

Abstract
Mineralocorticoid receptor (MR) activation by aldosterone may regulate vascular function in health or contribute to vascular dysfunction in cardiovascular disease. Whether the effects are beneficial or detrimental to vascular function appear to be dependent on the integrity of the vascular endothelium and whether the responses are short-term or chronic. Acute modulation of MR activation has resulted in conflicting outcomes on vascular function in young healthy adults. Little is known about the vascular role of aldosterone and MR activation in healthy human aging. The primary objective of this study was to examine whether acute inhibition of MR by the selective antagonist eplerenone, influences vascular function in healthy older adults. We performed a randomized, double-blind, placebo-controlled crossover study in 22 adults (61±1 y; mean ± SE, 53–79 y) who were free from overt clinical cardiovascular disease. We measured brachial artery flow-mediated endothelium-dependent dilation and endothelium-independent dilation to sublingual nitroglycerin (0.4mg) following eplerenone (100 mg/dose, 2 doses, 24 hours between doses) or placebo. In response to acute MR antagonism, flow-mediated dilation decreased by 19% (from 6.9±0.5 to 5.6±0.6 %, P=0.02; placebo vs. eplerenone). Endothelial nitric oxide synthase (eNOS) activity also decreased following MR antagonism based on the ratio of phosphorylated eNOSSer1177 to total eNOS (1.53±0.08 vs. 1.29±0.06, P=0.02). Nitroglycerin-induced dilation and blood pressure were unaffected (nitroglycerin-induced dilation: 21.9±1.9 vs. 21.0±1.5 %, P=0.5 and systolic/diastolic blood pressure: 135/77±4/2 vs. 134/77± 4/2 mmHg, P ≥0.6). In conclusion, acute MR antagonism impairs vascular endothelial function in healthy older adults without influencing vascular smooth muscle responsiveness to exogenous nitric oxide or blood pressure.
Keywords: aging, vascular function, mineralocorticoid receptor, eplerenone
1. Introduction
The mineralocorticoid receptor (MR) is an important regulator of blood pressure by modifying renal sodium retention (Williams 2005). In addition to the classical role of MR in renal epithelial cells, substantial new data support the presence of functional MR in human vascular endothelial and smooth muscle cells and animal studies reveal a role for vascular MR in regulating vessel function in vivo (Barrett Mueller et al. 2015; Caprio et al. 2008; Jaffe and Mendelsohn 2005; McCurley et al. 2012). Activation of extra-renal MRs has been shown to lead to direct vascular effects that may contribute to normal vascular function in health and also to vascular dysfunction in cardiovascular disease (reviewed in (McCurley and Jaffe 2012)). Both aldosterone and cortisol can activate the MR but vascular tissues likely respond to aldosterone due to the expression and function of the cortisol-inactivating enzyme 11-beta-hydroxysteroid dehydrogenase type 2 (11βHSD2), expressed in vascular endothelial and smooth muscle cells (Caprio et al. 2008; Jaffe and Mendelsohn 2005; McCurley et al. 2012).
Data on the acute vascular effects of MR activation by aldosterone in humans are conflicting (McCurley and Jaffe 2012; Toda et al. 2013). The discrepant findings may be explained by the health or disease state of the population under study due to the presence or absence of vascular damage and pre-existing vascular oxidative stress and inflammation. Differences in study design might also be contributing to the divergent results including the vascular bed under investigation, physiological versus pathological doses of aldosterone administration, and concomitant use of other drugs. The effect of MR activation also depends on whether the responses under investigation are short-term or chronic. One approach to study the role of aldosterone activation of MR in vascular function is to experimentally increase aldosterone levels (e.g., aldosterone infusion or fludrocortisone-induced aldosterone excess). However, results based on acute elevations in aldosterone, in the absence of a physiological stimulus to induce these elevations, may not represent what normally occurs under physiological conditions. A second experimental approach is to inhibit MR activation using an MR antagonist. This may be more physiologically relevant because it permits determination of the tonic influence of MR activation on vascular function.
Endothelial dysfunction is one of the earliest signs of atherosclerosis and is characterized by impairment in endothelium-dependent vasodilation (Davignon and Ganz 2004). Healthy endothelial cells produce vasodilators which diffuse to medial smooth muscle cells and activate signaling pathways that induce smooth muscle cells to relax thereby resulting in vasodilation (Davignon and Ganz 2004). Nitric oxide is a major mediator of endothelial-dependent vasorelaxation that is produced by healthy endothelial cells when the enzyme endothelial nitric oxide synthase (eNOS) is activated by phosphorylation at the serine residue 1177 (Ser1177) (Reviewed in (Huang 2003; Landmesser and Drexler 2007)). In the setting of cardiovascular disease, there are extensive data demonstrating that MR contributes to endothelial dysfunction (Abiose et al. 2004; Farquharson and Struthers 2000; Keidar et al. 2004; Rajagopalan et al. 2002; Sartorio et al. 2007; Thai et al. 2006). However, in young healthy adults, the results are inconsistent with some studies demonstrating that aldosterone activation of MR promotes endothelium-dependent nitric oxide-mediated vasodilation (Nietlispach et al. 2007), while other studies demonstrate that MR activation impairs endothelium-dependent dilation (Farquharson and Struthers 2002) or has no effect (Schmidt et al. 2003).
Little is known about the role of vascular MR activation in healthy human aging. We have recently demonstrated that chronic (1 month) administration of an MR antagonist did not significantly change flow-mediated vasodilation but rather influenced endothelial function in an adiposity-dependent manner in healthy older adults (Hwang et al. 2013b). The acute vascular effects of MR antagonism in healthy older adults, have never been explored. Therefore, we conducted a randomized double-blind placebo-controlled crossover study using short-term oral administration of the selective MR antagonist eplerenone (2 doses, 100 mg/dose) in older men and women free from overt clinical cardiovascular disease. The primary objective of this study was to investigate whether inhibition of MR activation acutely influences vascular function in healthy human aging. Endothelial dysfunction may be due to decreased nitric oxide production or to increased nitric oxide degradation due to the presence of oxidative stress (Vita 2011). Endothelial dysfunction also leads to vascular inflammation that contributes to cardiovascular disease (Vita 2011). Thus, to gain preliminary mechanistic insight, we also examined the effect of acute MR antagonism on the levels of blood markers of oxidative stress and inflammation and on the expression levels of endothelial cell proteins that are known to influence vascular function.
2. Methods
2.1. Subjects
Twenty-two older adults (8 men and 14 women), 53 to 79 years of age, were enrolled in this study. Subject recruitment was completed by using flyers and by advertising in newspapers and on the radio. Research volunteers who met one or more of the following criteria were excluded from study participation: age < 50 years, evidence of diabetes, cardiovascular, liver or renal disease, use of antihypertensive or vasoactive medications or hormone replacement therapy, use of antioxidant supplements, use of tobacco products, history of alcohol abuse, recent hospitalization, weight loss or gain greater than 5 pounds in the prior 3 months, participation in exercise training >30 min ≥ 3 times per week and for female volunteers, pre- or peri-menopausal status. To reduce the risk of hyperkalemia that is associated with eplerenone use, subjects were also excluded from the study if their baseline serum potassium was greater than 5.5 mmol/L, serum creatinine was greater than 1.6 mg/dL, or creatinine clearance was less than 30 mL/min.
Based on these exclusion criteria, the enrolled subjects were sedentary, non-smokers and were free from overt cardiovascular and other clinical diseases (e.g., diabetes, liver and renal disease) as assessed by medical history, physical examination, resting ECG, urinalysis, blood chemistries and hematological evaluation. All subjects demonstrated normal ECG and blood pressure responses to a graded exercise test on a treadmill. The exercise testing protocol has previously been described (Christou et al. 2005). Briefly, after a 6–10 minute warm-up, subjects walked at a comfortable speed that corresponded to 70 to 80% of their age-predicted maximal heart rate. The treadmill grade was increased 2.5% every two minutes until volitional exhaustion.
This study was carried out in accordance with the ethical standards of the Declaration of Helsinki and was approved by the Institutional Review Boards of the University of Florida, Texas A&M University, and Scott & White Health System. The purpose, nature, and risks of the study were explained to the volunteers and their written informed consent was obtained prior to participation. No subjects withdrew from the intervention after randomization.
2.2. Study design and general experimental procedures
The current investigation followed a randomized double-blind placebo-controlled crossover design using the MR antagonist eplerenone. Eplerenone and placebo (Consolidated Midland Corporation, Brewster, New York) were administered orally. Based on the 3–6 hour elimination half-life of eplerenone (Ravis et al. 2005), at least 1 week washout was allowed between eplerenone and placebo. We chose to use eplerenone due to its higher selectivity for MR and fewer side effects compared to spironolactone, the other FDA-approved MR antagonist. We selected to use a dose of 100 mg for the following reasons: 1) this dose was reported to be more effective at inhibiting the MR than 25 or 50 mg but equally effective to 200 mg eplerenone (White et al. 2003); 2) it was associated with similar side effects to placebo including similar serum potassium levels (White et al. 2003); and 3) it has previously been administered without titration (Cook et al. 2003; Ravis et al. 2005).
Following oral administration of 100 mg eplerenone, peak plasma concentration is reached within approximately 2 hours and is maintained for several hours (Ravis et al. 2005). In order to study vascular function during peak eplerenone levels, we scheduled the endothelial function test 3 hours after eplerenone administration. However, we decided to administer one more dose of 100 mg of eplerenone the day prior to data collection to ensure that there was adequate time for potential upregulation or downregulation of endothelial cell protein expression levels in response to eplerenone. The timing of the doses was based on previous in vitro and animal studies which demonstrated that within 24 hours of aldosterone administration, endothelial cell protein levels of NADPH oxidase (a major source of superoxide anions), nitrotyrosine (a marker of oxidative stress), eNOS and phosphorylated eNOS (eNOSSer1177), and superoxide dismutase (SOD; an endogenous antioxidant enzyme) changed and that these changes were inhibited by MR antagonism (Gromotowicz et al. 2011; Nagata et al. 2006; Taye and Morawietz 2011).
Therefore, in the current study a single dose of eplerenone (100 mg) was administered at 7 am on the day prior to data collection followed 24 hours later by a second dose (100 mg) also at 7 am. On day 1, subjects remained in the laboratory for monitoring for 1 hour following pill ingestion. On day 2, vascular function measures, endothelial cell biopsies and blood collection were performed at 10 am in a semi-darkened temperature-controlled room after a minimum of 30 minutes of supine quiet rest. Prior to data collection subjects completed a 12-hour overnight fast including abstinence from caffeine and alcohol consumption.
2.2.1. Large conduit artery vascular function
The brachial artery was imaged using an ultrasound/Doppler system equipped with a 7.5 MHz linear vascular transducer (Aplio XV, Toshiba) as previously described by our group (Christou et al. 2012; Hwang et al. 2013b). Briefly, the subject’s right arm was abducted and fixed in position at heart level. A duplex ultrasound image of the brachial artery (i.e., 2D image and spectral Doppler waveforms) was obtained approximately midway between the antecubital fossa and the shoulder. Blood velocity was assessed with the Doppler angle of insonation set ≤60 degrees. To ensure that the same segment of the brachial artery was imaged under both the eplerenone and placebo conditions, the location of the transducer was recorded and a digital photograph of the arm position was obtained. To prevent movement during data collection, the vascular transducer was clamped in place (Flexbar, Flexbar Machine Corporation, Islandia, NY).
Vascular endothelial function was assessed non-invasively using post-ischemic hyperemia-induced flow-mediated dilation following established guidelines (Corretti et al. 2002; Thijssen et al. 2011) and as reported by our group (Hwang et al. 2013b). A rapid inflation/deflation pressure cuff (E20 and AG 101, D. E. Hokanson, Bellevue, WA) was placed around the widest part of the subject’s forearm and was used to induce reactive hyperemia by inflating the cuff to 250 mmHg for 5 minutes followed by rapid deflation. ECG R-gated duplex ultrasound images of the brachial artery were digitally recorded (Vascular Imager, Medical Imaging Applications, LLC, Coralville, IA) for 1 min to establish end-diastolic pre-occlusion baseline diameter and for 2 min post-occlusion to determine the maximum brachial artery diameter.
A commercially available edge-detection wall-tracking software (Brachial Analyzer, Medical Imaging Applications, LLC, Coralville, IA) was used to analyze the diameters. A moving average using a 3-R-gated image bin was applied prior to identifying the peak diameter. Flow-mediated dilation was expressed as absolute change in mm (maximum diameter − baseline diameter) and as % change ([(maximum − baseline diameter)/baseline diameter] × 100). To quantify the hyperemic response, blood velocity was measured by analyzing the first 15 post-occlusion spectral Doppler envelopes and 15 baseline spectral Doppler envelopes using the Toshiba ultrasound system software. Shear stress (dyne/cm2) was calculated as 8 × μ × mean blood velocity/baseline diameter, where μ was blood viscosity, which was assumed to be 0.035 dyne/cm2 (Mitchell et al. 2004). Ultrasound images were analyzed by the same investigator who was blinded to treatment.
Vascular smooth muscle responsiveness to exogenous nitric oxide donor, a measure of endothelium-independent dilation, was assessed as we have previously reported (Christou et al. 2012). Briefly, brachial artery ECG-gated ultrasound images were acquired, as described above, at baseline and for 10 minutes following sublingual nitroglycerin (0.4 mg) administration. Maximum brachial vasodilatory response to nitroglycerin was expressed as % change ([(maximum − baseline diameter)/baseline diameter] × 100).
2.2.2. Endothelial cell biopsies and protein levels
Endothelial cells were biopsied as established by Colombo et al. (Colombo et al. 2002; Shenouda et al. 2011; Silver 2007; Silver et al. 2010) and more recently published by our group (Hwang et al. 2013b; Silver 2007; Silver et al. 2010). Endothelial cell biopsies were performed in all subjects for whom 18-gauge intravenous catheterization was successful (N=16). Two sterile J-shaped guidewires (Daig, Inc., Minnetonka, MN) were sequentially advanced ~ 10 cm through the catheter and retracted. Cells were recovered by washing the wires with a dissociation buffer and centrifugation. Cells were fixed with 4% paraformaldehyde (USB corporation, Cleveland, OH), washed thoroughly with PBS, plated on poly-L-lysine coated slides (Sigma Chemical, St. Louis, MO), and stored at −80°C until the immunofluorescence staining was performed.
For immunofluorescence staining, fixed vascular endothelial cells were rehydrated with PBS containing 50 mmol/L glycine and non-specific sites were blocked with 5% donkey serum (Jackson Immunoresearch, West Grove, PA). Slides were incubated with one of the following primary antibodies followed with corresponding secondary antibody with Alexa Fluor 488 (Invitrogen, Carlsbad, CA): eNOS, an enzyme responsible for nitric oxide production (BD Biosciences, San Jose, CA); phosphorylated eNOSSer1177, the activated form of eNOS (Calbiochem, Inc., San Diego, CA); NADPH oxidase subunit p47phox, the major source of vascular superoxide production (Millipore, Inc., Billerica, MA); nitrotyrosine, a marker of oxidative damage (Abcam, Inc., Cambridge, MA), cytosolic and mitochondrial SOD (CuZnSOD and MnSOD, respectively; Millipore, Inc., Billerica, MA), and pro-inflammatory factors tumor necrosis factor alpha (TNF-α, Millipore, Inc., Billerica, MA) and nuclear factor kappa B (NF-κB, Millipore, Inc., Billerica, MA). Slides were also incubated with a primary antibody for vascular endothelial (VE)-cadherin (Abcam, Inc., Cambridge, MA) and corresponding secondary antibody with Alexa Fluor 555 (Invitrogen, Carlsbad, CA) for identification of endothelial cells. Finally, slides were mounted with Vectashield containing the nuclear stain DAPI (Vector Laboratories, Inc., Burlingame, CA). Because of the large number of slides, staining was performed in several batches, but each subject’s slides from the eplerenone and placebo visits were included in the same batch to avoid the influence of day-to-day variability in staining. To minimize the potential confound of inter-batch variability in staining, 2 slides of human umbilical venous endothelial cells (HUVEC) were stained in each batch and intensity for each protein of interest was expressed relative to the average HUVEC intensity in that batch.
For analysis, cells were examined with a fluorescence microscope (Eclipse 80i, Nikon Instruments, Inc., Melville, NY) at 100x magnification using the same exposure time. Images of endothelial cells with intact nuclei were digitally captured by a coolSNAP ES2 camera (Photometrics, Tuscon, AR). Endothelial cells were identified by the presence of VE-cadherin staining and nuclear integrity was confirmed by DAPI staining. Vascular endothelial cell protein levels were measured with NIS Elements software (version 3.2, Nikon Instruments, Inc., Melville, NY) by quantifying Alexa Fluor 488 intensity while correcting for background fluorescence. Vascular endothelial cell protein levels are reported as intensity per HUVEC intensity.
2.2.3. Blood measures
Standard blood chemistries and hematological evaluation including white blood cell counts were performed by a clinical laboratory using conventional assays. Plasma oxidized low density lipoprotein, an indirect measure of oxidative stress, was measured using a commercially available ELISA kit (Mercodia) (Hwang et al. 2013a). Plasma F2-isoprostanes, another marker of oxidative stress, were measured by the Vanderbilt University Eicosanoid Core Laboratory using gas chromatography-mass spectrometry, as previously described (Hwang et al. 2013b; Milne et al. 2007). Plasma adiponectin (hexameric and high-molecular-weight forms) was measured using a commercially-available ELISA kit (Mercodia, Sweden) (Yoo et al. 2015).
2.2.4. Resting blood pressure
Resting blood pressures were recorded over the brachial artery with a semi-automated oscillometric device (Dinamap, GE, Salt Lake City, UT, USA).
2.2.5. Height, weight, and body mass index
Height was measured to the nearest mm using a stadiometer. Body weight was measured to the nearest 0.1 kg with an electronic scale (Tanita, Arlington Heights, IL, USA) while subjects were barefoot and dressed in light clothing. Body mass index was determined as weight divided by height squared (kg/m2).
2.3. Statistical analyses
Statistical analyses were performed using SPSS, version 22. Statistical significance for all analyses was set at P< 0.05. Data are presented as mean ± SE. Paired t-tests were used to compare the differences between eplerenone and placebo conditions. Pearson product moment correlation coefficients were calculated to evaluate bivariate relations of interest.
3. RESULTS
Baseline characteristics for the study population are given in Table 1. Mean values and ranges are presented revealing a mean age of 61 years (range 53–79).
Table 1.
Subject characteristics.
| Mean±SE | Min-Max | |
|---|---|---|
| Age, years | 61.1±1 | 53–79 |
| Weight, kg | 83.5±3.7 | 51.8–121.8 |
| Body mass index, kg/m2 | 29.1±1.0 | 22.6–38.6 |
| Total cholesterol, mg/dL | 189±8 | 119–246 |
| LDL cholesterol, mg/dL | 119±7 | 56–166 |
| HDL cholesterol, mg/dL | 50±3 | 28–76 |
| Triglycerides, mg/dL | 102±10 | 45–232 |
| Fasting glucose, mg/dL | 90±2 | 73–110 |
LDL, low density lipoprotein; HDL, high density lipoprotein.
3.1. Cardiovascular responses
Acute inhibition of MR activation with eplerenone reduced flow-mediated vascular dilation by 19% (P≤0.03; Figure 1 and Table 2). Baseline vessel diameter (P=0.1), blood velocity (P=0.7) and shear stress (P=0.6), however, remained unchanged by MR antagonism (Table 2). Blood velocity and shear stress increased to the same extent during reactive hyperemia with MR antagonism and placebo (P=0.9; Table 2). MR antagonism treatment for this short duration did not change heart rate or systolic and diastolic blood pressure compared with placebo (P≥0.6; Table 2). To determine whether the change in flow-mediated dilation in response to eplerenone was due to changes in endothelial function or to alterations in the smooth muscle cell signaling pathways that mediate dilation, we also measured vasodilation in response to nitroglycerin (a direct nitric oxide donor). Endothelium-independent (nitroglycerin-induced) dilation was not influenced by MR antagonism (P≥0.4; Figure 2 and Table 2) indicating that smooth muscle cell function is unchanged by acute MR blockade. These data suggest that MR inhibition acutely decreases endothelium-dependent relaxation in older patients without cardiovascular disease.
Figure 1.
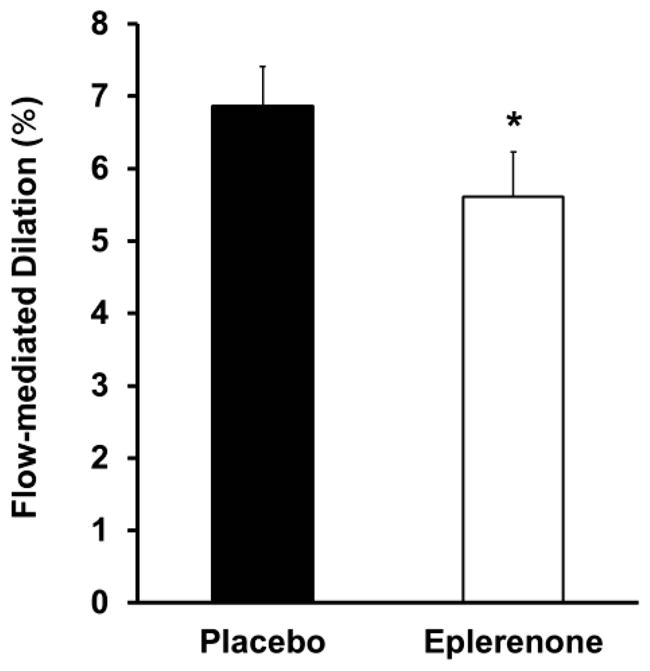
Acute mineralocorticoid receptor antagonism with eplerenone decreased brachial artery flow-mediated dilation. *P=0.02 vs. placebo.
Table 2.
Cardiovascular measurements after acute mineralocorticoid receptor antagonism with eplerenone.
| Placebo | Eplerenone | |
|---|---|---|
| Heart rate, bpm | 60±1 | 60±1 |
| Systolic blood pressure, mmHg | 135±4 | 134±4 |
| Diastolic blood pressure, mmHg | 77±2 | 77±2 |
| Baseline diameter, mm | 3.64±0.15 | 3.71±0.15 |
| Baseline blood velocity, cm/min | 24.7±2.4 | 23.9±1.8 |
| Baseline shear stress, dyne/cm2 | 19.7±2.0 | 19.1±1.9 |
| Hyperemic blood velocity, cm/min | 106.5±6.3 | 105.2±8.7 |
| Hyperemic shear stress, dyne/cm2 | 85.6±6.4 | 85.1±9.8 |
| Hyperemia, % | 358±20 | 349±24 |
| Flow-mediated dilation, mm | 0.25±0.02 | 0.21±0.02* |
| Nitroglycerin-induced dilation, mm | 0.82±0.06 | 0.78±0.04 |
Data are mean±SE.
P=0.03 vs. placebo.
Figure 2.

Acute mineralocorticoid receptor antagonism with eplerenone did not influence brachial artery nitroglycerin (NTG)-induced dilation.
3.2. Blood markers and vascular endothelial cell protein levels
Endothelium-dependent vasodilation is predominantly determined by the amount of nitric oxide produced by eNOS in endothelial cells and also by the bioavailability of nitric oxide to act on the smooth muscle as determined by the amount of vascular oxidative stress. Thus, we next examined the effect of eplerenone treatment on the degree of systemic and endothelial oxidative stress and on endothelial eNOS activity. Serum levels of oxidized LDL, F2-isoprostanes, and adiponectin remained unchanged in response to MR antagonism (63.3±5.6 vs. 59.1±3.8 U/L, placebo vs. eplerenone, P=0.5, 7.8±1.8 vs. 5.2±0.5 pg/mL and 10.5±1.1 vs. 10.1±1.2 μg/mL, respectively, P ≥0.2). MR antagonist treatment also did not significantly change the levels of endothelial cell expression of NADPH oxidase, a major source of superoxide production, or of superoxide dismutases (SOD; CuZnSOD and MnSOD), endogenous antioxidant defenses (P≥0.7; Table 3). Similarly, downstream markers of oxidative stress and vascular damage including nitrotyrosine, a marker of oxidative damage, and inflammation factors TNF-α and NF-κB, were unchanged in endothelial cells in response to MR antagonism (P≥0.4; Table 3) despite variable individual responses. Importantly, MR antagonism significantly decreased the level of activated eNOS in biopsied endothelial cells as measured by the ratio of active phosphorylated eNOSSer1177 to total eNOS (P=0.02; Figure 3). Taken together, these data reveal that acute MR inhibition in healthy older adults caused endothelial dysfunction associated with decreased eNOS activity without significant changes in systemic or endothelial oxidative stress or in endothelial levels of inflammatory markers.
Table 3.
Vascular endothelial cell protein levels following mineralocorticoid receptor antagonism with eplerenone.
| Placebo | Eplerenone | |
|---|---|---|
| eNOS | 0.23±0.01 | 0.24±0.02 |
| P-eNOS Ser1177 | 0.34±0.02 | 0.31±0.03 |
| NADPH p47phox | 0.65±0.06 | 0.64±0.07 |
| Nitrotyrosine | 0.54±0.04 | 0.55±0.07 |
| CuZnSOD | 0.47±0.03 | 0.47±0.04 |
| MnSOD | 0.53±0.03 | 0.56±0.06 |
| TNFα | 0.35±0.02 | 0.36±0.01 |
| NF-κB | 0.43±0.04 | 0.41±0.02 |
Data are mean±SE. Values are reported as intensity/HUVEC intensity based on immunofluorescence. eNOS, endothelial nitric oxide synthase; P-eNOS Ser1177, eNOS phosphorylated at Ser1177; NADPH p47phox, NADPH oxidase subunit p47phox; CuZnSOD, cytosolic superoxide dismutase; MnSOD, mitochondrial superoxide dismutase; TNFα, tumor necrosis factor alpha; NF-κB, nuclear factor kappa B.
Figure 3.
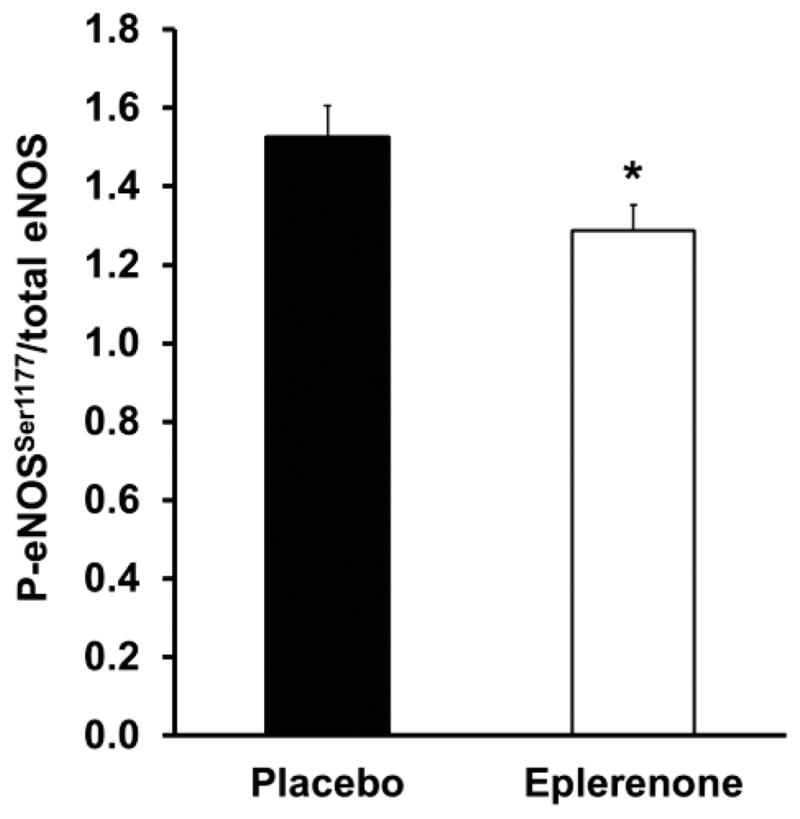
Acute mineralocorticoid receptor antagonism with eplerenone decreased the amount of activated eNOS as measured by the ratio of phosphorylated eNOS at Ser1177 (P-eNOSSer1177) to total eNOS. *P=0.02 vs. placebo.
3.3. Correlations between changes in flow-mediated dilation and biomarkers of oxidative stress and inflammation
We next examined whether there were baseline parameters that influenced the vascular responsiveness to acute MR blockade. Greater reductions in flow-mediated dilation in response to MR antagonism were associated with lower baseline white blood cell count (r=0.49, P=0.02; Figure 4A) and with lower baseline neutrophil levels (r=0.57, P=0.006; Figure 4B). There were no significant correlations between the change in flow-mediated dilation in response to MR antagonism and oxidized LDL, F2-isoprostanes or adiponectin levels (P≥0.3).
Figure 4.

The change in flow-mediated dilation (FMD) in response to mineralocorticoid receptor antagonism was positively related to baseline white blood cell (Panel A) and neutrophil (Panel B) counts.
3.4. Correlations between changes in flow-mediated dilation and vascular endothelial cell protein levels
We also examined whether the vascular response was associated with specific changes in expression of endothelial cell proteins. Greater reductions in flow-mediated dilation in response to MR antagonism were associated with higher baseline levels of endothelial cytosolic SOD (r=−0.54, P=0.03; Figure 5), and greater increases in endothelial cell mitochondrial SOD with MR antagonism (r=−0.85, P<0.0001; Figure 6). Furthermore, greater increases in eNOS and phosphorylated eNOSSer1177 were related to greater increases in endothelial cell nitrotyrosine levels (r=0.85 and 0.89, respectively, P<0.0001; Figure 7). There were no other significant correlations between the changes flow-mediated dilation in response to MR antagonism and the other vascular endothelial cell proteins that were measured (P>0.05).
Figure 5.

The change in flow-mediated dilation (FMD) in response to mineralocorticoid receptor antagonism was inversely related to baseline endothelial cell levels of cytosolic superoxide dismutase (CuZnSOD).
Figure 6.

The change in flow-mediated dilation (FMD) in response to mineralocorticoid receptor antagonism was inversely related to the change in endothelial cell levels of mitochondrial superoxide dismutase (MnSOD).
Figure 7.

The change in endothelial nitric oxide synthase (eNOS, Panel A) and phosphorylated eNOS at Ser1177 (P-eNOSSer1177, Panel B) in response to mineralocorticoid receptor antagonism were positively related to the change in endothelial cell levels of nitrotyrosine.
4. DISCUSSION
This study is the first to examine whether acute inhibition of the MR influences vascular function in older adults free from cardiovascular and other clinical disease. This randomized double-blind placebo-controlled crossover study provides evidence that acute inhibition of MR activation with eplerenone impairs vascular dilation by specifically impairing vascular endothelial function without affecting blood pressure or vascular smooth muscle responsiveness to exogenous nitric oxide. Acute MR blockade decreased eNOS activation in endothelial cells supporting that MR activation plays a physiologically important and beneficial role in regulating eNOS activity to promote endothelium-dependent dilation in healthy aging. Our study also reveals new correlations between MR modulation of vascular function and inflammatory cells and regulators of the balance between nitric oxide production and vascular oxidative stress.
4.1. Implications for the role of MR in vascular endothelial function
In vascular smooth muscle cells, MR has recently been shown to contribute to vasoconstriction (Galmiche et al. 2014; McCurley et al. 2012; Skott et al. 2006). In endothelial cells, however, aldosterone activation of MR stimulates PI3 kinase, Akt, and eNOS phosphorylation leading to production of nitric oxide (Skott et al. 2006) by a rapid mechanism that does not involve gene transcription and is thus termed non-genomic (Mihailidou and Funder 2005). Nitric oxide then diffuses to the vascular smooth muscle cells and counteracts MR-mediated vasoconstriction. The presence of cardiovascular disease risk factors that contribute to vascular inflammation, eNOS inhibition or uncoupling, or vascular oxidative stress would attenuate the endothelial benefits of MR activation (Jaffe and Jaisser 2014). Thus, the integrity of the vascular endothelium may determine the net effect of MR activation or inhibition on vascular relaxation (Skott et al. 2006). This hypothesis is supported by this study and by others in the literature. Indeed, in healthy young adults, aldosterone infusion enhances endothelium-dependent vasodilation (Nietlispach et al. 2007). Our data extend these findings by demonstrating that MR activation also contributes to endothelium-dependent dilation in healthy older adults. In our study, acute inhibition of MR led to an absolute reduction in flow-mediated dilation of 1.3% compared with placebo. A change of 1% in flow-mediated dilation has been associated with a 10% change in the risk for cardiovascular disease (Ras et al. 2012). Thus, vascular MR activity may contribute substantially to normal endothelial function in healthy aging.
In a previous study, we demonstrated that chronic (1 month) MR antagonism with eplerenone did not alter flow-mediated vasodilation in healthy older adults (Hwang et al. 2013b). One explanation for these seemingly conflicting findings is that MR antagonism initially results in impairment in flow-mediated dilation in healthy older adults by rapidly decreasing eNOS activation, but when MR antagonism persists chronically, then flow-mediated dilation is restored to baseline levels by mechanisms that require longer term treatment. MR is a hormone-activated transcription factor and most of its long term effects, including renal regulation of blood pressure, are mediated by its genomic actions to regulate gene transcription. Chronic MR antagonism modulates expression of genes that can offset or reverse the initial impairment in endothelium-dependent dilation including activation of anti-oxidant and anti-inflammatory pathways in the endothelium (Reviewed in (Moss and Jaffe 2015)).
In contrast to all of the data in healthy adults, there is extensive evidence that MR activation has detrimental vascular effects in patients with cardiovascular disease and in animal models that mimic cardiovascular diseases in which there is vascular endothelial damage, oxidative stress and inflammation (Abiose et al. 2004; Farquharson and Struthers 2000; Keidar et al. 2004; Rajagopalan et al. 2002; Sartorio et al. 2007; Thai et al. 2006). Indeed in our chronic study of eplerenone in older adults, those with obesity had improved vascular function in response to MR blockade (Hwang et al. 2013b) and similarly in a mouse model of diet-induced obesity, deletion of endothelial MR prevented obesity-associated endothelial dysfunction (Schafer et al. 2013). Thus, whether MR inhibition has beneficial or detrimental effects may depend on the presence and severity of vascular disease, on the balance between nitric oxide and oxidative stress, and on the duration of treatment. Although aging is associated with impaired endothelial function, oxidative stress and low-grade inflammation, the severity of these conditions is less than that occurring in atherosclerosis or heart failure. It appears that in the absence of additional vascular injury, MR has a beneficial physiological role in endothelial function by contributing to enhanced eNOS activity in healthy older adults.
4.2. Changes in flow-mediated dilation in response to acute MR inhibition are independent of changes in vascular smooth muscle cell function and blood pressure
In our study, vascular smooth muscle responsiveness to exogenous nitric oxide was not influenced by acute MR antagonism. This is consistent with most studies in which chronic activation or inhibition of vascular MR does not contribute to smooth muscle cell relaxation (McCurley and Jaffe 2012). Indeed, in an animal model in which the MR has been specifically deleted only from smooth muscle cells, there is no difference in endothelium-independent vasodilation to the nitric oxide donor sodium nitroprusside (McCurley et al. 2012). There are a few conflicting studies describing acute effects of aldosterone infusion in healthy young adults, with one study demonstrating enhanced vasodilation to exogenous nitric oxide (Nietlispach et al. 2007) and another showing attenuated vasodilation to sodium nitroprusside (Schmidt et al. 2003). These diverse effects might be due to differences in experimental design or the populations tested.
As expected, blood pressure did not change in response to acute MR antagonism in our study. This is an important strength of our experimental approach because it demonstrates the effect of MR antagonism on vascular endothelial function is independent of changes in blood pressure. Aldosterone activation of MR in renal epithelial cells is known to control blood pressure by genomic mechanisms resulting in modulation of ion channel expression, sodium and water retention, and in turn, plasma volume. This effect of MR antagonism is known to require longer term MR inhibition. Indeed, we have previously reported that 1 month treatment using the same dose of eplerenone as in the present study, resulted in significant decreases in blood pressure in healthy older adults (Hwang et al. 2013b).
4.3 Interactions between MR effects on flow-mediated dilation and circulating inflammatory cells
Our investigation cannot directly explain the mechanisms responsible for the vascular effects of MR in healthy aging. However, we found some intriguing associations between the vascular responses to MR antagonism and circulating inflammatory cells that may provide preliminary insight. We found that greater reductions in flow-mediated dilation in response to MR antagonism were associated with lower baseline white blood cell and neutrophil counts. These inflammatory markers have previously been linked with impaired endothelial function (i.e., flow-mediated dilation) (Walker et al. 2010). Neutrophils adhere to endothelial cells and migrate through the endothelial layer where they accumulate and contribute to endothelial dysfunction and vascular inflammation. There is evidence of functional MR and 11βHSD2 in neutrophils (Bergmann et al. 2010). Activation of MR in neutrophils inhibits NF-κB, an established pro-oxidant and pro-inflammatory transcription factor linked to suppression of endothelium-dependent dilation in older adults (Donato et al. 2008). Conversely, MR antagonism has been reported to block the anti-inflammatory effect of aldosterone in neutrophils (Bergmann et al. 2010). Based on these data, we raise the possibility that acute MR antagonism might have disrupted the MR-mediated suppression of neutrophil activation. Activation of neutrophils in this way would be expected to lead to impaired flow-mediated dilation in our study. Of course such correlations do not provide causation and additional studies would be needed to test this hypothesis.
4.4. The role of oxidative stress and the reactive oxygen species (ROS)/superoxide dismutase (SOD) balance
Based on our endothelial cell protein measures, we found that acute MR antagonism leads to decreased eNOS activity as measured by the decreased ratio of phosphorylated eNOSSer1177 to total eNOS. The reduction in eNOS activity could be contributing to lower nitric oxide levels and reduced flow-mediated dilation following MR antagonism. In addition to acting as a vasodilator on smooth muscle cells, nitric oxide is a buffer for vascular oxidative stress by interacting with the reactive oxygen species superoxide to produce peroxynitrite. Thus decreased nitric oxide would be associated with locally increased oxidative stress. Interestingly, greater reductions in flow-mediated dilation in response to MR antagonism were related to greater increases in SOD levels. We speculate that SOD levels might be increasing to compensate for the reductions in nitric oxide and the associated increase in vascular oxidative stress. In addition, we found that higher baseline levels of SOD were associated with greater reductions in flow-mediated dilation with MR antagonism. Again we can only postulate that in individuals with increased vascular oxidative stress at baseline and thus elevated basal SOD expression, MR may be contributing more to endothelial function by activating eNOS. If this positive influence were acutely abrogated by MR antagonism, it may be associated with greater impairment in endothelial function in these individuals. Finally, we found that greater increases in eNOS and phosphorylated eNOSSer1177 following eplerenone were related to higher endothelial cell levels of nitrotyrosine. Nitrotyrosine is a product of nitration of tyrosine residues by reactive nitrogen species including peroxynitrite which is elevated when nitric oxide is increased in the setting of oxidative stress. Thus, the correlation between eNOS activation and nitrotyrosine might be anticipated if some of the increased nitric oxide production is buffering vascular oxidative stress.
4.5. Study strengths and limitations
Our study has several strengths including: 1) the use of a randomized double-blind placebo-controlled crossover design; 2) exclusion of subjects with overt cardiovascular disease or other clinical disease such as diabetes and liver disease; 3) exclusion of subjects using medications that may modulate vascular function; 4) novelty of findings as this is the first study of acute effects of MR blockade in healthy aging; and 5) application of rigorous procedures to ensure high quality data. However, our study also has some important limitations that should be kept in mind when interpreting or applying the results. Our study was restricted to healthy older adults. Although, several studies have investigated the vascular effects of aldosterone administration in healthy young adults, the effect of acute MR antagonism in healthy young adults remains unknown. This needs to be directly examined in future investigations. Another potential limitation in our study is the lack of measures of renin and angiotensin II levels in response to acute MR antagonism. If levels increased then they could have secondary effects on vascular function and other endpoints. The sample size is small thus it is critical to reproduce the findings in larger cohorts that would also allow for secondary analyses based on sex and other factors. Some interesting correlations are identified in this study, however, as with all such correlations, they reveal only association and not causation. In addition, while speculations can be made based on the literature about potential mechanisms for these associations, mechanistic studies are needed to directly test such hypotheses. Finally, this study was specifically designed to examine acute MR inhibition in healthy adults without cardiovascular disease to try to understand the underlying role of MR activation in that population. MR antagonists are predominantly used in the treatment of hypertension and heart failure, conditions that are very common in the elderly. A growing literature supports that MR activation may be beneficial to endothelial function in healthy individuals, and this study extends that to older adults. However, in disease states, MR inhibition appears to have vascular benefit. Thus, further studies are needed to determine the role of MR in vascular function in older adults with cardiovascular disease.
5. CONCLUSIONS
Our results demonstrate that in older adults free from overt cardiovascular disease acute inhibition of the MR leads to impairments in vascular endothelial function. These findings enhance our current understanding of the vascular effects of MR activation and suggest that the MR has an important beneficial physiological function in regulating eNOS activity and flow-mediated endothelium-dependent dilation in healthy aging. Vascular smooth muscle responsiveness to exogenous nitric oxide is not influenced by acute MR antagonism in this population. Similarly, acute MR antagonism does not affect systemic blood pressure or circulating and endothelial cell markers of oxidative stress and inflammation.
Highlights.
In healthy older adults, acute MR antagonism impaired endothelial function.
Acute MR antagonism decreased endothelial nitric oxide synthase activity.
MR antagonism did not change endothelium-independent dilation or blood pressure.
MR antagonist effects may depend on vascular health, oxidative stress and treatment duration.
Acknowledgments
This work was supported by NIH AG 032067 and AHA 0865117F grants to DDC and NIH HL119290 to IZJ. The authors would like to thank Ms. Sharon Greer, R.N., Mr. Creighton Wilson R.N. and the Division of Cardiology and Radiology at the Scott and White Clinic at College Station, Texas, for their contributions. The authors would also like to thank Ms. Molly Cernosek for technical assistance and the study participants for their time and efforts.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abiose AK, Mansoor GA, Barry M, Soucier R, Nair CK, Hager D. Effect of spironolactone on endothelial function in patients with congestive heart failure on conventional medical therapy. Am J Cardiol. 2004;93:1564–1566. doi: 10.1016/j.amjcard.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Barrett Mueller K, Bender SB, Hong K, Yang Y, Aronovitz M, Jaisser F, Hill MA, Jaffe IZ. Endothelial Mineralocorticoid Receptors Differentially Contribute to Coronary and Mesenteric Vascular Function Without Modulating Blood Pressure. Hypertension. 2015;66:988–997. doi: 10.1161/HYPERTENSIONAHA.115.06172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann A, Eulenberg C, Wellner M, Rolle S, Luft F, Kettritz R. Aldosterone abrogates nuclear factor kappaB-mediated tumor necrosis factor alpha production in human neutrophils via the mineralocorticoid receptor. Hypertension. 2010;55:370–379. doi: 10.1161/HYPERTENSIONAHA.109.141309. [DOI] [PubMed] [Google Scholar]
- Caprio M, Newfell BG, la Sala A, Baur W, Fabbri A, Rosano G, Mendelsohn ME, Jaffe IZ. Functional mineralocorticoid receptors in human vascular endothelial cells regulate intercellular adhesion molecule-1 expression and promote leukocyte adhesion. Circulation research. 2008;102:1359–1367. doi: 10.1161/CIRCRESAHA.108.174235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christou DD, Gentile CL, DeSouza CA, Seals DR, Gates PE. Fatness is a better predictor of cardiovascular disease risk factor profile than aerobic fitness in healthy men. Circulation. 2005;111:1904–1914. doi: 10.1161/01.CIR.0000161818.28974.1A. [DOI] [PubMed] [Google Scholar]
- Christou DD, Pierce GL, Walker AE, Hwang MH, Yoo JK, Luttrell M, Meade TH, English M, Seals DR. Vascular smooth muscle responsiveness to nitric oxide is reduced in healthy adults with increased adiposity. Am J Physiol Heart Circ Physiol. 2012;303:H743–750. doi: 10.1152/ajpheart.00394.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo PC, Ashton AW, Celaj S, Talreja A, Banchs JE, Dubois NB, Marinaccio M, Malla S, Lachmann J, Ware JA, Le Jemtel TH. Biopsy coupled to quantitative immunofluorescence: a new method to study the human vascular endothelium. J Appl Physiol. 2002;92:1331–1338. doi: 10.1152/japplphysiol.00680.2001. [DOI] [PubMed] [Google Scholar]
- Cook CS, Berry LM, Bible RH, Hribar JD, Hajdu E, Liu NW. Pharmacokinetics and metabolism of [14C]eplerenone after oral administration to humans. Drug Metab Dispos. 2003;31:1448–1455. doi: 10.1124/dmd.31.11.1448. [DOI] [PubMed] [Google Scholar]
- Corretti MC, Anderson TJ, Benjamin EJ, Celermajer D, Charbonneau F, Creager MA, Deanfield J, Drexler H, Gerhard-Herman M, Herrington D, Vallance P, Vita J, Vogel R International Brachial Artery Reactivity Task F. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force.[erratum appears in J Am Coll Cardiol 2002 Mar 20;39(6):1082] Journal of the American College of Cardiology. 2002;39:257–265. doi: 10.1016/s0735-1097(01)01746-6. [DOI] [PubMed] [Google Scholar]
- Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:III27–32. doi: 10.1161/01.CIR.0000131515.03336.f8. [DOI] [PubMed] [Google Scholar]
- Donato AJ, Black AD, Jablonski KL, Gano LB, Seals DR. Aging is associated with greater nuclear NF kappa B, reduced I kappa B alpha, and increased expression of proinflammatory cytokines in vascular endothelial cells of healthy humans. Aging cell. 2008;7:805–812. doi: 10.1111/j.1474-9726.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farquharson CA, Struthers AD. Spironolactone increases nitric oxide bioactivity, improves endothelial vasodilator dysfunction, and suppresses vascular angiotensin I/angiotensin II conversion in patients with chronic heart failure. Circulation. 2000;101:594–597. doi: 10.1161/01.cir.101.6.594. [DOI] [PubMed] [Google Scholar]
- Farquharson CA, Struthers AD. Aldosterone induces acute endothelial dysfunction in vivo in humans: evidence for an aldosterone-induced vasculopathy. Clin Sci (Lond) 2002;103:425–431. doi: 10.1042/cs1030425. [DOI] [PubMed] [Google Scholar]
- Galmiche G, Pizard A, Gueret A, El Moghrabi S, Ouvrard-Pascaud A, Berger S, Challande P, Jaffe IZ, Labat C, Lacolley P, Jaisser F. Smooth muscle cell mineralocorticoid receptors are mandatory for aldosterone-salt to induce vascular stiffness. Hypertension. 2014;63:520–526. doi: 10.1161/HYPERTENSIONAHA.113.01967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromotowicz A, Szemraj J, Stankiewicz A, Zakrzeska A, Mantur M, Jaroszewicz E, Rogowski F, Chabielska E. Study of the mechanisms of aldosterone prothrombotic effect in rats. J Renin-Angio-Aldo S. 2011;12:430–439. doi: 10.1177/1470320310397405. [DOI] [PubMed] [Google Scholar]
- Huang PL. Endothelial nitric oxide synthase and endothelial dysfunction. Current hypertension reports. 2003;5:473–480. doi: 10.1007/s11906-003-0055-4. [DOI] [PubMed] [Google Scholar]
- Hwang MH, Yoo JK, Luttrell M, Kim HK, Meade TH, English M, Nichols WW, Christou DD. Role of mineralocorticoid receptors in arterial stiffness in human aging. Exp Gerontol. 2013a;48:701–704. doi: 10.1016/j.exger.2013.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang MH, Yoo JK, Luttrell M, Kim HK, Meade TH, English M, Segal MS, Christou DD. Mineralocorticoid receptors modulate vascular endothelial function in human obesity. Clin Sci (Lond) 2013b;125:513–520. doi: 10.1042/CS20130200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe IZ, Jaisser F. Endothelial cell mineralocorticoid receptors: turning cardiovascular risk factors into cardiovascular dysfunction. Hypertension. 2014;63:915–917. doi: 10.1161/HYPERTENSIONAHA.114.01997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe IZ, Mendelsohn ME. Angiotensin II and aldosterone regulate gene transcription via functional mineralocortocoid receptors in human coronary artery smooth muscle cells. Circulation research. 2005;96:643–650. doi: 10.1161/01.RES.0000159937.05502.d1. [DOI] [PubMed] [Google Scholar]
- Keidar S, Kaplan M, Pavlotzky E, Coleman R, Hayek T, Hamoud S, Aviram M. Aldosterone administration to mice stimulates macrophage NADPH oxidase and increases atherosclerosis development: a possible role for angiotensin-converting enzyme and the receptors for angiotensin II and aldosterone. Circulation. 2004;109:2213–2220. doi: 10.1161/01.CIR.0000127949.05756.9D. [DOI] [PubMed] [Google Scholar]
- Landmesser U, Drexler H. Endothelial function and hypertension. Current opinion in cardiology. 2007;22:316–320. doi: 10.1097/HCO.0b013e3281ca710d. [DOI] [PubMed] [Google Scholar]
- McCurley A, Jaffe IZ. Mineralocorticoid receptors in vascular function and disease. Mol Cell Endocrinol. 2012;350:256–265. doi: 10.1016/j.mce.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, Chambon P, Hill MA, Dorrance AM, Mendelsohn ME, Jaffe IZ. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nature medicine. 2012;18:1429–1433. doi: 10.1038/nm.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihailidou AS, Funder JW. Nongenomic effects of mineralocorticoid receptor activation in the cardiovascular system. Steroids. 2005;70:347–351. doi: 10.1016/j.steroids.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Milne GL, Sanchez SC, Musiek ES, Morrow JD. Quantification of F2-isoprostanes as a biomarker of oxidative stress. Nat Protoc. 2007;2:221–226. doi: 10.1038/nprot.2006.375. [DOI] [PubMed] [Google Scholar]
- Mitchell GF, Parise H, Vita JA, Larson MG, Warner E, Keaney JF, Jr, Keyes MJ, Levy D, Vasan RS, Benjamin EJ. Local shear stress and brachial artery flow-mediated dilation: the Framingham Heart Study. Hypertension. 2004;44:134–139. doi: 10.1161/01.HYP.0000137305.77635.68. [DOI] [PubMed] [Google Scholar]
- Moss ME, Jaffe IZ. Mineralocorticoid Receptors in the Pathophysiology of Vascular Inflammation and Atherosclerosis. Frontiers in endocrinology. 2015;6:153. doi: 10.3389/fendo.2015.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata D, Takahashi M, Sawai K, Tagami T, Usui T, Shimatsu A, Hirata Y, Naruse M. Molecular mechanism of the inhibitory effect of aldosterone on endothelial NO synthase activity. Hypertension. 2006;48:165–171. doi: 10.1161/01.HYP.0000226054.53527.bb. [DOI] [PubMed] [Google Scholar]
- Nietlispach F, Julius B, Schindler R, Bernheim A, Binkert C, Kiowski W, Brunner-La Rocca HP. Influence of acute and chronic mineralocorticoid excess on endothelial function in healthy men. Hypertension. 2007;50:82–88. doi: 10.1161/HYPERTENSIONAHA.107.088955. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S, Duquaine D, King S, Pitt B, Patel P. Mineralocorticoid receptor antagonism in experimental atherosclerosis. Circulation. 2002;105:2212–2216. doi: 10.1161/01.cir.0000015854.60710.10. [DOI] [PubMed] [Google Scholar]
- Ras RT, Streppel MT, Draijer R, Zock PL. Flow-mediated dilation and cardiovascular risk prediction: A systematic review with meta-analysis. Int J Cardiol. 2012 doi: 10.1016/j.ijcard.2012.09.047. [DOI] [PubMed] [Google Scholar]
- Ravis WR, Reid S, Sica DA, Tolbert DS. Pharmacokinetics of eplerenone after single and multiple dosing in subjects with and without renal impairment. Journal of clinical pharmacology. 2005;45:810–821. doi: 10.1177/0091270005275894. [DOI] [PubMed] [Google Scholar]
- Sartorio CL, Fraccarollo D, Galuppo P, Leutke M, Ertl G, Stefanon I, Bauersachs J. Mineralocorticoid receptor blockade improves vasomotor dysfunction and vascular oxidative stress early after myocardial infarction. Hypertension. 2007;50:919–925. doi: 10.1161/HYPERTENSIONAHA.107.093450. [DOI] [PubMed] [Google Scholar]
- Schafer N, Lohmann C, Winnik S, van Tits LJ, Miranda MX, Vergopoulos A, Ruschitzka F, Nussberger J, Berger S, Luscher TF, Verrey F, Matter CM. Endothelial mineralocorticoid receptor activation mediates endothelial dysfunction in diet-induced obesity. European heart journal. 2013;34:3515–3524. doi: 10.1093/eurheartj/eht095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt BM, Oehmer S, Delles C, Bratke R, Schneider MP, Klingbeil A, Fleischmann EH, Schmieder RE. Rapid nongenomic effects of aldosterone on human forearm vasculature. Hypertension. 2003;42:156–160. doi: 10.1161/01.HYP.0000083298.23119.16. [DOI] [PubMed] [Google Scholar]
- Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, Hamburg NM, Frame AA, Caiano TL, Kluge MA, Duess MA, Levit A, Kim B, Hartman ML, Joseph L, Shirihai OS, Vita JA. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. 2011;124:444–453. doi: 10.1161/CIRCULATIONAHA.110.014506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver A, Beske SD, Christou DD, Donato AJ, Moreau KL, Eskurza I, Gates PE, Seals DR. Overweight and obese humans demonstrate increased vascular endothelial NAD(P)H Oxidase-p47phox expression and evidence of endothelial oxidative stress. Circulation. 2007;115 doi: 10.1161/CIRCULATIONAHA.106.657486. [DOI] [PubMed] [Google Scholar]
- Silver AE, Christou DD, Donato AJ, Beske SD, Moreau KL, Magerko KA, Seals DR. Protein expression in vascular endothelial cells obtained from human peripheral arteries and veins. J Vasc Res. 2010;47:1–8. doi: 10.1159/000231715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skott O, Uhrenholt TR, Schjerning J, Hansen PB, Rasmussen LE, Jensen BL. Rapid actions of aldosterone in vascular health and disease--friend or foe? Pharmacology & therapeutics. 2006;111:495–507. doi: 10.1016/j.pharmthera.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Taye A, Morawietz H. Spironolactone inhibits NADPH oxidase-induced oxidative stress and enhances eNOS in human endothelial cells. Iranian journal of pharmaceutical research : IJPR. 2011;10:329–337. [PMC free article] [PubMed] [Google Scholar]
- Thai HM, Do BQ, Tran TD, Gaballa MA, Goldman S. Aldosterone antagonism improves endothelial-dependent vasorelaxation in heart failure via upregulation of endothelial nitric oxide synthase production. J Card Fail. 2006;12:240–245. doi: 10.1016/j.cardfail.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Thijssen DH, Black MA, Pyke KE, Padilla J, Atkinson G, Harris RA, Parker B, Widlansky ME, Tschakovsky ME, Green DJ. Assessment of flow-mediated dilation in humans: a methodological and physiological guideline. Am J Physiol Heart Circ Physiol. 2011;300:H2–12. doi: 10.1152/ajpheart.00471.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda N, Nakanishi S, Tanabe S. Aldosterone affects blood flow and vascular tone regulated by endothelium-derived NO: therapeutic implications. British journal of pharmacology. 2013;168:519–533. doi: 10.1111/j.1476-5381.2012.02194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vita JA. Endothelial function. Circulation. 2011;124:e906–912. doi: 10.1161/CIRCULATIONAHA.111.078824. [DOI] [PubMed] [Google Scholar]
- Walker AE, Seibert SM, Donato AJ, Pierce GL, Seals DR. Vascular endothelial function is related to white blood cell count and myeloperoxidase among healthy middle-aged and older adults. Hypertension. 2010;55:363–369. doi: 10.1161/HYPERTENSIONAHA.109.145870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White WB, Carr AA, Krause S, Jordan R, Roniker B, Oigman W. Assessment of the novel selective aldosterone blocker eplerenone using ambulatory and clinical blood pressure in patients with systemic hypertension. Am J Cardiol. 2003;92:38–42. doi: 10.1016/s0002-9149(03)00461-2. [DOI] [PubMed] [Google Scholar]
- Williams GH. Aldosterone biosynthesis, regulation, and classical mechanism of action. Heart Fail Rev. 2005;10:7–13. doi: 10.1007/s10741-005-2343-3. [DOI] [PubMed] [Google Scholar]
- Yoo JK, Hwang MH, Luttrell MJ, Kim HK, Meade TH, English M, Segal MS, Christou DD. Higher levels of adiponectin in vascular endothelial cells are associated with greater brachial artery flow-mediated dilation in older adults. Exp Gerontol. 2015;63:1–7. doi: 10.1016/j.exger.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]