EZview anti-Myc or anti-HA affinity gel (Sigma) or Pan mouse IgG dynabeads (Invitrogen).
Lysis/500 mL NaCl buffer: 18 mL 5 M NaCl, 232 mL of lysis/IP buffer.
IP wash buffer: 10 mM Tris–HCl, 0.25 M LiCl, 0.5 % NP-40, 0.5 % sodium deoxycholate, 1 mM EDTA.
1× TE: 50 mM Tris–HCl, 10 mM EDTA pH 8.0.
1× TE/1 % SDS solution.
1× TE/0.67 % SDS solution.
Rocking mixer.
20 mg/mL proteinase K.
45 and 65 °C water baths or heat blocks.
100 and 70 % ethanol.
20 mg/mL glycogen.
Pellet paint Co-Precipitant (Novagen).
5 M LiCl.
MinElute PCR purification kit (Qiagen), including EB buffer and MinElute column.
Nanodrop spectrophotometer (Thermo Scientific) or Qubit fluorometer (Invitrogen).
15 and 50 mL Falcon conical tubes.
5 mL Falcon snap-cap conical tubes.
2 mL screw-cap microcentrifuge tubes.
1.5 mL microcentrifuge tubes.
Reagents for generation of barcoded sequencing libraries.
-
High-throughput DNA sequencer and its data analysis suite.
For ChIP-Seq analysis on the Illumina Genome Analyzer IIx:
Annealing buffer: 10 mM Tris–HCl pH 7.5, 50 mM NaCl, 1 mM EDTA.
Agarose gel electrophoresis apparatus.
Clean scalpel or razor blades or 18 cm × 18 cm glass cover slips.
Gibco RNase-free DNase-free water.
End-It DNA end repair kit (Epicentre), including End-repair enzyme mix, 10× End-repair buffer, 10 mM ATP, 2.5 mM dNTP mix.
QIAquick PCR purification kit (Qiagen), including EB buffer and QIAquick column.
Klenow fragment of DNA polymerase I (3′→5′ exonuclease activity minus) and 10× NEBuffer2 (New England Biolabs).
100 mM dATP.
96-Well PCR plates (ABgene).
Microseal A Film sealing microfilms (MJ Research).
Illumina genomic DNA adapter oligonucleotides augmented by a short index (or barcode).
LigaFast T4 DNA ligase and 2× DNA ligase buffer (Promega).
Track-It 50 bp DNA ladder and Track-It Cyan/Orange loading buffer (Invitrogen).
2 % agarose single-comb 12-well E-Gel, stained with ethidium bromide or SYBR safe (Invitrogen).
E-Gel electrophoresis apparatus and visualization system (Invitrogen).
Illumina genomic DNA primers 1.1 and 2.1.
2× Phusion master mix with HF buffer (New England Biolabs).
MinElute and QIAquick gel extraction kits (Qiagen).
Illumina Genome Analyzer sequencer, with reagents, flowcell, and cluster station.
Genome Analyzer Pipeline (Illumina) and other DNA sequencing analysis software and tools.
3 Methods
3.1 Chromatin Immunoprecipitation
Tag the transcription factor of interest by fusing a Myc or HA epitope at the C-terminus (see Note 1).
Grow 500 mL cultures of yeast cells in the appropriate media to exponential phase (OD600 between 0.6 and 1.0). This represents about 4.5 × 109 cells per sample. Perform experiments in biological triplicates and process in parallel samples from epitope-tagged TF strains and untagged strains as an experimental control. Note 2 lists several modifications or alternative manipulations to the ChIP protocol described within Subheading 3.1.
Cross-link DNA and proteins by adding 14 mL of 37 % formaldehyde for 15 min, with vigorous swirling every 5 min (see Note 3).
Add 27 mL of 2.5 M glycine for 10 min to quench the reaction. Swirl at least twice vigorously.
Collect cells by filtration in 1 L filter unit. Wash cells twice with 100 mL nuclease-free water.
Transfer cells from the filter to a 50 mL Falcon conical tube with 10 mL sterile water and repeat this step. Centrifuge cells at room temperature for 10 min at 3,500 RPM in tabletop centrifuge and discard the supernatant.
Resuspend the pelleted cells in 1 mL sterile water and transfer them to 2 mL screw-cap tubes. Repeat this step. Both tubes should contain similar volumes.
Spin down cells at full speed (~16,000 × g) in a microfuge for 3 min and aspire the supernatant out. Weigh cells. Each tube should contain about 0.2–0.3 g of cells. Add approximately 1 mL of zirconium/silica beads. At this point, cell pellets can be frozen at −70°C for subsequent use.
Combine 50 mL lysis/IP buffer solution with 1 Roche Complete protease inhibitor tablet. Mix well until tablet is resuspended. Prior to use, add 0.5 mL 100 mM PMSF and keep on ice. 50 mL of this mixture can be used for six ChIP samples.
Add 1 mL of the lysis/IP solution from step 9 to each cell pellet.
Lyse cells with the FastPrep-24 machine (see Note 4). Perform lysis using five 1-min bursts at a speed of 6.0 m/s. Keep cells in an ice-water bath during the 5-min rest period.
Pierce the bottom of the 2 mL screw-cap tube with a needle and place in a cap-less 5 mL snap-cap Falcon tube. Recover lysates by centrifuging for 3 min at 400 × g in tabletop centrifuge with swinging bucket rotor. Add 0.5 mL of lysis/IP solution from step 9 and spin again. Collect the lysate from the other 2 mL tube originating from the same replicate using the same procedure.
Transfer lysates to a 15 mL Falcon conical tube. Rinse the 5 mL tube with 1 mL lysis/IP solution and pool to the same 15 mL tube. The volume prior to sonication should be slightly above 4 mL.
Shear chromatin by sonicating lysates five times using a Branson Digital 450 sonifier (see Note 5). Set the amplitude at 50 % and time ON and OFF at 30 s. Keep the sonicator tip 0.5–1.0 cm from the bottom of the tube to reduce foaming. Perform sonication for groups of biological replicates together (e.g., alternate between the three untagged replicates for all cycles and then alternate between the three tagged replicates). Put samples on ice for at least 2 min between sonication rounds.
Clarify sonicated lysates by spinning at 4 °C for 5 min at 1,620 × g in tabletop centrifuge and transfer each replicate to three 1.5 mL microcentrifuge tubes.
Clarify lysates at 16,000 × g in microfuge for 10 min at 4 °C and pool all supernatants to a 15 mL conical tube. Make sure to avoid cell debris at the bottom of the tube. Add 2 mL of lysis/IP buffer. The total volume of each sample should be around 6 mL.
Set aside 250 μL of clarified, sonicated lysate before immunoprecipitation to process as input DNA, a reference sample for ChIP-Seq enriched for open chromatin (see Note 6) [14, 15].
Wash the entire bottle of Sigma EZview anti-Myc or anti-HA affinity gel with 1 mL lysis/IP buffer. Transfer antibody-coupled beads to a 15 mL Falcon tube using a broadened 1 mL pipette or large orifice tips. Wash the bottle with 1 mL lysis/IP solution three times and transfer to the 15 mL tube. Vortex and centrifuge for 2 min at 720 × g at 4 °C in tabletop centrifuge. Discard supernatant. Add 5 mL lysis/IP buffer and repeat vortexing, centrifugation, and removal of supernatant three times. Resuspend beads in 1 mL fresh lysis/IP solution.
Pipet 150–300 μL of pre-washed antibody bead solution to each clarified sonicated lysate from the end of step 16. Perform immunoprecipitation at 4 °C on a rocking mixer overnight (between 12 and 16 h) for Myc- or HA-tagged TF strains and their untagged control strains (see Note 7).
After immunoprecipitation, fill Falcon tubes with lysis/IP buffer, pellet antibody-coupled beads by centrifugation at 720 × g for 5 min at 4 °C, and remove supernatant.
Resuspend pelleted beads in 600 μL of lysis/IP buffer and transfer to a 1.5 mL microfuge tube using a broadened 1 mL pipette. Repeat procedure and pool in the same tube. Mix on a rocker in the cold room for 5 min and discard carefully the supernatant, with a small pipette attached to an aspirator.
Perform the following washes sequentially with 1 mL of the appropriate buffer for 5–10 min on a rocking mixer at 4 °C, followed by a 1-min, 845 × g centrifugation at 4 °C and gentle removal of the supernatant by aspiration. Wash twice in lysis/IP buffer, once in lysis/500 mM NaCl buffer, twice in IP/wash buffer, and once in 1× TE buffer. Remove completely the small amount of TE from the beads.
Add 100–150 μL of 1× TE/1 % SDS solution to the immunoprecipitate to allow elution of chromatin complexes from the beads and mix. Incubate in a 65 °C water bath for 15 min, mixing briefly after 10 min (see Note 8). Pellet beads at 16,000 × g for 1 min and transfer supernatant (the eluate) to a 1.5 mL tube.
Repeat elution of the immunoprecipitate by adding 150–200 μL of 1× TE/0.67 % SDS solution and mix. Incubate in a 65 °C water bath for 10 min. Perform centrifugation and eluate transfer as described in step 23.
Spin down the eluate at 16,000 × g in microfuge for 2 min to send any residual antibody-coupled bead to the bottom of the tube and transfer the supernatant to a 2 mL screw-cap tube. Great care should be taken to avoid the last 5–20 μL fraction that may contain antibody beads (see Note 8).
Incubate eluates at 65 °C at least for 6–8 h or overnight. Cross-link reversal by heat treatment is crucial for isolating DNA from covalently bound DNA-protein complexes.
Briefly chill samples on ice for a few seconds. Dilute proteinase K solution in 1× TE to a final concentration of 0.4 mg/mL. Add 250 μL of the proteinase K/TE solution and incubate samples in a 42 °C water bath (37–50 °C works as well) for 2–4 h to ensure sufficient digestion of proteins.
After proteinase K treatment, add 3 μL pellet paint, 3 μL of 20 mg/mL glycogen, and 45 μL 5 M LiCl. Mix briefly. Add 1 mL of 100 % ethanol (more if it fits in 2 mL tube) and mix very well. Precipitate DNA overnight or several hours at −20 °C.
Finish DNA precipitation by placing samples at −70 °C for 1 h and centrifuge for 20 min at 16,000 × g at 4 °C in microfuge. A pinkish-white pellet should be visible. Discard supernatant.
Wash pellet with 1 mL 70 % ethanol for 5 min. Spin for 10 min at 16,000 × g at 4 °C and remove ethanol thoroughly.
Air-dry for 10 min or vacuum dry for 3–4 min. Resuspend completely in 100 μL TE.
Purify ChIP samples through MinElute columns. Each ChIP DNA sample is split between two MinElute columns. Elute with 21 μL EB and pool eluates from the same sample to a single tube.
Measure DNA concentration using a Nanodrop spectrophotometer (see Note 9).
Perform qPCR analysis of ChIP samples to ensure that the ChIP procedure worked properly, prior to the generation of a sequencing library and further computational analysis of ChIP-Seq samples (see Note 10).
3.2 Generation of Illumina DNA Sequencing Libraries from ChIP Samples
Use at least 100 ng of ChIP DNA from step 32 of previous protocol (or input DNA isolated from step 17 and processed as described in Note 6) to generate a sequencing library for a given sample. 150–250 ng of ChIP DNA (equivalent to about a quarter to half of the total volume of ChIP sample), or input DNA, is sufficient for preparation of high-quality Illumina sequencing libraries. The amount of starting DNA can be increased if previous generation of a sequencing library failed.
Optional: Isolate by agarose gel electrophoresis a ChIP DNA smear between 100 and 700 bp (see Note 11). We run samples on a 2 % gel at 100–110 V for about 20 min to minimize gel volume. Purify DNA using a QIAquick gel extraction procedure. Elute DNA on the QIAquick column with 34 μL EB.
End-repair ChIP DNA using the End-It DNA end repair kit, by combining 34 μL ChIP DNA, 5 μL 10 × End-It repair buffer, 5 μL 10 mM ATP, 5 μL 2.5 mM dNTP mix, and 1 μL End-It repair enzyme (see Note 12). Incubate at room temperature for 45 min.
After the incubation period, purify end-repaired DNA through a QIAquick column using the QIAquick PCR purification kit protocol. Elute in 34 μL EB.
Incorporate a single “A” nucleotide to the 3′ ends of end-repaired ChIP fragments using the Klenow fragment of DNA polymerase I (3′ −5′ exo minus), by mixing in a PCR plate 34 μL DNA from previous step, 5 μL 10 × NEBuffer2, 10 μL 1 mM dATP, and 1 μL Klenow (3′ →5′ exo minus) (see Note 13). Seal with a microfilm and incubate at 37 °C for 30 min in a PCR machine, without a heated lid (see Note 13).
After the reaction is completed, purify DNA through a MinElute column using the MinElute PCR purification kit procedure. Elute in 12.5 μL EB.
Dilute standard Illumina genomic DNA adapter oligonucleotide mix 1:40 (for ChIP samples, this is particularly important, use 1 μL of ~0.025–0.050 μM annealed barcoded adapters). For custom-made barcoded adapters for multiplex high-throughput sequencing, dilute annealed barcoded adapters to the working concentration used for standard Illumina genomic DNA adapters [13] (see Note 14 for barcoded adapter design, annealing, and determination of oligo concentration).
Ligate diluted adapters to each sample, with the following components: 12.5 μL DNA from step 6, 15 μL 2 × DNA ligase buffer, 1 μL diluted barcoded adapter oligo mix, and 1.5 μL LigaFast T4 DNA ligase enzyme. Incubate for 15 min at room temperature.
Remove enzyme and buffers using the MinElute PCR purification kit and elute in 12 μL EB.
Size-select DNA between 150 and 500 bp, getting rid of adapter-adapter dimers. Pre-run a 2 % agarose E-Gel, leaving the combs in, if needed. Make a 1:10 dilution of the Track-It 50 bp ladder and Track-It loading buffer. Add 3 μL of diluted Track-It loading buffer to each sample from the previous step. Remove E-Gel comb and load 20 μL diluted 50 bp ladder. Pipet samples containing the loading dye in individual wells, separating them by at least 1 or 2 empty rows and avoiding the last row. Load 15 μL of Gibco nuclease-free water in all empty wells. Perform gel electrophoresis for 18–20 min. Visualize gel and mark the location of 150–500 bp fragments (see Note 15). Open E-Gel with a large spatula and excise each gel fragment with a clean disposable scalpel blade. Extract DNA using the QIAquick gel extraction kit (see Note 15) and elute in 28 μL EB.
PCR-amplify ChIP DNA with ligated adapters using Illumina genomic DNA primers 1.1 and 2.1. Combine and mix well 28 μL DNA from previous step, 1 μL of 1:1 diluted Illumina genomic DNA primer 1.1, 1 μL of 1:1 diluted Illumina genomic DNA primer 2.1, and 30 μL 2 × Phusion HF polymerase master mix, and then transfer to a PCR plate. Amplify for 15–17 cycles using the following PCR parameters (see Note 16): 1 cycle of initial denaturation for 30 s at 98 °C, 15–17 cycles of amplification (denaturation for 10 s at 98 °C, annealing for 30 s at 65 °C, and extension for 30 s at 72 °C), a final extension for 5 min at 72 °C, and a cool down held indefinitely at 4 °C.
Purify PCR products through a MinElute column and elute in 12–15 μL EB.
Perform size selection of the final Illumina DNA sequencing library between 150 and 350 bp, using a 2 % agarose E-gel and the gel loading and band excision procedures described in step 10. Run the gel for 18–20 min. A bright DNA smear should appear from 150 bp to slightly below 500 bp (see Note 17 for additional details). Extract DNA from agarose gel band with a MinElute gel extraction kit and elute in 20–25 μL EB, respecting gel extraction tips given in Note 15.
Measure DNA concentration using a Nanodrop spectrophotometer or a Qubit fluorometer (see Note 18). After this step, store sequencing libraries at − 70 °C if sample submission to a sequencing facility will occur later.
Mix barcoded sequencing libraries in equimolar ratios, using DNA concentrations from previous step, to ensure appropriate representation of samples sequenced simultaneously in the same Illumina flowcell lane (see Note 18).
Run library on Bioanalyzer, and submit library, comprising a mixture of individual barcoded libraries, for multiplex high-throughput DNA sequencing on the Illumina Genome Analyzer IIx platform (see Note 19). Sequence the amplified, adapter-ligated ChIP DNA, which will be preceded by a unique short identifier (barcode).
3.3 ChIP-Seq Data Analysis
For Illumina Genome Analyzer IIx, run the Genome Analyzer Pipeline software, which comprised three modules. First, Firecrest performs image analysis and fine-tune cluster locations. Next, Bustard is the base caller: it calculates the occurrence probability of a nucleotide at a given cluster, considering the pixel intensities of four images taken during each sequencing cycle (A, C, G, and T). A final sequence read of 32–36 bp is generated, consisting of the sequence of nucleotides that were called with maximal likelihood. Of particular importance are quality scores generated by Bustard to assess the quality of individual reads and exclude reads for downstream analysis. Finally, Gerald uses the ELAND algorithm to align reads against the reference genome, allowing up to two mismatches. Prior to barcode parsing and their subsequent removal, one should not pay attention to alignment metrics.
For barcoded samples, parse sequence reads from the ELAND query file according to the index, into distinct bins. Discard sequence reads without an intact barcode into a separate bin, but include their number in the calculation of mapping values. After reads have been separated by barcode, remove this short tag. Align the residual bases (~26–30) against the latest version of the yeast genome, using ELAND’s stand-alone mode and allowing up to two mismatches (see Note 20 for typical mapping values). Open-source programs for short-read alignments often contain additional capabilities, such as SNP calling (MAQ [29]) or short gapped alignments (SOAP [30]). The popular short-read aligners Bowtie [31] and BWA [32] are part of open-source high-throughput sequencing program suites that can perform various tasks, such as analysis of gene expression, SNP calling, isoform assembly, or TF-binding site peak calling (see Note 21).
Visualize ChIP-Seq profiles in the Integrated Genome Browser [33]. First, convert the eland_results file to a suitable file type (.sgr or .wig is common, .bam can be loaded too). This operation determines the number of sequence reads mapping to every nucleotide position, thus generating a signal file. Select the correct S. cerevisiae genome version from the pull-down menu or import a genome sequence file. Annotations and other genome features from the Saccharomyces Genome Database (SGD) can be loaded as well. Explore and compare ChIP-Seq profiles (experimental TF-tagged strains vs. control untagged strains, or other relevant control) to RNA-Seq data, and open chromatin regions and other omics data that can be loaded in IGB (see Note 22).
Determine transcription factor-binding sites using a peak scoring algorithm. These programs were designed for determination of ChIP-Seq-binding regions across mammalian genomes, but simple modifications of key parameters can usually enable yeast-specific ChIP-Seq analysis (see Note 23). Distinctions between different algorithms usually concern how background is modeled or extracted from ChIP-Seq data, as well as the consideration of nonrandom artifacts and systematic biases. Given their different statistical treatment of random background and distinct criteria and thresholds for peak detection, the performance of peak-calling algorithm varies when dealing with identical samples [34]. In yeast, experimental ChIP-Seq data from epitope-tagged TF strains are scored either against a set of matched control ChIP-Seq experiments from untagged strains [16, 27], against a mock IP control if using an antibody directly against the TF of interest, or against Sono-Seq/Input-Seq experiments generated from input DNA, representing non-immunoprecipitated, cross-linked, sheared chromatin [14, 15, 24]. Here is a standard procedure to call transcription factor-binding sites using the PeakSeq algorithm [35]. Generate a mappability map of the yeast reference genome using PeakSeq and modify the following parameters to account for the smaller genome size: window size of 10 kb during the normalization step and bin size of 1 kb during the linear regression step. Considering only uniquely mapping reads, pool all replicates from a control ChIP-Seq experiment, such as ChIP in an untagged strain, into a scoring set (see Note 24). For a particular TF grown in the same medium, run the PeakSeq algorithm to score individual biological replicates against the scoring set, and to score pooled biological replicates against the same control. Score Sono-Seq/Input-Seq sample against the appropriate scoring set, generating a reference sample marking open chromatin. This will generate PeakSeq output files that contain significant regions.
Filter output files with a P-value or Q-value threshold to obtain statistically significant transcription factor-binding sites. For visualization of binding sites in IGB, convert the peak-calling output file into a.bed file of the following format: chromosome, chromosomal start position of peak, and chromosomal end position of peak. Inspect binding regions in IGB (see Note 25) and increase thresholds if needed. A minimal Q-value or P-value threshold of 0.05 should be used, although it is very common and recommended to use more stringent thresholds, such as 0.01, 0.001, or 10−5 [24]. To filter out binding sites that might be significant at the Q-value level only due to an extremely elevated number of sequence reads, albeit presenting low enrichment experimental vs. control reads and therefore likely not biologically relevant, set up other filtering criteria of the PeakSeq output file, such as the difference between the number of experimental and control (background) PeakSeq sequence reads, the ratio between experimental and control PeakSeq reads, or the number of background (or experimental) reads [13, 35, 36]. This procedure usually removes sequencing artifacts as well.
Compare ranked lists of binding sites from biological duplicates. If the response between replicates is expected to be similar and non-stochastic, >90 % overlap is expected, depending on the number of binding sites in each list [35]. Performing ChIP-Seq experiments in biological duplicates is usually enough when reproducibility between replicates is high; otherwise, ChIP-Seq experiments should be done in biological triplicates. Target agreement plots are great diagnostic tools to determine if duplicates have sufficient reproducibility. A target agreement plot determines the fraction of overlapping targets when comparing similar fractions of ranked target lists from individual replicates. When comparing multiple transcription factors, it is often simpler to overlap binding sites from two individual replicates and combine them into a single peak list for comparative analysis.
If possible and when available, compare binding sites obtained by ChIP-Seq to previously published ChIP-chip regions. In our experience, ChIP-Seq analysis recovers 62–74 % of ChIP-chip peaks and identifies numerous novel binding regions, even doubling the number of binding sites or more for a particular TF.
If needed, validate a selection of novel binding sites by qPCR, as discussed in Note 10.
Perform high-level bioinformatics analysis on the ChIP-Seq data and final target lists. Such analyses include annotation of binding sites to neighboring genes, discovery of TF binding motifs enriched among significant ChIP-Seq-binding regions, generation of ChIP-Seq signal aggregation plots around specific genomic features, determination of enriched biological processes by gene ontology (GO) analysis, and various multivariate statistical analyses (clustering, discriminant analysis, principal component analysis, linear regressions, etc.). The R package ChIPpeakAnno [37] enables several analyses, including target list overlap, annotation of binding region to nearest or overlapping genes, importing sequence data from genomic coordinates, and GO analysis. CisGenome is a biologist-friendly open-source analysis suite with a nice graphical user interface (GUI) and working under Windows [38]. CisGenome can identify peaks from standard alignment output files (ELAND or bowtie) with its built-in peak-calling algorithm seqpeak, load genome annotations, get DNA sequences from a target list, determine the genomic location of binding regions in comparison to transcription start sites, intergenic regions, and others, discover new TF consensus site motifs, and search for known TF-binding site motifs. A directory of available bioinformatics tools for high-throughput sequencing analysis can be found at the following web page: http://seqanswers.com/wiki/Software/list.
Set up and perform functional analyses, which may be additional biochemical assays, microscopy work, genetic manipulations, or other experimental validation.
Table 1.
Oligonucleotide sequences for barcoded ChIP-Seq, with 4-plex [13] and 12-plex [44] sequencing designs
| Barcode | Forward/reverse | Sequence (5′ → 3′) |
|---|---|---|
| ACGT | Forwarda Reverseb |
ACACTCTTTCCCTACACGACGCTCTTCCGATCTACGT CGTAGATCGGAAGAGCTCGTATGCCGTCTTCTGCTTG |
| CATT | Forwarda Reverseb |
ACACTCTTTCCCTACACGACGCTCTTCCGATCTCATT ATGAGATCGGAAGAGCTCGTATGCCGTCTTCTGCTTG |
| GTAT | Forwarda Reverseb |
ACACTCTTTCCCTACACGACGCTCTTCCGATCTGTAT TACAGATCGGAAGAGCTCGTATGCCGTCTTCTGCTTG |
| TGCT | Forwarda Reverseb |
ACACTCTTTCCCTACACGACGCTCTTCCGATCTTGCT GCAAGATCGGAAGAGCTCGTATGCCGTCTTCTGCTTG |
| ATCACGT | Forwarda Reverseb |
ACACTCTTTCCCTACACGACGCTCTTCCGATCTATCACGT CGTGATAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG |
| CGATGTT | Forwarda Reverseb |
ACACTCTTTCCCTACACGACGCTCTTCCGATCTCGATGTT ACATCGAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG |
| TTAGGCT | Forwarda Reverseb |
ACACTCTTTCCCTACACGACGCTCTTCCGATCTTTAGGCT GCCTAAAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG |
| TGACCAT | Forwarda Reverseb |
ACACTCTTTCCCTACACGACGCTCTTCCGATCTTGACCAT TGGTCAAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG |
| ACAGTGT | Forwarda Reverseb |
ACACTCTTTCCCTACACGACGCTCTTCCGATCTACAGTGT CACTGTAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG |
| GCCAATT | Forwarda Reverseb |
ACACTCTTTCCCTACACGACGCTCTTCCGATCTGCCAATT ATTGGCAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG |
| CAGATCT | Forwarda Reverseb |
ACACTCTTTCCCTACACGACGCTCTTCCGATCTCAGATCT GATCTGAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG |
| ACTTGAT | Forwarda Reverseb |
ACACTCTTTCCCTACACGACGCTCTTCCGATCTACTTGAT TCAAGTAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG |
| GATCAGT | Forwarda Reverseb |
ACACTCTTTCCCTACACGACGCTCTTCCGATCTGATCAGT CTGATCAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG |
| TAGCTTT | Forwarda Reverseb |
ACACTCTTTCCCTACACGACGCTCTTCCGATCTTAGCTTT AAGCTAAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG |
| GGCTACT | Forwarda Reverseb |
ACACTCTTTCCCTACACGACGCTCTTCCGATCTGGCTACT GTAGCCAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG |
| CTTGTAT | Forwarda Reverseb |
ACACTCTTTCCCTACACGACGCTCTTCCGATCTCTTGTAT TACAAGAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG |
Phosphorothioate bond between second-to-last and last (final “T” overhang) nucleotides
5′ ends are phosphorylated
Footnotes
We recommend using the 3-HA or 9-Myc epitopes from the PCR toolkit [39]. Correct TF-epitope fusions should be tested by sequencing the TF-epitope junctions and/or by western blot analysis to confirm the tag and the expression and/or by functional assays (cell viability, growth defects, etc.).
This note describes variations and alternatives to certain steps, or different manipulations, potentially useful if a lesser amount of starting material is used. At step 2, grow 50 mL of cells to collect 35–40 OD units of cells. In steps 5–8, spin in a 50 mL Falcon tube for 1–2 min at 2,880 × g, discard supernatant, resuspend in 1 mL of water, transfer to a 2 mL screw-capped tube, spin for 15 s at 16,000 × g, pipet out water, and flash freeze in liquid nitrogen. After step 8, pellets can be frozen at −70 °C for later use. At step 11, given the lesser amounts, three FastPrep bursts of at least 20 s are enough. At step 12, to recover lysates from the 2 mL screw-cap tubes, transfer into a 2 mL Eppendorf tube by spinning for 1 min with a ramp from 100 to 600 RPM; the additional wash is optional. There should be about 1–1.2 mL of lysate after transfer. A 3-min total sonication, using amplitude 20 % and time ON and OFF at 1 s, can be performed at step 14. At step 17, save only 10 μL input DNA. For the immunoprecipitation at step 18, use about 35 μL bead slurry per IP. If using a blocking procedure, first block the anti-Myc or anti-HA beads for 1 h at 4 °C with 0.5 μL 10 mg/mL BSA and 30 μL 10 mg/mL sonicated salmon sperm DNA. Using similarly blocked protein A beads, pre-clear lysates for 30 min using those blocked protein A beads, which is followed by the immunoprecipitation for 2 h at 4 °C, adding the blocked anti-Myc or anti-HA beads and 25 μL of 10 mg/mL BSA. At step 23, a gel loading tip that the beads do not fit in might be used for transferring the eluates. In addition, if using smaller elution volumes (for example, 100 and 90 μL TE/SDS solution), eluates can be transferred to a 0.2 mL PCR tube at step 23. In the latter case, for reversal of cross-links and proteinase K treatment (steps 26 and 27), add 10 μL of 20 mg/mL proteinase K and incubate in the PCR machine for at least 2 h at 37–50 °C and then overnight at 65 °C; skip to step 32 instead of performing the precipitation described in steps 28–31.
Cross-linking times could be increased if the protein of interest is indirectly binding DNA, through other proteins. Too much cross-linking might reduce the epitope availability, thus hindering the ChIP procedure. Too little cross-linking might not allow the recovery of enough material for ChIP analysis. In this case, it is possible to add DMA (dimethyladipimidate dihydrochloride) at 10 μM in 0.25 % DMSO for 10–45 min to increase cross-linking. Optimal cross-linking times could be about 10–15 min for histones and histone marks, 15–20 min for TFs, and 15 min up to 45 min for chromatin remodelers. Cross-linking can also be performed on a platform shaker.
This procedure using the FastPrep machine lyses over 95 % of yeast cells. Lysis can be monitored using regular light microscopy. Alternatively, a paint shaker can be used for cell lysis, but only 40 % of cells are typically lysed after a 30-min treatment.
Five cycles of sonication per replicate should shear chromatin to a median size of 450–500 bp. To optimize this procedure, samples with various numbers of sonication cycles should be clarified as described in steps 15–16. Take 250 μL of this sonicated lysate to degrade proteins, reverse cross-links, and purify DNA, as described in steps 26–31. Load on a 2 % agarose gel. A smear should appear between 100 and 1,000 bp, with a stronger intensity zone around the expected median size (450–500 bp). Using a Branson Analog 250 sonicator, 5–7 cycles of 30 s at constant (100 % duty cycle) and power setting about 6 should be equivalent.
Input DNA can be isolated for peak scoring purposes, but can also be as a reference sample as it marks open chromatin. Input DNA represents non-IP, cross-linked, sonicated chromatin. It is prepared similarly to ChIP DNA, skipping IP and IP washes. Briefly, add 250 μL of 1 × TE/1 % SDS to the sample from step 17, perform steps 26–31, perform an RNase A treatment at 37 °C for 30 min by adding 2 μL 10 mg/mL RNase A, purify DNA through a MinElute column, and elute in 21 μL EB.
Antibody quantities could be optimized through preliminary IP experiments. It is possible to perform a two-step IP on untagged strains if ChIP-grade antibodies are available. In this case, incubate overnight samples with 5–200 μL of primary antibody, pre-wash in lysis/IP buffer Protein G or Protein A or Protein A/G agarose bead slurry, add 250 μL of 50 % agarose slurry for 1–2 h at 4 °C, and perform washes described in step 22 in 15 mL Falcon tube at 4 °C with 10 mL of the appropriate buffer.
The temperature of the water bath must be above 65 °C, as determined with a thermometer. We sometimes have to set up the temperature to 67.5 or 68 °C to ensure that water reaches the appropriate temperature. Do not carry over residual beads prior to the reversal of cross-links. Residual beads might reduce the efficiency of this step and decrease the final amount of DNA recovered after the ChIP procedure.
This procedure yields 100–500 ng of ChIP DNA, but greater (up to 1 μg) or lower amounts (50 ng) are possible. Our range most often lies between 200 and 350 ng. DNA quantification could also be determined using the PicoGreen dsDNA assay (Invitrogen). To use Nanodrop, wash the probe with water and EB buffer, blank with 2 μL EB centered on the probe, and measure the DNA concentration and A260/280 ratio with 2 μL ChIP sample.
ChIP-qPCR can be useful to verify the ChIP efficiency prior to investing time and money in further experiments, if previously characterized binding sites exist. Setting up reactions in triplicates, biological replicates of experimental (tagged) and control (untagged) strains are analyzed by qPCR to determine the enrichment at 2–4 known binding regions vs. enrichment around at least one negative control region where the TF of interest is not expected to bind. A dilution series of genomic DNA should be used as a normalization factor for PCR efficiency [40].
This optional gel extraction is recommended for the exclusion of very short and longer ChIP fragments that are outside the size range acceptable on Illumina’s Genome Analyzer sequencer. At this step, input DNA should appear as a bright smear while the smear from ChIP DNA is usually visible but much fainter. During gel extraction, after agarose has been dissolved in QG buffer, samples should be chilled on ice for a few seconds (less than 30) to reduce GC bias in Illumina sequencing. Alternatively, one might perform a cold dissolution of agarose at room temperature to 37 °C instead of 50 °C, to eliminate such biases [41].
End repair blunts DNA fragments. It also ensures that all 5′ DNA ends are phosphorylated.
To prevent degradation of dATPs, cycles of freeze-thaw should be avoided. 20–25 μL aliquots of 1 mM dATP are prepared from the stock solution and frozen until single use. This step, given the low concentration of dATP, incorporates a single “A” nucleotide to the 3′ ends of blunted ChIP fragments for subsequent ligation of Illumina-specific adapters.
Level of multiplexing should be determined computationally by simulations to ensure that sufficient reads for each barcoded sample will be obtained. This can be achieved by taking into account the number of expected binding sites, the nature of the DNA-binding factor (point TF vs. broader binding observed for RNA polymerase II or histone modifications), and the relative enrichment of the binding site in the ChIP sample [13]. Ideally, at least a million reads per biological replicate should be obtained, for about a 2.5-fold whole-genome coverage. Multiplex Illumina sequencing relies on the introduction of short, 3–6 bp index (or barcode) on Illumina genomic DNA adapters, directly following the Illumina adapter sequence, which is required for later steps of PCR amplification [13, 42, 43]. Barcoded adapters contain a final “T” overhang for ligation to end-repaired ChIP DNA, to which had been incorporated a 3′ “A” overhang. Examples of oligonucleotides, with 4 bp barcodes for 4-plex sequencing [13] or with 7 bp for 12-plex sequencing [44], are shown in Table 1. The forward primer contains the index with the final “T,” at the 3′ end. It is highly advisable to add a phosphorothioate bond for this 3′ “T” overhang, although we have successfully generated barcoded libraries without this modification. The reverse primer has a 5′ phosphate group and the reverse-complement of the barcode at the 5′ end. In the design of barcodes, two criteria should be respected. First, according to the manufacturer’s guidelines, barcoded adapters should have a balanced nucleotide design. Second, barcodes should be unique enough so that sequencing errors (especially one- or two-base) would not misassign a sequencing read to the wrong sample. When performing analysis, only reads with intact barcodes should be considering. After sequencing, all sequence reads will start with the short barcode, for identification to the original sample, followed by ChIP DNA. After sorting reads by barcode, barcode removal, and read alignment, individual ChIP-Seq profiles will be generated for each barcoded sample. Recent studies have taken advantage of the increase in the number of sequencing reads by sequencing 20 ChIP samples simultaneously in a single flowcell lane [27]. More multiplexing would be possible if comparing ChIP-Seq profiles of point TFs with few binding sites when sequencing on the Illumina HiSeq machine. Here are a few steps to anneal and generate functional barcoded adapters: Synthesize HPLC-purified oligonucleotides (similar to those listed in Table 1) on the 0.05 μmol scale, resuspend each oligo in annealing buffer to a final concentration of 200 μM, mix the pair of forward and reverse barcoded oligos 1:1, denature in a wet heat block for 5 min at 95 °C, remove heat block to cool down at room temperature for 45 min, keep on ice for 30 min, and store annealed adapters at − 20 °C. When ready to use, dilute adapters to the working concentration of Illumina genomic DNA adapters generating successful input DNA or ChIP DNA libraries. To determine adapter concentrations quickly, just compare DNA concentration measurements obtained on a Nanodrop spectrophotometer for Illumina genomic DNA adapter mix and custom-made adapters.
This gel extraction get rids of adapter-adapter dimers. These byproducts are amplified by PCR preferentially and are visible on the gel as a strong intensity, compact band around 100–130 bp. PCR amplification of adapter-adapter dimers can compete with amplification of the regular adapter-ChIP DNA fragments, resulting in the partial or complete loss of the smear characterizing the generation of a successful sequencing library. Great care should be taken to avoid the isolation of DNA fragments around ~100 bp or lower at this step, hence the isolation in step 10 of fragments between 150 and 500 bp. At this step, it commonly occurs that ChIP DNA is not visible, without incidence on the successful generation of a high-quality library. If adapter-adapter dimers still remain present after the library purification or are visible on the Bioanalyzer run, smaller DNA fragments, such as those dimers, can be discarded using AMPure beads (Agencourt) at a 0.8–1.0 bead-to-DNA ratio.
The amplification of adapter-ligated ChIP fragments must remain linear to prevent overrepresentation and underrepresentation of fragments in the final sequencing library that do not reflect biological phenomena. Illumina recommends a maximum of 18 PCR cycles at this step, with the most common range between 13 and 17 cycles. The number of sequence reads would likely be greater for overrepresented fragments due to PCR artifact, potentially leading to the identification of a false-positive binding site.
The final library should appear as a bright smear between 150 and 500 bp, of medium-to-high intensity. For ideal cluster generation and subsequent DNA sequencing of these clusters, DNA fragments between 150 and 350 bp should be isolated. According to Illumina’s guidelines, the median size should be ~230 bp for optimal cluster generation. There should not be any band below 150 bp. If there is a faint band around 100–120 bp, it consists of adapter-adapter dimers and should be completely and carefully avoided. Presence of adapter-adapter dimers in the sequencing library will reduce the number of sequence reads passing quality filters and mapping to the reference genome.
Measure DNA concentration and A260/280 ratio using Nanodrop. In our experience, concentrations of ChIP DNA sequencing libraries are at least above 8.0 ng/μL, and are at least above 15.0 ng/μL for input DNA. Good-quality libraries have A260/280 ratios between 1.7 and 2.0. All libraries with lower DNA concentrations than 5.0 ng/μL are discarded, along with those with low A260/280 ratios indicative of poor quality. We submit to the sequencing facility a minimum of 10 μL of sequencing library at a DNA concentration equal or greater than 5.0 ng/μL. When mixing libraries for multiplex sequencing, similar quantities of DNA should be mixed (equimolar ratios) to obtain a proper representation of all barcoded samples during sequencing. The PicoGreen dsDNA assay could be used to determine DNA concentrations more precisely. However, using Nanodrop, we rarely get more than a twofold difference between the number of sequence reads from the least and most abundant barcoded samples. The Qubit fluorometer tends to perform better than the Nanodrop at lower DNA concentrations and higher salt concentrations.
Illumina sequencing is a microfluidics-based sequencing-by-synthesis approach with two main steps: cluster generation on a cluster station and sequencing per se on the Genome Analyzer. During cluster generation, individual molecules from the sequencing library are attached onto a flowcell containing a lawn of complementary oligonucleotides. After initial bridge PCR amplification, the initial template is washed away and the flowcell-bound template replica is submitted to multiple rounds of bridge amplification. Each cluster contains ~1,000 molecules. Accurate size selection of the library is crucial for proper sequencing. Longer fragments would form fewer clusters of larger size, resulting in a lesser number of sequencing reads. On the other hand, shorter DNA fragments would generate too many clusters of smaller size that will not pass quality filters, hence decreasing the overall mapping values. Once the flowcell is installed on the Genome Analyzer, a sequencing primer is first annealed and four fluorescently labeled nucleotides with a reversible terminator are added. Four cycles of imaging, one for each nucleotide, are performed to determine the identity of the incorporated nucleotide. Next, the reversible terminator is cleaved off and followed by the addition of four modified nucleotides as previously described. This process is repeated up to the desired number of cycles. About 20–40 million short sequence reads are generated per lane of the Genome Analyzer IIx, while this number can easily reach 60–140 million reads per lane on the HiSeq sequencer.
Several statistics are used to determine if the sequencing run was successful, including the number of clusters passing quality filters, the error percentage, the total number of reads, various mapping values, and the cluster density. Multiplex sequencing runs have typically an elevated error rate compared to non-barcoded ones. Raw mapping values before barcode removal should not be used given the presence of the index. The percentage of uniquely mapping reads is usually reported as sequencing metric, along with the total number of reads. For yeast barcoded ChIP-Seq experiments, we observe around 60 % or more total uniquely mapping reads and around 10–15 % total multiple genome matches. Presence of adapter-adapter dimers in the sequencing library could reduce overall mapping to only 10–20 %.
Bowtie [31] and Burrows-Wheeler Aligner (BWA) [32] are fast and accurate short read aligners widely used in the high-throughput sequencing field. Output files are directly used by downstream applications from their software suite, or other computational programs, to perform specific bioinformatics tasks (RNA-Seq, isoform discovery, genome re-sequencing, ChIP-Seq, SNP calling, cloud computing, etc.). When using Bowtie for the yeast genome, we use the following parameters for read alignment: -n 2 -l 26 (2 mismatches on 26 bp).
Sometimes ChIP-Seq profiles for a particular chromosome or for a whole sample do not appear in the IGB browser. By scrolling down in the chromosome selection menu, other chromosome labels might appear and contain the actual profile. Rename files to use the correct chromosome label and be consistent. For chromosome 1, one might notice the following names: “chr1,” “chr01,” “chrI,” “I,” and “1.”
Many peak scoring algorithms exist, including PeakFinder [8], FindPeaks [45], SISSRs [46], QuEST [47], PeakSeq [35], MACS [48], U-Seq [49], CisGenome [38], E-RANGE [50], spp R package [51], HPeak [52], SoleSearch [53], PeakRanger [54], and many others. CisGenome has a nice GUI and is very user friendly, requiring less programming skills [38]. First, the alignment file (ELAND or bowtie) is converted to a .aln file. Then, load the yeast genome and perform two-sample peak calling. Important parameters should be optimized, including the bin size B, the read extension length E, the window statistical cutoff C, and the half window size W. For W, point TF would have narrower peaks, thus a smaller W (<500 bp) than factors binding broader regions (RNA polymerase II, histone modifications). For E, in general, it should be determined with the following formula: E = fragment size—read length, to extend the fragment in the 3′ direction. Another popular algorithm is MACS, which uses a model-based approach [48]. It can be easily adapted to smaller genome contexts by modifying the effective genome size, and the bandwidth related to half of the estimated sonicated fragment size [48]. Higher confidence regions can be obtained by decreasing the P-value cutoff and modifying the model fold enrichment value (mfold) [24].
ChIP-Seq scoring samples in yeast include input DNA, ChIP on untagged strains, and, in the case of two-step IPs, agarose bead slurry only or ChIP on normal IgG (mock IP). Earlier ChIP-Seq efforts used mostly input DNA for scoring purposes, while recent yeast studies are using mock IPs as control samples. After pooling scoring replicates, an ~10 M reads scoring set is usually sufficient for proper algorithm performance.
It is critical to inspect binding regions in IGB to get a sense of the generated target list. The number of binding sites might be too high or too low. More importantly, the length of the binding site might be inaccurate and will constitute a problem, in particular if the fraction of very long binding regions is too high. We have seldom experienced cases in which target lists from point transcription factors contained many binding sites of length over 5 kb. Parameters of the peak scoring algorithm and/or filtering thresholds should be changed. When comparing the location of the called binding site to the actual peak from the ChIP-Seq signal, the significant region might include only the shoulders of the peak and not the center. If this phenomenon occurs regularly, the parameters of the scoring algorithm should be modified.
Contributor Information
Philippe Lefrançois, Department of Molecular, Cellular and Developmental Biology, Yale University, New Haven, CT, USA.
Jennifer E. G. Gallagher, Department of Biology, West Virginia University, Morgantown, WV, USA
Michael Snyder, Department of Genetics, Stanford University School of Medicine, Stanford, CA, USA.
References
- 1.Costanzo MC, Hogan JD, Cusick ME, et al. The yeast proteome database (YPD) and Caenorhabditis elegans proteome database (WormPD): comprehensive resources for the organization and comparison of model organism protein information. Nucleic Acids Res. 2000;28:73–76. doi: 10.1093/nar/28.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prakash A, Tompa M. Assessing the discordance of multiple sequence alignments. IEEE/ACM Trans Comput Biol Bioinform. 2009;6:542–551. doi: 10.1109/TCBB.2007.70271. [DOI] [PubMed] [Google Scholar]
- 3.Borneman AR, Gianoulis TA, Zhang ZD, et al. Divergence of transcription factor binding sites across related yeast species. Science. 2007;317:815–819. doi: 10.1126/science.1140748. [DOI] [PubMed] [Google Scholar]
- 4.Gilmour DS, Lis JT. Detecting protein-DNA interactions in vivo : distribution of RNA polymerase on specific bacterial genes. Proc Natl Acad Sci U S A. 1984;81:4275–4279. doi: 10.1073/pnas.81.14.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orlando V, Strutt H, Paro R. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods. 1997;11:205–214. doi: 10.1006/meth.1996.0407. [DOI] [PubMed] [Google Scholar]
- 6.Horak CE, Snyder M. ChIP-chip: a genomic approach for identifying transcription factor binding sites. Methods Enzymol. 2002;350:469–483. doi: 10.1016/s0076-6879(02)50979-4. [DOI] [PubMed] [Google Scholar]
- 7.Harbison C, Gordon DB, Lee TI, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson DS, Mortazavi A, Myers RM, et al. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 9.Robertson G, Hirst M, Bainbridge M, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 10.Euskirchen GM, Rozowsky JS, Wei CL, et al. Mapping of transcription factor binding regions in mammalian cells by ChIP: comparison of array- and sequencing-based technologies. Genome Res. 2007;17:898–909. doi: 10.1101/gr.5583007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Birney E, Stamatoyannopoulos JA, Dutta A, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Celniker SE, Dillon LA, Gerstein MB, et al. Unlocking the secrets of the genome. Nature. 2009;459:927–930. doi: 10.1038/459927a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lefrancois P, Euskirchen GM, Auerbach RK, et al. Efficient yeast ChIP-Seq using multiplex short-read DNA sequencing. BMC Genomics. 2009;10:37. doi: 10.1186/1471-2164-10-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teytelman L, Ozaydin B, Zill O, et al. Impact of chromatin structures on DNA processing for genomic analyses. PLoS One. 2009;4:e6700. doi: 10.1371/journal.pone.0006700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Auerbach RK, Euskirchen G, Rozowsky J, et al. Mapping accessible chromatin regions using Sono-Seq. Proc Natl Acad Sci U S A. 2009;106:14926–14931. doi: 10.1073/pnas.0905443106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Preti M, Ribeyre C, Pascali C, et al. The telomere-binding protein Tbf1 demarcates snoRNA gene promoters in Saccharomyces cerevisiae. Mol Cell. 2010;38:614–620. doi: 10.1016/j.molcel.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 17.Huber A, French SL, Tekotte H, et al. Sch9 regulates ribosome biogenesis via Stb3, Dot6 and Tod6 and the histone deacetylase complex RPD3L. EMBO J. 2011;30:3052–3064. doi: 10.1038/emboj.2011.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zill OA, Scannell D, Teytelman L, et al. Co-evolution of transcriptional silencing proteins and the DNA elements specifying their assembly. PLoS Biol. 2010;8:e1000550. doi: 10.1371/journal.pbio.1000550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Dijk EL, Chen CL, d’Aubenton-Carafa Y, et al. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature. 2011;475:114–117. doi: 10.1038/nature10118. [DOI] [PubMed] [Google Scholar]
- 20.Batta K, Zhang Z, Yen K, et al. Genome-wide function of H2B ubiquitylation in promoter and genic regions. Genes Dev. 2011;25:2254–2265. doi: 10.1101/gad.177238.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou X, O’Shea EK. Integrated approaches reveal determinants of genome-wide binding and function of the transcription factor Pho4. Mol Cell. 2011;42:826–836. doi: 10.1016/j.molcel.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cai L, Sutter BM, Li B, et al. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell. 2011;42:426–437. doi: 10.1016/j.molcel.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eaton ML, Galani K, Kang S, et al. Conserved nucleosome positioning defines replication origins. Genes Dev. 2010;24:748–753. doi: 10.1101/gad.1913210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng W, Zhao H, Mancera E, et al. Genetic analysis of variation in transcription factor binding in yeast. Nature. 2010;464:1187–1191. doi: 10.1038/nature08934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haynes BC, Skowyra ML, Spencer SJ, et al. Toward an integrated model of capsule regulation in Cryptococcus neoformans. PLoS Pathog. 2011;7:e1002411. doi: 10.1371/journal.ppat.1002411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith KM, Phatale PA, Sullivan CM, et al. Heterochromatin is required for normal distribution of Neurospora crassa CenH3. Mol Cell Biol. 2011;31:2528–2542. doi: 10.1128/MCB.01285-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Venters BJ, Wachi S, Mavrich TN, et al. A comprehensive genomic binding map of gene and chromatin regulatory proteins in Saccharomyces. Mol Cell. 2011;41:480–492. doi: 10.1016/j.molcel.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li R, Li Y, Kristiansen K, et al. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24:713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 31.Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nicol JW, Helt GA, Blanchard SG, Jr, et al. The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics. 2009;25:2730–2731. doi: 10.1093/bioinformatics/btp472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilbanks EG, Facciotti MT. Evaluation of algorithm performance in ChIP-seq peak detection. PLoS One. 2010;5:e11471. doi: 10.1371/journal.pone.0011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rozowsky J, Euskirchen G, Auerbach RK, et al. PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat Biotechnol. 2009;27:66–75. doi: 10.1038/nbt.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Euskirchen GM, Auerbach RK, Davidov E, et al. Diverse roles and interactions of the SWI/SNF chromatin remodeling complex revealed using global approaches. PLoS Genet. 2011;7:e1002008. doi: 10.1371/journal.pgen.1002008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu LJ, Gazin C, Lawson ND, et al. ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics. 2010;11:237. doi: 10.1186/1471-2105-11-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ji H, Jiang H, Ma W, et al. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat Biotechnol. 2008;26:1293–1300. doi: 10.1038/nbt.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Janke C, Magiera MM, Rathfelder N, et al. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast. 2004;21:947–962. doi: 10.1002/yea.1142. [DOI] [PubMed] [Google Scholar]
- 40.Pfaffl M. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quail MA, Kozarewa I, Smith F, et al. A large genome center’s improvements to the Illumina sequencing system. Nat Methods. 2008;5:1005–1010. doi: 10.1038/nmeth.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cronn R, Liston A, Parks M, et al. Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Res. 2008;36:e122. doi: 10.1093/nar/gkn502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Craig DW, Pearson JV, Szelinger S, et al. Identification of genetic variants using bar-coded multiplexed sequencing. Nat Methods. 2008;5:887–893. doi: 10.1038/nmeth.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wong KH, Struhl K. The Cyc8-Tup1 complex inhibits transcription primarily by masking the activation domain of the recruiting protein. Genes Dev. 2011;25:2525–2539. doi: 10.1101/gad.179275.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fejes AP, Robertson G, Bilenky M, et al. FindPeaks 3.1: a tool for identifying areas of enrichment from massively parallel short-read sequencing technology. Bioinformatics. 2008;24:1729–1730. doi: 10.1093/bioinformatics/btn305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jothi R, Cuddapah S, Barski A, et al. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008;36:5221–5231. doi: 10.1093/nar/gkn488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Valouev A, Johnson DS, Sundquist A, et al. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat Methods. 2008;5:829–834. doi: 10.1038/nmeth.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nix DA, Courdy SJ, Boucher KM. Empirical methods for controlling false positives and estimating confidence in ChIP-Seq peaks. BMC Bioinformatics. 2008;9:523. doi: 10.1186/1471-2105-9-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 51.Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol. 2008;26:1351–1359. doi: 10.1038/nbt.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qin ZS, Yu J, Shen J, et al. HPeak: an HMM-based algorithm for defining read-enriched regions in ChIP-Seq data. BMC Bioinformatics. 2010;11:369. doi: 10.1186/1471-2105-11-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blahnik KR, Dou L, O’Geen H, et al. Sole-Search: an integrated analysis program for peak detection and functional annotation using ChIP-seq data. Nucleic Acids Res. 2010;38:e13. doi: 10.1093/nar/gkp1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feng X, Grossman R, Stein L. PeakRanger: a cloud-enabled peak caller for ChIP-seq data. BMC Bioinformatics. 2011;12:139. doi: 10.1186/1471-2105-12-139. [DOI] [PMC free article] [PubMed] [Google Scholar]