

Abstract
An expanded family of ruthenium metathesis catalysts bearing cyclic alkyl amino carbene (CAAC) ligands is reported. These catalysts exhibited exceptional activity in the ethenolysis of the seed oil derivative methyl oleate. In many cases, TONs >100,000 were achieved, at only 3 ppm catalyst loading. Remarkably, the most active catalyst system was able to achieve a TON of 340,000, at only 1 ppm catalyst loading. This is the first time a series of metathesis catalysts has exhibited such high performance in cross metathesis reactions employing ethylene gas, with activities sufficient to render ethenolysis applicable towards the industrial scale production of linear alpha-olefins (LAOs) and other terminal olefin products.
Keywords: olefin metathesis, ethenolysis, cyclic alkyl amino carbenes, terminal olefins, seed oil derivatives
Graphical Abstract
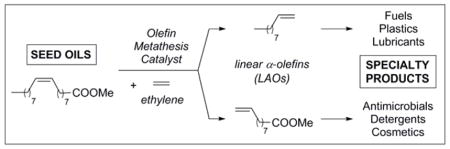
A new series of cyclic alkyl amino carbene (CAAC)-containing olefin metathesis cataysts was synthesized, and were found to exhibit unprecedented activity in the ethenolysis of methyl oleate. This work advances the state-of-the-art of the ethenolysis reaction, and is expected to find particular use in large scale industrial applications.
The transformation of small molecule chemical feedstocks to high-value chemicals has been a long-standing challenge that has received a significant resurgence of interest in the chemical sciences. This is a result of recently introduced programs promoting the use of greener chemistry practices, as well as the rising costs associated with the production of fine chemicals from petrochemicals. Consequently, the ability to access high-demand products from renewable sources such as oleochemicals presents a cost-effective and environmentally friendly alternative.[1]
Olefin metathesis reactions, such as cross-metathesis (CM), ring-closing metathesis (RCM), and ring-opening metathesis polymerization (ROMP), all of which generate a new internal olefin, have enjoyed widespread popularity in both academic and industrial settings as a result of their general applicability, ease of use, and Non-prohibitive costs.[2] Ruthenium-based metathesis catalysts are ideal for such transformations as a result of their generally robust nature, which enables handling in air, and imparts good tolerance to a variety of functional groups and trace impurities. All of these are necessary prerequisites when subjected to raw materials or biomass.
Many renewable or bio-based materials, such as fatty acids originating from seed oils and their derivatives, contain at least one unit of unsaturation, providing a synthetic handle for derivatization by olefin metathesis catalysts. The CM reaction with ethylene (2), commonly referred to as ethenolysis, has significant potential as a clean, scalable, and sustainable solution for the production of linear alpha olefins (LAOs) (eg. 3 and 4) from the natural oils found in oleochemicals such as methyl oleate (MO, 1) (Scheme 1). LAOs are direct precursors to a variety of commodity chemicals with applications as fuels, surfactants, lubricants, waxes, perfumes, antimicrobial agents, and thermoplastics. In addition, LAOs can be rapidly elaborated to more expensive products such as agrochemicals, insect pheromones, and pharmaceuticals.[3]
Scheme 1.

Ethenolysis of the seed oil derivative methyl oleate (1).
The production of terminal olefins from the ethenolysis of seed oil derivatives using metathesis catalysts has been previously demonstrated. However, the high catalyst loadings required (10 – 100 ppm) to achieve an acceptable yield of terminal olefins render these reported procedures cost prohibitive on an industrial scale.[4],[5],[6] In general, catalyst turnover numbers (TONs) of at least 35,000 and 50,000 are recommended in the manufacturing of specialty and commodity chemicals, respectively.[4] In the ethenolysis of the benchmark substrate MO (1), standard ruthenium-based metathesis catalysts such as 5 – 8 afforded TONs of only 2,000 – 5,000. This stands in contrast to the extremely high activity normally exhibited by these catalysts in CM with terminal or internal olefins. For example, TONs as high as 470,000 have been achieved with 8 in the CM of MO and 2-butene.[7] The most active catalyst for ethenolysis in the literature to date is cyclic alkyl amino carbene (CAAC) complex 10 (Scheme 2), which has been previously reported to generate a TON of 35,000 in the CM of MO with ethylene.[4],[5] As a result of the lack of a catalyst sufficiently active to produce teminal olefins using ethylene gas, industrial scale ethenolysis is currently accomplished using higher olefins as ethylene surrogates.[6b] Catalyst 7, for example, is able to achieve the in situ ethenolysis of MO with a TON as high as 192,900 with propylene gas.[4a] However, there is a need to develop catalysts capable of achieving high activity in ethenolysis reactions when ethylene gas is utilized directly. Whereas CM with higher olefins necessarily results in a substantial amount of undesired internal olefins being produced as byproducts, the only products derived from CM with ethylene are terminal olefins. This intrinsic advantage promotes both increased yield and ease of purification of the desired terminal olefin products, and is a particularly important consideration for bio-refinery feedstocks in which multiple downstream products are produced.[3]
Scheme 2.

Synthesis of CAAC complexes under study (isolated yield in brackets).
Herein, we report the discovery of the most active ethenolysis catalysts to date. In many cases, TONs surpassing 100,000 were achieved for the ethenolysis of MO, using ethylene gas. Remarkably, certain catalyst systems even exhibited TONs approaching 200,000, with the highest TON achieved being 330,000. This represents the first time that reaction conditions have been developed in order for ethenolysis to proceed efficiently on an industrial scale.
Despite the promising results previously exhibited by 10 in the ethenolysis of MO,[4],[5] CAAC ligated ruthenium complexes have yet to be investigated in detail. In particular, it was envisioned that more in-depth structure/activity relationship (SAR) studies would facilitate the development of new, more efficient catalysts. Thus, a variety of new catalysts were prepared through modifications of exisiting literature procedures (Scheme 2).[5],[ 8 ] Known CAAC catalysts (10, 13, 18, 25) were screened alongside the new catalysts, in order to ensure accurate SAR comparisons within the series.
Initially, derivatives of the previous benchmark catalyst 10 were targeted, in which only the ortho substituents of the N-aryl ring were varied (9 – 13). It is worthy to note that the syntheses of 9 and 10 are low yielding (ca. 20%), and purification is cumbersome. Furthermore, complexes bearing even smaller ortho substitution such as N-mesityl or N-2-isopropylphenyl, were unable to be accessed in even small amounts. In contrast, 11 – 13 can be produced in high yield (78 – 86%) and isolated without difficulty. This is hypothesized to be related to the stability of the free carbene intermediate that is generated in situ, which might otherwise be expected to decompose rapidly in the absence of steric protection. It was envisioned that this decomposition pathway could be circumvented through the installment of larger substituents at either R4 or R5. Indeed, this strategy proved to be successful, and we were able to readily access a variety of new complexes in moderate to high yield (29 – 82%). Several of the new backbone containing catalysts included N-aryl substitution that was previously inaccessible (14, 15, 17, 21), meanwhile others (16, 18 – 20, 22 – 25) were synthesized in order to provide a more thorough SAR study. Single-crystal X-ray diffraction of 11 and 24 revealed distorted square-pyramidal geometries, and structural parameters, including bond lengths and angles, were consistent with those found previously for 10 and 13 (Figure 2). Moreover, catalyst 24 exhibits a CAAC ligand featuring a chirogenic center as well as two different ortho N-aryl substituents. Accordingly, the single crystal of 24 revealed both N-aryl rotamers (24a and 24b), in a ratio of 64:36 respectively.9
Figure 2.

Solid-state structures of 11 and 24 (24 crystallized as 24a and 24b, in a ratio of 64:36), with thermal ellipsoids drawn at 50% probability.14 For clarity, hydrogen atoms have been omitted. Selected bond lengths (Å) for 11: C1-Ru 1.928(6), C19-Ru 1.824(6), O1-Ru 2.301(4), Cl1-Ru 2.3374(16), Cl2-Ru 2.3165(17); for 24a: C1-Ru 1.940(7), C24-Ru 1.836(9), O1-Ru 2.332(8), Cl1-Ru 2.3356(18), Cl2-Ru 2.3271(13); for 24b: C1-Ru 1.931(12), C24-Ru 1.828(18), O1-Ru 2.325(15), Cl1- 2.335(4), Cl2-Ru 2.307(3).
Once in hand, catalysts 9 – 25 were examined in the ethenolysis of 1 using ethylene (Table 1). Reaction conditions were adapted from those that were previously reported in the literature, and were initially re-optimized using benchmark catalyst 10 (neat MO, 40 °C, 150 psi ethylene).[4] The only deviation from the published procedures was the use of higher purity ethylene (99.95%) than previously reported (99.9%). We were pleased to find that this simple modification appeared to already result in a substantial increase in activity: 10 ppm loading of catalyst 10 resulted in a TON of 67,000, whereas the benchmark value for 10 published in the literature is a TON 35,000.[4b],[10].11[12],[13] We were delighted to find that the TON of catalyst 10 further increased to 120,000 upon reduction of the catalyst loading to 3 ppm. Thus, all subsequent reactions were run at 3 ppm catalyst loading, which was also expected to provide greater differentiation in activity between promising catalysts than at 10 ppm.
Table 1.
Ethenolysis of methyl oleate (1) using catalysts 9 – 25.a
| catalyst | conversion (%)b,c | selectivity (%)b,d,e | yield (%)f | TONg |
|---|---|---|---|---|
| 9 | 37 | 86 | 32 | 110,000 |
| 10 | 42 | 88 | 37 | 120,000 |
| 11 | 59 | 92 | 54 | 180,000 |
| 12 | 18 | 94 | 17 | 57,000 |
| 13 | 19 | 97 | 18 | 60,000 |
| 14 | 22 | 63 | 14 | 47,000 |
| 15 | 26 | 86 | 22 | 73,000 |
| 16 | 42 | 92 | 39 | 130,000 |
| 17 | 19 | 78 | 14 | 47,000 |
| 18 | 13 | 97 | 13 | 43,000 |
| 19 | 16 | 97 | 15 | 50,000 |
| 20 | <5% | --- | --- | --- |
| 21 | 41 | 83 | 34 | 110,000 |
| 22 | 46 | 85 | 39 | 130,000 |
| 23 | 48 | 88 | 43 | 140,000 |
| 24 | 57 | 94 | 54 | 180,000 |
| 25 | 47 | 98 | 46 | 150,000 |
Reaction conditions: catalyst (3 ppm), C2H4 (150 psi, 99.95% purity), 40 °C, 3 h.
Determined via GC, using dodecane as an internal standard.
Conversion = 100 – [(final moles 1) X 100/[initial moles 1)]
Selectivity for ethenolysis products (3 and 4) over self-metathesis products (3a and 4a).
Selectivity = 100 X (moles 3 + 4)/[(moles 3 + 4) + (2 X moles 3a + 4a)].
Yield = Conversion X Selectivity/100.
TON = Yield X (initial moles 1/moles catalyst)/100.
Remarkably, at 3 ppm catalyst loading, most catalysts surpassed a TON of 100,000! Specifically, catalysts 11 and 24 emerged as the most efficient, with TONs of 180,000. Catalyst activity correlates with N-aryl substitution, with larger substituents at R1 and R2 generally resulting in higher TONs, although this can also have a deleterious effect (as in 12 and 13, compared to 9 – 11). The ideal combination thus far appears to be when R1 is small and R2 is large, as in catalysts 11 and 24 (R1 = Me, R2 = iPr). Interestingly, substitution at R5 by a phenyl ring resulted in an overall improvement of activity, especially for N-2,6-diisopropylphenyl catalyst 25. Replacement of R4 and/or R5 by ethyl, propyl, or cyclohexyl did not result in a significant change in the TON (as in 15 – 19), although an adamantyl substituent (20) resulted in complete loss of activity. Interestingly, while consumption of MO appears to be the most important determining factor in the overall yield of ethenolysis products, increased N-aryl substitution on the catalyst appears to strongly favour selectivity for terminal olefins. These trends in selectivity and TON are most evident in the series of catalysts bearing a phenyl ring on the backbone at R5 (21 – 25).
A plausible explanation for the high activity exhibited by CAAC catalysts in ethenolysis transformations might be a result of increased stabilization of the ruthenium methylidene intermediate generated in the presence of ethylene gas. Ruthenium methylidenes are known to decompose rapidly via insertion of the N-aryl substituent into the methylidene carbene, which subsequently generates various ruthenium hydrides that are inactive in metathesis transformations.[15] CAAC ligands are known to be more electron donating than their N-heterocyclic carbene (NHC) counterparts.[16] Thus, when used in place of NHCs, it is expected that the increased electron density at ruthenium might somewhat stabilize the otherwise highly reactive and electron deficient methylidene intermediate.[15b,c], [17] Substitution of the ortho N-aryl substituents with a larger sterically encumbered group would also be expected to significantly decrease the rate of termination by insertion into the [Ru]=CH2 bond. However, increased substitution can also hinder coordination of olefins to the ruthenium metal center. Diminished reactivity with increasing N-aryl substitution was indeed noted when initiation rates of selected catalysts (9 – 14) were measured following exposure to n-butylvinylether.[18] When initiation rates are compared to TONs, it is clear that both the slowest initiating catalysts (12, 13) and the fastest initiating catalyst (14) exhibit the lowest TONs (Figure 3). This is likely a result of diminished catalytic rate for the former group and an increased susceptibility to decomposition for the latter. This study illustrates the importance of this delicate balance, as reflected in the superior TONs exhibited by catalysts 11 and 24, possessing asymmetric N-aryl substituents. In these systems, the smaller substituent (R1 = Me) faciltates rapid coordination of the incoming olefin substrate, whereas the larger substituent (R2 = iPr) prevents decomposition of the methylidene intermediate. This asymmetry might be expected to exhibit a greater effect in CAAC ligated catalysts, as the steric interaction of the ortho substituents on the N-aryl ring with the two adjacent geminal methyl substituents would be expected to influence the conformation of the N-aryl ring. If the larger substituent resides closer to the ruthenium metal center, the methyl group which is inherently more susceptible to CH insertion would be directed away from the reactive ruthenium methylidene.
Figure 3.

Comparison of initiation rates and TON for catalysts 9 – 14.
It has been postulated that high activity in ethenolysis might be correlated to the tendency of a catalyst to undergo degenerative metathesis events, through the preferential formation of 2,4-metallacycles rather then 2,3-metallacycles, which would result in increased selectivity for terminal olefins in the product distribution.[19] This is a powerful design principle in the context of achieving high kinetic selectivity for terminal olefins, when employing higher olefins such as propene and 1-butene gas as ethylene surrogates. However, when ethylene gas is employed, it is more likely that the lower selectivities for terminal olefins exhibited by previous generations of catalysts (5 – 8) is primarily a result of rapid catalyst death in the presence of ethylene. This would translate to products reflecting the kinetic distribution of rapid unselective cross metathesis reactions of 5 – 8 with both ethylene and terminal olefins.[20] A lthough CAAC-ligated ruthenium-based catalysts have been demonstrated to engage in metathesis reactions more slowly than phosphine or NHC-ligated catalysts,[5a],[18] they also appear to persist for a much longer time in the presence of ethylene.[21] This would allow for the ethenolysis reaction to proceed closer to completion in order to achieve the equilibrium ratio of terminal olefin products, and provides a feasible explanation for the notable increase in activity exhibited by this family of catalysts.[22]
Finally, given the notable dependance on the TON with respect to the purity of the ethylene employed, as seen earlier for catalyst 10, we briefly explored the effect of utilizing an even higher purity ethylene source (99.995% vs. 99.95%) at different loadings of catalyst 11 (Table 2).23 A dramatic increase in TON was noted at 1 ppm catalyst loading. To the best of our knowledge, this represents the highest value available in the literature for any ethenolysis catalyst to date (TON 340,000).
Table 2.
Effect of C2H4 (2) source on activity of catalyst 11.a
| Loading | purity of 2 | yield (%) | TON |
|---|---|---|---|
| 3 | 99.95% | 54 | 180,000 |
| 3 | 99.995% | 53 | 180,000 |
| 2 | 99.95% | 48 | 240,000 |
| 2 | 99.995% | 49 | 245,000 |
| 1 | 99.95% | 13 | 130,000 |
| 1 | 99.995% | 34 | 340,000 |
Reaction conditions, and calculations, are as listed in Table 1.
In summary, a new series of ruthenium metathesis catalysts, bearing CAAC ligands, is presented that displays exceptional activity in ethenolysis reactions. In the cross metathesis reaction of the seed oil derivative methyl oleate (1) and ethylene gas (2), TONs >100,000 are generated in many cases, which surpasses the minimum value of 50,000 required to be considered economically sustainable on an industrial scale. Furthermore, even higher TONs (180,000 – 340,000) were obtained in some cases. These are the highest values recorded in the literature to date for an ethenolysis reaction, and the only reported TONs >50,000 using ethylene gas specifically. As a result, it is envisioned that this work will find substantial application in the continued development of new methodologies and processes directed towards the economically and environmentally sustainable production of LAOs, as well as other valuable terminal olefins, especially through the transformation of seed oils and their derivatives.
Supplementary Material
Figure 1.

Selected ruthenium metathesis catalysts previously studied for ethenolysis.
Footnotes
Mr. Lawrence M. Henling is acknowledged for X-ray crystallography analysis. Dr. David VanderVelde is thanked for assistance with NMR experiments. Thay Ung, Dr. Daryl P. Allen, and Dr. Richard L. Pederson (Materia Inc.) are thanked for helpful discussions regarding initial experimentation and setup. This work was financially supported by the NIH (5R01GM031332, 7R01GM068825), the NSF (CHE-1212767), the DOE (DE-FG02-13ER16370) and NSERC (fellowship to VMM).
Supporting information for this article is available on the WWW under http://www.angewandte.org.
Contributor Information
Vanessa M. Marx, Department of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA, 91125 (USA)
Alexandra H. Sullivan, Department of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA, 91125 (USA)
Mohand Melaimi, UCSD-CNRS Joint Research Laboratory (UMI 3555), Department of Chemistry and Biochemistry, University of California, San Diego, San Diego, CA, 92093 (USA).
Scott C. Virgil, Email: svirgil@caltech.edu, Center for Catalysis and Chemical Synthesis, California Institute of Technology, Pasadena, CA, 91125 (USA)
Benjamin K. Keitz, Department of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA, 91125 (USA)
David S. Weinberger, UCSD-CNRS Joint Research Laboratory (UMI 3555), Department of Chemistry and Biochemistry, University of California, San Diego, San Diego, CA, 92093 (USA)
Guy Bertrand, Email: guybertand@ucsd.edu, UCSD-CNRS Joint Research Laboratory (UMI 3555), Department of Chemistry and Biochemistry, University of California, San Diego, San Diego, CA, 92093 (USA).
Robert H. Grubbs, Email: rhg@caltech.edu, Department of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA, 91125 (USA)
References
- 1.a) Biermann U, Bornscheuer U, Meier MAR, Metzger JO, Schäfer HJ. Angew Chem. 2011;123:3938. doi: 10.1002/anie.201002767. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2011;50:3854. [Google Scholar]; b) Montero de Espinosa L, Meier MAR. Eur Polym J. 2011;47:837. [Google Scholar]; c) Marshall A-L, Alaimo PJ. Chem Eur J. 2010;16:4970. doi: 10.1002/chem.200903028. [DOI] [PubMed] [Google Scholar]; d) Meier MAR, Metzger JO, Schubert US. Chem Soc Rev. 2007;36:1788. doi: 10.1039/b703294c. [DOI] [PubMed] [Google Scholar]; e) Sheldon RA. Green Chem. 2007;9:1273. [Google Scholar]; f) Sheldon RA, Arends I, Hanefeld U, editors. Green Chemistry and Catalysis. Wiley-VCH; Weinheim: 2007. [Google Scholar]; g) Corma A, Iborra S, Velty A. Chem Rev. 2007;107:2411. doi: 10.1021/cr050989d. [DOI] [PubMed] [Google Scholar]
- 2.a) Cossy J, Arseniyadis S, Meyer C, editors. Metathesis in Natural Product Synthesis. Wiley-VCH; Weinhem: 2010. [Google Scholar]; b) Lozano-Vila AM, Monsaert S, Bajek A, Verpoort F. Chem Rev. 2010;110:4865. doi: 10.1021/cr900346r. [DOI] [PubMed] [Google Scholar]; c) Vougioukalakis G, Grubbs RH. Chem Rev. 2010;110:1746. doi: 10.1021/cr9002424. [DOI] [PubMed] [Google Scholar]; d) Samojlowicz C, Bieniek M, Grela K. Chem Rev. 2009;109:3708. doi: 10.1021/cr800524f. [DOI] [PubMed] [Google Scholar]; e) Schrodi Y, Pederson RL. Aldrichim Acta. 2007;40:45. [Google Scholar]; f) Nicolaou KC, Bulger PG, Sarlah D. Angew Chem. 2005;117:4564. doi: 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:4490. [Google Scholar]; g) Grubbs RH, editor. Handbook of Metathesis. Vol. 2. Wiley-VCH; Weinhem: 2003. [Google Scholar]; h) Schrock RR, Hoveyda AH. Angew Chem. 2003;115:4740. doi: 10.1002/anie.200300576. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2003;42:4592. [Google Scholar]; i) Schrock RR. Chem Rev. 2002;102:145. doi: 10.1021/cr0103726. [DOI] [PubMed] [Google Scholar]; j) Trnka TM, Grubbs RH. Acc Chem Res. 2001;34:18. doi: 10.1021/ar000114f. [DOI] [PubMed] [Google Scholar]; k) Fürstner A. Angew Chem. 2000;112:3140. [Google Scholar]; Angew Chem Int Ed. 2000;39:3012. [Google Scholar]
- 3.a) Chikkali S, Meckling S. Angew Chem. 2012;124:5902. doi: 10.1002/anie.201107645. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2012;51:5802. [Google Scholar]; b) Montero de Espinosa L, Meier MAR. Top Organomet Chem. 2012;39:1. [Google Scholar]; c) Raluca M, Dixneuf PH. In Green Metathesis Chemistry. Vol. 185. Springer Netherlands; 2010. [Google Scholar]; c) Mol JC. Green Chem. 2002;4:5. [Google Scholar]
- 4.a) Nickel A, Ung T, Mkrtumyan G, Uy J, Lee C-W, Stoianova D, Papazian J, Wei W-H, Mallari A, Schrodi Y, Pederson RL. Top Catal. 2012;55:518. [Google Scholar]; b) Schrodi Y, Ung T, Vargas A, Mkrtumyan G, Lee C-W, Champagne TM, Pederson RL, Hong SH. Clean. 2008;36:669. [Google Scholar]
- 5.a) Anderson DR, Ung T, Mkrtumyan G, Bertrand G, Grubbs RH, Schrodi Y. Organometallics. 2008;27:563. doi: 10.1021/om7008028. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Anderson DR, Lavallo V, O’Leary DJ, Bertrand G, Grubbs RH. Angew Chem. 2007;119:7400. doi: 10.1002/anie.200702085. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:7262. [Google Scholar]
- 6.a) Zhang J, Song S, Wang X, Jiaoa J, Shi M. Chem Commun. 2013;49:9491. doi: 10.1039/c3cc45823g. [DOI] [PubMed] [Google Scholar]; b) van der Klis F, Le Nôtre J, Blaauw R, van Haveren J, van Es DS. Eur J Lipid Sci Technol. 2012;114:911. [Google Scholar]; c) Cohen SA, Luetkens ML, Balakrishnan C, Snyder R. WO2011/046872 A2. Elevance Renewable Sciences. 2011; d) Thomas RM, Keitz BK, Champagne T, Grubbs RH. J Am Chem Soc. 2011;133:7490. doi: 10.1021/ja200246e. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Hagadorn JR, Holtcamp MW, Bedoya MS. WO2011100022 A3. Exxonmobile. 2011; f) Mignani G, Pevere V, Olivier-Bourbigou H, Vallee C, Berthod M, Citadelle C. WO2010/010290 A1. Rhodia Operations, IFP. 2010; g) Marinescu SC, Schrock RR, Müller P, Hoveyda AH. J Am Chem Soc. 2009;131:10840. doi: 10.1021/ja904786y. [DOI] [PubMed] [Google Scholar]; h) Schrodi Y, Pederson RL, Kaido H, Tupy MJ. WO2008/046106 A2. Elevance Renewable Sciences. 2008; i) Burdett KA, Harris LD, Margl P, Maughon BR, Mokhtar-Zadeh T, Saucier PC, Wasserman EP. Organometallics. 2004;23:2027. [Google Scholar]
- 7.Patel J, Mujcinovic S, Jackson WR, Robinson AJ, Serelis AK, Such C. Green Chem. 2006;8:450. [Google Scholar]
- 8.Jazzar R, Dewhurst RD, Bourg J-B, Donnadieu B, Canac Y, Bertrand G. Angew Chem. 2007;119:2957. doi: 10.1002/anie.200605083. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:2899. [Google Scholar]
- 9.We also observed multiple conformational and rotational isomers in the NMR spectra of each of the catalysts 21 – 24. Ratios varied with respect to N-aryl substitution (see ESI for details).
- 10.Intuitively, this marked effect might be expected. Ethylene gas contains small amounts of impurities, such as carbon monoxide and acetylene, known catalyst poisons for ruthenium-based metathesis catalysts (see ref. 11). At such low catalyst loadings, even trace amounts of these substances would exhibit a greater effect.
- 11.a) Nelson DJ, Manzini S, Urbina-Blanco CA, Nolan SP. Chem Commun. 2014;50:10355. doi: 10.1039/c4cc02515f. [DOI] [PubMed] [Google Scholar]; b) Diver ST. Coord Chem Rev. 2007;251:671. doi: 10.1016/j.ccr.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Methyl oleate (Nu-Check-Prep, >99%, see ESI) was purified through filtration over alumina (see ESI). As disclosed previously this purification step is essential for high TON (see ref. 4). For example, when methyl oleate purchased from Nu-Check-Prep (>99%) or Sigma Aldrich (>99.0%, Fluka analytical standard) was used without further purification, TONs were only 8,500 and 230 respectively for catalyst 11 (3 ppm).
- 13.Interestingly, a similar increase was not noted for catalyst 5. For example, under the optimized reaction conditions 5 (100 ppm) generated a TON of only 4600 (cf. literature value of 5400, see ref. 4b).
- 14.CCDC 1025991 (11) and 1025992 (24) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- 15.a) Hong SH, Wenzel AG, Salguero TT, Day MW, Grubbs RH. J Am Chem Soc. 2007;129:7961. doi: 10.1021/ja0713577. [DOI] [PubMed] [Google Scholar]; b) Sanford MS, Love JA, Grubbs RH. J Am Chem Soc. 2001;123:6543. doi: 10.1021/ja010624k. [DOI] [PubMed] [Google Scholar]; c) Lehman SB, Wagener KB. Organometallics. 2005;24:1477. [Google Scholar]; d) Ulman M, Grubbs RH. J Org Chem. 1999;64:7202. [Google Scholar]
- 16.a) Back O, Henry-Ellinger M, Martin CD, Martin D, Bertrand G. Angew Chem. 2013;125:3011. doi: 10.1002/anie.201209109. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2013;52:2939. [Google Scholar]; b) Lavallo V, Canac Y, Prasang C, Donnadieu B, Bertrand G. Angew Chem. 2005;117:5851. doi: 10.1002/anie.200501841. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:5705. [Google Scholar]; c) Melaimi M, Soleilhavoup M, Bertrand G. Angew Chem. 2010;122:8992. doi: 10.1002/anie.201000165. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2010;49:8810. [Google Scholar]
- 17.Occhipinti G, Jensen VR. Organometallics. 2011;30:3522. [Google Scholar]
- 18.See ESI for further details.
- 19.Stewart IC, Keitz BK, Kuhn KM, Thomas RM, Grubbs RH. J Am Chem Soc. 2010;132:8534. doi: 10.1021/ja1029045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.For similar correlations regarding catalyst lifetime and approach to equilibrium for E/Z diastereoselectivity see: Lee C-W, Grubbs RH. Org Lett. 2000;2:2145. doi: 10.1021/ol006059s.
- 21.Keitz BK, Grubbs RH. J Am Chem Soc. 2011;133:16277. doi: 10.1021/ja207252r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.More detailed studies are currently underway and will be reported in due course.
- 23.Ethylene gas was purchased and used as received from Matheson, and was either Ultra High Purity (99.95%) or Matheson Purity (99.995%). Matheson Purity ethylene (99.995%) is certified to contain less than 4 ppm acetylene, and less than 2 ppm carbon monoxide.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.