

Abstract
Purpose
The analysis of MET gene copy number (CN) has been considered to be a potential biomarker to predict the response to MET-targeted therapies in various cancers. However, the current standard methods to determine MET CN are SNP 6.0 in the genomic DNA of cancer cell lines and fluorescence in situ hybridization (FISH) in tumor models, respectively, which are costly and require advanced technical skills and result in relatively subjective judgments. Therefore, we employed a novel method, droplet digital PCR (ddPCR), to determine the MET gene copy number with high accuracy and precision.
Methods
The genomic DNA of cancer cell lines or tumor models were tested and compared with the MET gene CN and MET/CEN-7 ratio determined by SNP 6.0 and FISH, respectively.
Results
In cell lines, the linear association of the MET CN detected by ddPCR and SNP 6.0 is strong (Pearson correlation = 0.867). In tumor models, the MET CN detected by ddPCR was significantly different between the MET gene amplification and non-amplification groups according to FISH (mean: 15.4 vs 2.1; P = 0.044). Given that MET gene amplification is defined as MET CN >5.5 by ddPCR, the concordance rate between ddPCR and FISH was 98.0%, and Cohen's kappa coefficient was 0.760 (95% CI, 0.498–1.000; P <0.001).
Conclusions
The results demonstrated that the ddPCR method has the potential to quantify the MET gene copy number with high precision and accuracy as compared with the results from SNP 6.0 and FISH in cancer cell lines and tumor samples, respectively.
Introduction
In normal physiological functions, MET is expressed in cells of epithelial origin, where it plays an essential role in cell growth and homeostasis [1]. However, aberrant MET signaling has been observed in multiple human cancers, including hepatic and gastric cancers [2–6]. MET gene amplification has been reported to be correlated with poor prognosis in patients with GC [7–9] and may be used as a potential biomarker to estimate the disease prognosis or predictive response to MET inhibitors in clinical trials. Thus, it is important to develop an accurate and optimized platform to determine the MET gene copy number prior to MET-targeted therapy. In routine studies, SNP 6.0 and FISH are the standard methods to evaluate the MET copy number of cancer cell lines in vitro and tumor samples in vivo, respectively. However, these methods have many limitations, including the advanced technical skills required high costs and the need for experienced experts to analyze the tumor samples, which results in variable results between labs. For example, FISH analysis is usually performed on formalin-fixed, paraffin-embedded (FFPE) tissues, which requires a number of complex processes, such as fixation and immunohistological staining, and may cause genomic DNA damage and fracture. These issues increase the probability of false negative results because of the low quality and quantity of DNA. Therefore, the detection of gene amplifications is challenging because of the low sensitivity of these approaches.
The droplet digital PCR (ddPCR) is a method that can absolutely quantify the MET copy number without need for standard curves. In a typical digital PCR, the sample is randomly distributed into discrete partitions such that some contain no nucleic acid template and others contain one or more template copies. The partitions are PCR amplified to end point and then read using a droplet reader to determine the fraction of positive partitions based on fluorescence amplitude, from which the absolute concentration of the target or reference DNA is estimated statistically by modeling as a Poisson distribution. Therefore, ddPCR is an end-point measurement that enables to quantify nucleic acids without the need for standard curves, external calibrators and endogenous controls [10].
In this study, we sought to employ ddPCR assays to absolutely quantify the MET copy number in cancer cell lines and tumor samples and compared our results with those obtained using SNP 6.0 and FISH for the same samples.
Materials and Methods
Cell lines, PDX samples, and extraction of genomic DNA
Eight human GC and thirty-eight HCC cell lines were purchased from five organizations: the American Type Culture Collection (ATCC), Japanese Collection of Research Bioresources (JCRB), Korean Cell Line Bank (KCLB), Shanghai Institutes of Biological Sciences, CAS (SIBS), and Zhongshan Hospital Fudan University (ZHFU) (see Table 1). The cell lines were routinely cultured in 96-well plates in ATCC’s recommended growth medium at 37°C, 5% CO2 and 95% humidity. Genomic DNA was extracted from the cancer cell lines with the TIANampGenomic DNA Kit (Tiangen, Cat: DP304) according to the manufacturer’s instructions. One hundred and fifty-six FFPE of patient-derived xenograft (PDX) models (including 116 GC and 39 HCC models) were obtained from the Shanghai LIDE Biotech Company. For the PDX model samples, genomic DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen, Cat#: 69509) according to the manufacturer’s protocol. The DNA concentration was determined with a NanoDrop spectrophotometer (Thermo), and digested with the HaeⅢ enzyme (NEB, Cat#: R0108S) at 37°C for 1 h. The digested DNA sample was diluted 10-fold with autoclaved Millipore H2O and stored in a -20°C freezer.
Table 1. Comparison of MET gene amplification measured by ddPCR versus FISH in gDNA of FFPE tissue.
| gDNA of FFPE samples | ddPCR | ||
|---|---|---|---|
| FISH | N | P | Total |
| N | 147 | 3 | 150 |
| P | 0 | 5 | 5 |
| Total | 147 | 8 | 155 |
DDPCR for the determination of MET CN
The TaqMan PCR reaction mixture was assembled in a final volume of 20 μL with 2x Supermix, 20x primers and 20x probes and 20 ng of the genomic DNA as the template. Each reaction mixture was then loaded into a DG8 cartridge (Bio-Rad) with 70 μL of droplet generation oil to generate a droplet. The droplets from each well were then transferred into a 96-well PCR plate. The plates were heat-sealed and then thermally cycled under the following conditions: 95°C for 10 min (one cycle); 40 PCR cycles of 95°C for 15 seconds and 60°C for 1 min; and a hold at 4°C. After PCR, the plates were placed on a QX200 droplet reader (Bio-Rad) that analyzed the droplets of each well of the plate and quantified the target DNA. The PCR data were analyzed using QuantaSoft version 1.7.4.0917 (Bio-Rad) to determine the copy number variation (CNV). The 20x reference assay for AP3B1 included primers targeting the centromere loci on Chromosome 5 and a probe labeled with a HEX fluorescent signal (Bio-Rad). The 20 x CNV PCR assay for MET included primers targeting the region from intron 20-exon 21 on Chromosome 7, and the probe was labeled with a FAM fluorescent signal (Life Technologies, cat #Hs02884964_cn). Each well was replicated for all of the samples. The MET copy number was calculated as the ratio of the concentrations of MET and AP3B1 and multiplied by two. Each data for MET CN using ddPCR represents two or three merged technical replicate wells with no template control (NTC) as a negative control.
SNP 6.0 assay
Among the 46 cell lines, the SNP 6.0 raw data for 35 of the cell lines were downloaded from the CCLE project (http://www.broadinstitute.org/ccle/home), and the raw data for the other 11 cell lines were collected using the Affymetrix Genome-Wide Human SNP Array 6.0 platform, including BEL7402, HCCC-810, NOZ, OCUG-1, OZ, QGY7701, QGY7703, SMMC7721, SNU354, SNU368, and SNU739. All of the raw data were processed using PICNIC software and presented as MET copies.
MET FISH
MET gene amplification was analyzed by FISH using the Dako MET/CEN-7 IQISH Probe Mix (RUO). The CEN-7 centromere probe was used as a reference control, according to the manufacturer’s instructions. The formalin-fixed, paraffin-embedded specimens (cell pellets) were sectioned and then subjected to deparaffinization and rehydration. H&E staining was performed first. The heat pretreatment was performed in pretreatment solution in a microwave oven for 3 min and 50 sec, cooled to RT over 15 min, and then washed. The sections were digested in RTU pepsin for 6 min at 37°C (the time can be adjusted for different section thicknesses). After washing, the sections were completely dehydrated. The sections containing the probe were denatured at 66°C for 10 min and then hybridized at 45°C for 1–2 h. After stringent washing, the sections were dehydrated, mounted in medium with DAPI, and stored in the dark at 4°C for 15 min before reading. A fluorescence microscope and appropriate filters were used to scan and identify the relevant tumor area. The signals of the red MET gene and green CEN-7 gene were counted in 20 tumor nuclei in a minimum of two areas to determine the MET/CEN-7 ratio. The FISH scoring was defined as follows: ratio ≤2.0: MET amplification was not observed; ratio >2.0: MET amplification was observed. If the ratio was borderline (1.8–2.2), an additional 20 nuclei were counted, and the ratios were recalculated.
Statistical analysis
The valid HCC and GC measurements for MET CN were combined for the statistical analyses. Pearson’s correlations and linear regression were used to evaluate the relationship between ddPCR and SNP 6.0 or FISH. To compare the differences in MET CN analyzed by ddPCR between the gene amplification and non-amplification groups analyzed by FISH, t-tests were used. The consistency of gene amplification based on ddPCR and FISH was evaluated according to the concordance rate and Cohen's kappa coefficient. The analyses were performed with SAS 9.4 software.
Results
Comparison of the MET CN test between ddPCR and SNP 6.0 in cancer cell lines
Genomic DNA was obtained from 8 GC and 38 HCC cell lines for MET copy number detection, and the results of the ddPCR assay were compared to those of SNP 6.0 to determine whether ddPCR can replace the standard molecular biology techniques. The results were shown in S1 Table. We determined the MET copy numbers of cell lines in vitro using an Affymetrix microarray. Next, we used ddPCR to test the MET copy number in the same samples by normalizing to the AP3B2 reference gene. We observed that the cell lines with high MET copy numbers according to SNP 6.0 also had high ddPCR measurements. The linear association for MET copy number measurements between ddPCR and SNP 6.0 is strong based on Pearson’s correlation (r = 0.867; P <0.001). Moreover, the slope and intercept of MET CN by SNP 6.0 to ddPCR in the linear regression were significant (P <0.001) with a value equal to 1.996 (standard errors of estimates = 0.177) and -4.159 (standard errors of estimates = 0.752), respectively (Fig 1).
Fig 1. Measurement of MET copy number in cancer cell lines using ddPCR versus SNP 6.0.

The data is based on a linear regression model that adjusts for SNP 6.0 with intercept. Solid line indicates fitting curve; gray box represents 95% confidence limits; dashed line depicts 95% prediction limits. Each data for MET CN using ddPCR represents two or three merged technical replicate wells with no template control (NTC) as a negative control.
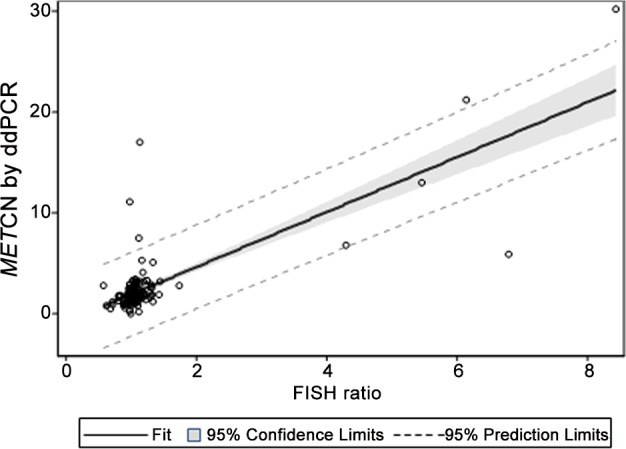
MET gene amplification analysis in GC and HCC PDX models
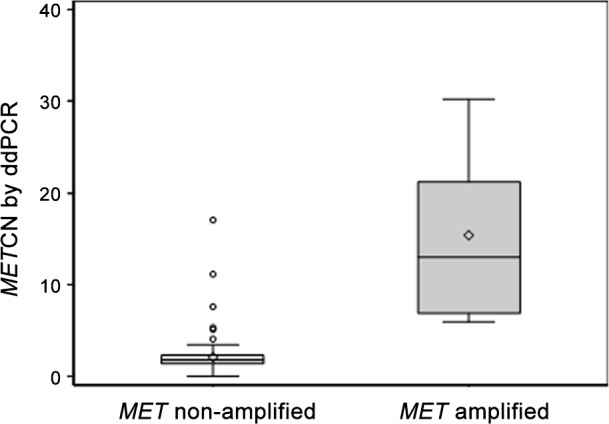
Because ddPCR could accurately evaluate the MET copy number in cancer cell lines, we then determined whether ddPCR could detect the presence of MET amplification and compared the results to those obtained from FISH. Many publications have reported the application of digital PCR measurement of gene copy number in FFPE tissues compared to FISH [11,12]. Here we analyzed 116 GC and 39 HCC PDX tumor models. The raw data are summarized in S2 Table. MET copy number analyzed by ddPCR is significantly elevated in the MET amplification group (mean, 15.4) compared to the MET non-amplification group (mean, 2.1) analyzed by FISH using a t-test with unequal variance (P = 0.044) (Fig 2). The linear association between the MET CN analyzed by ddPCR and the MET/CEN-7 ratio analyzed by FISH was relatively strong, as evaluated by Pearson’s correlation (r = 0.782; P <0.001). Furthermore, using linear regression, the slope and intercept of the ratio of FISH to MET CN by ddPCR were significant (P < 0.001), with a value equal to 2.723 (standard errors of estimates = 0.176) and -0.803 (standard errors of estimates = 0.272), respectively (Fig 3). Generally, the results suggested that the MET copy number values obtained by ddPCR could distinguish between the MET non-amplification and amplification groups defined by FISH and that ddPCR can be used to measure the MET copy number in PDX models.
Fig 2. Distribution of MET CN measured by ddPCR in MET amplification and non-amplification groups by FISH.
P-value was based on t-test assuming difference variances of each group. Each data for MET CN using ddPCR represents two or three merged technical replicate wells with no template control (NTC) as a negative control.
Fig 3. Measurement of MET copy number in PDX models using ddPCR versus FISH.
The data is based on a linear regression model that adjusts for FISH ratio with intercept. Solid line indicates fitting curve; gray box represents 95% confidence limits; dashed line depicts 95% prediction limits. Each data for MET CN using ddPCR represents two or three merged technical replicate wells with no template control (NTC) as a negative control.
The concordance rate and Cohen's kappa coefficient were used to observe the consistency of gene amplification between ddPCR and FISH and evaluate whether there was a good cut-off value for defining the ddPCR-based gene amplification compared to the FISH ratio >2.0. Among the cut-off candidates, MET CN >5.5 by ddPCR had the highest concordance rate (98%) and Cohen's kappa coefficient (κ = 0.760) (95% CI, 0.498–1.000; P <0.001). Notably, among the 155 valid PDX samples, although most of samples had the consistency of MET gene amplification between ddPCR and FISH, only 3 PDX models (GAPF528, GAPF509, and GAPF012) that were positive for MET CN by ddPCR did not show a detectable FISH ratio >2.0 (Table 1).
Discussion
Human genomes exhibit segmental copy number variation (CNV) at thousands of loci [13]. MET gene amplification has been described in gastric cancer (GC) and hepatocellular carcinoma (HCC), which results in dysregulation of MET signaling and is associated with clinical prognosis and poor outcome [6]. The aberrant MET signaling has been regarded as a robust target in the anti-cancer therapy. Thus, it is conceivable that a number of studies [7–9] have demonstrated MET gene amplification as a potential biomarker to estimate patients with cancers who will benefit from the treatment with selective MET inhibitors or antibodies. So far, there has been no standardized method to validate MET gene amplification or MET copy number.
SNP 6.0 and FISH are the methods that are conventionally used to evaluate MET copy number in cancer cell lines in vitro and tumor samples in vivo, respectively. However, a number of issues can complicate the detection of gene amplification. First, these methods are very costly and require advanced technical skills, and thus, informatics technicians or experienced pathologists must analyze the data and make subjective judgments. Second, in vivo tumor samples, such as FFPE tissues, are always tested by FISH and use long fragment probes, resulting in variations in the gene copy number detection depending on the probes targeting different gene loci. Thus, we sought to identify a method with simple technical requirements to more accurately detect the MET copy number.
In this study, we evaluated the feasibility of using a novel method of ddPCR to quantify the MET copy number or assess gene amplification from cancer samples consisting of cell lines or FFPE tumors. The results showed that ddPCR could determine the MET status in tumor samples. We found there was a positive correlation between the MET copy numbers detected by ddPCR and SNP 6.0 according to Pearson’s correlations (r = 0.867; P <0.001). Although microarray technologies are valuable approaches for CNV determination, they have limited dynamic range and are expensive for high-throughput screening in population studies [10]. Because of the limitations of SNP 6.0 approach in accurately measuring copy numbers greater than 4 and lack of sensitivity and resolution, ddPCR can be developed to measure high-copy CNV more accurately in a large numbers of samples, as described before [13]. Based on the assessment of MET amplification in 155 FFPE PDX tumors, ddPCR can reliably reflect the MET amplification status compared to FISH, with a 98% concordance rate. Moreover, there is significant difference of MET CNV detected by ddPCR between FISH-positive and FISH-negative groups, providing a highly significant proof of principle.
Notably, although in most of 155 FFPE PDX tumors, status of MET detected by ddPCR were comparable to those detected by FISH, three FFPE tumors whose MET copy number values were high (CN >5.5) in ddPCR did not show gene amplification by FISH (MET/CEN-7 ratio >2.0). The reason for the discordance of the MET amplification between ddPCR and FISH may be the underestimation of MET amplification by FISH or overestimation of MET amplification by digital PCR. On the one hand, the presence of MET gene amplification could be masked because of characteristic of incomplete hybridization of long fragment probes used in FISH, resulting in non-specific identification of MET amplification. In particular, the variation in the MET amplification could be ascribed to the probe targeting different genomic loci detected by FISH. On the other hand, the fixation process used for the FFPE tissues could cause DNA fragmentation, damage or fusion, thus leading to a false-negative result. Additionally, considering that loss of chromosome frequently occurred in many cancers can result in genomic chromosomal instability [14–16], it implies that the loss of the reference gene loci in the tumor may cause a false-positive elevated ratio of MET:AP3B1 in ddPCR, rather than high MET copy number.
Of note, MET CN of another PDX sample (GAPF101) is 0.00022 with no successful CN call by ddPCR, extremely lower than MET/CEN-7 ratio of 1 by FISH. It implies that the deletion or damage of MET might occur in partial region of MET which were targeted by different probes employed in ddPCR and FISH, thus resulting in the difference of MET status between FISH and ddPCR. It will be intriguing to explore the reasons for the discordance in the future research.
Otherwise, the analysis of tumor samples by FISH requires complex experimental technology and may depend on the subjective judgment of pathologists. The results can vary between different pathologists based on their experiences. In this study, we show that ddPCR is a quantitative method that can absolutely quantify the MET copy number in cancer samples either in vitro or in vivo, without the need for standard curves or endogenous control.
Overall, these data suggest that ddPCR has the potential to detect the MET copy number in cancer cell lines or FFPE DNA and highly correlated with SNP 6.0 or FISH, respectively. MET gene amplification has been described to be associated with tumorigenesis and metastatic progression and is regarded as a biomarker to predict benefit from MET-targeted therapy in various clinical studies [7–9]. Our findings provide the insight that the ddPCR platform may be able to estimate the relationship between the MET status and patient prognosis in clinical studies and help to better predict and screen the patients in the response to MET-targeted therapy.
Supporting Information
(PDF)
(PDF)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This study was funded by Amgen Inc. All authors are employees of Amgen Inc. The funder provided support in the form of salaries for YZ, ET, and ZD, but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific roles of these authors are articulated in the Author Contributions section.
References
- 1.Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol 2010; 11(12): 834–848. 10.1038/nrm3012 [DOI] [PubMed] [Google Scholar]
- 2.Inoue T, Kataoka H, Goto K, Nagaike K, Igami K, Naka D, et al. Activation of c-Met (hepatocyte growth factor receptor) in human gastric cancer tissue. Cancer Sci 2004; 95(10): 803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park WS, Dong SM, Kim SY, Na EY, Shin MS, Pi JH, et al. Somatic mutations in the kinase domain of the Met/hepatocyte growth factor receptor gene in childhood hepatocellular carcinomas. Cancer Res 1999; 59(2): 307–310. [PubMed] [Google Scholar]
- 4.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol 2003; 4(12): 915–925. [DOI] [PubMed] [Google Scholar]
- 5.Maulik G, Shrikhande A, Kijima T, Ma PC, Morrison PT, Salgia R. Role of the hepatocyte growth factor receptor, c-Met, in oncogenesis and potential for therapeutic inhibition. Cytokine Growth Factor Rev 2002; 13(1): 41–59. [DOI] [PubMed] [Google Scholar]
- 6.Smolen GA, Sordella R, Muir B, Mohapatra G, Barmettler A, Archibald H, et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc Natl Acad Sci U S A 2006; 103(7): 2316–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.An X, Wang F, Shao Q, Wang FH, Wang ZQ, Wang ZQ, et al. MET amplification is not rare and predicts unfavorable clinical outcomes in patients with recurrent/metastatic gastric cancer after chemotherapy. Cancer 2014; 120(5): 675–682. [DOI] [PubMed] [Google Scholar]
- 8.Lee HE, Kim MA, Lee HS, Jung EJ, Yang HK, Lee BL, et al. MET in gastric carcinomas: comparison between protein expression and gene copy number and impact on clinical outcome. Br J Cancer 2012; 107(2): 325–333. 10.1038/bjc.2012.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu YJ, Shen D, Yin X, Gavine P, Zhang T, Su X, et al. HER2, MET and FGFR2 oncogenic driver alterations define distinct molecular segments for targeted therapies in gastric carcinoma. Br J Cancer 2014; 110(5): 1169–1178. 10.1038/bjc.2014.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem 2011; 83(22): 8604–8610. 10.1021/ac202028g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang JT, Liu YJ, Wang J, Xu ZG, Yang Y, Shen F, et al. Next generation digital PCR measurement of hepatitis B virus copy number in formalin-fixed paraffin-embedded hepatocellular carcinoma tissue. Clin Chem 2015; 61(1): 290–296. 10.1373/clinchem.2014.230227 [DOI] [PubMed] [Google Scholar]
- 12.Heredia NJ, Belgrader P, Wang S, Koehler R, Regan J, Cosman AM, et al. Droplet Digital PCR quantitation of HER2 expression in FFPE breast cancer samples. Methods 2013; 59(1): S20–23. 10.1016/j.ymeth.2012.09.012 [DOI] [PubMed] [Google Scholar]
- 13.Handsaker RE, Van Doren V, Berman JR, Genovese G, Kashin S, Boettger LM, et al. Large multiallelic copy number variations in humans. Nat Genet 2015; 47(3): 296–303. 10.1038/ng.3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yakicier MC, Legoix P, Vaury C, Gressin L, Tubacher E, Capron F, et al. Identification of homozygous deletions at chromosome 16q23 in aflatoxin B1 exposed hepatocellular carcinoma. Oncogene 2001; 20(37): 5232–5238. [DOI] [PubMed] [Google Scholar]
- 15.Wong CM, Lee JM, Lau TC, Fan ST, Ng IO. Clinicopathological significance of loss of heterozygosity on chromosome 13q in hepatocellular carcinoma. Clin Cancer Res 2002; 8(7): 2266–2272. [PubMed] [Google Scholar]
- 16.Hong SJ, Jeon EJ, Oh JH, Seo EJ, Choi SW, Rhyu MG. The gene-reduction effect of chromosomal losses detected in gastric cancers. BMC Gastroenterol 2010; 10: 138 10.1186/1471-230X-10-138 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF)
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.