

Abstract
Ochratoxin A (OTA) is a mycotoxin harmful for animals and humans. Aspergillus carbonarius is the main responsible for OTA contamination of grapes and derived products. Gene transcriptional profiling of 4 A. carbonarius strains was carried out by RNA-Seq analysis to study transcriptome changes associated with OTA production. By comparing OTA inducing (OTAI) vs. non-inducing (OTAN) cultural conditions, a total of 3,705 differentially expressed genes (DEGs) (fold change > |2| and FDR ≤ 0.05) were identified. Several genes involved in primary metabolic processes, with particular regard to carbohydrate and amino acid metabolisms, secondary metabolic processes, transport, response to stress and sporulation were up-regulated by OTAI conditions at all the analysed sampling times (4, 6 and 8 DAI) or starting from 6 DAI. Highly up-regulated DEGs encoding enzymes involved in biosynthesis of secondary metabolites, oxidoreductases, transporters and transcription factors were examined for their potential involvement in OTA biosynthesis and related metabolic pathways. Differential expression of genes encoding polyketide synthases (pks), non-ribosomal peptide synthetases (nrps) and chloroperoxidase (cpo) was validated by RT-qPCR. Among clusters of co-regulated genes involved in SM biosynthesis, one putative OTA-gene cluster, including both pks and nrps genes, was detected in the A. carbonarius genome.
Introduction
Ochratoxin A (OTA) is a mycotoxin produced by fungal species belonging to the genera Aspergillus and Penicillium. It is responsible for several adverse effects on animals and humans, having neurotoxic, carcinogenic, immunotoxic, genotoxic, teratogenic properties, and is classified as possible human carcinogen (group 2B) by the International Agency for Research on Cancer [1]. OTA is a common contaminant of various foods and drinks [2,3,4,5,6], and was first reported in wine by Zimmerli and Dick [7]. After cereals, wine is the second source of daily OTA intake for humans [8]. Aspergillus carbonarius (Bainier) Thom is recognized as the main OTA-producing fungus on grapes and derived products [9,10,11].
The chemical structure of OTA, namely N-{[(3R)-5-chloro-8-hydroxy-3-methyl-1-oxo-3,4-dihydro-1H-isochromen-7-yl]carbonyl}-L-phenylalanine, consists of a pentaketide-derived dihydroisocoumarin moiety linked by an amide bond to a phenylalanine amino acid [12]. The chlorine atom attached to the dihydroisocoumarin aromatic ring confers a relatively high toxicity to the molecule in comparison to other ochratoxins [13]. The OTA biosynthetic pathway has not yet been fully clarified, even if many studies have been carried out in several OTA-producing fungi leading to the identification of key enzymes: a polyketide synthase (PKS) required for the biosynthesis of the dihydrocumarin’s precursor; a methyltransferase or a C-methylation (C-Met) domain included in the PKS, adding a methyl group to the C-7 of the newly formed molecule, and a P450 monooxygenase (CYP), oxidizing the methyl group to a carboxyl group; a non-ribosomal peptide synthetase (NRPS) catalyzing the formation of a peptide bond between the polyketide and a phenylalanine; and, finally, a chloroperoxidase adding a chlorine atom at the C-5 position [14].
Disruption of the pks gene in Aspergillus ochraceus proved its functional role in OTA biosynthesis [15]. A similar approach showed the involvement of the aoks1 gene in Aspergillus westerdijkiae [16] and the AcOTApks gene in A. carbonarius [17]. The role of NRPS, previously reported in Penicillium nordicum (encoded by the npsPN gene) and Penicillium verrucosum (npsPV) [18], has been demonstrated in A. carbonarius (AcOTAnrps) by gene knockout [19].
Several techniques, such as AFLP-based transcript profiling (cDNA-AFLP) [20] and suppression subtractive hybridization (SSH) [21], have been used to identify genes differentially expressed in OTA producer and non-producer A. carbonarius strains. Recently, the complete genome sequences of an OTA non-producer A. carbonarius strain has been released [22]. However, the knowledge on OTA biosynthetic pathway in the fungus, its activation and regulation remains incomplete.
High-throughput Next Generation Sequencing (NGS) technologies make nowadays possible to carry out detailed transcriptomic profiling. In this study, we applied the NGS-based RNA-Seq method to carry out a global transcriptional analysis on 4 OTA-producer strains of A. carbonarius grown under inducing and non-inducing conditions. Differential gene expression analysis was aimed at improving knowledge on OTA-related gene expression and at identifying genes directly or indirectly related to OTA biosynthetic pathway.
Materials and Methods
A. carbonarius strains and growing conditions
The OTA-producer strains of A. carbonarius used in this study, AC49, AC66, AC67 and AC70, were obtained from naturally infected grape berries. All strains were grown on potato dextrose agar (PDA; infusion from 200 g peeled and sliced potatoes kept at 60°C for 1 h, 20 g dextrose, adjusted at pH 6.5, 20 g agar Oxoid no. 3, per litre) at 25°C in the dark for 5 days. Conidia of each strain were inoculated (1x106 mL-1, final concentration) into 250 mL Erlenmeyer flasks containing 100 mL of minimal medium [MM; 10 mL solution A (10 g KH2PO4, 100 mL−1 water), 10 mL solution B (20 g NaNO3, 5 g KCl, 5 g MgSO4·7H2O, 0.1 g FeSO4, 100 mL−1 water), 1 mL of micronutritive solution [23], 20 g glucose, per litre] and incubated at 25°C in the dark, with or without shaking (150 rpm), corresponding to non-inducing (OTAN) or inducing (OTAI) conditions for OTA production, respectively.
Three replicated cultures were grown for each combination of strains and conditions. Culture broths for OTA quantification were obtained by filtration through Miracloth (Calbiochem, San Diego, California, USA) 0, 2, 4, 6 and 8 Days After Inoculation (DAI). The mycelium was collected at 4, 6 and 8 DAI, frozen in liquid nitrogen and stored at -80°C until use for RNA-Seq analysis.
OTA Quantification
For HPLC analysis of OTA, aliquots (100 μL) of culture broths were injected into the chromatographic apparatus made up by an isocratic pump (HP 1100, Agilent Technologies, Santa Clara, California, USA) equipped with an injection valve (mod. 7125, Rheodyne, Cotati, California, USA), a fluorometric detector (HP 1100, λex = 333 nm, λem = 460 nm) and a Chemstation Rev A.08.03 data system (Agilent Technologies). The analytical column was a reversed-phase Discovery C18 (15 cm x 4.6 mm, 5 mm particles) (Supelco, Bellefonte, Pennsylvania, USA) preceded by a SecurityGuard (Phenomenex, Torrance, California, USA). OTA in extracts was identified because having a retention time identical to that of the OTA standard (Supelco Sigma-Aldrich, St. Louis, Missouri, USA).
RNA extraction, RNA-Seq library preparation and sequencing
For RNA-Seq analysis, pooled mycelia from 3 replicated cultures were used to prepare an RNA sample per each strain, growing condition and sampling time. Frozen mycelium (100 mg) was powdered in liquid nitrogen. Total RNA was extracted and purified by using RNeasy Plant Mini Kit (Qiagen, Milan, Italy), following the manufacturer's protocol. RNA quantity and quality were determined with a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific Inc., Wilmington, Delaware, USA) and a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). cDNA libraries were prepared from 4 μg total RNA using TruSeq RNA Sample Preparation Kit v2 (Illumina, Inc., San Diego, California, USA) and validated according to Illumina’s low-throughput protocol. After normalization, cDNA libraries were pooled for multiplexing before loading onto a flow cell (8–9 samples per lane). The hybridization and cluster generation were performed on a cBot System using TruSeq SR Cluster Kit v3. Sequencing was performed on an Illumina HiScanSQ platform using TruSeq SBS kit v3 (Illumina, Inc.) to obtain Single Reads, 50 nt in length. Indexed raw sequencing reads from each library were de-multiplexed using the CASAVA v1.8 software (Illumina, Inc.).
RNA-Seq data analysis
The quality of the raw sequence reads was checked using FastX-tools integrated into the Galaxy platform (https://usegalaxy.org/; [24]). Filtered reads from each sample were then separately aligned to the reference genome of A. carbonarius (ITEM 5010, assembly v3; DOE Joint Genome Institute, http://genome.jgi-psf.org/Aspca3/Aspca3.home.html), using the Q-Seq module of the ArrayStar software v. 5.0.0 (DNASTAR, Madison, WI, USA) with default mapping parameters, and used to estimate the abundance of 11,624 gene transcripts, measured as Reads Per Kilobase per Million mapped reads (RPKM) [25]. RPKM > 0.5 was used as cut-off for gene expression. A multiple correlation test (Cross-R2 statistical test, Q-Seq) on RPKM values for all pairwise combinations was performed for preliminary batch comparisons of replicates and experimental conditions.
Differential gene expression analysis was performed with the statistical R package DESeq (http://bioconductor.org/packages/2.12/bioc/html/DESeq.html) using the total number of sequence reads from individual samples mapped on annotated transcripts, and the 4 A. carbonarius strains as biological replicates. Fold Change (FC) was calculated comparing the RPKM expression values under OTAI vs. OTAN conditions; an expression value of 0.05 RPKM was imposed to non-expressed genes. False Discovery Rate (FDR) was determined and genes with FC > |2| and FDR ≤ 0.05 in at least one of the sampling times were considered as differentially expressed genes (DEGs) and submitted to functional analysis. The expression profiles of DEGs at different DAI were analysed by hierarchical clustering and heat map of expression values (T-MeV 4.9.0 software [26]).
Functional analysis and clustering of DEGs
Blast2GO v2.6.4 [27] was used for functional annotation of A. carbonarius transcriptome, and Aspergillus-specific GO Slim (http://www.geneontology.org/ontology/subsets/goslim_aspergillus.obo) for summarising GO annotations. GO Enrichment analysis (Fisher's Exact Test) was used (p-value ≤ 0.05) to detect functional categories of biological processes and molecular functions over-represented, with statistical significance (FDR ≤ 0.05), in each DEG set as compared with the remaining non-differentially expressed genes (used as reference transcriptome). The distribution of GO terms in DEG sets was explored through Multilevel Pie charts.
The antiSMASH 2.0 platform (http://antismash.secondarymetabolites.org/ [28]) was used to predict gene clusters involved in biosynthetic pathways of secondary metabolites (SMs) in the A. carbonarius masked scaffolds and to perform a ClusterBlast analysis on available fungal genome sequences (http://genome.jgi-psf.org/programs/fungi/index.jsf). Co-expressed genes (CEGs) were identified using the quality threshold (QT) clustering (Pearson correlation ≥ 0.75, minimum 4 genes per cluster) in T-MeV 4.9.0 software and clustered in groups showing similar expression profiles and including maximum one non-correlating gene in between them. The average RPKM values of the CEGs included in each putative gene clusters were used to explore their co-regulation through correlation analysis.
Validation of RNA-Seq analysis by RT-qPCR
RT-qPCR analysis was carried out according to MIQE guidelines [29] to validate RNA-Seq results for ten selected DEGs. Total RNA was extracted from 2 A. carbonarius strains, AC49 and AC67, used as biological replicates, using TRI Reagent (Sigma-Aldrich) according to the manufacturer’s instructions. All primer pairs were designed with the Primer3 software (http://frodo.wi.mit.edu/primer3/ [30]) with the forward ones designed on the exon-junction sites of the target gene to amplify only cDNA and not possible contaminant genomic DNA (S1 Table). First strand cDNA was synthesized from 2 μg of RNA using M-MLV reverse transcriptase (Life Technologies, Milan, Italy) and random primers in a volume of 20 μL, according to the manufacturer’s instructions. qPCR was performed in a CFX96TM Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules CA, USA) in a volume of 25 μL containing 12.5 μL of iQ SYBR Green Super Mix (Bio-Rad Laboratories), 0.5 μM of each primer and 1 μL of the reverse transcription reaction. The conditions for amplification were as follows: 3 min denaturation at 95°C followed by 35 cycles of 95°C for 10 s and 60°C for 45 s. The absence of unwanted products was assessed though melting curve from 60°C to 95°C with 10-sec steps of 0.5°C increments. All samples were analysed in duplicate. Genes encoding β-tubulin (β-tub; ID 202852), calmodulin (cal; ID 205510), and ubiquitin-conjugating enzyme (ubc; ID 393986) from the A. carbonarius genome were selected as candidate references and BestKeeper algorithm was used to evaluate their gene expression stability [31]. Relative gene expression was calculated using CFX Manager Software (Bio-Rad Laboratories) and the 2-ΔΔCT method [32].
Results
OTA production under inducing and non-inducing conditions
The production of OTA was favoured in static cultures in MM (OTAI) and prevented in shaken cultures in MM (OTAN). Under OTAN conditions, OTA concentrations decreased or remained stable from 0 to 8 DAI. Under OTAI conditions, OTA concentrations increased from 2 to 8 DAI with a strain-dependent variation. The strain AC49 was the best OTA-producer followed, in the order, by AC70, AC67 and AC66 (Table 1).
Table 1. OTA concentrations (μg L-1) at different Days After Inoculation (DAI) in cultural broth of 4 A. carbonarius strains grown in liquid minimal medium (MM) under OTA inducing (OTAI, static culture) and non-inducing (OTAN, shaken culture) conditions.
Data are the mean values of 3 replicates and their standard errors.
| Strain | Cultural condition | DAI | ||||
|---|---|---|---|---|---|---|
| 0 | 2 | 4 | 6 | 8 | ||
| AC49 | OTAI | 171.5±4.4 | 174.9±5.7 | 221.4±10.2 | 754.3±45.1 | 1,603.4±212.5 |
| AC67 | OTAI | 115.7±3.9 | 111.1±2.6 | 143.7±6.8 | 479.7±92.8 | 558.5±129.7 |
| AC66 | OTAI | 19.6±0.8 | 19.6±0.3 | 19.8±1.0 | 150.8±40.4 | 425.3±97.5 |
| AC70 | OTAI | 5.8±0.2 | 5.9±0.2 | 6.1±0.2 | 177.6±69.7 | 1,343.9±180.8 |
| AC49 | OTAN | 180.6±7.2 | 144.0±10.1 | 86.4±9.6 | 80.7±12.2 | 80.9±27.2 |
| AC67 | OTAN | 113.6±5.0 | 96.4±11.7 | 50.2±2.1 | 47.3±3.5 | 36.6±3.8 |
| AC66 | OTAN | 19.8±1.0 | 20.9±0.5 | 15.7±2.9 | 15.6±4.3 | 20.3±1.7 |
| AC70 | OTAN | 5.5±0.3 | 6.2±0.2 | 7.0±0.3 | 7.0±0.5 | 5.5±1.9 |
RNA-Seq analysis
A summary of RNA-Seq data is reported in S2 Table. A total of 255,290,345 high-quality (QS≥30) short-sequence single reads (50 bp), yielding approximately 13 Gb of transcriptomic sequence data, was obtained from all the sequenced samples and deposited in the NCBI Sequence Read Archive (SRA) database under study accession number SRP059422. Approximately 82% of short reads, more than 6.6 million qualified reads per each biological replicate, were successfully mapped on annotated gene transcripts of the A. carbonarius genome, corresponding to a coverage of at least 14X of the fungal transcriptome. Average GC content in the sequenced transcriptomes was 54%. The number of total mapped reads increased (237,697,062, i.e. 93%) when they were aligned to masked scaffolds of the assembled A. carbonarius genome. No significant differences were observed in terms of total number of aligned reads between the tested strains (AC49, AC66, AC67 and AC70), conditions (OTAI and OTAN) or sampling times (4, 6 and 8 DAI). Most of the mapped reads were uniquely assigned to transcript sequences (54–73%), while the remaining reads were unmapped (12–34%) or showed multi-position matches (12–16%). A total of 10,479 and 10,257 genes, out of 11,624 predicted genes in the A. carbonarius genome, were expressed under OTAI and OTAN conditions, respectively. A high Person’s correlation coefficient (R2) (0.86) was observed between RPKM values of the 4 A. carbonarius strains, used as biological replicates, indicating that the approach was appropriate for further analysis (S3 Table). A high correlation (average R2 ≥ 0.82) was observed between sampling times in both OTAI or OTAN conditions; lower R2 values were observed when the two growing conditions were compared at 4 (average R2 = 0.82), 6 (R2 = 0.72) and 8 DAI (R2 = 0.77). Hence, sampling time caused fewer transcriptional changes than growing conditions.
Differential gene expression and functional analysis
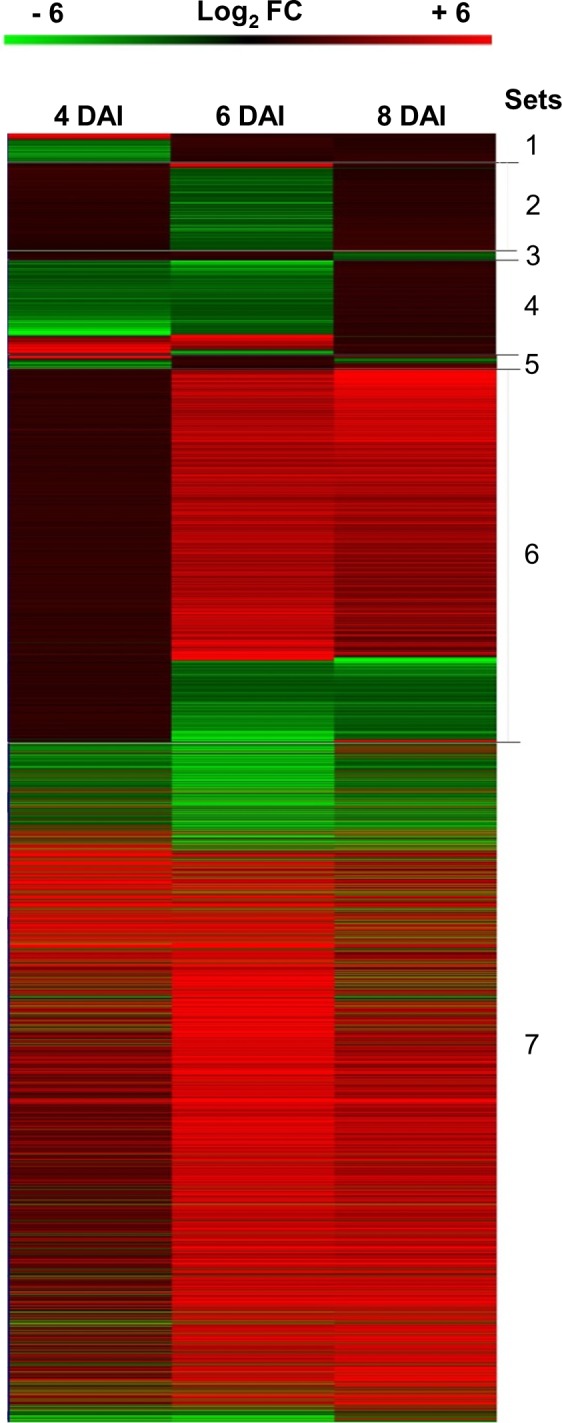
A total of 3,705 differentially expressed genes (DEGs; FC > |2|, FDR ≤ 0.05) were identified comparing OTAI vs. OTAN conditions and submitted to clustering and functional analysis. Relatively few genes (0.7–6.8%) were modulated only at single sampling times (Sets 1–3), whereas the majority of DEGs were modulated at 6 and 8 DAI (29%, Set 6) or at all times (53%, Set 7) (Fig 1). The expression profiles of DEG sets are reported in Fig 2. Sets 1 to 5 contained more down-regulated than up-regulated DEGs; as opposite, the Sets 6 and 7 contained prevalently up-regulated DEGs.
Fig 1. Venn diagram of DEGs (FC >|2|) comparing OTAI vs. OTAN conditions at 4, 6 and 8 DAI.

Fig 2. Heat-map showing expression profiles of DEG sets identified by the Venn diagram.
Distribution of GO-terms among up- and down-regulated DEGs is shown in S1 Fig. Some functional categories were related to both primary and secondary metabolic processes, fungal growth, cell cycle and reproduction. The term asexual sporulation was strictly associated with up-regulated DEGs, while sexual sporulation was prevalently associated with down-regulated DEGs. GO-terms, such as vesicle-mediated transport, lipid metabolic processes, protein catabolic process and response to stress, were more frequently associated to up- than to down-regulated DEGs.
Enrichment analysis for GO terms was carried out in order to determine processes and functions over-represented in DEG sets as compared to the reference transcriptome (Fig 3 and S4 Table). Among up-regulated DEGs, processes related to secondary metabolism (1.8%) and carbohydrate metabolism (8.4%) were significantly over-represented in OTAI conditions at 4 DAI; secondary metabolic processes were more represented, although with no statistical significance, at 6 DAI (0.7%), along with terms related to amino acid and protein metabolism, such as cellular amino acid metabolic process (5.5%), translation (6.5%), protein folding (1.6%) and ribosome biogenesis (1.5%). Among down-regulated DEGs, over-represented process categories were transport (22.5–24.0%) and sexual sporulation (0.8%) at 4 and 8 DAI, cellular respiration (1.2–1.7%) at 6 and 8 DAI, carbohydrate metabolic process (9.3%) and pathogenesis (0.8%) at 8 DAI. In particular, secondary metabolic processes were associated to DEGs such as pks, nrps, phenylalanine ammonia-lyase (PAL) and genes related to pigment biosynthesis and antibiotic biosynthetic pathways. Processes of carbohydrate metabolism were associated to up-regulated DEGs involved in catabolism, e.g. hydrolases, and to down-regulated DEGs involved in anabolism, e.g. synthases and transferases.
Fig 3. Enrichment analysis for biological process (a) and molecular function (b) for genes up- or down-regulated at different DAI (Days After Inoculation).

Asterisks indicate GO categories over-represented (FDR ≤ 0.05) among DEGs (blue) compared to the reference A. carbonarius transcriptome (red).
As to molecular functions is concerned, among up-regulated DEGs, oxidoreductase activity was significantly over-represented at 6 and 8 DAI (22.2–33.3%); additional terms at 6 DAI were RNA-binding (4.5%), structural molecular activity (4.2%) and isomerase activity (3.0%). Among down-regulated DEGs, over-represented functions were transporters (all times), signal transducers, hydrolases acting on ester bonds (6 DAI) and protein kinase activities (8 DAI).
Eight-hundred-ninety highly up-regulated (FC > 8, FDR ≤ 0.05) DEGs were selected. K-means clustering grouped them into 4 clusters sharing similar temporal expression patterns (S2 Fig). In this sets, we detected, prevalently in the clusters b and d, 151 DEGs of potential interest for their functions and temporal expression profiles coherent with OTA production (Table 2).
Table 2. Selected A. carbonarius genes highly up-regulated under OTAI vs. OTAN conditions.
| Transcript ID* | Functional category | FC** | ||
|---|---|---|---|---|
| 4 DAI | 6 DAI | 8 DAI | ||
| Secondary metabolic processes | ||||
| 5570/5640 | polyketide synthase | 6.7 | 59.0 | 59.0 |
| 56260 | polyketide synthase | 1.5 | 382.9 | 235.2 |
| 172075 | polyketide synthase (Alb1) | 36.0 | 10.7 | 6.2 |
| 505925 | polyketide synthase (Acpks) | 40.9 | 18.6 | 11.9 |
| 173482 | polyketide synthase (AcOTApks) | 2.1 | 6.3 | 2.1 |
| 132610 | non-ribosomal peptide synthetase (AcOTAnps) | 38.5 | 70.9 | 6.9 |
| 204544 | non-ribosomal peptide synthetase | 49.6 | 70.3 | 41.5 |
| 206989 | non-ribosomal peptide synthetase | 48.6 | 80.8 | 39.2 |
| 505182 | non-ribosomal peptide synthetase | 1.1 | 7.1 | 10.2 |
| 208824 | non-ribosomal peptide synthetase-like enzyme | 8.5 | 98.9 | 106.6 |
| 209543 | radH flavin-dependent halogenase | 39.2 | 247.5 | 24.6 |
| 205535 | cercosporin toxin biosynthesis protein | 6.5 | 29.9 | 38.4 |
| 127829 | steroid monooxygenase | 5.5 | 21.9 | 7.7 |
| 10909 | salicylate hydroxylase | 9.5 | 7.5 | 14.2 |
| 208828 | GPI anchored dioxygenase | 40.3 | 43.7 | 16.7 |
| 205273 | dienelactone hydrolase family protein (HDL) | 1.1 | 10.9 | 12.1 |
| 397762 | dienelactone hydrolase family protein (HDL) | 2.8 | 13.5 | 19.2 |
| 158065 | oxalacetate acetylhydrolase | 3.0 | 11.0 | 12.1 |
| 512610 | sesquiterpene cyclase | 12.7 | 49.0 | 14.2 |
| 132163 | Rab geranylgeranyl transferase, beta subunit | 3.5 | 10.9 | 10.3 |
| 209742 | conidial pigment biosynthesis oxidase (Abr1)/brown 1 | 23.7 | 17.6 | 11.3 |
| 8073 | pigment biosynthesis protein (Ayg1) | 66.2 | 12.6 | 6.8 |
| 5560/405938 | conidial pigment biosynthesis scytalone dehydratase (Arp1) | 6.2 | 47.1 | 48.0 |
| Transport | ||||
| 209684 | general substrate transporter-like protein | 14.6 | 45.1 | 26.4 |
| 173073 | efflux pump antibiotic resistance protein | 52.4 | 33.7 | 25.6 |
| 209281 | efflux pump antibiotic resistance protein | 1.5 | 9.1 | 8.8 |
| 173225 | MFS toxin efflux pump (aflT) | 13.8 | 5.1 | 2.2 |
| 49247/49992 | MFS transporter | 143.8 | 2,102 | 2,257 |
| 130550/130610 | MFS transporter | 7.1 | 29.5 | 94.3 |
| 173694 | MFS transporter | 2.5 | 11.9 | 9.8 |
| 209285 | MFS transporter | 35.5 | 23.7 | 12.2 |
| 135554 | MFS transporter (FLU1) | 6.8 | 12.2 | 5.7 |
| 211441 | ABC drug exporter (AtrF) | 2.9 | 6.0 | 14.5 |
| 171384 | ABC transporter | 3.9 | 46.5 | 27.9 |
| 177479 | Aquaporin | 1.3 | 12.3 | 11.8 |
| 39367 | dynamin family protein | 107.0 | 507.4 | 190.5 |
| 204836 | galactose-proton symporter | 5.3 | 14.1 | 8.3 |
| 132418 | formate/nitrite family transporter | 5.5 | 22.0 | 14.7 |
| 392642 | H+/nucleoside cotransporter | 10.3 | 14.0 | 7.5 |
| 126007 | purine-cytosine permease | 19.3 | 3.6 | 1.1 |
| 125662 | high affinity methionine permease | 17.9 | 18.5 | 28.3 |
| Fungal growth and asexual sporulation | ||||
| 396337 | solid-state culture specific protein | 8.2 | 42.2 | 25.6 |
| 205578 | plasma membrane protein Pth11-like protein | 18.5 | 15.8 | 7.0 |
| 211206 | cell wall protein | 4.8 | 10.8 | 9.9 |
| 131880 | cell wall protein | 56.7 | 11.5 | 4.5 |
| 208632 | F5/8 type C domain protein | 384.2 | 19.1 | 9.0 |
| 130752 | conidiation-specific protein 10 | 16.1 | 27.3 | 20.5 |
| 205405 | hydrophobic surface binding protein A | 8.1 | 8.2 | 4.8 |
| 208459 | conidial hydrophobin Hyp1/(RodA) | 446.5 | 11.1 | 5.4 |
| 205575 | hydrophobin/(RodB) | 19.0 | 45.2 | 12.7 |
| 130776 | small S protein-like | 12.8 | 26.5 | 17.7 |
| 506080 | extracellular matrix protein | 44.1 | 11.9 | 5.9 |
| 151218 | NACHT domain protein | 19.0 | 15.6 | 5.3 |
| 156387 | NACHT domain protein | 24.6 | 28.0 | 19.8 |
| Metabolic processes of carbohydrates, lipids, amino acids, proteins and other macromolecules | ||||
| 212042 | beta-1,6-glucanase | 17.4 | 13.6 | 10.9 |
| 510653 | exo-beta-1,3-glucanase | 14.5 | 14.5 | 10.5 |
| 167755 | glucose dehydrogenase | 8.0 | 7.5 | 2.6 |
| 38981 | alpha-1,2-mannosidase | 15.5 | 8.6 | 7.2 |
| 517301 | cell wall glycosyl hydrolase | 26.6 | 11.6 | 8.5 |
| 174174 | polygalacturonase, putative | 1.6 | 6.4 | 20.5 |
| 206965 | Chitinase | 4.5 | 11.1 | 5.1 |
| 157125 | extracellular serine-rich protein | 10.7 | 7.2 | 6.2 |
| 135028 | Lipase | 9.0 | 29.7 | 32.4 |
| 133260 | Cholinesterase | 10.0 | 125.0 | 146.5 |
| 510672 | triacylglycerol lipase | 8.2 | 10.3 | 15.5 |
| 132362 | Carboxylesterase | 3.0 | 14.4 | 22.9 |
| 46975 | dihydrodipicolinate synthetase family protein | 1.2 | 24.1 | 25.8 |
| 204357 | 3’-phosphoadenosine-5’-phosphosulfate reductase | 4.0 | 11.3 | 13.1 |
| 505924 | glucose-methanol-choline oxidoreductase | 66.3 | 320.6 | 361.9 |
| 7736 | acetylglutamate kinases | 4.4 | 4.1 | 4.1 |
| 206969 | N-acetyltransferase | 27.0 | 23.6 | 20.7 |
| 154116 | acetylglutamate kinase | 15.1 | 14.4 | 13.7 |
| 399389 | aminotransferase class-III | 4.8 | 46.5 | 23.0 |
| 212184 | NADP-specific glutamate dehydrogenase | 8.2 | 6.8 | 6.9 |
| 507488 | proline oxidase (Put1) | 16.2 | 34.0 | 27.8 |
| 518143 | Aspartokinase | 4.6 | 8.6 | 9.5 |
| 507815 | poly(aspartic acid) hydrolase | 4.4 | 13.9 | 11.3 |
| 507808 | X-Pro dipeptidyl-peptidase (S15 family) | 26.7 | 30.2 | 14.9 |
| 6644 | matrix metalloproteinase-11 | 11.5 | 37.2 | 39.1 |
| 134932 | nucleoside-diphosphate-sugar epimerase | 9.1 | 10.4 | 12.3 |
| 211990 | nucleoside-diphosphate-sugar epimerase | 4.7 | 13.8 | 3.7 |
| 210550 | nucleoside-diphosphate-sugar epimerase | 2.6 | 8.5 | 7.0 |
| 40537 | UDP-glucose 4-epimerase | 9.7 | 9.0 | 9.5 |
| 202711 | RNAse III | 15.3 | 55.2 | 30.6 |
| 208875 | uracil-DNA glycosylase | 2.2 | 12.4 | 7.1 |
| 204318 | metal dependent phosphohydrolase | 1.6 | 9.4 | 5.4 |
| 205142 | IMP cyclohydrolase | 2.6 | 8.3 | 6.0 |
| 208869 | nucleoside diphosphate kinase | 3.2 | 19.4 | 12.8 |
| Transferase activity | ||||
| 510988 | UMTA methyltransferase family protein (MT) | 122.6 | 61.6 | 41.6 |
| 56226 | O-methyltransferase (MT) | 1.0 | 207.0 | 139.1 |
| 133219 | O-methyltransferase (MT) | 12.6 | 519.2 | 31.5 |
| 131073 | O-methyltransferase (MT) | 75.2 | 38.3 | 17.9 |
| 50066 | O-methyltransferase (MT) | 26.8 | 10.0 | 6.0 |
| 492 | O-methyltransferase (MT) | 5.1 | 15.8 | 19.2 |
| 504341 | O-methyltransferase (MT) | 1.8 | 10.7 | 9.5 |
| 51018 | Acyltransferase | 6.5 | 8.1 | 10.6 |
| Response to stress | ||||
| 37752 | Catalase | 10.4 | 32.4 | 17.6 |
| 131650/516443 | Catalase | 38.5 | 83.8 | 52.6 |
| 9402 | Catalase | 23.5 | 224.5 | 77.9 |
| 518556 | spore-specific catalase A (AocatA) | 3.2 | 21.1 | 12.0 |
| 53994 | stress response protein (Rds1) | 14.3 | 20.0 | 7.3 |
| Oxidation-reduction processes | ||||
| 212238 | chloroperoxidase (CPO) | 74.6 | 13.2 | 7.2 |
| 399443 | benzoate 4-monooxygenase cytochrome P450 (CYP) | 2.8 | 219.8 | 22.5 |
| 208126 | cytochrome P450 monooxygenase (CYP) | 63.5 | 15.2 | 6.7 |
| 392816 | cytochrome P450 monooxygenase (CYP) | 3.4 | 9.2 | 13.4 |
| 517149 | cytochrome P450 monooxygenase (CYP) | 38.2 | 382.2 | 27.1 |
| 153077 | cytochrome P450 monooxygenase (CYP) | 1.2 | 9.1 | 10.7 |
| 53587 | cytochrome P450 monooxygenase (CYP) | 8.7 | 12.5 | 9.4 |
| 178693 | cytochrome P450 monooxygenase (CYP) | 2.9 | 14.6 | 13.5 |
| 516985 | cytochrome P450 monooxygenase (CYP) | 22.5 | 68.0 | 82.9 |
| 166660 | cytochrome P450 monooxygenase (CYP) | 56.5 | 46.2 | 33.8 |
| 56024 | monooxigenase (MOX) | 1.4 | 533.5 | 216.6 |
| 155617 | flavin-containing monooxygenase (MOX) | 35.2 | 16.3 | 7.4 |
| 45176 | FAD-dependent monooxygenase (MOX) | 48.4 | 27.3 | 4.6 |
| 206523 | FAD-dependent monooxygenase (MOX) | 14.1 | 78.5 | 31.7 |
| 53354 | FAD dependent oxidoreductase | 181.0 | 47.0 | 43.6 |
| 206408 | FAD binding oxidoreductase | 33.5 | 29.2 | 23.3 |
| 154209 | aldo-keto reductase | 12.5 | 213.9 | 134.2 |
| 204540 | sulfite reductase | 5.8 | 9.1 | 8.3 |
| 208167 | NADPH-dependent FMN reductase | 1.7 | 9.0 | 7.8 |
| 209570 | amid-like NADH oxidoreductase | 21.2 | 30.0 | 9.7 |
| 505939 | pyridine nucleotide-disulfide oxidoreductase | 30.7 | 3.4 | 4.8 |
| 207168 | UDP-glucose dehydrogenase (DHO) | 2.2 | 6.4 | 8.7 |
| 204657 | alcohol dehydrogenase (DHO) | 1.1 | 11.2 | 18.5 |
| 179224 | short-chain dehydrogenase/reductase (DHO) | 1.9 | 9.3 | 11.2 |
| 125746 | short-chain dehydrogenase/reductase (DHO) | 3.5 | 9.9 | 7.4 |
| 139967 | retinol dehydrogenase 12 | 10.7 | 27.5 | 10.8 |
| 519260 | multicopper oxidase | 4.2 | 194.7 | 325.3 |
| Regulation | ||||
| 202814 | fungal specific transcription factor | 21.4 | 8.8 | 5.1 |
| 212444 | C2H2 type conidiation transcription factor (BrlA) | 25.4 | 3.9 | 3.7 |
| 400774 | Zn(II)2Cys6 transcription factor | 3.5 | 9.4 | 6.2 |
| 209691 | plasma membrane ATPase proteolipid | 5.7 | 8.5 | 6.4 |
| 206402 | SesB-related regulatory protein | 39.2 | 74.2 | 74.8 |
| 135080 | BTB/POZ domain-containing protein | 11.1 | 16.0 | 17.9 |
| 175588 | protein phosphatase type 1 complex subunit Hex2/Reg1 | 9.5 | 9.9 | 4.9 |
| 178837 | NmrA-like family protein | 105.3 | 99.4 | 56.8 |
| 209900 | NmrA-like family protein | 10.9 | 35.3 | 21.1 |
| Protein folding | ||||
| 130540/130601 | chaperone protein HSP31 | 1.2 | 11.3 | 8.2 |
| Signal transduction | ||||
| 203881 | protein kinase domain-containing protein | 11.3 | 17.5 | 13.3 |
| 519130 | Ser/Thr protein phosphatase | 28.4 | 89.3 | 84.2 |
| Transcription | ||||
| 209896 | Nucleotidyltransferase | 107.4 | 24.1 | 10.7 |
| 142425 | WD repeat protein | 1.6 | 6.1 | 10.0 |
| Translation | ||||
| 404332 | translation elongation factor EF-1, α subunit | 1.7 | 8.6 | 7.0 |
| 208755 | tryptophanyl-tRNA synthetase | 2.8 | 13.9 | 9.8 |
| Ribosome biogenesis | ||||
| 211752 | snRNP and snoRNP protein | 2.5 | 8.8 | 9.2 |
*Transcript identification number (http://genome.jgi-psf.org/Aspca3/Aspca3.home.html).
**Figures in bold were up-regulated with statistically significance (FDR ≤ 0.05).
Five pks genes and 5 nrps genes, coding for enzymes involved in biosynthesis of secondary metabolites (SMs), were identified. In particular, the 2 pks DEGs 5570 and 5640 were identical sequences included in a ~200 kb repeated genomic region. The pks DEG 505925 contained the partial Acpks gene sequence [33] and the DEG 173482 corresponded to the AcOTApks gene of A. carbonarius. The nrps DEG 132610 corresponded to the AcOTAnps gene of the same fungus. Additional DEGs were putatively involved in biosynthesis of mycotoxins and other SMs, such as chloroperoxidases (CPO) (1), cytochrome P450 monooxygenases (CYP) (9), monooxygenases (MOX) (4), dehydrogenases (DHO) (4), hydrolases (HDL) (2) and methyltransferases (MT) (7). Moreover, the DEG 158065 was homologous to the A. niger ANI_1_92174 gene encoding oxalacetate acetylhydrolase, the enzyme converting oxalacetate to oxalate and acetate, which is a potential source of polyketide precursors.
The DEGs 172075, 8073, 209742 and 5560/405938, homologous to the A. fumigatus genes involved in melanin biosynthesis Alb1, Ayg1, Abr1 and Arp1, respectively, showed an early up-regulation. Unlikely A. fumigatus, in which the four genes are in a cluster, the homologous sequences in A. carbonarius were located in different genomic regions. Moreover, 2 DEGs (208459 and 205575) encoding hydrophobins, homologous to the A. fumigatus RodA and RodB genes, and the DEG 212444, homologous to the brlA gene of Aspergillus species, encoding a transcription factor (C2H2 type) activating genes involved in asexual reproduction, were detected.
Three DEGs (37752, 131650/516443 and 9402) encoded catalases involved in stress response and 1 (518556) was homologous to the A. ochraceus AocatA gene coding for a spore-specific catalase involved in the maintenance of cellular redox balance.
Eight DEGs involved in transport functions were membrane transporters belonging to the Major Facilitator Superfamily (MFS). They included 2 identical sequences (49247 and 49992), the DEG 173225, homologous to the aflT gene in the aflatoxin biosynthesis gene cluster of Aspergillus parasiticus and Aspergillus flavus, and the DEG 135554, corresponding to the FLU1 MFS transporter active in A. carbonarius OTA-producer strains. The ABC transporter 211441 was homologous to the A. fumigatus atrF gene causing multidrug resistance and toxicant extrusion.
Several DEGs were involved in amino acid metabolism, such as 399389 (aminotransferase class-III), 212184 (NADP-specific glutamate dehydrogenase) and 507488 (proline oxidase, Put1), catalysing catabolic reactions leading to glutamate production, and 206969 (N-acetyltransferase), 154116 and 7736 (acetylglutamate kinases), involved in the conversion of glutamate to ornithine.
Validation of RNA-Seq results by RT-qPCR
RT-qPCR was performed on selected DEGs: 5 pks (5570/5640, 56260, 172075, 173482 and 505925), 4 nrps (132610, 204544, 206989 and 505182) and 1 cpo (212238) DEGs. Three genes, β-tub, cal and ubc, were tested as candidate reference genes. The ubc gene showed the highest expression stability, according to the BestKeeper algorithm (p-value = 0.02; SD = 0.38; CV = 2.24), under the adopted experimental conditions, and it was hence used as reference gene. Gene expression patterns of the selected DEGS, assessed at 4, 6 and 8 DAI by RT-qPCR, were consistent with those obtained by RNA-Seq (Fig 4), confirming the reliability and accuracy of the NGS analysis.
Fig 4. Comparison of RT-qPCR (dotted lines) and RNA-Seq (histograms) results for the nrps, pks and cpo genes up-regulated under OTAI conditions.

Figures are the mean values of 2 biological replicates (strains AC49 and AC67) and bars represent standard error.
Co-expression analysis and putative OTA gene cluster
AntiSMASH 2.0 predicted 51 gene clusters involved in the synthesis of SMs in the A. carbonarius genome. Nine predicted clusters included a pks gene, 13 an nrps gene, 4 an hybrid pks/nrps gene, 1 included both pks and nrps genes, 5 were likely involved in terpene biosynthesis and 20 in other classes of end products (S5 Table).
Fifteen of 51 predicted gene clusters included 4 to 8 CEGs. Only the putative gene cluster 17 was down-regulated under OTAN conditions, while all the other putative gene clusters were up-regulated. These were divided in 2 groups on the ground of their expression profile: i) clusters with expression values increasing from 4 to 8 DAI (clusters 4, 13, 14, 19, 22, 37) and ii) clusters with expression values increasing from 4 to 6 DAI and decreasing later (24, 25, 26, 32, 38, 40, 42, 48) (S5 Table).
The putative gene cluster 38 displayed high homology with the OTA-gene cluster of A. niger and was made up by 6 CEGs. It contained both the nrps (132610) and pks (173482) DEGs up-regulated under OTAI conditions which were homologous to the pks and nrps genes playing a role in OTA biosynthesis in other Aspergillus species and in P. nordicum. In particular, the protein encoded by the pks gene in the cluster 4 showed 64% and 74% identities with the homologous proteins of A. ochraceus (AAP32477.1) and A. niger (CAK42679.1), respectively, and lower identities with OTA PKSs of A. westerdijkiae (AAT92023.1) (37%) and P. nordicum (AAP33839.2) (34%); while the nrps gene in the same cluster codes for a protein homologous to OTA NRPSs of A. niger (CAK42678.1) (60%) and P. nordicum (AAS98174.1) (39%). The cluster also included the cytochrome P450 monooxygenase (cyp) DEG 517149, showing homology in BlastP search (66% identity) with the p450-B03 gene of A. ochraceus suggested to be involved in OTA biosynthesis, in addition to a flavin-containing monooxygenase (fmo) DEG (209543), a hypothetical protein DEG (209537) and a bZIP type transcription factor (bZIP) DEG (7821) (S5 Table).
Most of the putative gene clusters, with the exception of the clusters 22, 32 and 42, were not detected in other species belonging to the Aspergillus section Nigri. The cluster 40, containing a nrps gene and the cluster 42, containing a pks gene, along with the clusters 24 and 48 showed to be significantly co-regulated (R2≥0.75) with the putative OTA gene cluster (38) (S6 Table).
Discussion and Conclusions
A. carbonarius is the main OTA-producing fungus in grape and derived products, especially in the Mediterranean areas [2,10]. OTA is a secondary metabolite derived from a polyketide precursor which is enzimatically modified to produce a chlorinated isocoumarin derivative linked to the amino acid L-phenylalanine. The OTA biosynthetic pathway has not yet been completely clarified in A. carbonarius and other Aspergillus species, and only few structural and regulatory genes have been suggested or proved to be involved [15,16,17,19,34].
In the present study, we applied RNA-Seq to study whole transcriptional changes associated to OTA production in 4 A. carbonarius strains grown under OTAI and OTAN conditions, aiming at getting a more global and accurate picture of OTA biosynthetic pathway and regulatory mechanisms. The same approach has been successfully applied to investigate several aspects in other Aspergillus species (e.g., [35]).
About 86% of 50-bp reads generated from RNA-Seq mapped to the reference A. carbonarius genome and allowed us to quantify transcript abundance for over 93% of annotated genes. About 13% of reads were within intergenic or intronic genomic regions and ∼14% were unmapped, presumably because derived from till unrecognized transcripts or transcript isoforms, polymorphic regions or sequence aberrations in the genomes of the tested isolates [36]. The large RNA-Seq data sets obtained (∼13 Gb) might be useful for improving the quality of annotation of the A. carbonarius genome and for getting deeper insights into other growing-dependent biological processes, such as secondary metabolism and fungal development (e.g., conidiogenesis).
Culture media and growing conditions greatly affect fungal growth and metabolite production. Several authors reported that static cultures in MM or other media induce OTA production in Aspergillus and Penicillium species ([18,33] [15]). We confirmed that OTA production in A. carbonarius is induced in static cultures and prevented in shaken cultures in MM. Four A. carbonarius strains were grown in the same medium to compare their transcriptomes under these two basic growing conditions and avoiding interferences of other nutritional and environmental factors. Both RNA-Seq analysis and OTA quantification in cultural broths were carried out at different sampling times, in order to investigate on relationships between gene transcription and OTA biosynthesis and secretion.
OTAI conditions caused a significant transcriptional changes in a large set of genes. Analysis of transcriptional profiles showed that, generally, the genes early modulated, until 6 DAI, were down-regulated, genes modulated later or always were mainly up-regulated. Functional analysis and enrichment of GO terms in DEG sets revealed that processes related to regulation, translation and protein folding, associated with functions such as RNA binding, isomerases and structural molecular activities were affected, indicating that profound changes in the fungal metabolism occurred in OTAI as compared to OTAN conditions.
Up-regulated DEGs included several genes with hydrolase activity in carbohydrate metabolism. Hydrolysis of carbohydrates may contribute to generate malonate and acetate which are substrates used by PKS to generate dihydrocumarin [37]. Moreover, other primary metabolic pathways could be also responsible for generating polyketide precursors. The GO category amino acid metabolism was significantly over-represented among up-regulated genes. The existence of a relationship between mycotoxin biosynthesis (e.g., aflatoxin and OTA) and amino acid metabolism has been reported in different fungal species. It has been suggested that amino acid catabolism increases intracellular concentration of acetyl-CoA, used as carbon source in polyketide biosynthesis [38]. Some DEGs identified by RNA-Seq were involved in the metabolism of amino acids, such as proline, glutamic and aspartic acids. Proline and glutamic acids in liquid media are known to promote OTA production in A. ochraceus and Penicillium viridicatum [39] and aspartic acid and other amino acids promote aflatoxin production in Aspergillus spp. [38]. Furthermore, glutamic acid is incorporated in both the isocoumarin ring and phenylalanine of OTA by A. ochraceus [40].
Genes up-regulated by OTAI conditions were mainly involved in secondary metabolic processes and the oxidoreductase activity was one of the more represented molecular functions. The two categories included DEGs such as 2 PAL, 5 pks and 5 nrps genes in addition to several methyltransferases, P450 monooxygenases, dehydrogenases, hydrolases and 1 cloroperoxidase, all genes involved in the biosynthesis of mycotoxins and other SMs [14].
RNA-seq analysis identified several DEGs associated with transport and secretion activities, including MFS and ABC transporters, in addition to transporters of other compounds (amino acids, antibiotics, etc.). Transporters, acting as efflux pumps, play a role in avoiding auto-toxic effects by extruding toxic metabolites endogenously produced by micro-organisms [41]. Two MFS transporters (FLU1 and Atr1) putatively involved in OTA transport have been reported in A. carbonarius [21]. FLU1 (DEG 135554) was always significantly up-regulated in OTAI conditions while Atr1 (DEG 49724) was not differentially expressed in our experimental conditions. The MFS transporter 173225, up-regulated under OTAI conditions, was homologous to the aflt gene within the aflatoxin biosynthetic gene cluster in A. flavus and A. parasiticus [42]. MFS transporters are involved in the transport of mycotoxins, such as trichothecene and cercosporine, in several fungal species (e.g., [43]) and the otatraPN gene of P. nordicum is responsible for OTA secretion [44]. The up-regulated ABC transporter 211441 showed homology with the atrF gene of A. fumigatus which is involved in cell drug extrusion [45].
The involvement of PKS and NRPS in mycotoxin biosynthesis has been reported in several fungal species. PKS catalyses reactions leading to the isocoumarin ring and NRPS is responsible for the peptide bond formation between the isocoumarin nucleus and an L-phenylalanine [14]. Comparative RNA-Seq analysis between OTAI and OTAN conditions corroborates previous results. Three pks genes (Acpks1, Acpks3 and Acpks4) [33] were not differentially expressed, while the Acpks (DEG 505925) and AcOTApks (173482) genes were significantly up-regulated under OTAI conditions. The latter gene has been proved involved in OTA biosynthetic pathway of A. carbonarius by gene disruption [17,33]). In addition, 4 novel putative pks genes were identified among those up-regulated under OTAI conditions. The pks DEG 172075 was homologous to the Alb1 gene of A. fumigatus involved in melanin biosynthesis [46]. The two pks DEGs, 5570 and 5640, with identical sequences, as well as the pks DEG 56260 did not display any sequence homology to previously characterized genes and their functional role remains to be established. RNA-Seq confirmed up-regulation by OTAI conditions of the AcOTAnrps gene (DEG 132610), whose role in OTA biosynthesis has been recently demonstrated in A. carbonarius by gene disruption [19]. A single cpo gene (212238), encoding a chloroperoxidase, was significantly up-regulated under OTAI conditions and its expression levels increased during the time. This gene might be responsible for the addition of a chlorine atom to an OTA precursor, in the last step of the OTA biosynthetic pathway, although its role should be demonstrated with specific gene knockout studies. For all the up-regulated pks, nrps and cpo genes, RT-qPCR yielded results consistent with the gene expression patterns revealed by RNA-Seq, thus evidencing the validity of the approach.
A large number of DEGs were involved in SM biosynthesis suggesting that OTAI conditions adopted in our experiments induced not only OTA biosynthesis but also other metabolic pathways. Genes responsible for biosynthesis of SMs in filamentous fungi are frequently in clusters and transcriptionally co-regulated [47]. Clusters often include genes encoding proteins involved in metabolite secretion as well as regulatory proteins containing DNA-binding sequences. For instance, in A. nidulans a 60-kb genomic region contains a cluster of 25 co-regulated genes needed for the biosynthesis of sterigmatocystin [48]. Organization into gene clusters has been also reported for genes involved in the biosynthesis of aflatoxins in A. flavus and A. parasiticus [42], deoxynivalenol and fumonisin in Fusarium spp. [49,50]. An OTA gene cluster has been described in P. nordicum and contains 5 genes: an alkaline serine protease (aspPN), a pks (otapksPN), an nrps (otanpsPN), an MFS transporter (otatraPN), responsable for OTA extrusion, and a chloroperoxidase (otachlPN) [44].
The availability of assembled sequences of the A. carbonarius genome and the RNA-Seq data made it possible to investigate gene clusters potentially involved in SM biosynthetic pathway, with particular reference to OTA. Cluster prediction using antiSMASH 2.0 led to identify a large number of putative SM gene clusters in the genome like it occurs in other Aspergillus species [51]. QT clustering of RNA-Seq data was then applied to select CEGs in the predicted clusters. This approach, along with gene functional analysis, led to identify 1 gene cluster down-regulated and 14 gene clusters up-regulated under OTAI conditions, encoding enzymatic or regulatory proteins potentially involved in the biosynthesis of mycotoxins or other SMs.
Most of the putative gene clusters identified in this study contain genes encoding enzymes involved in the synthesis of unknown products and are therefore candidates of novel SM pathways. Only the putative clusters 22, 32 and 42 are conserved in the genomes of several species of Aspergillus section Nigri (S5 Table), while most clusters seem to be unique for the species A. carbonarius, and hence they might represent sources of exclusive metabolites useful for the specific lifestyle of the fungus.
The cluster 38, including both the pks and nrps genes involved in the OTA biosynthesis [17,19], contained 6 CEGs in our experimental conditions: i) the AcOTApks gene (DEG 173482); ii) the DEG 209537 coding for a hypothetical protein, iii) the AcOTAnps gene (DEG 132610), iv) the DEG 517149 encoding a cytochrome P450 monooxygenase (cyp), v) the DEG 209543 encoding a monooxygenase containing a FAD-binding domain (fmo), vi) the DEG 7821 encoding a bZIP type transcription factor (bZIP). Three of the included genes (pks, nrps and cyp) were conserved in other OTA-producer species of Aspergillus and Penicillium [15,16,17,18,19,34]. With the only exception of the hypothetical protein, homologous genes were found, similarly organized in a gene cluster, in the genome of an OTA-producer strain but not in that of an OTA not-producer strain of A. niger [52]. All these findings support the hypothesis that the cluster 38 is likely the OTA gene cluster in A. carbonarius. The cluster includes a bZIP (basic leucine zipper)-type transcription factor (TF). The family of eukaryotic bZIP transcription factors plays critical roles in environmental responses and are associated with production of SMs and in the regulation of mycotoxin biosynthesis in Aspergillus species [53].
SM gene clusters often contain a TF regulating specifically gene expression within the cluster [54]; TFs of the Zn(2)-Cys(6) family, were found in some of the identified gene clusters. A TF of the same type (aflR) activates transcription of gene clusters involved in aflatoxin and sterigmatocystin biosynthesis pathway in A. flavus, A. nidulans and A. parasiticus [55,56,57]. SM biosynthesis is responsive to environmental cues, including carbon and nitrogen source, temperature, light, and pH. Different regulation pathways can mediate these environmental signals activating one or more gene clusters [54], and indeed we found 4 putative gene clusters significantly co-regulated with the putative OTA gene cluster. The key genes for these clusters encoded: antibiotic synthetase (cluster 24), NRPS (40), PKS (42) and NRPS-like enzyme (48).
OTAI conditions induced an abundant conidiation and some up-regulated genes were indeed involved in asexual sporulation, while genes involved in sexual sporulation were more represented among down-regulated genes. We identified an up-regulated TF (DEG 212444) having homology with the brlA gene, conserved in several Aspergillus species, and playing a key role in the activation of conidiogenesis [58,59]. Two A. carbonarius TFs (VeA and LaeA) regulating conidiation and OTA biosynthesis in response to light [60] were not modulated under our experimental conditions. Genetic links between SM biosynthesis and fungal development have been widely studied [54]. A connection between mycotoxin biosynthesis, conidiation and ornithine metabolism has been established in aflatoxin-producer A. parasiticus [61] and proposed for A. carbonarius [62]. We identified at least 6 highly up-regulated DEGs related to ornitine metabolic pathway: DEGs 399389, 212184 and 507488 encoding enzymes responsible for glutamate production and DEGs 206969, 154116 and 7736 converting glutamate to ornithine. These findings suggest that they can be key genes in early steps of OTA biosynthesis and sporulation in A. carbonarius, although deeper investigations on the issue are needed.
In conclusion, RNA-Seq results herein discussed represent a comprehensive transcriptional profiling of genes induced under growing conditions promoting OTA biosynthesis using 4 A. carbonarius strains. Our results are consistent with previous findings, obtained with different approaches, including gene disruption [17,19], suppression subtractive hybridization and proteomic analysis [21,62], and provide further evidence of the involvement of specific OTA-related genes, the activation of major metabolic pathways leading to OTA biosynthesis and their possible connection with other biological processes (i.e., fungal growth, sporulation and pigmentation).
Supporting Information
(TIF)
The numbers in parentheses indicate gene numbers in the cluster. Blue lines are genes listed in Table 1, selected for their potential involvement in OTA production.
(TIF)
(DOC)
(DOC)
(DOC)
(DOC)
(XLS)
(DOC)
Acknowledgments
We thank Donato Perrelli and Patrizia Natale for technical assistance in chemical analysis of OTA.
Data Availability
All relevant data are within the paper and its Supporting Information files. In addition, sequencing data from cDNA libraries have been submitted in the NCBI Sequence Read Archive (SRA) database with study accession number SRP059422.
Funding Statement
The work was partially carried out in the framework of the Projects: “Laboratory network for the selection, characterization and conservation of germplasm and for preventing the spread of economically-relevant and quarantine pests (SELGE) No. 14”, founded by the Apulia Region, PO FESR 2007–2013—Axis I, Line of intervention 1.2., Action 1.2.1; “Promotion of ECO-friendly processes for the enhancement of quality of apulian food productions” granted by the “Distretto Agroalimentare Regionale – D.A.Re. s.c.ar.l.. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.IARC. Ochratoxin A. In: IARC monographs on the evaluation of carcinogenic risks to humans Some Naturally Occurring Substances: Food Items and Constituents, Heterocycles Azoaromatic Amines and Mycotoxins, Vol 56 Lyon: International Agency for Research on Cancer; 1993. pp. 489–521. [Google Scholar]
- 2.Battilani P, Giorni P, Pietri A. Epidemiology of toxin-producing fungi and ochratoxin A occurrence in grape. Eur J Plant Pathol. 2003;109:715–722. [Google Scholar]
- 3.Medina Á, Jiménez M, Gimeno-Adelantado JV, Valle-Algarra FM, Mateo R. Determination of ochratoxin A in beer marketed in Spain by liquid chromatography with fluorescence detection using lead hydroxyacetate as a clean-up agent. J Chromatogr A. 2005;1083:7–13. [DOI] [PubMed] [Google Scholar]
- 4.Ozden S, Akdeniz AS, Alpertunga B. Occurrence of ochratoxin A in cereal-derived food products commonly consumed in Turkey. Food Control. 2012;25:69–74. [Google Scholar]
- 5.Pardo E, Marin S, Ramos AJ, Sanchis V. Occurrence of ochratoxigenic fungi and ochratoxin A in green coffee from different origins. Food Sci Technol Int. 2004;10:45–49. [Google Scholar]
- 6.Shundo L, de Almeida AP, Alaburda J, Lamardo LC, Navas SA, Ruvieri V, et al. Aflatoxins and ochratoxin A in Brazilian paprika. Food Control. 2009;20:1099–1102. [Google Scholar]
- 7.Zimmerli B, Dick R. Ochratoxin A in table wine and grape-juice: occurrence and risk assessment. Food Addit Contam. 1996;13:655–668. [DOI] [PubMed] [Google Scholar]
- 8.Mateo R, Medina Á, Mateo EM, Mateo F, Jiménez M. An overview of ochratoxin A in beer and wine. Int J Food Microbiol. 2007;119:79–83. [DOI] [PubMed] [Google Scholar]
- 9.Abarca ML, Accensi F, Bragulat MR, Castellá G, Cabañes FJ. Aspergillus carbonarius as the main source of ochratoxin A contamination in dried vine fruits from the Spanish market. J Food Protect. 2003;66:504–506. [DOI] [PubMed] [Google Scholar]
- 10.Cabañes FJ, Accensi F, Bragulat MR, Abarca ML, Castellá G, Minguez S, et al. What is the source of ochratoxin A in wine? Int J Food Microbiol. 2002;79:213–215. [DOI] [PubMed] [Google Scholar]
- 11.Pollastro S, Dongiovanni C, Abbatecola A, Tauro G, Natale P, Pascale M, et al. Wine contamination by ochratoxin A in South Italy: causes and preventive actions. J Plant Pathol. 2003;85:281. [Google Scholar]
- 12.Marquardt RR, Frohlich AA. A review of recent advances in understanding ochratoxicosis. J Anim Sci. 1992;70:3968–3988. [DOI] [PubMed] [Google Scholar]
- 13.el Khoury A, Atoui A. Ochratoxin A: General Overview and Actual Molecular Status. Toxins. 2010;2:461–493. 10.3390/toxins2040461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huffman J, Gerber R, Du L. Recent advancements in the biosynthetic mechanisms for polyketide-derived mycotoxins. Biopolymers. 2010;93:764–776. 10.1002/bip.21483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Callaghan J, Caddick MX, Dobson ADW. A polyketide synthase gene required for ochratoxin A biosynthesis in Aspergillus ochraceus. Microbiology. 2003;149:3485–3491. [DOI] [PubMed] [Google Scholar]
- 16.Bacha N, Atoui A, Mathieu F, Liboz T, Lebrihi A. Aspergillus westerdijkiae polyketide synthase gene "aoks1" is involved in the biosynthesis of ochratoxin A. Fungal Genet Biol. 2009;46:77–84. 10.1016/j.fgb.2008.09.015 [DOI] [PubMed] [Google Scholar]
- 17.Gallo A, Knox BP, Bruno KS, Solfrizzo M, Baker SE, Perrone G. Identification and characterization of the polyketide synthase involved in ochratoxin A biosynthesis in Aspergillus carbonarius. Int J Food Microbiol. 2014;179:10–17. 10.1016/j.ijfoodmicro.2014.03.013 [DOI] [PubMed] [Google Scholar]
- 18.Karolewiez A, Geisen R. Cloning a part of the ochratoxin A biosynthetic gene cluster of Penicillium nordicum and characterization of the ochratoxin polyketide synthase gene. Syst Appl Microbiol. 2005;28:588–595. [DOI] [PubMed] [Google Scholar]
- 19.Gallo A, Bruno KS, Solfrizzo M, Perrone G, Mulè G, Visconti A, et al. New insight into the ochratoxin A biosynthetic pathway through deletion of a non-ribosomal peptide synthetase gene in Aspergillus carbonarius. Appl Environ Microb. 2012;78:8208–8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Botton A, Ferrigo D, Scopel C, Causin R, Bonghi C, Ramina A. A cDNA-AFLP approach to study ochratoxin A production in Aspergillus carbonarius. Int J Food Microbiol. 2008;127:105–115. 10.1016/j.ijfoodmicro.2008.06.037 [DOI] [PubMed] [Google Scholar]
- 21.Crespo-Sempere A, Gonzáles-Candelas L, Martínez-Culabras PV. Genes differentially expressed by Aspergillus carbonarius strains under ochratoxin A producing conditions. Int J Food Microbiol. 2010;142:170–179. 10.1016/j.ijfoodmicro.2010.06.019 [DOI] [PubMed] [Google Scholar]
- 22.Cabañes FJ, Sanseverino W, Castellá G, Bragulat MR, Cigliano RA, Sánchez A. Rapid genome resequencing of an atoxigenic strain of Aspergillus carbonarius. Sci Rep. 2015;5:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanderson KE, Srb AM. Heterokaryosis and parasexuality in the fungus Ascochyta imperfecta. Am J Bot. 1965;52:72–81. [PubMed] [Google Scholar]
- 24.Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, et al. Galaxy: a web-based genome analysis tool for experimentalists In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors., Current Protocols in Molecular Biology Chapter 19 Unit 19.10. Hoboken, NJ: John Wiley & Sons, Inc; 2010. pp. 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. 10.1038/nmeth.1226 [DOI] [PubMed] [Google Scholar]
- 26.Howe EA, Sinha R, Schlauch D, Quackenbush J. RNA-Seq analysis in MeV. Bioinformatics 2011;27;3209–3210. 10.1093/bioinformatics/btr490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conesa A, Götz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. [DOI] [PubMed] [Google Scholar]
- 28.Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, et al. antiSMASH 2.0—a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013;41:W204–W212. 10.1093/nar/gkt449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–622. 10.1373/clinchem.2008.112797 [DOI] [PubMed] [Google Scholar]
- 30.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods in Mol Biol. 2000;132:365–86. [DOI] [PubMed] [Google Scholar]
- 31.Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol Lett. 2004;26:509–515. [DOI] [PubMed] [Google Scholar]
- 32.Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 33.Gallo A, Perrone G, Solfrizzo M, Epifani F, Abbas A, Dobson ADW, et al. Characterisation of a pks gene which is expressed during ochratoxin A production by Aspergillus carbonarius. Int J Food Microbiol. 2009;129:8–15. 10.1016/j.ijfoodmicro.2008.10.022 [DOI] [PubMed] [Google Scholar]
- 34.O’Callaghan PC, Stapleton MX, Dobson ADW. Ochratoxin A biosynthetic genes in Aspergillus ochraceus are differentially regulated by pH and nutritional stimuli. Fungal Genet Biol. 2006;43:213–221. [DOI] [PubMed] [Google Scholar]
- 35.Novodvorska M, Hayer K, Pullan ST, Wilson R, Blythe MJ, Stam H, et al. Transcriptional landscape of Aspergillus niger at breaking of conidial dormancy revealed by RNA-sequencing. BMC Genomics. 2013;14:246 10.1186/1471-2164-14-246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morozova O, Hirst M, Marra MA. Applications of new sequencing technologies for transcriptome analysis. Annu Rev Genom Hum. G. 2009;10:135–151. [DOI] [PubMed] [Google Scholar]
- 37.Anli E, Alkis IM. Ochratoxin A and brewing technology: a review. J Inst Brew. 2010;116:23–32. [Google Scholar]
- 38.Buchanan RL, Bennett JW. Nitrogen regulation of polyketide mycotoxin production In: Sanchez-Esquival S, editor. Nitrogen source control of microbial processes. Boca Raton, FL: CRC Press; 1988. pp. 137–149. [Google Scholar]
- 39.Häggblom PE, Ghosh J. Postharvest production of ochratoxin A by Aspergillus ochraceus and Penicillium viridicatum in barley with different protein levels. Appl Environ Microbiol. 1985;49:787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bacon CW, Robbins JD, Burdick D. Metabolism of glutamic acid in Aspergillus ochraceus during the biosynthesis of ochratoxin A. Appl Microbiol. 1975;29:317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martín JF, Casqueiro J, Liras P. Secretion systems for secondary metabolites: how producer cells send out messages of intercellular communication. Curr Opin Microbiol. 2005;8:282–293. [DOI] [PubMed] [Google Scholar]
- 42.Yu J, Chang PK, Ehrlich KC, Cary JW, Bhatnagar D, Cleveland TE, et al. Clustered pathway genes in aflatoxin biosynthesis. Appl Environ Microbiol. 2004;70:1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alexander NJ, McCormick SP, Hohn TM. TRI12, a trichothecene efflux pump from Fusarium sporotrichioides: gene isolation and expression in yeast. Mol Genet Genomics 1999;261:977–984. [DOI] [PubMed] [Google Scholar]
- 44.Geisen R, Schmidt-Heydt M, Karolewiez A. A gene cluster of the ochratoxin A biosynthetic genes in Penicillium. Mycotoxin Res. 2006;22;134–141. 10.1007/BF02956777 [DOI] [PubMed] [Google Scholar]
- 45.Slaven JW, Anderson MJ, Sanglard D, Dixon GK, Bille J, Roberts IS, et al. Increased expression of a novel Aspergillus fumigatus ABC transporter gene, atrF, in the presence of itraconazole in an itraconazole resistant clinical isolate. Fungal Genet Biol. 2002;36:199–206. [DOI] [PubMed] [Google Scholar]
- 46.Krijgsheld P, Bleichrodt R, van Veluw GJ, Wang F, Müller WH, Dijksterhuis J, et al. Development in Aspergillus. Stud. Micol. 2013;74:1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brakhage AA, Schroeckh V. Fungal secondary metabolites–strategies to activate silent gene clusters. Fungal Genet. Biol. 2011;48:15–22. 10.1016/j.fgb.2010.04.004 [DOI] [PubMed] [Google Scholar]
- 48.Brown DW, Yu JH, Kelkar HS, Fernandes M, Nesbitt TC, Keller NP, et al. Twenty-five coregulated transcripts define a sterigmatocystin gene cluster in Aspergillus nidulans. P Natl A Sci USA. 1996;93:1418–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alexander NJ, Proctor RH, McCormick SP. Genes, gene clusters, and biosynthesis of trichothecenes and fumonisins in Fusarium. Toxin Rev. 2009;28:198–215. [Google Scholar]
- 50.Brown DW, Butchko RA, Busman M, Proctor RH. The Fusarium verticillioides FUM gene cluster encodes a Zn (II) 2Cys6 protein that affects FUM gene expression and fumonisin production. Eukaryot Cell 2007;6:1210–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Inglis DO, Binkley J, Skrzypek MS, Arnaud MB, Cerqueira GC, Shah P, et al. Comprehensive annotation of secondary metabolite biosynthetic genes and gene clusters of Aspergillus nidulans, A. fumigatus, A. niger and A. oryzae. BMC Microbiol. 2013;13:91 10.1186/1471-2180-13-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Andersen MR, Salazar MP, Schaap PJ, van de Vondervoort PJ, Culley D, Thykaer J, et al. Comparative genomics of citric-acid-producing Aspergillus niger ATCC 1015 versus enzyme-producing CBS 513.88. Genome Res. 2011;21:885–897. 10.1101/gr.112169.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reverberi M, Gazzetti K, Punelli F, Scarpari M, Zjalic S, Ricelli A, et al. Aoyap1 regulates OTA synthesis by controlling cell redox balance in Aspergillus ochraceus. Appl Microbiol Biotechnol. 2012;95:1293–1304. 10.1007/s00253-012-3985-4 [DOI] [PubMed] [Google Scholar]
- 54.Calvo AM, Wilson RA, Woo Bok J, Keller NP. Relationship between secondary metabolism and fungal development. Microbiol Mol Biol R. 2002;66:447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang PK, Ehrlich KC, Yu J, Bhatnagar D, Cleveland TE. Increased expression of Aspergillus parasiticus aflR, encoding a sequence-specific DNA-binding protein, relieves nitrate inhibition of aflatoxin biosynthesis. Appl Environ Microb. 1995;61:2372–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ehrlich KC, Montalbano BG, Cary JW. Binding of the C6-zinc cluster protein, AFLR, to the promoters of aflatoxin pathway biosynthesis genes in Aspergillus parasiticus. Gene 1999;230:249–257. [DOI] [PubMed] [Google Scholar]
- 57.Fernandes M, Keller NP, Adams TH. Sequence‐specific binding by Aspergillus nidulans AflR, a C6 zinc cluster protein regulating mycotoxin biosynthesis. Mol Microbiol. 1998;28:1355–1365. [DOI] [PubMed] [Google Scholar]
- 58.Ogawa M, Tokuoka M, Jin FJ, Takahashi T, Koyama Y. Genetic analysis of conidiation regulatory pathways in koji-mold Aspergillus oryzae. Fungal Genet Biol. 2010;47:10–18. 10.1016/j.fgb.2009.10.004 [DOI] [PubMed] [Google Scholar]
- 59.Yu J- H. Regulation of Development in Aspergillus nidulans and Aspergillus fumigatus. Mycobiology. 2010;38:229–237. 10.4489/MYCO.2010.38.4.229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crespo-Sempere A, Marín S, Sanchis V, Ramos AJ. VeA and LaeA transcriptional factors regulate ochratoxin A biosynthesis in Aspergillus carbonarius. Int J Food Microbiol. 2013;166:479–486. 10.1016/j.ijfoodmicro.2013.07.027 [DOI] [PubMed] [Google Scholar]
- 61.Guzmán-de-Peña D, Ruiz-Herrera J. Relationship between aflatoxin biosynthesis and sporulation in Aspergillus parasiticus. Fungal Genet Biol. 1997;21:198–205. [DOI] [PubMed] [Google Scholar]
- 62.Crespo-Sempere A, Gil JV, Martínez-Culebras PV. Proteome analysis of the fungus Aspergillus carbonarius under ochratoxin A producing conditions. Int J Food Microbiol. 2011;147:162–169. 10.1016/j.ijfoodmicro.2011.03.021 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
The numbers in parentheses indicate gene numbers in the cluster. Blue lines are genes listed in Table 1, selected for their potential involvement in OTA production.
(TIF)
(DOC)
(DOC)
(DOC)
(DOC)
(XLS)
(DOC)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files. In addition, sequencing data from cDNA libraries have been submitted in the NCBI Sequence Read Archive (SRA) database with study accession number SRP059422.