

The coordinated action of the actin and microtubule cytoskeletal networks is required for Golgi ribbon assembly. The novel formin FHDC1 accumulates on the Golgi-derived microtubule network, where it acts to regulate Golgi ribbon assembly in an actin- and microtubule-dependent manner.
Abstract
The Golgi apparatus is the central hub of intracellular trafficking and consists of tethered stacks of cis, medial, and trans cisternae. In mammalian cells, these cisternae are stitched together as a perinuclear Golgi ribbon, which is required for the establishment of cell polarity and normal subcellular organization. We previously identified FHDC1 (also known as INF1) as a unique microtubule-binding member of the formin family of cytoskeletal-remodeling proteins. We show here that endogenous FHDC1 regulates Golgi ribbon formation and has an apparent preferential association with the Golgi-derived microtubule network. Knockdown of FHDC1 expression results in defective Golgi assembly and suggests a role for FHDC1 in maintenance of the Golgi-derived microtubule network. Similarly, overexpression of FHDC1 induces dispersion of the Golgi ribbon into functional ministacks. This effect is independent of centrosome-derived microtubules and instead likely requires the interaction between the FHDC1 microtubule-binding domain and the Golgi-derived microtubule network. These effects also depend on the interaction between the FHDC1 FH2 domain and the actin cytoskeleton. Thus our results suggest that the coordination of actin and microtubule dynamics by FHDC1 is required for normal Golgi ribbon formation.
INTRODUCTION
The Golgi apparatus is the central controller of subcellular trafficking found in all eukaryotes. In most cells, the Golgi occurs as a series of cisternae tethered together as stacks organized from cis to trans. Incoming traffic arrives at the cis face of the Golgi and moves through the medial and trans cisternae before exiting through the trans-Golgi network. At the same time, retrograde trafficking returns cargo proteins to the endoplasmic reticulum (ER) and maintains the localization of Golgi-resident enzymes as the cisternae mature (Rizzo et al., 2013). In higher organisms, these stacks are tethered laterally to form a Golgi ribbon typically found in tight association with the centrosome. The cis-to-trans stacking of the Golgi is found in almost all eukaryotes and is believed to facilitate cargo processing and modification. The additional requirement for the ribbon is unclear, but it has been suggested that ribbon formation may aid in the trafficking of very large cargo and reflects the more complex role of the Golgi in higher organisms (Lavieu et al., 2014). Current models suggest that the assembly of the Golgi ribbon is an actin- and microtubule (MT)-dependent process and that proper positioning and maintenance of the Golgi is required for polarized cellular trafficking and normal cell motility (Dippold et al., 2009; Miller et al., 2009; Yadav et al., 2009; Zhu and Kaverina, 2013; Deakin and Turner, 2014; Guet et al., 2014; Gurel et al., 2014).
Assembly of the perinuclear Golgi ribbon relies on two cytoplasmic MT networks, one originating at the centrosome and one originating at the Golgi itself (Rogalski and Singer, 1984; Miller et al., 2009; Rivero et al., 2009). Golgi-derived MTs are nucleated at the cis-Golgi through the GM130-mediated recruitment of AKAP450, myomegalin, and the γ-TuRC MT-nucleating complex (Chabin-Brion et al., 2001; Rivero et al., 2009; Roubin et al., 2013; Wang et al., 2014). This complex is absent from the trans-Golgi, and it has been proposed that, as the nascent MTs emerge at the cis-Golgi, they are likely captured by CLASP proteins, which are themselves recruited to the trans-cisternae by the Golgin GCC185 (Efimov et al., 2007; Sutterlin and Colanzi, 2010; Vinogradova et al., 2012; Zhu and Kaverina, 2013). Treatment with nocodazole or other MT-disrupting drugs causes the Golgi to disperse into ministacks that redistribute to ER-exit sites (Thyberg and Moskalewski, 1985; Cole et al., 1996). After removal of the drug, reassembly of the ribbon proceeds in two stages (Miller et al., 2009). In the G stage, the Golgi-derived MTs collect the dispersed ministacks into larger assemblies, which provide a bigger target for capture by MTs originating from the centrosome. In the C stage of assembly, the captured ministacks are transported along centrosome-derived MTs toward the centrosome, where they are tethered into the perinuclear Golgi ribbon (Corthesy-Theulaz et al., 1992; Burkhardt, 1998; Miller et al., 2009; Vinogradova et al., 2012; Maia et al., 2013). Disruption of the Golgi-derived MT network causes the G stage of reassembly to fail, resulting in Golgi dispersion or formation of compact Golgi circles located proximal to the centrosome (Miller et al., 2009; Maia et al., 2013).
The asymmetric subcellular positioning of the Golgi ribbon results in the polarized orientation of the Golgi-derived MT network. This asymmetry is required for directional cell migration and is believed to facilitate polarized secretion to the leading edge (Preisinger et al., 2004; Bisel et al., 2008; Vinogradova et al., 2009; Yadav et al., 2009; Hurtado et al., 2011). Golgi-derived MTs are enriched for acetylated α-tubulin (Jasmin et al., 1990; Thyberg and Moskalewski, 1993; Chabin-Brion et al., 2001), which likely affects both MT dynamics and selective recruitment of motor proteins (Reed et al., 2006; Janke, 2014). We previously found a close association between MT acetylation and the function of the novel MT-binding formin FHDC1 (Young et al., 2008; Thurston et al., 2012), raising the possibility that FHDC1 activity might be connected to the Golgi-derived MT network.
FHDC1 (previously known as INF1), like other formins, contains formin homology (FH) 1 and FH2 actin regulatory domains (Higgs, 2005; Young et al., 2008). It is distinguished from other formin family members by the presence of a unique C-terminal MT-binding domain (MTBD) and the absence of any of the regulatory domains present in other formins (Young et al., 2008). The full-length protein is constitutively active, and expression of full-length FHDC1 is sufficient to induce actin stress fiber formation, MT stabilization, and MT acetylation. Thus it is distinct from other formins in that its activity is not regulated by autoinhibition (Chesarone et al., 2010). Conversely, knockdown of FHDC1 expression inhibits the accumulation of acetylated MTs (Young et al., 2008), suggesting that FHDC1 participates in the normal regulation of this process. The effects of FHDC1 on actin dynamics are FH2 dependent, as expected. In contrast, expression of either the isolated FH1-FH2 or isolated MTBD is sufficient to induce MT stabilization and acetylation (Young et al., 2008; Thurston et al., 2012). Indeed, the ability to regulate MT modification and stabilization is a common feature of FH2 domains (Bartolini et al., 2008; Bartolini and Gundersen, 2010; Thurston et al., 2012).
As with the Golgi ribbon, FHDC1 is an evolutionary innovation in vertebrates (Young et al., 2008), and there is a growing appreciation that normal Golgi assembly and maintenance is both actin and MT dependent (Egea et al., 2006; Campellone et al., 2008; Dippold et al., 2009; Farber-Katz et al., 2014). Given the association between FHDC1 and MT acetylation, as well as its ability to regulate both actin and MT dynamics, we investigated the role of FHDC1 in Golgi ribbon formation. We show here that a pool of endogenous FHDC1 protein accumulates at the Golgi and likely has a preferential affinity for the Golgi-derived MT network. FHDC1 overexpression disperses the Golgi into functional ministacks, and FHDC1-induced Golgi dispersion is both FH2 and MTBD dependent. The ability of FHDC1 to induce Golgi dispersion did not correlate with its ability to induce MT acetylation, suggesting that the effects of FHDC1 on Golgi assembly are separate consequences of FHDC1 expression. Consistent with these observations, small interfering RNA (siRNA)–mediated knockdown of FHDC1 expression also disrupts Golgi ribbon formation. Together our results suggest that FHDC1 acts at the Golgi-derived MT network to coordinate actin and MT dynamics and facilitate formation of the Golgi ribbon.
RESULTS
A pool of FHDC1 associates with the Golgi ribbon
We first noted a connection between FHDC1 and the Golgi ribbon while investigating a potential role for FHDC1 in cell polarity. In these experiments, the subcellular localization of endogenous FHDC1 protein was determined in NIH 3T3 fibroblasts migrating in a scratch-wound model of directional cell migration (Figure 1A). The position of the Golgi ribbon was used as a marker of cell polarity and visualized with the cis-Golgi marker GM130; endogenous FHDC1 protein was detected with anti-FHDC1 antibody (Young et al., 2008). Two hours after wounding, cells at the wound’s edge reoriented and began to migrate into the wound. Both at the wound’s edge and away from the wound, endogenous FHDC1 is associated with the microtubule (MT) network and is concentrated in a perinuclear region corresponding to the position of the Golgi ribbon. A similar distribution of FHDC1 protein was also observed in randomly migrating cells (Figure 1B). Colocalization of FHDC1 with GM130 was further confirmed by a pixel intensity plot, showing the extensive overlap of the FHDC1 and GM130 signals (Figure 1B′). We also observed preferential association of endogenous FHDC1 with the Golgi ribbon in spreading cells (Supplemental Figure S1). In newly attached cells, the Golgi and centrosome are not connected (Kisurina-Evgen’eva and Onishchenko, 2004). Under these conditions, FHDC1 accumulates at and around the Golgi and not with the distinct population of cytoplasmic MTs at the cell periphery. These observations prompted us to further investigate the connections between FHDC1, the Golgi ribbon, and Golgi-derived MTs.
FIGURE 1:

A pool of FHDC1 accumulates at the Golgi ribbon. (A) NIH 3T3 cells migrating in a scratch-wound assay (wound to left) were fixed 2 h after wounding and stained with rabbit anti-FHDC1 antibody to detect endogenous FHDC1 (red) and mouse anti-GM130 antibody to detect the Golgi marker GM130 (green). FHDC1 localizes to cytoplasmic microtubules and concentrates at the Golgi ribbon in cells migrating into the wound, as well as in cells away from the wound’s edge. (B) FHDC1 associates with the Golgi ribbon. As in A, endogenous FHDC1 protein is distributed on cytoplasmic microtubules and concentrates at the Golgi in randomly polarized, cycling cells. Scale bar, 10 μm. (B′) Pixel intensity plot to assess the colocalization of FHDC1 with the Golgi marker GM130 in the area highlighted in B. The pixel intensity histogram is plotted for the area indicated by the white line in the left image and shows extensive colocalization of the FHDC1 and GM130 signals (Pearson’s r = 0.59 ± 0.04 [SEM]).
FHDC1 accumulates at Golgi-derived microtubules
Golgi ribbon assembly and integrity is MT dependent (Rogalski and Singer, 1984; Chabin-Brion et al., 2001; Vinogradova et al., 2012). Because FHDC1 is a MT-binding protein, we wanted to determine whether the accumulation of FHDC1 at the Golgi is MT dependent. We treated NIH 3T3 cells with nocodazole to dissolve MTs and induce Golgi dispersion (Miller et al., 2009). In untreated cells, FHDC1 is bound to MTs and concentrated at the Golgi as before (Figure 2A). After nocodazole treatment, FHDC1 punctae are distributed diffusely throughout the cytoplasm, and the Golgi is dispersed into ministacks. We did not observe any obvious association of FHDC1 with the Golgi remnants in nocodazole-treated cells (Figure 2B). After nocodazole washout, FHDC1 can be seen to take on a more filamentous distribution as the Golgi and MTs recover (Figure 2, C–F). We also found that in recovering cells, FHDC1 protein concentrates at cytoplasmic Golgi clusters (Figure 2, C′–E′) that form as the Golgi-derived MT network is reestablished and are likely to correspond to the late G phase of Golgi assembly (Efimov et al., 2007; Miller et al., 2009; Vinogradova et al., 2012). Two hours after removal of nocodazole, both Golgi and FHDC1 distributions have fully recovered, and a clear association of FHDC1 with the perinuclear Golgi ribbon is observed (Figure 2, F and F′). This suggests that FHDC1 accumulates at the Golgi in an MT-dependent manner.
FIGURE 2:

FHDC1 localizes to cytoplasmic sites of Golgi reassembly after nocodazole treatment. (A) In untreated NIH 3T3 fibroblasts, FHDC1 (red) localizes primarily to the microtubule network and concentrates at the Golgi ribbon (GM130, green). (B) FHDC1 punctae are distributed diffusely throughout the cytoplasm of cells treated with 2.5 μg/ml nocodazole for 120 min; the Golgi is dispersed into ministacks in treated cells. (C) At 20 min and (D, E) 40 min after removal of nocodazole, FHDC1 clusters at cytoplasmic sites of Golgi reassembly (C′, D′, E′). (F) At 120 min after removal of nocodazole, FHDC1 subcellular localization is fully recovered, and the protein is distributed on microtubules and concentrated at the perinuclear Golgi ribbon (F′). Scale bar, 10 μm.
Cytoplasmic MTs are nucleated at both the centrosome and the Golgi (Petry and Vale, 2015). The centrosome can be specifically depleted from NIH 3T3 fibroblasts by extended treatment with the PLK4 inhibitor centrinone (Wong et al., 2015). Surprisingly, this does not block division of the treated cells or affect normal Golgi assembly. We cultured cells in the presence of centrinone for 14 d to completely eliminate centrosome formation and determined the effect on Golgi assembly and endogenous FHDC1 subcellular localization (Figure 3). In control cells, FHDC1 was present on the MT network, and the centrosome was readily detected with an anti–γ-tubulin antibody (Figure 3A). In centrinone-treated cells, the centrosome could no longer be detected, the Golgi remained intact, and FHDC1 maintained its association with cytoplasmic MTs (Figure 3, B and C).
FIGURE 3:

Endogenous FHDC1 distribution is not affected by centrinone-induced centrosome depletion. NIH 3T3 cells were treated with the Plk4 inhibitor centrinone for 14 d to induce centrosome depletion. The effects of this treatment on the distribution of the endogenous FHDC1 protein were assessed by immunofluorescence. (A) Vehicle-treated control cells were stained with anti-FHDC1 (red) and anti–γ-tubulin (green) antibodies. The centrosome is easily detected as a prominent perinuclear spot. (B) Centrinone-treated cells were stained with anti-FHDC1 (red) and anti–γ-tubulin (green) antibodies. The centrosome is no longer detected in treated cells, whereas FHDC1 maintains its filamentous distribution. (C) Centrinone-treated cells were stained with anti-FHDC1 (red) and anti-GM130 (green) antibodies. The accumulation of a pool of FHDC1 protein at the Golgi ribbon is unaffected in centrinone-treated cells. Scale bar, 10 μm.
The centrinone results prompted us to consider that FHDC1 may have a preferential affinity for the Golgi-derived microtubule network. Golgi-derived MTs are enriched for acetylated MTs (Thyberg and Moskalewski, 1993; Chabin-Brion et al., 2001), and we previously observed a close association between FHDC1 activity and MT acetylation (Young et al., 2008; Thurston et al., 2012). To test the idea that FHDC1 associates with Golgi-derived MTs, we treated cells with brefeldin A (BFA) to disrupt the Golgi ribbon and Golgi-derived MTs while leaving centrosome-derived MTs intact. In vehicle-treated cells, FHDC1 is predominantly associated with the MT network and Golgi ribbon as before (Figure 4A). BFA treatment quickly induces Golgi ribbon disassembly, leaving behind a remnant Golgi matrix as revealed by GM130 staining (Nakamura et al., 1995). We found that with prolonged BFA treatment (45 min), the accumulation of FHDC1 at the Golgi was lost, and the filamentous distribution of FHDC1 was replaced with a more punctate staining pattern in the majority of cells (Figure 4, B and E). On washout of BFA, the filamentous distribution of FHDC1 was quickly regained, concomitant with the reassembly of the Golgi ribbon (Figure 4, C and D). These results suggest a preferential association of FHDC1 with the Golgi-derived MT network. This hypothesis was further tested by examining the subcellular distribution of endogenous FHDC1 in cells allowed to recover from nocodazole-induced MT disruption in the presence or absence of BFA. As before, FHDC1 was primarily associated with the MT network and enriched at the Golgi in untreated cells (Figure 5A). In cells treated with nocodazole and BFA, the Golgi is dispersed, and FHDC1 was redistributed as a diffuse array of cytoplasmic punctae (Figure 5B). Two hours after nocodazole washout, in the absence of BFA, the perinuclear Golgi ribbon had reassembled and FHDC1 had completely regained its MT association (Figure 5D). In cells recovered from nocodazole treatment in the presence of BFA, however, the Golgi remained dispersed, and FHDC1 failed to regain its filamentous distribution and remained primarily in cytoplasmic punctae (Figure 5C). We also compared the distribution of endogenous FHDC1 with that of CLASP2, an MT-binding protein and marker for the Golgi-derived MT network. Using antibodies against endogenous FHDC1 and endogenous CLASP2, we found that a pool of both proteins accumulated on MTs at the Golgi ribbon and colocalized on a subset of cytoplasmic MTs (Figure 6). Accordingly, in cells recovering from nocodazole treatment in the presence of BFA, CLASP2, like FHDC1, no longer accumulates on MTs at the Golgi ribbon (Supplemental Figure S2). Together these data point to a preferential association of FHDC1 with the Golgi-derived MT network.
FIGURE 4:

Brefeldin A treatment decreases FHDC1 microtubule localization. (A) In untreated cells, FHDC1 is found primarily on the microtubule network and in close association with the Golgi. (B) After 45 min of treatment with 5.0 μg/ml brefeldin A, FHDC1 subcellular localization in many cells is less filamentous and has a more punctuate and diffuse cytoplasmic distribution, as typified by the indicated cell (arrow). (C) At15 min after removal of brefeldin A, the Golgi ribbon has begun to reassemble; FHDC1 is regaining a more filamentous distribution and is concentrated at the Golgi. (D) In fully recovered cells, FHDC1 is primarily filamentous and preferentially associated with the Golgi. Scale bar, 10 μm. (E) Quantification of data shown in A–D. In untreated cells, FHDC1 is primarily filamentous (blue bars). In contrast, after 45 min of treatment with brefeldin A, FHDC1 exhibits a more punctuate distribution. N = 3, >100 cells counted per experiment. Error bars indicate SEM.
FIGURE 5:

Brefeldin A blocks FHDC1 recruitment to microtubules during recovery from nocodazole treatment. (A) In untreated cells, FHDC1 (red) is concentrated on microtubules associated with the Golgi (GM130; green). (B) Cells were treated for 2 h with 2.5 μg/ml nocodazole and 5.0 μg/ml brefeldin A. The Golgi is dispersed throughout the cytoplasm; FHDC1 has a punctate cytoplasmic distribution. (C) Cells were allowed to recover after nocodazole washout for 120 min in the presence of brefeldin A. FHDC1 does not recover its filamentous staining pattern and maintains its punctate cytoplasmic distribution. (D) Cells were allowed to recover for 120 min in the absence of BFA, with FHDC1 regaining its association with the microtubule network. Scale bar, 10 μm.
FIGURE 6:

FHDC1 and CLASP2 localize to an overlapping microtubule network. (A) NIH 3T3 cells were stained with rabbit anti-FHDC1 antibody to detect endogenous FHDC1 (red) and rat anti-CLASP2 antibody to detect endogenous CLASP2 (green), and mouse anti-GM130 (white) was used as a Golgi marker. FHDC1 and CLASP2 colocalize (yellow) to an overlapping set of cytoplasmic microtubules and concentrate at the Golgi ribbon. Boxes 1 and 2 highlight areas of FHDC1 and CLASP2 colocalization. The white arrows indicate MT-associated FHDC1 and CLASP2 codistribution. Cells were fixed in 4% paraformaldehyde in PHEM buffer for optimal CLASP2 staining. Scale bar, 10 μm. (B) A pixel intensity plot used to assess the colocalization of FHDC1 and CLASP2 in the region indicated by the white line in B′. The pixel intensity histogram reveals extensive colocalization of the FHDC1 and CLASP2 signals.
FHDC1 induces Golgi dispersion
We next sought to investigate what role FHDC1 might play in the assembly and positioning of the Golgi. We first chose to address this question by testing the effects of FHDC1 overexpression on Golgi morphology. We expressed a full-length FHDC1 derivative with an N-terminal mCherry tag (FHDC1.mCherry) in NIH 3T3 cells by transient transfection. mCherry-transfected cells served as a negative control. The cells were fixed 24 h posttransfection and the effects of FHDC1 expression on Golgi assembly and localization were assessed by immunofluorescence. In mCherry-expressing controls, the Golgi was organized as a perinuclear ribbon in nearly 90% of transfected cells (Figure 7, A and C). In contrast, in >90% of FHDC1-expressing cells, the Golgi ribbon was completely dispersed (Figure 7, B and C). The Golgi fragments, while sometimes found in a perinuclear halo, had no consistent, specific subcellular localization (see later discussion of Figures 8B, 9, B, E, and H, 10C, 11A, and 12C for additional examples). This striking phenotype is remarkably robust and is reminiscent of the effects caused by loss or overexpression of other regulators of Golgi assembly (Campellone et al., 2008; Dippold et al., 2009; Yadav et al., 2009, 2012; Farber-Katz et al., 2014). It also suggests that FHDC1 might play an important role in this process.
FIGURE 7:

FHDC1 expression induces Golgi dispersion. (A) mCherryFP was transiently expressed in NIH 3T3 cells. The Golgi ribbon (giantin; green) and microtubule network (white) are unaffected in Cherry-expressing cells (red). (B) Expression of FHDC1 (red) induces cellular elongation and concomitant reorganization of the microtubule network. FHDC1 expression also induces dispersion of the Golgi ribbon into small cytoplasmic punctae. Scale bar, 10 μm. (C) Quantification of the data in B. Golgi classified as dispersed were fragmented, and the distribution of the fragments had no obvious polarity. Fragmented Golgi with an obvious polarized perinuclear distribution or nonpolarized Golgi ribbons were classified as intermediate. N = 3, >100 transfected cells counted per experiment. Error bars indicate SEM.
FIGURE 8:

FHDC1-induced Golgi dispersion is FH2 and MTBD dependent. (A) mCherry, (B) full-length FHDC1, (C) FHDC1.I180A, (D) FHDC1 958N, and (E) FHDC1 958C (left; red) were transiently expressed in NIH 3T3 cells. Expression of full-length FHDC1 induced Golgi dispersion (giantin; green), whereas expression of FHDC1 derivatives lacking FH2 activity (FHDC1.I180A, 958C) or the MTBD (958N) did not. Microtubules are shown in white. Scale bar, 10 μm. (F) Quantification of the data in A–E. N = 3, >100 cells counted per experiment. Error bars, SEM. (G) Schematic of FHDC1 derivatives. Boxes 1–5 represent regions of homology conserved between FHDC1 homologues of different species.
FIGURE 9:

FHDC1-induced Golgi dispersion does not block VSV-G trafficking. NIH 3T3 cells were transiently transfected with plasmids encoding VSVG-ts045.GFP (green) and mCherry (A, D, G), full-length FHDC1 (B, E, H), or FHDC1.I180A (C, F, I; left, red). Transfected cells were held at 42°C for 6 h; VSVG-ts045 is trapped in the ER in all transfected cells (A–C). The cells were shifted to 32°C for 30 min; VSVG-ts045 is trafficked to the Golgi (center left; white) in all transfected cells (D–F). After 90 min at 32°C, VSVG-ts045 is trafficked to the plasma membrane in all transfected cells (G–I). Scale bar, 10 μm.
FIGURE 10:
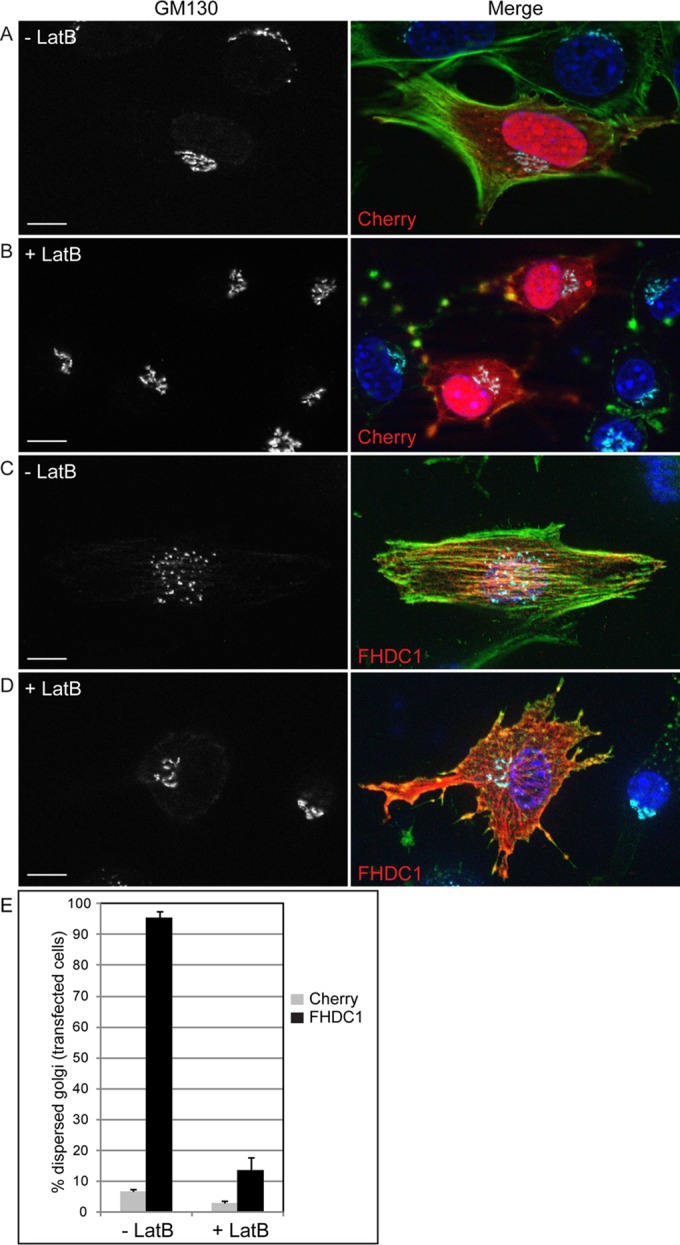
Latrunculin B inhibits FHDC1-induced Golgi dispersion. mCherry (red in merged image)-expressing cells were treated with (A) vehicle (–LatB) or (B) 2 μM latrunculin B (+LatB) for 2 h to disrupt the F-actin network as detected by phalloidin staining (green in merged image). The effects on Golgi morphology were assessed by immunofluorescence using anti-GM130 (cyan in merge). (C) FHDC1 expression (red in merge) induces actin stress fiber formation and Golgi dispersion in vehicle-treated cells. (D) LatB treatment reverses the effects of FHDC1 expression and induces the formation of perinuclear Golgi clusters similar to control and untransfected cells. (E) Quantification of data in A–D. Scale bar, 10 μm.
FIGURE 11:

FHDC1-induced Golgi dispersion is unaffected in centrinone-treated cells. NIH 3T3 cells were treated with the Plk4 inhibitor centrinone (300 nM) for 14 d, and the effects of this treatment on FHDC1-induced Golgi dispersion were assessed by immunofluorescence. (A) mCherryFP-tagged FHDC1 (red) was expressed by transient transfection in control cells that were subsequently stained with anti-giantin (white) and anti–γ-tubulin (green) antibodies. (B) mCherryFP-tagged FHDC1 (red) was expressed by transient transfection in centrinone-treated cells that were subsequently stained with anti-giantin (white) and anti–γ-tubulin (green) antibodies. FHDC1 expression is sufficient to induce Golgi dispersion in both cases, even though the centrosome can no longer be detected in centrinone-treated cells. Scale bar, 10 μm. (C) Quantification of data in A and B. Centrinone treatment did not modify the effects of FHDC1 and FHDC1.I180A expression on Golgi dispersion. N = 3, >100 cells counted per experiment; error bars indicate SEM.
FIGURE 12:

FHDC1-induced Golgi dispersion is inhibited in GM130-depleted cells. NIH 3T3 cells were transfected with (A) control siRNA duplexes or (B) duplexes targeting GM130 to knock down GM130 expression. Distribution of endogenous FHDC1 protein (red) is not affected in control cells but is less filamentous and more punctate in GM130-knockdown cells (green). Full-length FHDC1 (red) was transiently expressed in (C) control and (D) GM130-knockdown cells. FHDC1-induced Golgi dispersion is unaffected in control cells but is inhibited in GM130-depleted cells. Scale bar, 10 μm. (E) Quantification of the data in C and D. GM130 knockdown did not modify the effects of FHDC1.958N expression on Golgi morphology. N = 3, >100 cells counted per experiment. Error bars, SEM. (F) Whole-cell lysates from GM130-knockdown, control transfected cells and untransfected cells were immunoblotted for GM130, FHDC1, and α-tubulin. Endogenous FHDC1 expression is not affected by GM130 depletion.
FHDC1-induced Golgi dispersion is FH2 and MTBD dependent
FHDC1 binds both actin filaments and MTs (Young et al., 2008), and Golgi ribbon assembly and perinuclear positioning are both actin and MT dependent (Rogalski and Singer, 1984; Magdalena et al., 2003; Lazaro-Dieguez et al., 2006; Dippold et al., 2009; Gurel et al., 2014). We therefore sought to determine which aspect of FHDC1 function might be required for its effects on Golgi dispersion. To this end, we generated the FHDC1.I180A derivative to selectively inactivate FH2. In this mutant, residue 180 in the FHDC1 FH2 is mutated from isoleucine to alanine to abolish the ability of FH2 to bind to the barbed end of F-actin (Xu et al., 2004). We confirmed that this mutant no longer affects actin dynamics but is still able to bind MTs and induce MT acetylation in cell-based assays (Supplemental Figure S3). The I180A mutant and a series of FHDC1 deletion derivatives defective in either FH2 or MTBD activity were tested for their effects on Golgi morphology. As before, expression of FHDC1 is sufficient to induce Golgi dispersion in >90% of transfected cells, and the tagged protein is bound to the MT network. The FHDC1.I180A derivative also associated with MTs and was enriched at the Golgi, but expression of FHDC1.I180A failed to induce Golgi dispersion (Figure 8, C and F). This suggests that FHDC1 effects on Golgi are likely FH2 and actin dependent. Similarly, expression of the isolated FHDC1 MT-binding domain (958C) also had no effect on perinuclear Golgi ribbon assembly (Figure 8, E and F), despite its marked effects on MT bundling and network organization (Young et al., 2008). In the same way, expression of an MTBD-deleted FHDC1 derivative (958N) also failed to induce Golgi dispersion to the same extent as the full-length protein, with a normal perinuclear ribbon present in >60% of transfected cells (Figure 8, D and F). Thus the effects of FHDC1 on Golgi dispersion are both FH2 and MTBD dependent. We also compared the effects of FHDC1 expression to the effects of expressing constitutively active derivatives of other formins believed to participate in the regulation of Golgi dynamics (Colon-Franco et al., 2011; Ramabhadran et al., 2011; Zilberman et al., 2011). In cell-based assays, these proteins (FMNL1, INF2, mDia1) are all potent inducers of F-actin accumulation and MT acetylation (Thurston et al., 2012). As previously observed, expression of constitutively active FH1 + FH2–containing derivatives of FMNL1, INF2, and mDia1 had some effect on Golgi morphology but failed to induce dispersion in the majority of cells (Supplemental Figure S4). These results demonstrate the specificity and striking robustness of the effects of full-length FHDC1 on Golgi assembly.
FHDC1-dispersed Golgi fragments support subcellular trafficking
To gain insight into the nature of the FHDC1-dispersed Golgi remnants, we sought to determine whether the Golgi fragments are still capable of supporting intracellular trafficking. To do this, we took advantage of the temperature-sensitive vesicular stomatitis virus G (VSVG) ts045–green fluorescent protein (GFP) trafficking reporter (Presley et al., 1997). At the nonpermissive temperature (40°C), the mutant VSVG protein is trapped in the ER and is released only upon a shift to the permissive temperature of 32°C. Before this shift, cycloheximide is added to inhibit protein synthesis to facilitate the tracking of the VSVG reporter once it is released from the ER. Once shifted to the permissive temperature, VSVG proceeds from the ER to the Golgi and from there to the plasma membrane. We coexpressed VSVG ts045-GFP in NIH 3T3 cells with FHDC1, FHDC1.I180A, or mCherry and followed trafficking of the reporter at nonpermissive and permissive temperatures. After 6 h at 40°C, the VSVG-GFP protein has accumulated in the ER of mCherry-, FHDC1.I180A-, and FHDC1-expressing cells (Figure 9, A–C). Thirty minutes after the shift to 32°C, in the presence of cycloheximide, VSVG-GFP had left the ER and accumulated in the perinuclear Golgi ribbon of mCherry- and FHDC1.I180A-expressing cells (Figure 9, D and F); it could also be clearly seen to have trafficked to the Golgi fragments present in FHDC1-expressing cells (Figure 9E). After 90 min at 32°C, VSVG-GFP had moved from the Golgi to the plasma membrane in all cells, including those with FHDC1-dispersed Golgi (Figure 9, G–I). We also examined distribution of the lysosome marker LAMP1-RFP and the ER marker calreticulin-GFP in untransfected and FHDC1-expressing cells and found no obvious differences in their distribution (Supplemental Figure S5). Thus the FHDC1-induced Golgi fragments were still able to support trafficking from the Golgi to the plasma membrane, the lysosome, and the ER.
FHDC1-induced Golgi dispersion is F-actin and MT dependent
Golgi assembly and positioning relies on the coordinated action of both the actin and MT networks, and we observed that FHDC1-induced dispersion is both FH2 and MTBD dependent. The FHDC1 FH2 domain, however, is able to regulate both actin and MT dynamics (Young et al., 2008; Thurston et al., 2012). To determine whether the effects of FHDC1 expression on Golgi assembly are actin dependent, we tested the effects of the actin-disrupting drug latrunculin B (LatB) on FHDC1-induced Golgi dispersion. FHDC1-expressing cells were fixed after 120 min of treatment with 2 μM LatB, and the effects on actin assembly and Golgi morphology were determined by immunofluorescence. In control mCherry-expressing cells, LatB treatment altered ribbon morphology and induced the formation of perinuclear Golgi clusters (Figure 10, A and B), as observed previously (Lazaro-Dieguez et al., 2006; Zilberman et al., 2011). Similar effects were observed in FHDC1-expressing cells, for which LatB treatment reversed FHDC1-induced Golgi dispersion and resulted in the reassembly of perinuclear Golgi clusters (Figure 10, D and E). This suggests that the effects of FHDC1 are actin dependent, consistent with the results obtained with the I180A mutant.
The data in Figures 2–6 suggest that FHDC1 may preferentially associate with Golgi-derived MTs; however, the FHDC1 overexpression phenotype could also be caused by disrupting the connection between the Golgi ribbon and the centrosome-derived MT network (Miller et al., 2009; Yadav et al., 2009). We thus wished to determine whether or not either of these two networks is required for the effects of FHDC1 on Golgi assembly. To determine whether FHDC1-induced dispersion was dependent on the centrosome-derived MT network, we expressed FHDC1 in centrinone-treated cells and assessed the effects on Golgi assembly. In both control and centrosome-depleted cells, FHDC1 induced Golgi dispersion in >90% of transfected cells (Figure 11), suggesting that the effects of FHDC1 are not centrosome dependent.
Assembly of the Golgi-derived MT network requires the GM130-dependent recruitment of AKAP450 to the cis-Golgi, which in turn recruits the γ-TuRC MT-nucleating complex to allow assembly of Golgi-derived MTs (Rivero et al., 2009; Roubin et al., 2013; Rios, 2014; Petry and Vale, 2015). Knockdown of GM130 expression should therefore selectively inhibit MT nucleation at the cis-Golgi while leaving the Golgi ribbon and the centrosome-derived MT network largely intact. We therefore examined the effects of GM130 depletion on FHDC1 function and subcellular localization. We transfected cells with GM130-targeted siRNA or control duplex and assessed the efficiency of GM130 depletion by immunoblotting and immunofluorescence (Figure 12, D and F, and Supplemental Figure S6). In control cells, endogenous FHDC1 was associated with cytoplasmic MTs and concentrated at the Golgi as before (Figure 12A). The depletion of GM130, however, caused a distinct reduction in filamentous FHDC1 staining despite the presence of abundant cytoplasmic MTs (Figure 12B and Supplemental Figure S6, A and B). Indeed, the FHDC1 distribution was very similar to that seen in cells recovering from nocodazole treatment in the presence of BFA (Figure 4 and Supplemental Figure S2). GM130 depletion did not affect FHDC1 expression levels as assessed by immunoblotting (Figure 12F and Supplemental Figure S6G).
Depletion of GM130 also inhibited the ability of FHDC1 expression to induce Golgi dispersion. In control cells, as before, FHDC1 expression induced Golgi dispersion, and intact perinuclear Golgi ribbons were observed in <10% of FHDC1-expressing cells (Figure 12, C and E). In GM130-depleted cells, giantin staining was used to visualize the Golgi. In these cells, the Golgi appeared somewhat fragmented but retained its perinuclear position (Figure 12D). Despite this fragmentation, knockdown of GM130 expression actually protected the Golgi from the effects of FHDC1; in GM-130 depleted cells, normal Golgi ribbons were observed in >30% of transfected cells expressing FHDC1. GM130 knockdown had no effect on the ability of FHDC1 to induce MT acetylation (Supplemental Figure S6, C–E), consistent with previous reports that loss of the Golgi-derived MT network does not affect acetylation of non-Golgi MTs (Rivero et al., 2009). This suggests that FHDC1 activity was not generally inhibited in GM130-depleted cells and that the centrosome-derived MT network remains intact (Figure 12B). We also found that GM130 depletion neither enhanced nor suppressed the effects on Golgi dispersion of an FHDC1 derivative lacking the MTBD (FHDC1.958N; Figure 12E). This is consistent with the protective effects of GM130 depletion being dependent on the loss of Golgi-derived MTs. Together these data point to a model in which the ability of FHDC1 to affect Golgi morphology depends on its ability to bind to the Golgi-derived MT network and to actin filaments. In support of this model, we find that the full-length FHDC1 protein is able to bind directly to both F-actin and MTs in vitro (Supplemental Figure S7).
FHDC1 depletion inhibits Golgi assembly
Given the dramatic effect that FHDC1 expression has on Golgi morphology, we sought to determine whether FHDC1 activity is required for Golgi ribbon formation. We transfected NIH 3T3 cells with control siRNA duplexes or duplexes targeting FHDC1. The efficiency of FHDC1 knockdown was assessed by immunoblotting (Figure 13F), and the effects on Golgi morphology were determined by immunofluorescence, using GM130 as a Golgi marker. As expected, a normal perinuclear Golgi ribbon was observed in the majority of control cells (Figure 13, A and E). In contrast, major defects in Golgi assembly were observed in >80% of cells transfected with the FHDC1 siRNA duplex. The defects fell into three categories: cytoplasmic Golgi dispersion (Figure 13B), perinuclear compact Golgi circles (Figure 13C), and a diverse group of fragmented Golgi structures (Figure 13D). Knockdown of FHDC1 with a second siRNA duplex yielded similar results (Figure 13E). Each of these phenotypes is consistent with defects in the function of the Golgi-derived MT network during Golgi assembly (Miller et al., 2009; Vinogradova et al., 2012), which suggests that FHDC1 plays an important role in this process.
FIGURE 13:

Knockdown of FHDC1 expression disrupts Golgi ribbon assembly. NIH 3T3 cells were transfected with (A) control siRNA duplexes or (B–D) duplexes targeting FHDC1 to knock down FHDC1 expression. (A) In control cells, the Golgi (GM130; green) forms a perinuclear ribbon, and FHDC1 (red) is found on microtubules concentrated at the Golgi. (B) FHDC1 depletion induced Golgi dispersion in nearly 50% of cells. (C) Loss of FHDC1 expression also induced the formation of compact, circular Golgi in ∼25% of cells. (D) The remaining FHDC1- knockdown cells had either fragmented Golgi ribbons (∼10%) or normal perinuclear ribbons (15%). Scale bar, 10 μm. (E) Quantification of data in A–D and for a second siRNA duplex targeting FHDC1. N = 3, >100 cells counted per experiment. Error bars, SEM. (F) Whole-cell lysates from FHDC1-knockdown, control transfected, and untransfected cells were immunoblotted for FHDC1 and α-tubulin. In the gels shown, duplex 1 resulted in a 63% knockdown of FHDC1 protein levels relative to untransfected cells, and duplex 2 resulted in a 51% knockdown.
DISCUSSION
Our findings indicate that FHDC1 likely acts at the Golgi-derived MT network to regulate Golgi assembly. Golgi-derived MTs are highly acetylated (Thyberg and Moskalewski, 1993), and, as noted previously, FHDC1 preferentially associates with acetylated MTs (Young et al., 2008). We find that the endogenous FHDC1 protein accumulates on MTs in close association with the Golgi ribbon. This association is also observed during recovery from nocodazole treatment at cytoplasmic Golgi clusters. These clusters likely represent late G phase of Golgi assembly as Golgi-derived MTs are reformed (Miller et al., 2009), suggesting that FHDC1 is recruited to nascent Golgi-derived MTs. Similar results were obtained in spreading cells, where FHDC1 first accumulates at the Golgi and is largely absent from the abundant cytoplasmic microtubules present in newly attached cells. Conversely, in BFA-treated cells, the association of FHDC1 with MTs is reduced, and BFA treatment largely prevents the reassociation of FHDC1 with MTs during recovery from nocodazole treatment. Consistent with the effects of BFA on MT binding by FHDC1, we find that endogenous FHDC1 is efficiently recruited to MTs in centrosome-depleted cells, but MT binding is reduced in GM130-knockdown cells, where Golgi-derived MT formation should be inhibited (Rivero et al., 2009; Rios, 2014). Two major phenotypes were observed in FHDC1-depleted cells: Golgi dispersion and formation of compact Golgi circles. Both of these are consistent with defects in the G phase of Golgi assembly as observed either during recovery from nocodazole treatment (Miller et al., 2009) or during postmitotic Golgi reassembly (Vinogradova et al., 2012; Maia et al., 2013).
As with FHDC1 depletion, FHDC1 overexpression also has major effects on Golgi structure. Expression of the full-length FHDC1 protein induces Golgi dispersion. The dispersed Golgi fragments most likely represent Golgi ministacks and are still able to support most aspects of Golgi trafficking. It is unlikely that FHDC1-induced dispersion arises as a secondary effect of FHDC1-induced changes in MT dynamics. First, there is no correlation between the ability of FHDC1 to induce MT acetylation and the ability to induce Golgi dispersion. All of the tested FHDC1 derivatives induce MT acetylation to a similar extent (Young et al., 2008; Thurston et al., 2012), but only the full-length protein induces dispersion. Second, dispersion is unrelated to effects on MT stability: the isolated FHDC1 MTBD is a much more effective inducer of MT stabilization than the full-length protein (Young et al., 2008), yet expression of the isolated MTBD does not induce Golgi dispersion.
We also found that FH2 activity is required for the effects of FHDC1 on Golgi assembly and that these effects are actin dependent. It is not clear how FHDC1-induced changes in actin dynamics induce dispersion directly. Previous studies suggested that F-actin formation would be expected to induce Golgi compaction (Lazaro-Dieguez et al., 2006) rather than the dispersion we observe with FHDC1 expression. In addition, expression of either full-length FHDC1 or the 958N deletion derivative is sufficient to induce extensive thin stress fiber formation (Young et al., 2008), but 958N expression has only minimal effects on Golgi dispersion.
We propose that FHDC1-induced Golgi dispersion is not a simple consequence of disrupting normal actin or MT dynamics. Instead, FHDC1 expression disrupts a specific connection between Golgi, actin filaments, and the Golgi-derived MT network. This is consistent with the protective effect that GM130 knockdown has on FHDC1-induced Golgi dispersion. GM130 depletion should inhibit the nucleation of Golgi-derived MTs and would therefore remove the proposed site of FHDC1 action. In agreement with this model, we find that loss of GM130 does not modify the effects of FHDC1.958N expression on Golgi morphology. The 958N derivative lacks the MTBD, and therefore its activity should be unaffected by loss of Golgi-derived MTs, precisely as observed. It is likely that the overexpressed FHDC1 protein is able to bind both Golgi- and centrosome-derived MTs (Young et al., 2008), and our data do not exclude additional FHDC1 functions at centrosome-derived MTs, which may also be affected by GM130 knockdown (Kodani and Sutterlin, 2008; Kodani et al., 2009). GM130 depletion does not affect the ability of FHDC1 to induce MT acetylation, and thus not all aspects of FHDC1 activity are affected by the loss of GM130. Given that the loss of Golgi-derived MTs does not affect the acetylation of centrosome-derived MTs (Rivero et al., 2009) this suggests that the centrosome-derived MT network is still intact in GM130-depleted cells but does not contribute significantly to the effects of FHDC1 on Golgi assembly. This is entirely consistent with the observation that centrosome depletion does not affect FHDC1-induced Golgi dispersion.
Our results therefore point to a specific function for FHDC1 at the Golgi-derived MT network during Golgi assembly. We favor a model in which FHDC1 acts as a cytoskeletal bridging factor required to promote the stability of Golgi-derived MTs by coordinating actin and MT dynamics (Rodriguez et al., 2003; Gundersen et al., 2004; Dippold et al., 2009). In this scenario, FHDC1 would be recruited to the Golgi-derived MT network at the cis-Golgi, where it acts in an actin-dependent manner to stabilize the nascent MTs as they are handed over from the GM130/AKAP450/γ-TuRC complex at the cis-Golgi to GCC185/CLASP proteins at the trans-Golgi (Millarte and Farhan, 2012; Zhu and Kaverina, 2013). Our results suggest that FHDC1 would then remain bound to the Golgi-derived MTs as they extend into the cytoplasm and track along actin filaments. This model accounts for the accumulation of FHDC1 with cis-Golgi markers, the association of FHDC1 with the Golgi in spreading cells, and the ability of brefeldin A treatment or GM130 depletion to inhibit MT binding by FHDC1. It also accounts for the FH2/F-actin and MTBD/MT-dependent effects of FHDC1 expression on Golgi assembly and is consistent with the Golgi phenotypes observed in FHDC1-depleted cells.
In conclusion, the assembly of a perinuclear Golgi ribbon is a vertebrate evolutionary innovation whose function has still not been clearly defined (Nakamura et al., 2012). FHDC1, like the Golgi ribbon, is also unique to the vertebrate lineage. It was proposed recently that ribbon assembly is required to facilitate the efficient transport of large cargo such as extracellular matrix (ECM) proteins (Lavieu et al., 2014). We reported previously that FHDC1 is highly expressed in fibroblasts (Young et al., 2008). It is therefore interesting to speculate that FHDC1 activity may be of particular importance for maintaining Golgi structure in these cells so as to assist in their primary function of generating and secreting ECM proteins. Our results identify a new role for FHDC1 in the assembly of the Golgi ribbon and suggest that FHDC1 plays an important role in the function of the Golgi-derived MT network during this process. How endogenous FHDC1 is preferentially recruited to Golgi-derived MTs is being investigated.
MATERIAL AND METHODS
Reagents and plasmids
Full-length FHDC1 and the 958N and 958C derivatives, as well as the mDia1, FMNL1, and INF2 F1F2 + C derivatives, were previously described (Young et al., 2008; Thurston et al., 2012). The FHDC1.I180A mutation was generated by overlapping PCR to convert the codon encoding isoleucine at position 180 to alanine. The VSV-G reporter gene (11912; Addgene, Cambridge, MA) was previously described (Presley et al., 1997). The following antibodies were used in this study: mouse anti–α-tubulin (T5168; Sigma-Aldrich, Oakville, Canada), mouse anti-GM130 (610822; BD Transduction Laboratories, Mississauga, Canada), rabbit anti-giantin (ab24586; Abcam, Toronto, Canada), mouse anti–acetylated tubulin (clone 6-11B-1; T-6793; Sigma-Aldrich), mouse anti-FLAG (F7425; Sigma-Aldrich), rabbit affinity-purified anti-FHDC1 (Young et al., 2008), rat anti-CLASP2 (MAB9738; Abnova, Walnut, CA), and Alexa Fluor 488–phalloidin (A12379; Molecular Probes, Burlington, Canada). Nocodazole (M1404; Sigma-Aldrich) and brefeldin A (B7651; Sigma-Aldrich) were used at the indicated concentrations. Centrinone was kindly provided by Tim Gahman (Small Molecule Discovery Program, Ludwig Cancer Research, San Diego, CA).
Cell culture, transfections, and treatments
NIH 3T3 fibroblasts were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and cultured according to the supplied guidelines in DMEM (Wisent, St. Jean Baptiste, Canada) supplemented with 10% donor bovine serum (DBS; ATCC) in 5% CO2. Transient transfections were performed using polyethylenimine (PEI) as described previously (Young et al., 2008). Briefly, 1.5 μg of total plasmid DNA was diluted in 50 μl of OptiMEM, 5 μl of 1 mg/ml PEI was added, and the mixture was incubated for 25–30 min at room temperature. The DNA/PEI mix was added to cells in 1 ml of OptiMEM and left for 5 h under normal culture conditions. At the end of 5 h, the medium was replaced with 2 ml of the appropriate culture medium. siRNA-mediated knockdown was performed as previously described (Young et al., 2008) with Dicer substrate siRNA duplexes from Integrated DNA Technologies and Dharmafect1 transfection reagent according to the supplier’s protocol. We used FHDC1 siRNA duplex 1, 5′-GCUAUAGCACCAAAGAGAAAUUCCT-3′ and 5′-AGGAAUUUCUCUUUGGUGCUAUAGCAU-3′ (Young et al., 2008); duplex 2, 5′-CCAUCGUAGAGGAUAUCUATT-3′ and 5′-UAGAUAUCCUCUACGAUGGAC-3′; and GM130 siRNA duplex, 5′-GCAUGUCAAGAAAGAGCUAGCCCGG-3′ and 5′-CCGGGCUAGCUCUUUCUUGACAUGCUG-3′. Efficient knockdown of FHDC1 was obtained by performing the initial transfection, followed by a repeat transfection 48 h later. Cells were fixed 48 h after the second transfection. For FHDC1 expression in GM130 knockdown cells, the cells were transfected with siRNA duplex, replated after 48 h, and transfected with the appropriate plasmid the next day. Cells were fixed 96 h after the initial siRNA transfection.
Cells were treated with nocodazole for 2 h at 37°C at a final concentration of 2.5 μg/ml. After treatment, cells were washed five times in ice-cold complete medium and 37°C medium and allowed to recover for 20, 40, 60, and 120 min before fixation in methanol. Cells were treated with brefeldin A for 5, 10, 15, 10, and 45 min at a final concentration of 5.0 μg/ml, recovered by washing twice in complete medium, and fixed 5, 15, 30, 60, 90, and 120 min after inhibitor removal. In combined nocodazole/brefeldin A treatments, cells were incubated with nocodazole (2.5 μg/ml) and brefeldin A (5.0 μg/ml) for 2 h and then washed with ice-cold medium with or without brefeldin A, allowed to recover at 37°C, and then fixed in methanol at 20, 40, 60, and 120 min after removal of nocodazole. For latrunculin B treatment, cells were treated with the drug (2 μM) for 120 min before fixation. Cells were treated with 300 nM centrinone for 14 d to induce centrosome depletion, as previously described (Wong et al., 2015).
Cell-based assays
Scratch wounds were created in confluent cells using a sterile P200 pipette tip. Three wounds were created per coverslip, and cells were fixed in methanol at 0, 1, 2, 4, and 6 h after wounding. For spreading-cell experiments, cells were trypsinized, reseeded at 19,000 cells/cm2, and fixed in methanol 1, 2, and 4 h after plating.
The Golgi was scored as “dispersed” when the ribbon was fragmented into >30 units with an average fragment size of ≤1 μm and there was loss of polarized distribution of the fragments. The Golgi was scored as “intermediate” if either it was fragmented but still had an asymmetrical perinuclear position or the ribbon was intact (≤25 subunits, ≥2 μm in size) but perinuclear polarity was lost. An intact ribbon obviously located to one side of the nucleus was scored as “normal.” To control for cell-cycle effects on Golgi morphology, we included a separate mCherry-transfected control sample as a comparator. The initial quantification of the effects of FHDC1 overexpression on Golgi morphology were confirmed by a second observer working blind with respect to the identity of the sample.
SRF reporter gene assays were performed as in Copeland et al. (2007). Briefly, 50 ng of the SRF reporter p3D.ALuc and 0.25 μg of the transfection control reporter pMLV-LacZ were transfected for each sample. Cells transfected with 50 ng of pEF-SRF.VP16 were included as a positive control standard and set to 100% in each reporter gene experiment. After transfection, cells were incubated overnight in DMEM plus 0.5% DBS and harvested in reporter gene lysis buffer (Promega).
To monitor VSV-G trafficking, cells were transfected with 0.25 μg of pCDM8.1-VSVG.ts045-GFP (Presley et al., 1997) as described. After 16 h, transfected cells were shifted to 42°C for 6 h. Cycloheximide (20 μg/ml final concentration) was added 5 min before the shift to 32°C. The temperature shift was performed by changing to 32°C medium also containing cycloheximide. Cells were fixed in methanol at 30, 60, and 90 min after the 32°C shift.
Immunofluorescence
Cells were prepared for immunofluorescence as in Young et al. (2008). Briefly, cells cultured on acid-washed glass coverslips were fixed for 10 min directly in 2 ml of ice-cold methanol or 4% paraformaldehyde freshly prepared in PHEM (60 mM PIPES, 21 mM HEPES, 10 mM EGTA, 2 mM MgCl2, pH 7.0) buffer (Schliwa and van Blerkom, 1981). After fixation, the cells were permeabilized and blocked for 20 min in 0.3% Triton X-100 and 5% DBS in 1× phosphate-buffered saline (PBS). The coverslips were washed in 1× PBS and incubated with the appropriate primary antibody in 0.03% Triton X-100 and 5% DBS in 1× PBS for 1 h at room temperature. The coverslips were washed three times in 1× PBS and then incubated with secondary antibody in the same solution for 1 h at room temperature. After being washed in 1× PBS, the coverslips were mounted in Vectashield with 4′,6-diamidino-2-phenylindole (DAPI) and sealed with nail polish.
Microscopy
All microscopy was performed on a Zeiss AXIO Imager.Z1 with a Zeiss Apotome.2 structured illumination system for optical sectioning and using a 63× (numerical aperture [NA] 1.4) oil immersion lens or a 40× (NA 0.75) dry objective and a Zeiss AxioCam HRm camera (60N-C 1′′ 1.0X 426114) controlled with AxioVision (release 4.8.2; Zeiss). Coverslips were mounted in Vectashield (Vector Labs) with or without DAPI. Cy5 and Alexa Fluor 488 and 594 secondary antibodies were from Jackson ImmunoResearch. Figures were prepared in Adobe Photoshop and Adobe Illustrator.
Supplementary Material
Acknowledgments
We thank Thea Worthylake, Jocelyn-Anne Beckstead, and Chelsea Smallwood for technical assistance with preliminary experiments. We also thank Paul Melançon, Laurence Pelletier, Angela Andersen, and Laura Trinkle-Mulcahy for support and useful advice. This work was initiated with support from Operating Grant G-13-0003059 from the Heart and Stroke Foundation of Canada and continued with support from Grant 102616 from the Canadian Institutes of Health Research.
Abbreviations used:
- BFA
brefeldin A
- ER
endoplasmic reticulum
- FH1
formin homology 1
- FH2
formin homology 2
- GFP
green fluorescent protein
- MT
microtubules
- MTBD
microtubule-binding domain.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-02-0070) on November 12, 2015.
REFERENCES
- Bartolini F, Gundersen GG. Formins and microtubules. Biochim Biophys Acta. 2010;1803:164–173. doi: 10.1016/j.bbamcr.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolini F, Moseley JB, Schmoranzer J, Cassimeris L, Goode BL, Gundersen GG. The formin mDia2 stabilizes microtubules independently of its actin nucleation activity. J Cell Biol. 2008;181:523–536. doi: 10.1083/jcb.200709029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisel B, Wang Y, Wei JH, Xiang Y, Tang D, Miron-Mendoza M, Yoshimura S, Nakamura N, Seemann J. ERK regulates Golgi and centrosome orientation towards the leading edge through GRASP65. J Cell Biol. 2008;182:837–843. doi: 10.1083/jcb.200805045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt JK. The role of microtubule-based motor proteins in maintaining the structure and function of the Golgi complex. Biochim Biophys Acta. 1998;1404:113–126. doi: 10.1016/s0167-4889(98)00052-4. [DOI] [PubMed] [Google Scholar]
- Campellone KG, Webb NJ, Znameroski EA, Welch MD. WHAMM is an Arp2/3 complex activator that binds microtubules and functions in ER to Golgi transport. Cell. 2008;134:148–161. doi: 10.1016/j.cell.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabin-Brion K, Marceiller J, Perez F, Settegrana C, Drechou A, Durand G, Pous C. The Golgi complex is a microtubule-organizing organelle. Mol Biol Cell. 2001;12:2047–2060. doi: 10.1091/mbc.12.7.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesarone MA, DuPage AG, Goode BL. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat Rev Mol Cell Biol. 2010;11:62–74. doi: 10.1038/nrm2816. [DOI] [PubMed] [Google Scholar]
- Cole NB, Sciaky N, Marotta A, Song J, Lippincott-Schwartz J. Golgi dispersal during microtubule disruption: regeneration of Golgi stacks at peripheral endoplasmic reticulum exit sites. Mol Biol Cell. 1996;7:631–650. doi: 10.1091/mbc.7.4.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colon-Franco JM, Gomez TS, Billadeau DD. Dynamic remodeling of the actin cytoskeleton by FMNL1gamma is required for structural maintenance of the Golgi complex. J Cell Sci. 2011;124:3118–3126. doi: 10.1242/jcs.083725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland SJ, Green BJ, Burchat S, Papalia GA, Banner D, Copeland JW. The diaphanous inhibitory domain/diaphanous autoregulatory domain interaction is able to mediate heterodimerization between mDia1 and mDia2. J Biol Chem. 2007;282:30120–30130. doi: 10.1074/jbc.M703834200. [DOI] [PubMed] [Google Scholar]
- Corthesy-Theulaz I, Pauloin A, Pfeffer SR. Cytoplasmic dynein participates in the centrosomal localization of the Golgi complex. J Cell Biol. 1992;118:1333–1345. doi: 10.1083/jcb.118.6.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deakin NO, Turner CE. Paxillin inhibits HDAC6 to regulate microtubule acetylation, Golgi structure, and polarized migration. J Cell Biol. 2014;206:395–413. doi: 10.1083/jcb.201403039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dippold HC, Ng MM, Farber-Katz SE, Lee SK, Kerr ML, Peterman MC, Sim R, Wiharto PA, Galbraith KA, Madhavarapu S, et al. GOLPH3 bridges phosphatidylinositol-4-phosphate and actomyosin to stretch and shape the Golgi to promote budding. Cell. 2009;139:337–351. doi: 10.1016/j.cell.2009.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efimov A, Kharitonov A, Efimova N, Loncarek J, Miller PM, Andreyeva N, Gleeson P, Galjart N, Maia AR, McLeod IX, et al. Asymmetric CLASP-dependent nucleation of noncentrosomal microtubules at the trans-Golgi network. Dev Cell. 2007;12:917–930. doi: 10.1016/j.devcel.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egea G, Lazaro-Dieguez F, Vilella M. Actin dynamics at the Golgi complex in mammalian cells. Curr Opin Cell Biol. 2006;18:168–178. doi: 10.1016/j.ceb.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Farber-Katz SE, Dippold HC, Buschman MD, Peterman MC, Xing M, Noakes CJ, Tat J, Ng MM, Rahajeng J, Cowan DM, et al. DNA damage triggers Golgi dispersal via DNA-PK and GOLPH3. Cell. 2014;156:413–427. doi: 10.1016/j.cell.2013.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guet D, Mandal K, Pinot M, Hoffmann J, Abidine Y, Sigaut W, Bardin S, Schauer K, Goud B, Manneville JB. Mechanical role of actin dynamics in the rheology of the Golgi complex and in Golgi-associated trafficking events. Curr Biol. 2014;24:1700–1711. doi: 10.1016/j.cub.2014.06.048. [DOI] [PubMed] [Google Scholar]
- Gundersen GG, Gomes ER, Wen Y. Cortical control of microtubule stability and polarization. Curr Opin Cell Biol. 2004;16:106–112. doi: 10.1016/j.ceb.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Gurel PS, Hatch AL, Higgs HN. Connecting the cytoskeleton to the endoplasmic reticulum and Golgi. Curr Biol. 2014;24:R660–R672. doi: 10.1016/j.cub.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs HN. Formin proteins: a domain-based approach. Trends Biochem Sci. 2005;30:342–353. doi: 10.1016/j.tibs.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Hurtado L, Caballero C, Gavilan MP, Cardenas J, Bornens M, Rios RM. Disconnecting the Golgi ribbon from the centrosome prevents directional cell migration and ciliogenesis. J Cell Biol. 2011;193:917–933. doi: 10.1083/jcb.201011014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C. The tubulin code: molecular components, readout mechanisms, and functions. J Cell Biol. 2014;206:461–472. doi: 10.1083/jcb.201406055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasmin BJ, Changeux JP, Cartaud J. Compartmentalization of cold-stable and acetylated microtubules in the subsynaptic domain of chick skeletal muscle fibre. Nature. 1990;344:673–675. doi: 10.1038/344673a0. [DOI] [PubMed] [Google Scholar]
- Kisurina-Evgen’eva OP, Onishchenko GE. Organelle redistribution during PK cell spreading in normal conditions and in the presence of sodium azide [in Russian] Tsitologiia. 2004;46:967–978. [PubMed] [Google Scholar]
- Kodani A, Kristensen I, Huang L, Sutterlin C. GM130-dependent control of Cdc42 activity at the Golgi regulates centrosome organization. Mol Biol Cell. 2009;20:1192–1200. doi: 10.1091/mbc.E08-08-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodani A, Sutterlin C. The Golgi protein GM130 regulates centrosome morphology and function. Mol Biol Cell. 2008;19:745–753. doi: 10.1091/mbc.E07-08-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavieu G, Dunlop MH, Lerich A, Zheng H, Bottanelli F, Rothman JE. The Golgi-ribbon structure facilitates anterograde transport of large cargoes. Mol Biol Cell. 2014;25:3028–3036. doi: 10.1091/mbc.E14-04-0931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaro-Dieguez F, Jimenez N, Barth H, Koster AJ, Renau-Piqueras J, Llopis JL, Burger KN, Egea G. Actin filaments are involved in the maintenance of Golgi cisternae morphology and intra-Golgi pH. Cell Motil Cytoskeleton. 2006;63:778–791. doi: 10.1002/cm.20161. [DOI] [PubMed] [Google Scholar]
- Magdalena J, Millard TH, Etienne-Manneville S, Launay S, Warwick HK, Machesky LM. Involvement of the Arp2/3 complex and Scar2 in Golgi polarity in scratch wound models. Mol Biol Cell. 2003;14:670–684. doi: 10.1091/mbc.E02-06-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia AR, Zhu X, Miller P, Gu G, Maiato H, Kaverina I. Modulation of Golgi-associated microtubule nucleation throughout the cell cycle. Cytoskeleton (Hoboken) 2013;70:32–43. doi: 10.1002/cm.21079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millarte V, Farhan H. The Golgi in cell migration: regulation by signal transduction and its implications for cancer cell metastasis. ScientificWorldJournal. 2012;2012:498278. doi: 10.1100/2012/498278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller PM, Folkmann AW, Maia AR, Efimova N, Efimov A, Kaverina I. Golgi-derived CLASP-dependent microtubules control Golgi organization and polarized trafficking in motile cells. Nat Cell Biol. 2009;11:1069–1080. doi: 10.1038/ncb1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis TE, Warren G. Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol. 1995;131:1715–1726. doi: 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Wei JH, Seemann J. Modular organization of the mammalian Golgi apparatus. Curr Opin Cell Biol. 2012;24:467–474. doi: 10.1016/j.ceb.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petry S, Vale RD. Microtubule nucleation at the centrosome and beyond. Nat Cell Biol. 2015;17:1089–1093. doi: 10.1038/ncb3220. [DOI] [PubMed] [Google Scholar]
- Preisinger C, Short B, De Corte V, Bruyneel E, Haas A, Kopajtich R, Gettemans J, Barr FA. YSK1 is activated by the Golgi matrix protein GM130 and plays a role in cell migration through its substrate 14–3-3zeta. J Cell Biol. 2004;164:1009–1020. doi: 10.1083/jcb.200310061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- Ramabhadran V, Korobova F, Rahme GJ, Higgs HN. Splice variant-specific cellular function of the formin INF2 in maintenance of Golgi architecture. Mol Biol Cell. 2011;22:4822–4833. doi: 10.1091/mbc.E11-05-0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed NA, Cai D, Blasius TL, Jih GT, Meyhofer E, Gaertig J, Verhey KJ. Microtubule acetylation promotes kinesin-1 binding and transport. Curr Biol. 2006;16:2166–2172. doi: 10.1016/j.cub.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Rios RM. The centrosome-Golgi apparatus nexus. Philos Trans R Soc Lond B Biol Sci. 2014;369 doi: 10.1098/rstb.2013.0462. 20130462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivero S, Cardenas J, Bornens M, Rios RM. Microtubule nucleation at the cis-side of the Golgi apparatus requires AKAP450 and GM130. EMBO J. 2009;28:1016–1028. doi: 10.1038/emboj.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo R, Parashuraman S, Mirabelli P, Puri C, Lucocq J, Luini A. The dynamics of engineered resident proteins in the mammalian Golgi complex relies on cisternal maturation. J Cell Biol. 2013;201:1027–1036. doi: 10.1083/jcb.201211147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez OC, Schaefer AW, Mandato CA, Forscher P, Bement WM, Waterman-Storer CM. Conserved microtubule-actin interactions in cell movement and morphogenesis. Nat Cell Biol. 2003;5:599–609. doi: 10.1038/ncb0703-599. [DOI] [PubMed] [Google Scholar]
- Rogalski AA, Singer SJ. Associations of elements of the Golgi apparatus with microtubules. J Cell Biol. 1984;99:1092–1100. doi: 10.1083/jcb.99.3.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roubin R, Acquaviva C, Chevrier V, Sedjai F, Zyss D, Birnbaum D, Rosnet O. Myomegalin is necessary for the formation of centrosomal and Golgi-derived microtubules. Biol Open. 2013;2:238–250. doi: 10.1242/bio.20123392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliwa M, van Blerkom J. Structural interaction of cytoskeletal components. J Cell Biol. 1981;90:222–235. doi: 10.1083/jcb.90.1.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutterlin C, Colanzi A. The Golgi and the centrosome: building a functional partnership. J Cell Biol. 2010;188:621–628. doi: 10.1083/jcb.200910001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston SF, Kulacz WA, Shaikh S, Lee JM, Copeland JW. The ability to induce microtubule acetylation is a general feature of formin proteins. PLoS One. 2012;7:e48041. doi: 10.1371/journal.pone.0048041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyberg J, Moskalewski S. Microtubules and the organization of the Golgi complex. Exp Cell Res. 1985;159:1–16. doi: 10.1016/s0014-4827(85)80032-x. [DOI] [PubMed] [Google Scholar]
- Thyberg J, Moskalewski S. Relationship between the Golgi complex and microtubules enriched in detyrosinated or acetylated alpha-tubulin: studies on cells recovering from nocodazole and cells in the terminal phase of cytokinesis. Cell Tissue Res. 1993;273:457–466. doi: 10.1007/BF00333700. [DOI] [PubMed] [Google Scholar]
- Vinogradova T, Miller PM, Kaverina I. Microtubule network asymmetry in motile cells: role of Golgi-derived array. Cell Cycle. 2009;8:2168–2174. doi: 10.4161/cc.8.14.9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradova T, Paul R, Grimaldi AD, Loncarek J, Miller PM, Yampolsky D, Magidson V, Khodjakov A, Mogilner A, Kaverina I. Concerted effort of centrosomal and Golgi-derived microtubules is required for proper Golgi complex assembly but not for maintenance. Mol Biol Cell. 2012;23:820–833. doi: 10.1091/mbc.E11-06-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhang C, Qi RZ. A newly identified myomegalin isoform functions in Golgi microtubule organization and ER-Golgi transport. J Cell Sci. 2014;127:4904–4917. doi: 10.1242/jcs.155408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YL, Anzola JV, Davis RL, Yoon M, Motamedi A, Kroll A, Seo CP, Hsia JE, Kim SK, Mitchell JW, et al. Cell biology. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science. 2015;348:1155–1160. doi: 10.1126/science.aaa5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Moseley JB, Sagot I, Poy F, Pellman D, Goode BL, Eck MJ. Crystal structures of a Formin Homology-2 domain reveal a tethered dimer architecture. Cell. 2004;116:711–723. doi: 10.1016/s0092-8674(04)00210-7. [DOI] [PubMed] [Google Scholar]
- Yadav S, Puri S, Linstedt AD. A primary role for Golgi positioning in directed secretion, cell polarity, and wound healing. Mol Biol Cell. 2009;20:1728–1736. doi: 10.1091/mbc.E08-10-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav S, Puthenveedu MA, Linstedt AD. Golgin160 recruits the dynein motor to position the Golgi apparatus. Dev Cell. 2012;23:153–165. doi: 10.1016/j.devcel.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KG, Thurston SF, Copeland S, Smallwood C, Copeland JW. INF1 is a novel microtubule-associated formin. Mol Biol Cell. 2008;19:5168–5180. doi: 10.1091/mbc.E08-05-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Kaverina I. Golgi as an MTOC: making microtubules for its own good. Histochem Cell Biol. 2013;140:361–367. doi: 10.1007/s00418-013-1119-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman Y, Alieva NO, Miserey-Lenkei S, Lichtenstein A, Kam Z, Sabanay H, Bershadsky A. Involvement of the Rho-mDia1 pathway in the regulation of Golgi complex architecture and dynamics. Mol Biol Cell. 2011;22:2900–2911. doi: 10.1091/mbc.E11-01-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.