

Abstract
Memory CD8+ T cells are critical for host defense upon reexposure to intracellular pathogens. We found that interleukin 10 (IL-10) derived from CD4+ regulatory T cells (Treg cells) was necessary for the maturation of memory CD8+ T cells following acute infection with lymphocytic choriomeningitis virus (LCMV). Treg cell–derived IL-10 was most important during the resolution phase, calming inflammation and the activation state of dendritic cells. Adoptive transfer of IL-10-sufficient Treg cells during the resolution phase ‘restored’ the maturation of memory CD8+ T cells in IL-10-deficient mice. Our data indicate that Treg cell–derived IL-10 is needed to insulate CD8+ T cells from inflammatory signals, and reveal that the resolution phase of infection is a critical period that influences the quality and function of developing memory CD8+ T cells.
Memory CD8+ T cells are a principal component of immunity to intracellular pathogens such as viruses. They are distinguished by their ability to survive long term and to undergo rapid and robust proliferation and acquisition of effector function upon reexposure to antigen1. Despite the utility of memory CD8+ T cells in protection against pathogens (such as human immunodeficiency virus) that rapidly mutate to elude neutralizing antibodies, the development of T cell–based vaccines has proven problematic2. This failure has been largely due to an incomplete understanding of the signals and cell types that operate at different stages of the immune response to influence the quantity and quality of developing memory CD8+ T cells.
The T cell response to an acute infection can typically be divided into the following three phases: expansion, contraction and memory. During the first phase, naive CD8+ T cells divide and differentiate into effector cells that acquire the ability to produce the pro-inflammatory cytokines interferon- γ (IFN-γ) and tumor-necrosis factor (TNF), as well as cytotoxic proteins such as granzymes and perforin3. This process by which cytotoxic T lymphocytes (CTLs) undergo differentiation and clonal expansion is governed by signaling via antigens, costimulation and cytokine receptors (including the receptors for IL-2, IL-12, IL-27 and type I interferons) that induce the expression of transcription factors such as Eomes, T-bet and Id2 (ref. 4). However, the strength and duration of these signals, particularly signaling via receptors for inflammatory cytokines, also regulate the long-term fates of these effector cells by influencing whether they differentiate into terminal effector cells (TECs) or maintain memory-cell potential and develop into memory precursor cells (MPCs). These cell fates are controlled by a coordinated set of changes in the expression of the transcription factors Id2, T-bet and Blimp-1, which promote TEC differentiation, and Foxo1, TCF-1, Eomes and Bcl-6, which promote MPC development5–10. Activation of the kinases mTOR and Akt downstream of signaling via antigens, costimulation and cytokine receptors provide central regulation of the proliferation and function of CTLs by controlling anabolic metabolism, but they also regulate the differentiation of TECs and MPCs by enhancing T-bet expression and repressing Foxo1 activity11,12.
Following clearance of the virus, the contraction and resolution phase ensues, in which the majority of the effector CD8+ T cells die and ~5–10% of the cells survive. The surviving cells enter the third stage, the ‘memory’ phase, and develop into central memory T cells (TCM cells), effector memory T cells and resident memory T cells that are maintained long term by IL-7 and IL-15 (ref. 4). Little is known about the signals that operate during the second stage (the contraction and resolution phase) to influence the types and protective capacity of developing memory CD8+ T cells. Although virus is typically cleared by this time point during an acute infection, tissues remain inflamed, and repair processes are initiated to resolve inflammation and retain tissue homeostasis13. Sustained exposure of effector CTLs to bystander inflammation impairs the formation of mature memory cells and their precursors14. CD4+ T cells are also required during the contraction phase for the formation of functional memory CD8+ T cells, but the mechanisms of their actions are unknown15. Additionally, the anti-inflammatory cytokine IL-10 is important for the optimal maturation of memory CD8+ T cells5,16, but the relevant physiological source of IL-10, as well as the phase in which IL-10 acts to regulate the formation of memory CD8+ T cells, remain ill defined.
Regulatory T cells (Treg cells) are necessary for resolving inflammation and achieving tissue homeostasis following infection, through multiple mechanisms, including expression of inhibitory cytokines such as IL-10 and transforming growth factor-β, regulation of nutrient and cytokine availability, and inhibition of the maturation and function of dendritic cell (DCs) and macrophages17. However, the importance of Treg cells in regulating the formation of memory CD8+ T cells is unclear, with some studies identifying their negative role in the development of memory CD8+ T cells18 and others suggesting the opposite19–21. Given the connection between the requirement for CD4+ T cells and that of IL-10 in promoting the formation of memory CD8+ T cells, we investigated whether Treg cells might be linked to this process. In doing so, we identified a previously unknown role for Treg cells in promoting the development of memory CD8+ T cells through their production of IL-10 during the resolution phase of an acute viral infection. This finding indicated that the resolution of inflammation mediated by Treg cells is critical not only for tissue homeostasis but also for the formation of immunological memory.
RESULTS
IL-10 is required during the resolution phase of infection
To confirm and extend published studies investigating the role of IL-10 in the development of memory T cells, we infected IL-10-deficient (Il10−/−) and wild-type (Il10+/+) C57BL/6 mice with the acutely infectious (Armstrong) strain of lymphocytic choriomeningitis virus (LCMV) and assessed the virus-specific CD8+ T cell response at memory time points (day 45–60 after infection)5,16. Although the magnitude of the LCMV-specific response to the immunodominant epitopes of LCMV glycoprotein residues 33–41 (gp(33–41)) and LCMV nucleoprotein residues 396–404 was similar for the two groups, there was a distinct difference in the composition of the splenic memory CD8+ T cell pool (Fig. 1a,b and Supplementary Fig. 1a,b). The majority of the CD8+ T cells from wild-type mice bore a KLRG1−CD127+ TCM cell phenotype, whereas the CD8+ T cells from Il10−/− mice were KLRG1+CD127− and had higher expression of granzyme B than that of their wild-type counterparts and therefore resembled TECs (Fig. 1a,b and Supplementary Fig. 1a,b). Furthermore, the formation of CXCR3+CD27+CD62L+KLRG1− TCM cells with higher expression of TCF-1 was impaired in Il10−/− mice relative to their formation in wild-type mice (Fig. 1a,b and Supplementary Fig. 1a,b). Thus, consistent with published work5,16, we found that IL-10 was important qualitatively for the development of mature memory CD8+ T cells.
Figure 1.
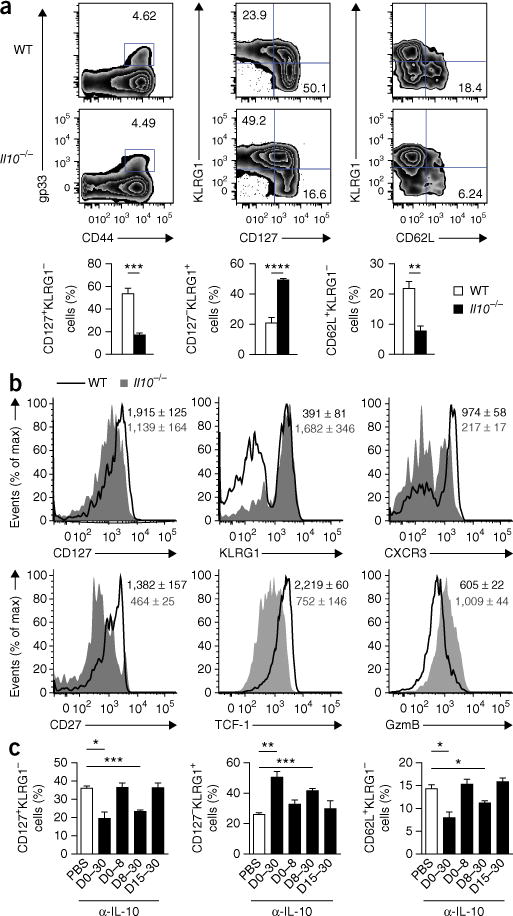
IL-10 is required during the resolution phase of infection to allow optimal maturation of memory CD8+ T cells. (a) Flow cytometry of cells from the spleen of wild-type (WT) and Il10−/− mice 60 d after infection with LCMV (Armstrong strain), assessing the response of T cells positive for the epitope gp(33–41) (gp33). Numbers in plots indicate percent gp33+ T cells (far left) or percent KLRG1−CD127+ cells (top left quadrant) or KLRG1+CD127− cells (bottom right quadrant) in the gp33+ T cell population (middle) or KLRG1−CD62L+ cells in the gp33+ T cell population (far right). Below, frequency of CD127+KLRG1− cells (left), CD127−KLRG1+ cells (middle) or CD62L+KLRG1− cells (right) among gp33+ T cells (assessed on days 45–60). (b) Expression of CD127, KLRG1, CXCR3, CD27, TCF-1 and granzyme B (GzmB) in wild-type and Il10−/− gp33+ T cells from mice as in a. Numbers in plots indicate mean fluorescence intensity (MFI) of marker (horizontal axis) in wild-type cells (top number) or Il10−/− cells (number below). (c) Frequency of CD127+KLRG1−, CD127−KLRG1+ and CD62L+KLRG1− cells among gp33+ T cells from mice infected with LCMV (Armstrong strain) (day 0) and given mock injection of PBS or injection of antibody to IL-10 (α-IL-10) on days (D) 0–30, 0–8, 8–30 or 15–30 (horizontal axis), assessed at day 30. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 (unpaired two-tailed Student’s t-test). Data are from one experiment representative of five experiments with at least four mice per group (a,b) or three experiments (c; mean and s.e.m.).
To determine when IL-10 was needed for the maturation of memory T cells, we infected wild-type mice with LCMV and treated them with neutralizing monoclonal antibody (mAb) to IL-10 during various phases of infection. As observed for Il10−/− mice, mice treated with mAb to IL-10 for the entirety of the infection displayed impaired formation of mature KLRG1loCD127hi and CD62LhiKLRG1lo TCM cells relative to that of their control counterparts treated with phosphate-buffered saline (PBS) (Fig. 1c and Supplementary Fig. 1c). This finding also suggested that alterations in the basal inflammatory state and/or the microbiota of Il10−/− mice were probably not responsible for the shift in the makeup of the memory CD8+ T cell pool22,23. Mice treated with mAb to IL-10 during the priming phase of infection (days 0–8) had little or no alteration in the composition of their memory CD8+ T cell population relative to that of their counterparts treated for the entirety of infection (Fig. 1c). In contrast, CD8+ T cells in mice in which IL-10 blockade was initiated at day 8 displayed a more TEC-like differentiation state than that of control (PBS-treated) mice (Fig. 1c), similar to mice treated with mAb to IL-10 for the entirety of infection. However, when the IL-10 blockade was begun at day 15, there was no defect in memory maturation (Fig. 1c). These data suggested that the presence of IL-10 during the resolution phase of infection (days 8–15) was critical for the maturation of memory CD8+ T cells.
Signaling through STAT3 is important in promoting the maturation of memory CD8+ T cells, which suggests that IL-10 might signal via this transcription factor and signal transducer5. To test this hypothesis, we generated P14 mice (which have transgenic expression of an LCMV-specific T cell antigen receptor) with T cell–specific deletion of the gene encoding the IL-10 receptor (Il10ra)(generating ‘Il10raf/fCd4-Cre’ mice, with loxP-flanked Il10ra alleles (Il10raf/f) deleted by Cre recombinase expressed from the T cell–specific gene encoding the coreceptor CD4 (Cd4-Cre)). We obtained T cells from those mice and transferred small numbers of these cells into congenically mismatched donors, followed by infection of the recipients with LCMV the following day. Although there was a substantially lower percentage and number of Il10raf/fCd4-Cre P14 cells than Il10raf/f P14 cells that survived to a memory time point (Supplementary Fig. 2a), consistent with published work examining Stat3-deficient CD8+ T cells5, Il10raf/fCd4-Cre P14 cells displayed no gross impairment in the formation of mature KLRG1−CD127+ or CD62L+KLRG1− populations, although they did display slightly more granzyme B protein and less TCF-1 protein than did Il10raf/f P14 cells (Supplementary Fig. 2a,b). Thus, although Il10ra expression seemed to be important for the survival of CD8+ T cells in a competitive setting, the phenotypes of the Il10ra-deficient memory CD8 T cells did not fully recapitulate those observed in the IL-10-deficient mouse. These experiments suggested that IL-10 signaling acted in both an intrinsic manner and an extrinsic manner to support the generation and maturation of a phenotypically mature memory CD8+ T cell population.
It is possible that IL-10 acts in a CD8+ T cell–extrinsic manner to support memory maturation by insulating the effector CD8+ T cells from exposure to pro-inflammatory signals, such as IFN-α, IFN-β, IL-2 and IL-12, which drive terminal effector differentiation8,24,25. To investigate this hypothesis, we infected wild-type and Il10−/− mice with LCMV and administered the dinucleotide CpG B, which is an agonist of Toll-like receptor 9, at day 8 or day 15 to induce further inflammation26. Wild-type mice were largely insulated from the excess inflammation, with negligible differences between the CpG B–treated group and the control (PBS-treated) group in memory CD8+ T cell composition (Fig. 2). However, Il10−/− mice treated with CpG B at day 8 displayed significantly greater loss of KLRG1−CD127+ and CD62L+KLRG1− TCM cells than did control (PBS-treated) Il10−/− mice. In contrast, CD8+ T cells in Il10−/− mice treated with CpG B at day 15 were indistinguishable from those in control (PBS-treated) Il10−/− mice. Thus, IL-10 seemed to be important for insulating effector CD8+ T cells from inflammatory cytokines present during the resolution of infection that could sustain the differentiation or survival of TECs.
Figure 2.
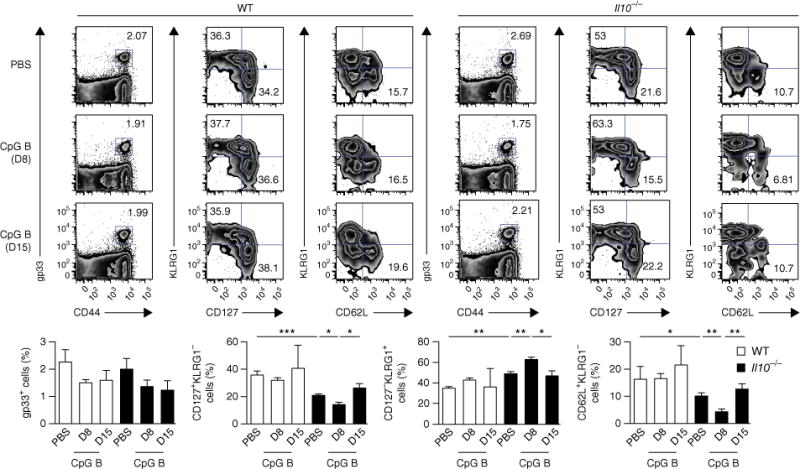
IL-10 is important in insulating CD8+ T cells from inflammatory signals following viral infection. Flow cytometry of cells from wild-type and Il10−/− mice infected with LCMV (Armstrong strain) and given mock injection of PBS or injection of CpG B at day 8 (D8) or day 15 (D15) after infection, assessed 60 d after infection (numbers in plots (top) as in Fig. 1a, top). Below, summary of results above (gp33+ cells among CD8+ T cells (far left) or as in Fig. 1a, bottom). *P < 0.05, **P < 0.01 and ***P < 0.001 (unpaired two-tailed Student’s t-test). Data are from one experiment representative of three experiments with at least four mice per group (mean and s.e.m.).
Treg cell–derived IL-10 is critical for CD8+ T cell memory
Multiple cell types, including DCs, monocytes-macrophages and T cells, secrete IL-10 following infection with LCMV27,28. To distinguish the physiologically relevant source of IL-10 necessary for the memory maturation of CD8+ T cells, we generated mice in which Il10 was deleted specifically in DCs (Il10f/fCd11c-Cre mice), myeloid cells (Il10f/fLyz2-Cre mice) or T cells (Il10f/fCd4-Cre mice). We then infected these mice with LCMV and killed them at memory time points (days 45–60). Although the deletion of Il10 in DCs or myeloid cells had little effect on the maturation of memory CD8+ T cells, deletion of Il10 in T cells resulted in the loss of mature KLRG1−CD127+ and CD62L+KLRG1− TCM cells, comparable to the phenotype seen in Il10−/− mice (Fig. 3a and Supplementary Fig. 3a).
Figure 3.
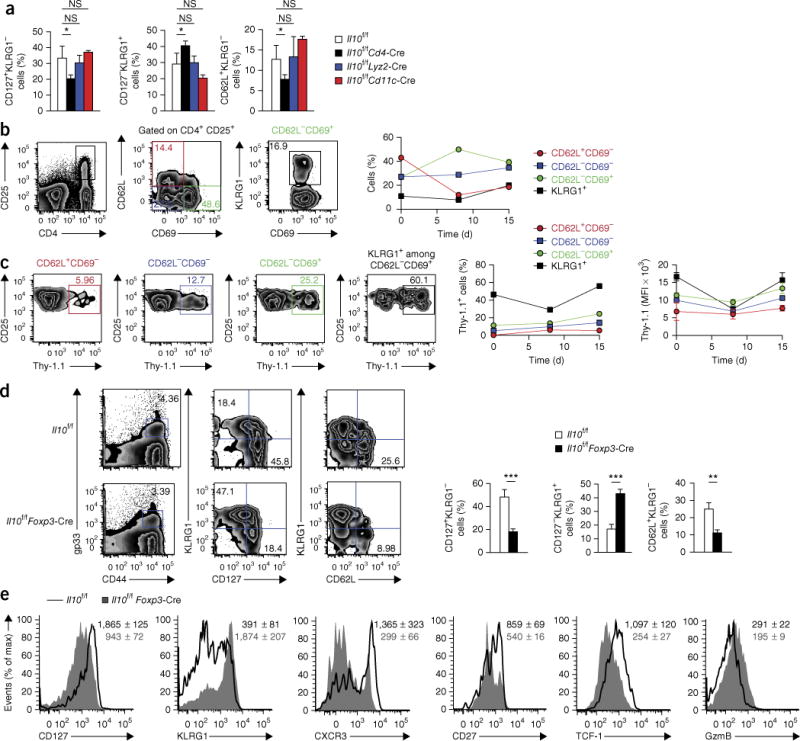
CD4+ Treg cell–derived IL-10 is critical for the maturation of memory CD8+ T cells. (a) Frequency of CD127+KLRG1−, CD127−KLRG1+ and CD62L+KLRG1− cells in the gp33+ T cell population from Il10f/f, Il10f/fCd4-Cre, Il10f/fLyz2-Cre, and Il10f/fCd11c-Cre mice 60 d after infection with LCMV (Armstrong strain). (b) Flow cytometry (top) of T cells from the spleen of 10BiT IL-10 reporter mice 15 d after infection with LCMV (Armstrong strain). Numbers in quadrants (middle) indicate percent CD62L+CD69− cells (top left), CD62L−CD69+ cells (bottom right) or CD62L−CD69+ cells (bottom left) among CD25+CD4+ cells gated at left (outlined area); number adjacent to outlined area (right) indicates percent KLRG1+CD69+ cells among CD62L−CD69+ cells gated at left (green outlined area). Right, frequency of CD62L+CD69−, CD62L−CD69− or CD62L−CD69+ cells among CD25+CD4+ cells (colors match middle plot at left), or KLRG1+ cells among CD62L−CD69+ cells, at days 0, 8 and 15 after infection as above. (c) Expression of IL-10 (assessed as Thy-1.1) by subsets (above plots) of cells from mice as in b (top). Right, frequency of Thy-1.1+ cells (left) and MFI of Thy-1.1 (right) among the Thy-1.1+ cells in those subsets (as in b) at days 0, 8 and 15 after infection as above. (d) Flow cytometry (left) of cells from Il10f/f and Il10f/fFoxp3-Cre mice at 60 d after infection with LCMV (Armstrong strain) (numbers in plots as in Fig. 1a, top). Right, summary of results at left (as in Fig. 1a, bottom). (e) Expression of CD127, KLRG1, CXCR3, CD27, TCF-1 and granzyme B in Il10f/f and Il10f/fFoxp3-Cre gp33+ T cells. Numbers in plots indicate MFI of marker (horizontal axis) in Il10f/f cells (top number) or Il10f/fFoxp3-Cre cells (number below). *P < 0.05, **P < 0.01 and ***P < 0.001 (unpaired two-tailed Student’s t-test). Data are from one experiment representative of three experiments with three to six mice per group (mean and s.e.m. in a–d).
The findings reported above suggested that effector CD8+ T cells, CD4+ T cells, and/or Treg cells might act as the physiological source of IL-10 needed for the memory maturation of CD8+ T cells. We assessed IL-10 expression in these populations in 10BiT reporter mice, following infection with LCMV. 10BiT reporter mice have a bacterial artificial chromosome transgene containing Il10, with replacement of the endogenous coding segment of Il10 exon 1 with cDNA encoding the alloantigen Thy-1.1 (containing a stop codon), such that cells transcribing mRNA from the transgene express Thy-1.1 on their surface29. Published work has confirmed that Thy-1.1+ cells produce IL-10 during infection with LCMV27. Although virus-specific CD4+ T cells and CD8+ T cells, as well as Treg cells, had high expression of Thy-1.1 at day 5 after infection, as reported before27, only Treg cells continued to express Il10 during the resolution phase of infection. The substantial increase in the number of IL-10-competent Treg cells occurring between day 8 and day 15 is likely due in part to the increase in total Treg cell numbers as type I interferons wane30 (Supplementary Fig. 3b). Furthermore, Treg cells present during this phase displayed a more activated phenotype than that of Treg cells present at earlier time points, as shown by their loss of expression of the memory marker CD62L and progressive acquisition of expression of the activation markers CD69 and KLRG1 (ref. 31) (Fig. 3b). The expression of Il10 (as inferred from the expression of Thy-1.1 protein) correlated strongly with the activation state of Treg cells, with KLRG1+ terminally differentiated Treg cells having higher Thy-1.1 expression than that of less-differentiated Treg cell subsets, both on a population basis and a per-cell basis (Fig. 3c). IL-10-competent Treg cells were largely protected from in vivo labeling with antibody to CD4 (Supplementary Fig. 3c), which suggested that these cells were located mainly in the white pulp of the spleen, a site that also contains lymphoid DCs and MPCs32,33.
The data above suggested that Treg cell–derived IL-10 might be important for the maturation of memory CD8+ T cells. To test this hypothesis directly, we generated mice with deletion of Il10 specifically in Treg cells expressing the transcription factor Foxp3 (Il10f/fFoxp3-Cre mice), as described34, infected these mice with LCMV and evaluated the formation of virus-specific CD8+ T cells at memory time points (days 45–60 after infection). Il10f/fFoxp3-Cre mice displayed considerable alteration in the maturation of memory CD8+ T cells but not the number of these cells, relative to that of their Il10f/f counterparts, as indicated by the large fraction of KLRG1+CD127− TEC-like CD8+ T cells (Fig. 3d and Supplementary Fig. 3d). Furthermore, Il10f/fFoxp3-Cre mice had significantly fewer CD62L+KLRG1− TCM cells, along with lower expression of the memory-associated markers CXCR3, CD27 and TCF-1 and higher expression of the effector-associated cytolytic factor granzyme B, relative to that of their Il10f/f counterparts (Fig. 3d,e). These results demonstrated that Treg cell–derived IL-10 was critical for the maturation of memory CD8+ T cells.
To further evaluate the importance of Treg cell–derived IL-10 in the maturation and function of memory CD8+ T cells, we infected Il10f/f and Il10f/fFoxp3-Cre mice with LCMV (Armstrong strain), isolated gp(33–41)-specific CD8+ T cells from these mice at a memory time point, and transferred an equal number of each genotype into congenically mismatched mice. We then challenged the recipient mice the next day with a recombinant strain of Listeria monocytogenes expressing the gp(33–41) epitope and, 4 d later, assessed the ability of the transferred cells to expand their populations and mediate bacterial control. Although the Il10f/f cells were able to robustly increase in number and diminish the bacterial burden, the cells transferred from Il10f/fFoxp3-Cre mice demonstrated substantial impairment in proliferation relative to the Il10f/f cells and failed to reduce bacterial loads from the levels seen in control mice not given cell transfer (Fig. 4). Therefore, Treg cell–derived IL-10 was critical for the development of protective memory CD8+ T cells in the circulation.
Figure 4.
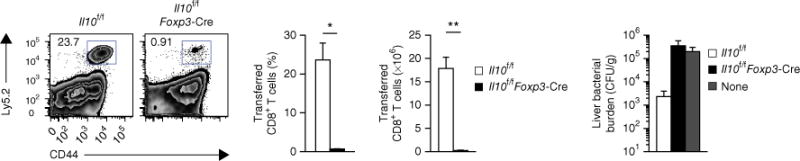
CD4+ Treg cell–derived IL-10 is necessary for the formation of protective memory CD8+ T cells. Flow cytometry (left) of splenic T cells from host mice (Ly5.1+) given no cells (None) or 1.5 × 105 gp33+ CD8+ T cells pooled from congenically mismatched (Ly5.2+) Il10f/f and Il10f/fFoxp3-Cre mice 45 d after infection of donor mice with LCMV (Armstrong strain), followed by challenge of the recipient mice with recombinant L. monocytogenes expressing the gp(33–41) epitope 1 d after cell transfer and analysis 4 d after challenge. Numbers adjacent to outlined areas (far left) indicate percent Ly5.2+ (donor) CD44+ cells. Middle, frequency (middle left) and abundance (middle right) of donor CD8+ T cells in the spleen of host mice. Far right, bacterial burden in the liver of host mice, presented as colony-forming units (CFU) per gram of liver. *P < 0.001 and **P < 0.0001 (unpaired two-tailed Student’s t-test). Data are from one experiment representative of two experiments with at least four mice per group (mean and s.e.m.).
Treg cell–derived IL-10 regulates exposure to inflammation
We next performed high-throughput sequencing for cDNA (RNA-seq) to evaluate the transcriptional profiles of virus-specific CD8+ T cells isolated from Il10f/fFoxp3-Cre and Il10f/f mice at day 15 after infection with LCMV (Fig. 5a and Supplementary Fig. 4a). Although the composition of virus-specific CD8+ T cells at this early time point seemed to be similar between the groups (Supplementary Fig. 4b), we hypothesized that CD8+ T cells might be already programmed to adopt a more terminally differentiated state as further development occurs. Indeed many of the 81 genes differentially expressed by Il10f/fFoxp3-Cre cells relative to their expression by Il10f/f cells (adjusted P value, <0.2) encoded products associated with the formation and function of memory T cells (Fig. 5a and Supplementary Fig. 5). A panel of select genes encoding biologically relevant products also showed dysregulation in CD8+ T cells from Il10f/fFoxp3-Cre mice relative to their regulation in CD8+ T cells from Il10f/f mice, with many of these genes being associated with pro- or anti-inflammatory gene sets (Fig. 5a). We used gene-set–enrichment analysis (GSEA) to detect genome-wide changes in expression and identified significant enrichment in the effector signature in CD8+ T cells isolated from Il10f/fFoxp3-Cre mice relative to results obtained for CD8+ T cells isolated from Il10f/f mice, which displayed robust enrichment for genes with high expression in the memory signature35,36 (Fig. 5b and Supplementary Fig. 6a). The transcriptional profile of CD8+ T cells isolated from Il10f/fFoxp3-Cre mice also showed significant enrichment for genes encoding products in pro-inflammatory pathways, with their transcriptional signature resembling that of cells exposed to pro-inflammatory cytokines such as IL-12 or type I interferons, or Toll-like receptor ligands such as the synthetic RNA duplex poly(I:C) or CpG37,38 (Fig. 5c,d and Supplementary Fig. 6b–d). Therefore, the progenitors of memory cells generated in Il10f/fFoxp3-Cre mice displayed a more TEC-like differentiation state than that of their counterparts generated in Il10f/f mice and a genetic signature suggestive of enhanced exposure to inflammatory signals.
Figure 5.

Virus-specific CD8+ T cells from mice lacking CD4+ Treg cell–derived IL-10 display a robust inflammatory gene signature. (a) RNA-seq analysis of selected biologically relevant genes among mRNA isolated from gp33+ CD8+ T cells and CD8+ T cells positive for the epitope nucleoprotein residues 396–404, pooled from Il10f/f and Il10f/fFoxp3-Cre mice 15 d after infection with LCMV (Armstrong strain), presented as expression (log2) in Il10f/fFoxp3-Cre cells relative to that in Il10f/f cells (key below; columns indicate paired replicates); left margin, grouping of genes by signature (key, bottom right), as determined by published gene sets8,35–38,50. (b) GSEA of gene sets from the Molecular Signatures Database of the Broad Institute, showing gene sets with significant enrichment (false-discovery rate, <10−5) and their enrichment score (where a positive score indicates ‘enrichment’ (higher expression) in the Il10f/fFoxp3-Cre sample relative to that in the Il10f/f sample), with members of the gene set presented in the ranked list of genes (‘bar code’ below) and the signal-to-noise ranking metric (bar at bottom), assessing effector signatures versus (vs) memory signatures in CD8+ T cells, based on published gene sets35,36. (c,d) GSEA of genes upregulated (Up) or downregulated (Down) after stimulation with IFN-α and IFN-β (collectively called ‘IFN-α/β’)) (c) or IL-12 (d), based on published gene sets37; presented as in b. Data are from three independent experiments with three mice per group pooled for each sample.
Transfer of Treg cells ‘rescues’ memory maturation of CD8+ T cells
The findings reported above indicated that Treg cell–derived IL-10 might be important during the resolution phase of infection in controlling the degree of inflammatory exposure to effector CD8+ T cells as they develop into memory T cells. IL-10 restricts the maturation of DCs and their production of the pro-inflammatory cytokines IL-6, IL-1β and TNF39. Therefore, we assessed the maturation status of CD45+CD11chiMHCII+ DCs in Il10f/fFoxp3-Cre mice at day 15 after infection with LCMV and found that the surface expression of markers associated with mature DCs, including CD86, CD80, PD-L1 and PD-L2, was significantly higher in those cells than in their counterparts from LCMV-infected Il10f/f mice (Fig. 6a). Additionally, at day 15 after infection, higher concentrations of pro-inflammatory cytokines associated with mature DCs, including IL-6, IL-1β, TNF and IL-12p70, were present in the serum of Il10f/fFoxp3-Cre mice than in that of Il10f/f mice (Fig. 6b). There was no difference between uninfected Il10f/fFoxp3-Cre mice and uninfected Il10f/f mice in their serum cytokines (Fig. 6b), which indicated that these systemic alterations in the inflammatory milieu did not occur until after infection.
Figure 6.
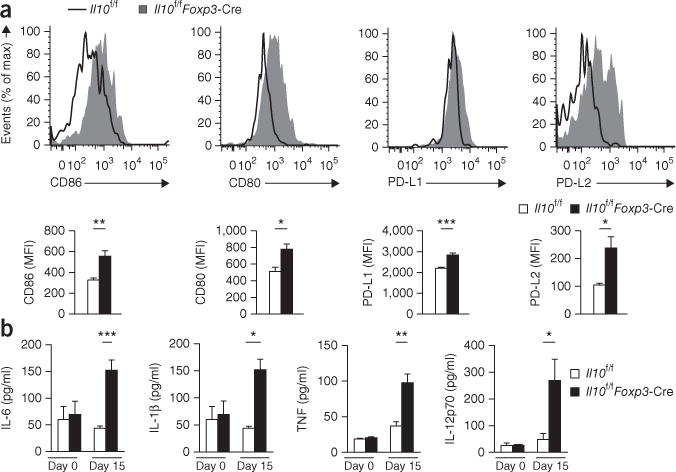
Enhanced maturation of DCs and inflammation in the absence of Treg cell–derived IL-10. (a) Flow cytometry (top) analyzing the expression of CD86, CD80, PD-L1 and PD-L2 in CD45+CD11chiMHCII+ DCs from Il10f/f and Il10f/fFoxp3-Cre mice 15 d after infection with LCMV (Armstrong strain). Bottom, summary of results above. (b) Immunoassay of IL-6, IL-1β, TNF and IL-12p70 in the serum of Il10f/f and Il10f/fFoxp3-Cre mice left uninfected (day 0) or at day 15 after infection with LCMV. *P < 0.05, **P < 0.01 and ***P < 0.001 (unpaired two-tailed Student’s t-test). Data are from one experiment representative of three experiments with at three to five mice per group (mean and s.e.m.).
We hypothesized that the production of IL-10 by Treg cells during the resolution phase might be critical for the suppression of inflammation and promotion of the memory maturation of CD8+ T cells. To further test this hypothesis, we used Foxp3GFP-DTR mice (which have cDNA encoding the human diphtheria toxin receptor (DTR) fused to sequence encoding green fluorescent protein (GFP) inserted into Foxp3) to ensure depletion of Treg cells (via treatment with diphtheria toxin) at various time points after infection40. Consistent with published work19,20, depletion of Treg cells at the time of infection with LCMV led to impairment in the maturation of CD8+ T cells at memory time points (days 45–60) (Fig. 7a and Supplementary Fig. 7a). However, when the depletion of Treg cells began at day 8 after infection, there was a similar defect in the formation of mature KLRG1−CD127+ memory CD8+ T cells and that of CD62L+KLRG1− memory CD8+ T cells. Mice that underwent depletion of Treg cells at day 15 after infection were indistinguishable from mice not treated with diphtheria toxin, which lent additional support to the model in which Treg cell–derived IL-10 was most critical during the immediate resolution phase in promoting the maturation of memory CD8+ T cells.
Figure 7.
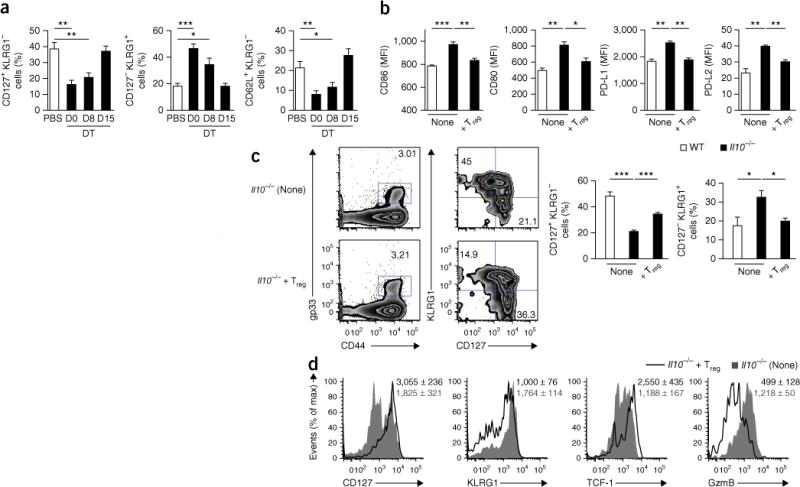
Transfer of IL-10-competent CD4+ Treg cells during the resolution phase of LCMV infection is sufficient to ‘rescue’ the defect in the maturation of memory CD8+ T cells in Il10−/− mice. (a) Frequency of CD127+KLRG1−, CD127−KLRG1+ and CD62L+KLRG1− cells in the gp33+ T cell population of Foxp3GFP-DTR mice infected with LCMV (Armstrong strain) and give mock injection of PBS or injection of diphtheria toxin (DT) on day 0 (D0), day 8 (D8) or day 15 (D15) after infection, followed by analysis 60 d after infection. (b) Expression of CD86, CD80, PD-L1 and PD-L2 in DCs from wild-type and Il10−/− host mice infected with LCMV (Armstrong strain) and, 8 d later, given no cells (None) or 3 × 105 Foxp3+ CD4+ Treg cells isolated from ‘infection-matched’ Foxp3GFP-DTR donor mice (+Treg), followed by analysis 15 d after infection of host mice. (c) Flow cytometry (left) of cells from Il10−/− mice as in b, analyzed 60 d after quadrants (near left) indicate percent KLRG1−CD127+ cells (bottom right) or KLRG1+CD127− cells (top left) in the gp33+ T cell population. Right, frequency of CD127+KLRG1−cells or CD127−KLRG1+ cells among gp33+ T cells. (d) Expression of CD127, KLRG1, TCF-1 and granzyme B in gp33+ T cells from Il10−/− mice as in b. Numbers in plots indicate MFI of marker (horizontal axis) in cells from Il10−/− mice given no cells (top number) or given CD4+ Treg cells (number below). *P < 0.05; **P < 0.01 and ***P < 0.001 (unpaired two-tailed Student’s t-test). Data are from one experiment representative of three experiments (a) or two experiments (b–d) with three to seven mice per group (mean and s.e.m.).
To further test that model, we purified IL-10-sufficient Foxp3+ CD4+ Treg cells from donor mice 8 d after infection with LCMV and transferred the cells into ‘infection-matched’ Il10−/− host mice. Transfer of Foxp3+ CD4+ Treg cells into Il10−/− mice was sufficient to restore the maturation state of CD45+CD11chiMHCII+ DCs at day 15 after infection to that found in Il10−/− mice that did not receive Treg cells (Fig. 7b). Additionally, Il10−/− mice that received IL-10-sufficient Treg cells displayed a significantly greater proportion of mature memory CD8+ T cells 45–60 d after infection and had higher expression of CD127 and TCF-1 along with lower expression of KLRG1 and granzyme B relative to that of Il10−/− mice that did not receive Treg cells (Fig. 7c,d and Supplementary Fig. 7b). Therefore, the presence of IL-10-sufficient Treg cells during the resolution phase of infection was sufficient to at least partially ‘rescue’ many of the defects in memory CD8+ T cell maturation observed in the absence of IL-10.
DISCUSSION
Understanding the signals that promote the formation of a protective memory CD8+ T cell population following viral infection is of crucial importance to furthering efforts in the design of T cell–based vaccines. Here we have described a process by which signals present during the resolution phase of infection promoted the formation of a mature memory CD8+ T cell pool. Early suppression of Treg cell function following infection allowed the activation and population expansion of effector T cells, but following eradication of the virus, IL-10-producing Treg cells accumulated to resolve inflammation and initiate tissue repair and simultaneously promoted the development and maturation of protective memory CD8+ T cells. Our results highlight the need to consider, in the design of T cell–based vaccines, not only the initial signals T cells encounter following vaccination but also the inflammatory environment present during the resolution phase.
Our findings indicated an important role for Treg cells in promoting the maturation of memory CD8+ T cells through calming of the activity of DCs (and probably other pro-inflammatory cells) via the production of IL-10. Our work is consistent with findings showing that DCs that cannot sense IL-10 secrete enhanced amounts of pro-inflammatory cytokines following activation39. Treg cells can also suppress DC activation through other mechanisms. For example, Treg cells can induce downregulation of the expression of CD80 and CD86 on DCs through a process dependent on the integrin LFA-1 (αLβ2) and the immunomodulatory receptor CTLA-4 (CD152)41. Additionally, Treg cells can inhibit the activation of DCs through the hydrolysis of extracellular ATP via expression of the ectoenzyme CD39, or through the induction of immunosuppressive tryptophan metabolism in DCs42,43. Thus, IL-10 production is probably one of multiple mechanisms by which Treg cells can promote the development of memory CD8+ T cells.
DCs can be categorized into distinct subsets by their surface expression of CD8, with CD8+ DCs (‘lymphoid DCs’) marked by enhanced expression of pro-inflammatory cytokines44. Lymphoid DCs are selectively located in the T cell zone in the spleen and are found mainly in the white pulp32. MPCs are also located mainly in the white pulp and, when they encounter large amounts of inflammatory signals, can further differentiate into TECs24,33. We found that IL-10-competent Treg cells were present mostly in the white pulp, which would position them optimally to regulate the inflammatory state of DCs and thus insulate CD8+ T cells from excess signals and preserve their MPC state. Our work suggests that tight regulation of the amount of inflammatory cytokines produced by DCs during the resolution phase is critical for the maturation of memory CD8+ T cells.
Treg cells are reported to have both negative roles18 and positive roles19–21 in regulating the development of CD8+ T cell memory. Studies indicating a role for Treg cells in the promotion of splenic CD8+ T cell memory have focused on the function of Treg cells during the priming phase of infection through regulation of T cell avidity or IL-2 availability. Here we have identified a previously unknown role for Treg cells, during the resolution phase following viral infection, in promoting the memory maturation of CD8+ T cells. The function of Treg cells during the resolution phase is distinct from that shown in previous work19–21, as Treg cell–derived IL-10 was not necessary during the priming phase for the optimal memory maturation of CD8+ T cells. Collectively, this indicates that Treg cells are capable of modulating T cells through discrete mechanisms at different time points and sites to allow the memory maturation of CD8+ T cells while simultaneously suppressing immunopathology and promoting the reestablishment of tissue homeostasis45,46.
Our study might also have implications for the ability to design vaccines against pathogens such as human immunodeficiency virus and influenza virus, which are intransigent to current vaccination approaches. Our work indicated that the presence of IL-10-competent Treg cells during the resolution phase was necessary for the maturation of memory CD8+ T cells. Thus, treatment with IL-10 during the resolution phase following vaccination might allow improved vaccine efficacy. Alternately, the administration of rapamycin during this phase might represent another approach, because rapamycin promotes the selective population expansion of Treg cells in vivo and enhances the quantity and quality of CD8+ T cell memory11,47. Treatment with rapamycin also enhances the formation of broadly neutralizing antibodies48 and thus might operate through multiple mechanisms to promote vaccine efficacy. Additionally, our work suggests that the use of adjuvants that elicit more IL-10 production might be more effective in eliciting a protective CD8+ T cell response than are adjuvants that induce small amounts of IL-10. Indeed, published work has found that the ability of adjuvants to elicit IL-10 positively correlates with their ability to generate memory CD8+ T cell upon immunization49. Thus, our study suggests a critical period of time during which therapeutic intervention might allow considerable enhancement of the ability of vaccines to promote a productive immune response, leading to long-lasting and protective immunological memory.
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
Mice, infections and bacterial titers
C57BL/6 mice were from the National Cancer Institute or The Jackson Laboratory. B6.129P2 Il10tm1Cgn/J (Il10−/−) mice, B6.129(cg)-Foxp3tm3(DTR/GFP)Ayr/J (Foxp3GFP–DTR) mice and B6.129 (Cg)-Foxp3tm4(YFP/cre)Ayr/J (Foxp3-Cre) mice were from The Jackson Laboratory51. 10BiT mice52, Il10f/f mice53 and Cd4-Cre, Lyz2-Cre, Cd11c-Cre and P14 mice54 have been described. Il10raf/f mice were generated by the Flavell laboratory. Mouse genomic DNA of Il10ra was isolated from the C57BL/6 bacterial artificial chromosome clone RP23-329F21 (BACPAC Resources Center, Children’s Hospital Oakland Research Institute). A targeting vector was constructed by cloning of three genomic fragments into the plasmid pEasy-Flirt. The construct was designed to target exon 3 of Il10ra, which contains the translation-initiation site of Il10ra, and thus allow abolishment of IL-10Rα protein after deletion by Cre recombinase. The vector pEasy-Flirt has both Flp recombinase for deletion of the neomycin cassette and Cre recombinase sites to target exon 3 of Il10ra. The linearized targeting vector was then transfected into mouse embryonic stem cells (JM8 line). Homologous recombinants were screened and identified by PCR analysis. Clones carrying the mutated alleles of Il10ra were injected into blastocycts that were then implanted into foster mothers. Chimeric mice were bred to C57BL/6 mice, and the F1 generation was screened for identification of germline transmission. All mice studied were 6–8 weeks of age.
For the generation of mice bearing LCMV-specific epitopes, splenocytes from P14 donor mice ‘normalized’ for 5 × 104 naive P14 CD8+ T cells (with expected engraftment of 10%; thus, 5 × 103 cells) were transferred into C57BL/6 mice by intravenous injection. Bacterial titers were quantified by lysing of whole livers in 0.5% Triton-X100 and plating of tenfold serial dilutions of bacteria overnight on brain-heart–infusion agar plates. All animal experiments were done with approval of the Yale Institutional Animal Care and Use Committee.
Infection and treatment
For primary infection, mice were given intraperitoneal administration of 2 × 105 plaque-forming units of LCMV, Armstrong strain. For rechallenge experiments, mice were given 2 × 104 colony-forming units of recombinant L. monocytogenes expressing the LCMV gp(33–41) epitope. For blockade of IL-10, mAb to IL-10 (JES5-2A5; provided by J.M.M. den Haan) was administered at a dose of 0.25 mg/ml every other day for the appropriate period of time for the experiment. For treatment with CpG B, mice were given intraperitoneal administration of a dose of 3.75 μg CpG B per mouse every other day starting on the day appropriate for the experiment and ending after three doses had been administered. Bacterial titers were quantified by lysing of whole livers in 0.5% Triton-X100 and plating of tenfold serial dilutions of bacteria on brain-heart–infusion agar plates, followed by incubation overnight. Diphtheria toxin was reconstituted according to manufacturer’s instructions (Sigma). Mice were given two intraperitoneal injections of diphtheria toxin at a dose of 50 μg per kg body weight on the appropriate day, as well as the following day, as described55.
Antibodies for surface and intracellular staining
The isolation of lymphocytes, along with surface and intracellular staining, was performed as described56. Staining of transcription factors was performed after permabilization with the FoxP3 Fixation and Permeabilization Kit (eBioscience). The following antibodies were used for flow cytometry staining: Brilliant Violet 421–anti-CD44 (103039), allophycocyanin–anti-KLRG1 (138412), phycoerythrin (PE)–indotricarbocyanine (Cy7)–anti-CD69 (104512), peridinin chlorophyll protein (PerCP)–anti-Thy-1.1 (202512), fluorescein isothiocyanate (FITC)–anti-CD80 (104706), PE-Cy7–anti-PDL1 (124314), allophycocyanin–anti-Ly5.2 (109814), Alexa Fluor 780–anti-CD8 (100714), allophycocyanin–anti-CXCR3 (126512) and Pacific Blue–conjugated antibody to major histocompatibility complex class II (107619) (all from BioLegend); PE-Cy7–anti-CD127 (25-1271-82), FITC–anti-CD27 (11-0271-82), PE–anti-PDL2 (12-5986-81), PE–anti-CD25 (12-0251-83) and allophycocyanin–anti-CD11c (17-0114-81) (all from eBioscience); FITC–anti-CD62L (553150) and PE–anti-CD86 (553692) (both from BD Biosciences); rabbit mAb to TCF1 (C63D9; Cell Signaling); and allophycocyanin–anti-GzmB (GRB05; Invitrogen). Major histocompatibility complex class I tetramers were generated as described54. Flow cytometry data were acquired on a BD LSRII with FACSDiva software and were analyzed with FlowJo software (TreeStar).
In vivo CD4+ T cell labeling
For intravascular staining of CD4+ T cells, mice were given injection of 3 μg biotin-conjugated antibody to CD4 (RM4-5; eBioscience) diluted in 300 μl sterile DPBS (Life Technologies) as described57 and were killed 5 min after this injection.
Isolation of lymphocytes from tissues
Spleens were homogenized with a cell strainer, and red blood cells were lysed with ACK lysing buffer (Quality Biologicals). Lymphocytes were then washed and counted.
Isolation of RNA and real-time quantitative PCR
For quantitative PCR, first, total RNA was isolated from sorted samples with the use of Qiazol and an RNeasy Mini kit (Qiagen), in which on-column treatment with DNase was included. Reverse transcription was then carried out with an iScript cDNA synthesis kit (Bio-Rad). Quantitative RT-PCR was performed on cDNA with iTaq Universal SYBR Green super mix (Bio-Rad) and the following primer sets: Ccr7 (forward, GTGGTGGCTCTCCTTGTCAT; reverse, AGTTCCGCACATCCTTCTTG), Pim1 (forward, GATCATCAAGGGCCAAGTGT; reverse, GATGGTTCCGGATTTCTTCA), Cx3cr1 (forward, AAGTTCCCTTCCCATCTGCT; reverse, GGACAGGAAGATGGTTCCAA), Zeb2 (forward, GAGCAGGTAACCGCAAGTTC; reverse, TGTTTCTCATTCGG) and HPRT (forward, GCTATAAATTCTTTGCTGACCTGCTG; reverse, AATTACTTTTATGTCCCCTGTTGACTGG). Differences in expression were calculated with expression of the control gene Hprt (encoding hypoxanthine guanine phosphoribosyl transferase) as a normalization constant.
RNA-seq library preparation and data analysis
Total RNA was purified with the use of a Qiazol and RNeasy Mini kit (Qiagen), in which on-column treatment with DNase was included. Purified RNA was submitted to the Yale Center for Genomic Analysis, where it was subjected to isolation of mRNA and library preparation. Libraries were pooled, six samples per lane, and were sequenced on an Illumina HISEQ 2500 (75–base pair paired-end reads), followed by alignment with TopHat2 software. A ‘count-based’ differential-expression protocol was adapted for this analysis58; data that could be mapped were ‘counted’ with the HTSeq package that provides infrastructure for data processing, then were imported into software of the R project for analysis of differential expression with DESeq2 software. A multi-factor design was used to take into the two conditions (Il10f/fFoxp3-Cre and Il10f/f) and account for pairwise groupings of the six samples. For the generation of the heat map, genes with a difference in expression (log2) of 1.5-fold or greater and a false-discovery rate of <0.2 were chosen for visualization of the overall consensus of rankings (via column z-score). Select biologically relevant genes were chosen for a subsequent presentation on a heat map, with genes in pro- and anti-inflammatory or effector-memory signatures designated as such according to gene-set definitions of the Molecular Signatures Database.
GSEA
Gene sets (1,911) from the immunological signatures collection (C7) of the Molecular Signatures Database were chosen for assessment of enrichment in the RNA-seq analysis of Il10f/fFoxp3-Cre cells versus Il10f/f cells. R-GSEA code was used for analysis of the data, with the signal-to-noise ranking metric and 1 × 105 permutations of the gene labels for testing of statistical significance. Results were visualized with software of the R project, with the running enrichment score, member position by the signal-to-noise ranking metric (‘bar code’), and ranking metric scale presented. Values in gene-set plots showing individual differences in expression (log2) ± standard error were derived from analysis of the RNA-seq data with DESeq2 software as described59.
Cytokine analysis
Serum was collected from mice at the appropriate time points and was stored at −80 °C until analysis. The expression of IL-6, IL-12p70, IL-1β and TNF was determined with Milliplex Cytokine kits according to the manufacturer’s instructions (Millipore) and results were ‘read’ on a Bio Plex System (Bio-Rad).
Statistical analysis
Statistical significance was determined by the unpaired two-tailed Student’s t-test. Statistical analyses were performed using Prism GraphPad software, version 6.0.
Supplementary Material
Acknowledgments
We thank J.M.M. den Haan (UV University Medical Center, Amsterdam) for mAb ES5-2A5; and all members of the Kaech and Craft laboratories for discussions and critical reading of the manuscript. Supported by the US National Institutes of Health (RO1AI066232 and R01AI074699 to S.M.K.; R01AR40072, P30AR053495 and R21AR063942 to J.C.; T32AI07019 and F31AG07777 to B.J.L.; and T32GM07205 to S.M.G.) and the Howard Hughes Medical Institute (S.M.K., B.J.L., R.A.F. and T.G.).
Footnotes
Accession codes. GEO: RNA-seq data, SRP058713.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
B.J.L., J.C., S.M.K. conceived of and designed the experiments, analyzed the data and wrote the manuscript; and B.J.L., W.C., R.A.A., S.M.G., T.G., Y.L., Y.K., S.H.K. and R.A.F. performed the experiments.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Ahmed R, Gray D. Immunological memory and protective immunity: understanding their relation. Science. 1996;272:54–60. doi: 10.1126/science.272.5258.54. [DOI] [PubMed] [Google Scholar]
- 2.Korber BT, Letvin NL, Haynes BF. T-cell vaccine strategies for human immunodeficiency virus, the virus with a thousand faces. J Virol. 2009;83:8300–8314. doi: 10.1128/JVI.00114-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaech SM, Ahmed R. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naïve cells. Nat Immunol. 2001;2:415–422. doi: 10.1038/87720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. 2012;12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cui W, Liu Y, Weinstein JS, Craft J, Kaech SM. An interleukin-21-interleukin-10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity. 2011;35:792–805. doi: 10.1016/j.immuni.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rutishauser RL, et al. Transcriptional repressor Blimp-1 promotes CD8+ T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity. 2009;31:296–308. doi: 10.1016/j.immuni.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim MV, Ouyang W, Liao W, Zhang MQ, Li MO. The transcription factor Foxo1 controls central-memory CD8+ T cell responses to infection. Immunity. 2013;39:286–297. doi: 10.1016/j.immuni.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joshi NS, et al. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang CY, et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol. 2011;12:1221–1229. doi: 10.1038/ni.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou X, et al. Differentiation and persistence of memory CD8+ T cells depend on T cell factor 1. Immunity. 2010;33:229–240. doi: 10.1016/j.immuni.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Araki K, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rao RR, Li Q, Gubbels Bupp MR, Shrikant PA. Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8+ T cell differentiation. Immunity. 2012;36:374–387. doi: 10.1016/j.immuni.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haring JS, Harty JT. Interleukin-18-related genes are induced during the contraction phase but do not play major roles in regulating the dynamics or function of the T-cell response to Listeria monocytogenes infection. Infect Immun. 2009;77:1894–1903. doi: 10.1128/IAI.01315-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stelekati E, et al. Bystander chronic infection negatively impacts development of CD8+ T cell memory. Immunity. 2014;40:801–813. doi: 10.1016/j.immuni.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol. 2004;5:927–933. doi: 10.1038/ni1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foulds KE, Rotte MJ, Seder RA. IL-10 is required for optimal CD8 T cell memory following Listeria monocytogenes infection. J Immunol. 2006;177:2565–2574. doi: 10.4049/jimmunol.177.4.2565. [DOI] [PubMed] [Google Scholar]
- 17.Vignali DAA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suvas S, Kumaraguru U, Pack CD, Lee S, Rouse BT. CD4+CD25+ T cells regulate virus-specific primary and memory CD8+ T cell responses. J Exp Med. 2003;198:889–901. doi: 10.1084/jem.20030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pace L, et al. Regulatory T cells increase the avidity of primary CD8+ T cell responses and promote memory. Science. 2012;338:532–536. doi: 10.1126/science.1227049. [DOI] [PubMed] [Google Scholar]
- 20.de Goër de Herve MG, Jaafoura S, Vallée M, Taoufik Y. FoxP3+ regulatory CD4 T cells control the generation of functional CD8 memory. Nat Commun. 2012;3:986. doi: 10.1038/ncomms1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graham JB, Da Costa A, Lund JM. Regulatory T cells shape the resident memory T cell response to virus infection in the tissues. J Immunol. 2014;192:683–690. doi: 10.4049/jimmunol.1202153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kühn R, Löhler J, Rennick D, Rajewsky K, Müller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 23.Maharshak N, et al. Altered enteric microbiota ecology in interleukin 10-deficient mice during development and progression of intestinal inflammation. Gut Microbes. 2013;4:316–324. doi: 10.4161/gmic.25486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stelekati E, Wherry EJ. Chronic bystander infections and immunity to unrelated antigens. Cell Host Microbe. 2012;12:458–469. doi: 10.1016/j.chom.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol. 2011;29:71–109. doi: 10.1146/annurev-immunol-031210-101312. [DOI] [PubMed] [Google Scholar]
- 26.Cui W, Joshi NS, Jiang A, Kaech SM. Effects of Signal 3 during CD8 T cell priming: Bystander production of IL-12 enhances effector T cell expansion but promotes terminal differentiation. Vaccine. 2009;27:2177–2187. doi: 10.1016/j.vaccine.2009.01.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parish IA, et al. Chronic viral infection promotes sustained Th1-derived immunoregulatory IL-10 via BLIMP-1. J Clin Invest. 2014;124:3455–3468. doi: 10.1172/JCI66108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brooks DG, et al. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med. 2006;12:1301–1309. doi: 10.1038/nm1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maynard CL, et al. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3− precursor cells in the absence of interleukin 10. Nat Immunol. 2007;8:931–941. doi: 10.1038/ni1504. [DOI] [PubMed] [Google Scholar]
- 30.Srivastava S, Koch MA, Pepper M, Campbell DJ. Type I interferons directly inhibit regulatory T cells to allow optimal antiviral T cell responses during acute LCMV infection. J Exp Med. 2014;211:961–974. doi: 10.1084/jem.20131556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng G, et al. IL-2 receptor signaling is essential for the development of Klrg1+ terminally differentiated T regulatory cells. J Immunol. 2012;189:1780–1791. doi: 10.4049/jimmunol.1103768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steinman RM, Pack M, Inaba K. Dendritic cells in the T-cell areas of lymphoid organs. Immunol Rev. 1997;156:25–37. doi: 10.1111/j.1600-065x.1997.tb00956.x. [DOI] [PubMed] [Google Scholar]
- 33.Jung YW, Rutishauser RL, Joshi NS, Haberman AM, Kaech SM. Differential localization of effector and memory CD8 T cell subsets in lymphoid organs during acute viral infection. J Immunol. 2010;185:5315–5325. doi: 10.4049/jimmunol.1001948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rubtsov YP, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 35.Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 2002;111:837–851. doi: 10.1016/s0092-8674(02)01139-x. [DOI] [PubMed] [Google Scholar]
- 36.Luckey CJ, et al. Memory T and memory B cells share a transcriptional program of self-renewal with long-term hematopoietic stem cells. Proc Natl Acad Sci USA. 2006;103:3304–3309. doi: 10.1073/pnas.0511137103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agarwal P, et al. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J Immunol. 2009;183:1695–1704. doi: 10.4049/jimmunol.0900592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amit I, et al. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science. 2009;326:257–263. doi: 10.1126/science.1179050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Girard-Madoux MJH, Kel JM, Reizis B, Clausen BE. IL-10 controls dendritic cell-induced T-cell reactivation in the skin to limit contact hypersensitivity. J Allergy Clin Immunol. 2012;129:143–150. e1–10. doi: 10.1016/j.jaci.2011.08.032. [DOI] [PubMed] [Google Scholar]
- 40.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 41.Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci USA. 2008;105:10113–10118. doi: 10.1073/pnas.0711106105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borsellino G, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–1232. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- 43.Fallarino F, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. 2003;4:1206–1212. doi: 10.1038/ni1003. [DOI] [PubMed] [Google Scholar]
- 44.Hochrein H, et al. Differential production of IL-12, IFN-α, and IFN-γ by mouse dendritic cell subsets. J Immunol. 2001;166:5448–5455. doi: 10.4049/jimmunol.166.9.5448. [DOI] [PubMed] [Google Scholar]
- 45.Moser EK, Hufford MM, Braciale TJ. Late engagement of CD86 after influenza virus clearance promotes recovery in a FoxP3+ regulatory T cell dependent manner. PLoS Pathog. 2014;10:e1004315. doi: 10.1371/journal.ppat.1004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liston A, Gray DHD. Homeostatic control of regulatory T cell diversity. Nat Rev Immunol. 2014;14:154–165. doi: 10.1038/nri3605. [DOI] [PubMed] [Google Scholar]
- 47.Zeiser R, et al. Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp3+ regulatory T cells compared with conventional CD4+ T cells. Blood. 2008;111:453–462. doi: 10.1182/blood-2007-06-094482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keating R, et al. The kinase mTOR modulates the antibody response to provide cross-protective immunity to lethal infection with influenza virus. Nat Immunol. 2013;14:1266–1276. doi: 10.1038/ni.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cui W, et al. TLR4 ligands lipopolysaccharide and monophosphoryl lipid a differentially regulate effector and memory CD8+ T Cell differentiation. J Immunol. 2014;192:4221–4232. doi: 10.4049/jimmunol.1302569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Best JA, et al. Transcriptional insights into the CD8+ T cell response to infection and memory T cell formation. Nat Immunol. 2013;14:404–412. doi: 10.1038/ni.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kühn R, Löhler J, Rennick D, Rajewsky K, Müller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 52.Maynard CL, et al. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3− precursor cells in the absence of interleukin 10. Nat Immunol. 2007;8:931–941. doi: 10.1038/ni1504. [DOI] [PubMed] [Google Scholar]
- 53.Roers A, et al. T cell-specific inactivation of the interleukin 10 gene in mice results in enhanced T cell responses but normal innate responses to lipopolysaccharide or skin irritation. J Exp Med. 2004;200:1289–1297. doi: 10.1084/jem.20041789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaech SM, et al. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 55.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 56.Joshi NS, et al. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Teijaro JR, et al. Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol. 2011;187:5510–5514. doi: 10.4049/jimmunol.1102243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anders S, et al. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc. 2013;8:1765–1786. doi: 10.1038/nprot.2013.099. [DOI] [PubMed] [Google Scholar]
- 59.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.