

Abstract
Environmental stress is among the most important contributors to increased susceptibility to develop psychiatric disorders, including anxiety and post-traumatic stress disorder. While even acute stress alters gene expression, the molecular mechanisms underlying these changes remain largely unknown. 5-hydroxymethylcytosine (5hmC) is a novel environmentally sensitive DNA modification that is highly enriched in post-mitotic neurons and is associated with active transcription of neuronal genes. Recently, we found a hippocampal increase of 5hmC in the glucocorticoid receptor gene (Nr3c1) following acute stress, warranting a deeper investigation of stress-related 5hmC levels. Here, we used an established chemical labeling and affinity purification method coupled with high-throughput sequencing technology to generate the first genome-wide profile of hippocampal 5hmC following exposure to acute restraint stress and a one-hour recovery. This approach found a genome-wide disruption in 5hmC associated with acute stress response, primarily in genic regions, and identified known and potentially novel stress-related targets that have a significant enrichment for neuronal ontological functions. Integration of these data with hippocampal gene expression data from these same mice found stress-related hydroxymethylation correlated to altered transcript levels and sequence motif predictions indicated that 5hmC may function by mediating transcription factor binding to these transcripts. Together, these data reveal an environmental impact on this newly discovered epigenetic mark in the brain and represent a critical step toward understanding stress-related epigenetic mechanisms that alter gene expression and can lead to the development of psychiatric disorders.
Keywords: acute stress, epigenetics, DNA methylation, 5-hydroxymethylcytosine, gene expression
Introduction
Environmental stress, particularly via its effects on limbic structures such as the hippocampus, is among the most important contributors to increased susceptibility to develop anxiety and depression related disorders1,2. An acute stress response involves activation of the hypothalamic pituitary adrenal (HPA) axis, which results in the sequential release of corticotrophin releasing factor (CRF), adrenocorticotropic hormone, and cortisol. The hippocampus is particularly important because receptors for both CRF and cortisol (e.g., the glucocorticoid receptor) are present in the hippocampus, which suggests that these stress mediators can directly influence neuronal activity and facilitate the onset of neuropsychiatric disorders3–5. The negative feedback inhibition produced upon binding of glucocorticoids to their receptors in the hippocampus is critical for a healthy stress response. Environmentally sensitive epigenetic modifications that are disrupted in response to traumatic experience and stress are emerging as important factors in the long-term biological trajectories leading to stress-related psychiatric disorders6. The hippocampus was the first brain region to show an epigenetic response to an exogenous stimulus when alterations in histone modifications and gene expression were found following a single acute restraint stress7,8. Elucidating the molecular mechanisms by which an acute stressor contributes to functional and molecular changes in the brain is needed to improve our understanding of complex behaviors and the origins of psychiatric disorders.
DNA methylation is an epigenetic modification with important roles in chromatin remolding, gene silencing, embryonic development, cellular differentiation, and the maintenance of cellular identity9–12. Traditional studies of DNA methylation have focused on the dynamic variation of a methyl group on cytosine (i.e., 5-methylcytosine; 5mC), which has been linked to neurological disorders as well as psychiatric disorders, including depression, anxiety, post- traumatic stress disorders (PTSD) and schizophrenia13–16. We and others have shown that this modification is a key factor in experience-dependent plasticity such as the expression of an anxious temperament disposition, an important risk factor for the development of anxiety and depression17,18. Despite these connections of 5mC to human psychiatric disorders, it was recently shown that 5mC can be successively oxidized by the ten-eleven translocation family of proteins (Tet1-3), which converts 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine, and 5-carboxylcytosine following exposure to environmental stimuli (e.g., oxidative stress). Since the detection of DNA methylation using traditional sodium bisulfite methodologies cannot distinguish 5mC and 5hmC, previous interpretations of DNA methylation in health and disease should be reconsidered in future studies. Initial studies found that 5hmC is enriched in neuronal cells and is associated with the regulation of neuronal activity19. Genomic studies revealed that 5hmC is located within distal cis-regulatory elements and in the gene bodies of synaptic plasticity-related loci, particularly at intron-exon boundaries, suggesting an important role for 5hmC in coordinating transcriptional activity19,20. These findings have prompted investigations into the potential role(s) of 5hmC in disease, where it appears to function in neurological disorders (e.g., Rett syndrome and Autism)21–23 and neurodegenerative diseases (e.g., Huntington’s and Alzheimer’s)24–27. Recently, we reported that acute stress increases 5hmC levels in the glucocorticoid receptor gene, Nr3c1, suggesting a role for 5hmC in stress28. Together, with recent links to learning and cognition29, studies of 5hmC function(s) have become a significant focus of neuroscience research.
In this study, an established chemical labeling and affinity purification method coupled with high-throughput sequencing technology was used to generate an unbiased genome-wide profile of hippocampal 5hmC following exposure to acute restraint stress. An overlay of these data with genome-wide gene expression data from the same mice found stress-related hydroxymethylation correlated to altered transcript levels. Together, these genomic surveys provide new insight into the potential for functional epigenetic contributions to a stress response and they are intended to serve as a conceptual basis that will facilitate the future study of cellular and brain regional dynamics of 5hmC, especially as it relates to stress and stress recovery. These results demonstrate the power of coupling methylome and transcriptome data to determine the molecular origins of stress-related psychiatric disorders, such as PTSD and anxiety disorders. Here we provide the first genome-wide map of 5hmC in the mouse hippocampus following an acute stress, which reveals known and potentially novel genes contributing to the stress response. These findings establish a role for 5hmC in acute stress and provide insights into the immediate genome-wide neuromolecular response to traumatic events.
Methods
Stress paradigm, Tissue acquisition, and DNA/RNA extraction
Mice were purchased from the Jackson laboratories (Bar Harbor, ME) and maintained on C57BL/6J background. The mice were housed under uniform conditions in a pathogen-free mouse facility with a 12-hour light/dark cycle. Food and water were available ad libitum. All experiments were approved by the University of Wisconsin – Madison Institutional Animal Care and Use Committee (M02529). On the day of the experiment seven-week old male C57BL/6 mice were randomly divided into experimental or control (naïve) groups (N = 6 and 5 per group, respectively). At the same time that the experimental mice were taken from the cage to begin the stress paradigm, the naïve mice also were taken from the cage for brief anesthesia (isoflurane) prior to sacrifice and tissue dissection. Following a published acute stress paradigm that resulted in alterations in histone modifications and gene expression7,8, the experimental mice were restrained for thirty minutes (2 hours after lights-on) using a 50 ml conical vial and then individually housed in a clean cage for one hour. Following this recovery period, experimental animals received brief anesthesia (isoflurane) prior to sacrifice and tissue dissection. Whole brains were extracted and immediately flash frozen in 2-methylbutane and dry ice. Whole hippocampal tissue was excised by micropunch (−0.95 to −3.79 mm posterior to bregma) and approximately 30 milligrams of tissue was homogenized with glass beads (Sigma) and DNA and RNA were extracted using AllPrep DNA/RNA mini kit (Qiagen).
5hmC Enrichment of Genomic DNA
Chemical labeling-based 5hmC enrichment was described previously30. Briefly, a total of 10ug of pooled hippocampal DNA (N=2 mice for all enrichments, except N=1 for one of the enrichment) was sonicated to 300 bp and incubated for 1 hour at 37°C in the following labeling reaction: 1.5 ul of N3-UDPG (2mM); 1.5ulβ of -GT (60uM); and 3ul of 10X β-GT buffer, in a total of 30ul. Biotin was added and the reaction was incubated at 37°C for 2 hours prior to capture on streptavidin-coupled dynabeads (Invitrogen, 65001). Enriched DNA was released from the beads during a 2-hour incubation at room temperature with 100mM DTT (Invitrogen, 15508013), which was removed using a Bio-Rad column (Bio-Rad, 732-6227). Capture efficiency was approximately 5–7% for each sample.
Library Preparation and high-throughput sequencing of genomic DNA
5hmC-enriched libraries were generated using the NEBNext ChIP-Seq Library Prep Reagent Set for Illumina sequencing, according to the manufacturer’s protocol. Briefly, 5hmC-enriched DNA fragments were purified after the adapter ligation step using AMPure XP beads (Agencourt A63880). An Agilent 2100 BioAnalyzer was used to quantify the amplified library DNA and 20-pM of diluted libraries (N=6) were used in each lane for sequencing, which yielded approximately 25 to 35 million uniquely mapped 50 bp reads from each library. 50-cycle single-end sequencing was performed by Beckman Coulter Genomics. Image processing and sequence extraction were done using the standard Illumina Pipeline.
Analysis of 5hmC data: sequence alignment and peak identification
Alignment of sequence data was described previously31. Briefly, FastQ files from each sequenced library were separately aligned to mouse NCBI37v1/mm9 references using Bowtie 0.12.9. Each uniquely mapped read (.bed files), with no more than two mismatches in the first 25 bp, was concatenated to separately achieve experimental and control 5hmC peaks of sequence reads. The MACS software was used to identify peaks of 5hmC content using the default parameters, except for the following: effective genome size = 2.7×109; tag size = 85; band width = 300; P-value cutoff = 1e-5.
DhMR identification and annotations
Data processing of mapped reads was conducted in R using the Bioconductor GenomicRanges and edgeR packages32,33. Read data from individual genomes were converted to GRangesObjects and were extended to 275 basepairs, which is roughly the average fragment length. Peaks under one condition (e.g., control) were defined as the regions that overlap with the peaks from at least two of the biological replicates under the same condition. The total peaks of both control and experimental groups were pooled together as the candidates for differentially hydroxymethylated regions (DhMRs). EdgeR was then used to perform differential enrichment analysis and a negative binomial distribution was used to determine the P-value for each candidate region. Multiple testing was corrected for using a Benjamini and Hochberg false discovery rate (FDR). The candidate regions with a FDR P-value < 0.05 were labeled as DhMRs. DhMR annotation to each chromosome, gene (within 10 kilobases), and genomic structure was performed on nebula.curie.fr. A Manhattan plot was generated using a modified function of the qqman package. DhMRs were then labeled as either hyper or hypo based on the average log fold-changes in normalized read depth between the two groups (calculated by edgeR). A DhMR was hyper if the experimental group has higher average normalized read depth than the control group, otherwise hypo. There were no cases where the two groups had exactly the same average normalized depth.
Gene ontologies
All Gene Ontology (GO) analyses were conducted in R using the “TopGO” package34. This package calculates the P-values for over-representation of a set of genes to specific GO terms, using a hypergeometric test to calculate the P-values given a set of genes and a gene universe (i.e., the super set from which the smaller gene set is drawn). The gene universe used for the GO analysis of the DhMR-associated genes was 13,742 genes. The gene universe used for the GO analysis of the differentially expressed genes was 15,042 genes.
RNA sequencing
Approximately 100ng of total RNA was used for sequence library construction following instructions of NuGen mRNA sample prep kit (cat# 0348). In brief, total RNA was copied into first strand cDNA using reverse transcriptase and random primers. This was followed by second strand cDNA synthesis using DNA Polymerase I and RNaseH. These cDNA fragments went through an end repair process, the addition of a single ‘A’ base, and then ligation of the adapters. These products were gel purified and enriched with PCR to create the final cDNA libraries. The library constructs were run on the bioanalyzer to verify the size and concentration before sequencing on the Illumina HiSeq2500 machine where 100-cycle paired-end sequencing was performed by the University of Wisconsin Biotechnology Center. In total, three libraries (N=2 genomes/library, except N=1 for one library; the same mice and combinations that were used to generate the 5hmC data) were sequenced for each experimental condition.
RNA sequence analysis
In order to elucidate differences in gene expression, we employed the Tuxedo Suite for RNA-seq analysis35. Each testing condition contained three samples each that contributed > 80 million reads with seed lengths of 101 bps per condition. Exon-intron splice-junctions were found using TopHat (v1.0.12) with all parameters set to default, with the exception of the following: the addition of a reference gene model annotation (obtained through the Illumina iGenomes website (https://support.illumina.com/sequencing/sequencing_software/igenome.html); the anchor length was reduced to 6 bp, and the minimum intron length was raised to 70 bp36,37. This analysis produced > 90,000 splice junctions and > 100 million alignments per condition with an average read depth of 2,384.
Aligned RNA-seq reads accepted by our TopHat parameters were then assembled into a set of transcripts and their relative abundances estimated using Cufflinks (v2.1.1)38. All parameters were set to default, except that the reads were quantitated against a reference gene model annotation instead of attempting to find novel transcripts. These testing parameters produced > 30,000 full transcripts per testing condition. Assembled transcriptomes were then merged into one, along with a reference gene model annotation, using Cuffmerge35. This produced a “master” transcriptome with 23,861 known annotated genes. Finally, to find significant differential gene expression between our two working conditions, accepted aligned reads for each condition were quantitated against our produced “master” transcriptome using Cuffdiff (v2.1.1) with all parameters set to default39.
Transcription factor binding site motifs
The DREME suite40 was used to identify enrichments of transcription factor sequence motifs in hyper- and hypo-DhMRs. Motifs that were not found in control-only 5hmC regions, experimental-only 5hmC regions, and all 5hmC regions and that had an E-value <10−3 were considered to be significant. Putative binding factors were predicted using SpaMo directly from the DREME suite software package and are listed in the tables shown with their putative transcription factors.
Statistical analysis
Over- and under-representation of DhMRs (6,909 peaks): notably, peaks were counted twice if associated with multiple genomic structures. For permutation testing, we randomly selected 6,909 peaks (from the full set of 216,650 peaks) and counted how many peaks were in each genomic structure. We termed this the “permuted number” and generated this number 10,000 times. The permutation P-value is the number of times the set of “permuted numbers” exceeded the “actual number,” divided by 10,000. Over- and under-representation of DhMRs on each chromosome was computed similarly by permutation as described for the genomic structures, except for each chromosome. To correct for multiple hypotheses (i.e., 19 chromosomes, X, and Y), the actual proportion of each chromosome was compared with the permuted proportions of any chromosome for each permutation. Permutations for gene structure: The overlap between the DhMRs and the peaks belonging to a particular gene region (e.g., TSS-1.5K, Exon, etc., called the “actual overlap”) was compared with those obtained by randomly selecting an equal number (i.e., equal in number to the DhMRs) and evaluating the overlap (“permuted overlap”). This was performed for all the DhMRs and the DhMRs that contain both hyper-DhMR and hypo-DhMR peaks.
For the comparison of DhMR-associated genes and known stress-related genes, we used a chi-square test on genes associated with the following terms: anxiety; stress; depression; psychosis; major depressive disorder; post traumatic stress disorder; bipolar; fear; schizophrenia; and trauma, which were identified from the GeneCards database; N = 4,348) and the DhMR-associated genes (N = 638). The gene universe consisted of all the genes associated with peaks from the sequence data of all the mice (N = 13,742).
Results
Disruption of the male hippocampal hydroxymethylome following a single acute stress
To determine the genome-wide 5hmC distribution in male mouse hippocampal tissue following acute stress, we randomly divided seven-week-old C57BL/6 male mice into experimental or control groups (N = 4 and 5 per group, respectively). The experimental mice were restrained for thirty minutes followed by a one-hour recovery period prior to tissue collection (Methods). 5hmC containing DNA sequences were enriched from whole hippocampal total DNA using an established chemical labeling and affinity purification method30. Enriched 5hmC containing fragments were sequenced using high-throughput sequencing technology. This approach yielded approximately 25 to 35 million uniquely mapped reads from each genome (Methods; Fig. S1a). Read density mapping showed no visible differences among the chromosomes, except depletion on chromosomes X and Y, which is consistent with previous observations (Fig. S1b)31.
Significant accumulations of uniquely mapped reads represent hydroxymethylated regions throughout the mouse genome. Differentially hydroxymethylated regions (DhMRs) associated with stress were identified in both experimental and control groups (Methods). DhMRs found in experimental mice were classified as hyper-DhMRs and DhMRs absent in experimental mice (present in control mice) were classified as hypo-DhMRs. A total of 458 hyper – and 174 hypo-DhMRs were identified and these loci were distributed across all chromosomes, again with noticeable depletion on X and Y (Fig 1a; Fig. S2; Dataset S1). These data indicate a >2.5-fold increase in 5hmC across the genome following acute stress. Since specific regions of the genome are differentially methylated based on the biological functions of the genes contained within the region, we used permutation testing to determine if the DhMRs are specific to certain regions of the genome by comparing the proportions of DhMRs to the proportions of total detected hydroxymethylated regions in all mice for each chromosome (Methods). This analysis revealed that chromosomes 1, 13 and 18 have disproportionately less DhMRs and that chromosomes 4, 7, 11, and 17 have disproportionately more DhMRs than expected by chance alone (permutation P-value < 0.02; Fig. S2). These data indicate that some chromosomes are more sensitive to hydroxymethylation changes following acute stress.
Fig. 1.
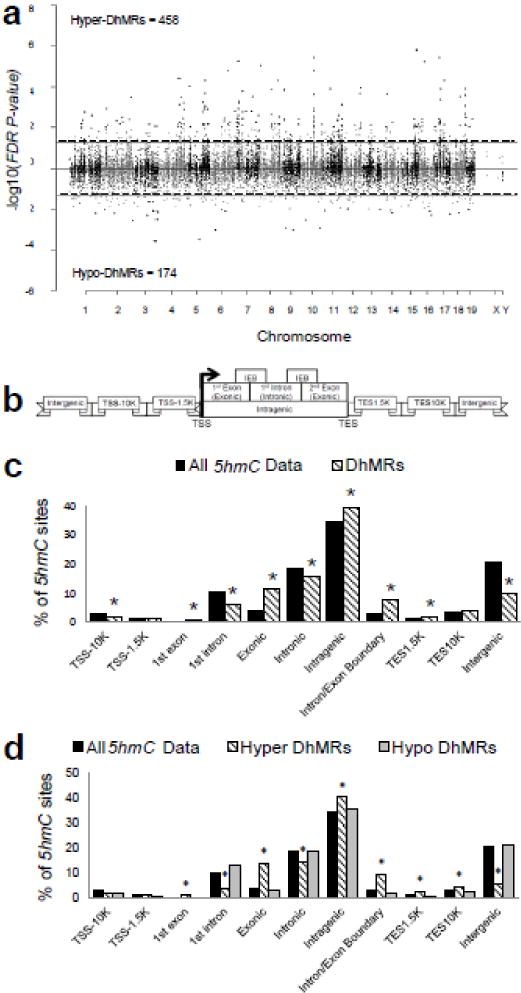
Characterization of 5hmC across standard genomic structures. (a) Modified Manhattan plot of acute stress-associated DhMRs from the mouse hippocampus reveals DhMRs to be distributed across the genome. Positively correlated DhMRs are displayed with a positive -log10 of the P-value and negatively correlated DhMRs are displayed with a negative -log10 of the P-value. Significant DhMRs are displayed outside the dashed lines (P-value < 0.05), while all DhMRs alternate between black and gray to indicate each chromosome. (b) Schematic of the standard regions of the genome. 5hmC and DhMR data were classified into the following genomic locations: 10 kilobases (kb) upstream of the transcription start site (TSS; TSS-10K); 1.5 kb upstream of the TSS (TSS-1.5K); 1st exon; 1st intron; Exonic; Intronic; Intragenic; Intron/Exon boundary (IEB); 1.5 kb downstream of the transcription end site (TES1.5K); 10 kb downstream of the TES (TES10K); or intergenic regions. Hydroxymethylation data were binned into these groups based on the density of sequence reads. (c) The percent distribution (y-axis) of all 5hmC peak data (black) and DhMRs (white) across each genomic region is shown. *Permutation P-value = 0.00723; 0.00011; < 1e-05; < 1e-05; 0.0041; 0.00021; < 1e-05; 0.01874; < 1e-05, respectively. (d) A breakdown of hyper- and hypo-DhMRs in each genomic region. The percent distribution (y-axis) of all 5hmC peak data (black), hyper-DhMRs (gray), and hypo-DhMRs (white) in each genomic region is shown. *Permutation P-value for hyper-DhMRs: = 1.00E-05; < 1e-05; < 1e-05; 0.00097; 7.00E-05; < 1e-05; 0.0032; 0.01369; < 1e-05, respectively.
We next annotated the DhMRs to the nearest gene structure, meaning the DhMRs were assigned to the following genomic elements: 10 kilobases (kb) upstream of the transcription start site (TSS-10K); 1.5 kb upstream of the TSS (TSS-1.5K); 1st exon; 1st intron; exonic; intronic; intragenic; intron/exon boundary (IEB); 1.5 kb downstream of the transcription end site (TES1.5K); 10 kb downstream of the TES (TES-10K); and intergenic regions (Fig. 1b). The distribution of the data indicates that the largest fraction of DhMRs (39.6%) is in the intragenic regions of the genome (Fig. 1c). To determine the relative abundance of DhMRs in each annotated gene region we compared, via permutation testing, the proportion of DhMRs that reside in or around each gene to the total number of hydroxymethylated regions identified by the enrichment method (i.e., all 5hmC data; Methods). This analysis found that several genic regions had a significant abundance of DhMRs, including the 1st exon, exonic, intragenic, intron/exon boundary, and TES1.5K (Fig. 1c; permutation P-value < 0.01). In contrast, the TSS-10K, 1st intron, intronic, and intergenic regions had fewer DhMRs than would be expected by chance alone (permutation P-value < 0.01; Fig 1c). When the abundance of the hyper and hypo-DhMRs was analyzed separately we found that these genomic region enrichments and depletions could be primarily attributed to hyper-DhMRs (Fig. 1d). For example, a significant abundance of hyper-DhMRs explained the overall abundance of DhMRs in the intragenic region and a significant depletion of hyper-DhMRs explained the deficit in the intronic and intergenic regions (permutation P-value < 0.01; Fig. 1d). These findings indicate that fluctuations in hyper-DhMRs cause the majority of significant changes in each genomic region following acute stress. Together, these data indicate that acute stress-related hydroxymethylation primarily resides in genic regions of the genome and suggest that acute stress-related changes are not randomly distributed throughout the genome.
Annotation of DhMRs to genes
Annotation of the DhMRs to genes revealed 638 genes with altered hydroxymethylation following acute stress, including 470 and 166 genes that have hyper- and hypo-DhMRs, respectively (Dataset S2). Thirty-one genes were found to have more than one DhMR and two genes were identified with both hyper- and hypo-DhMRs, meaning these genes contained regions with both increases (hyper) and decreases (hypo) in 5hmC following acute stress (Dataset S2). Notably, the average distance between inversely abundant DhMRs was >500 kb, suggesting that when found on the same gene hyper- and hypo-DhMRs are not located near each other and may have unique roles in response to acute stress.
DhMRs identify known and potentially novel stress-related genes
An initial observation of the 638 DhMR-associated genes suggested that they contained an enrichment of genes previously implicated in stress such as Notch pathway genes (Notch1, Notch2, and Notch3), Irs2, and Crebbp41–43. As a means to examine whether the DhMR-associated genes are enriched for stress related genes, we compared them to a list of known stress-related genes that was generated using the GeneCards database and terms associated with stress (e.g., psychosis, anxiety, fear, PTSD, and depression; N = 4,348 genes; Methods). This comparison found that a significant number of the DhMR-associated genes (N = 240 of 638) are known stress-related genes, which indicates that stress-related genes harbor DhMRs following exposure to acute stress and suggest that 5hmC has a molecular role in response to stress. Moreover, these findings also revealed potentially novel genes responding to acute stress (i.e., a subset of the non-stress-related genes (N = 398); Fig. 2a).
Fig. 2.
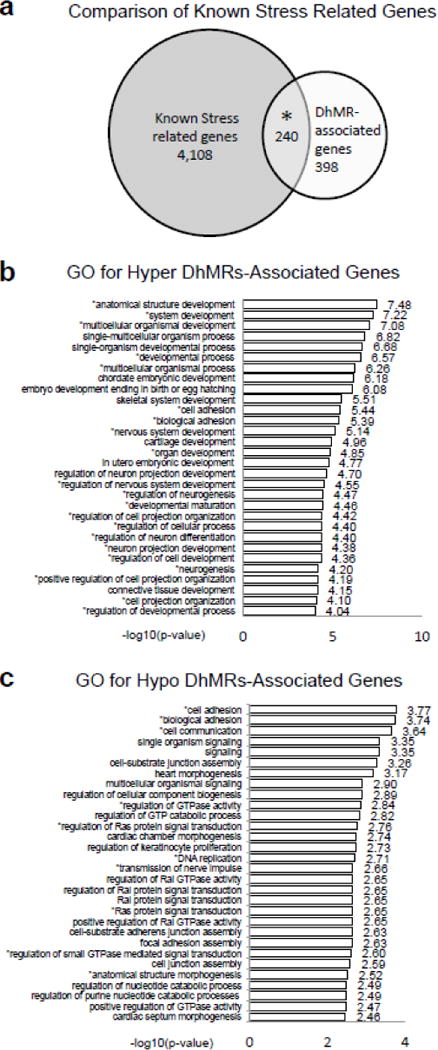
Annotation of DhMRs to genes. (a) Comparison of known stress related genes. Venn diagram showing the proportion and significant overlap of DhMR-associated genes (white; N = 3,361) and known stress related genes (grey: N = 4,348). *P-value = 0.0001. (b–c) Top 30 gene ontological (GO) biological processes associated with the hyper-(b) and hypo-(c) DhMR-associated genes. X-axis shows the −log10 of the P-value, determined by a Fischer test. * indicates neuronal related pathways, as previously determined64.
To further characterize the genes and pathways altered by acute stress, we next examined the gene ontologies (GO) of the 638 DhMR-associated genes and found a significant enrichment of neuronal ontological terms for both the hyper- and hypo-DhMR-associated genes. While the GO terms for hyper-DhMR-associated genes included nervous system development, regulation of neuron differentiation and projection, and regulation of cellular process (Fig. 2b; permutation P-value < 0.01; Table 1; Methods), the GO terms for hypo-DhMR-associated genes differed, including cell adhesion and communication, regulation of GTPase activity and Ras protein signal transduction (Fig. 2c; permutation P-value < 0.01; Table 2; Methods). Despite the differences among these terms, many of the hyper- and hypo-DhMR-associated genes that contributed to the overrepresentation of them have been previously implicated in stress-related disorders, including glutamate receptors (e.g., Grik3 and Grik5), Nrxn1, Irs2 (Tables 1 and 2)44–46. Together, these findings suggest that hippocampal DhMRs are associated with known stress-related neurodevelopmental pathways.
Table 1.
Neuronal enriched gene ontology of biological processes on Hyper DhMR-associated genes
| Gene Ontology Term | * P-value | Significant GO genes |
|---|---|---|
| anatomical structure development | 1.70E-05 | Akt1, Apba2, Apba3, Cebpb, Ctnnb1, Cyr61, Dusp3, Ell, Ercc2, Erf, Gdf1, Gli2, Ints1, Jag2, Kat2a, Krt19, Mgat1, Ncor2, Notch1, Pkd1, Rxra, Rxrb, Tab1, Tpm1, Vegfa, Wnt7b. Xab2. Zmiz1 |
| system development | 0.00019 | Carm1, Dvl1, Fkbp1a, Hdac4, Men1, Pax7, Pkd1, Plxnd1, Ptprf, Smarcd3, Ticam1, Wfikkn1, Zfpm1 |
| multicellular organismal development | 0.0004 | Akt1.Akt2, Irs2 |
| developmental process | 0.00057 | Akt1, Akt2, Irs1, lrs2 |
| multicellular organismal process | 0.0008 | Akt1, Akt2, Erbb3. Irs1, Irs2 |
| cell adhesion | 0.0011 | Dchs1, Fat4, Plxnb1, Thbs3 |
| biological adhesion | 0.00132 | Actn4, Nr1h2, Nr1h3 |
| nervous system development | 0.00132 | Ctnnb1, Notch1, Vegfa |
| organ development | 0.00145 | Gli2, Hdac4, Men1, Pth1r |
| regulation of nervous system development | 0.00189 | Akt1, Dazap1. Rxra, Rxrb |
| regulation of neurogenesis | 0.00197 | Amigo1, Celsr2, Dchs1, Dscaml1, Fat4, Pcdh1, Ptprf, Ret |
| developmental maturation | 0.00206 | Cyr61, Erf, Wnt7b |
| regulation of cell projection organization | 0.00206 | Ddr1, Dpp4, Fgfr4 |
| regulation of neuron differentiation | 0.00213 | Aatk, Akt1, Bcl11a, Limk1, Mapt, Ntn1, Ret, Tsc2, Vegfa |
| regulation of cellular process | 0.00244 | Adcy6, Akt1, Akt2, Ctnnb1, Esrra, Foxs1, Irs1, Irs2, Notch1, Nr1h2, Nr1h3, Ptprf, Rest, Rxra, Rxrb, Slc27a1,Slc9a1 |
| neuron projection development | 0.00244 | Sstr2, Tsc2, Celsr2, Col1a1, Cyr61, Ddr1, Dpp4, Dscaml1, Epha8. Erbb3, Jag2, Kif26b, Lama5, Notch1, Nuak1, Plxnb1, Prex1, R, et, Srcin1, Tpm1, Vegfa |
| regulation of cell development | 0.00274 | Ctnnb1, Fat4, Kif26b, Klf15, Lamb2, Wnt7b |
| neurogenesis | 0.003 | Aatk, Ddr1, Vegfa |
| positive regulation of cell projection organization | 0.00082 | Aatk, Adam11, Adcy6, Adora2a, Akt1, Akt2, Arfgap3, Arhgap22, Arhgap8, Arhgef17, Arhgef25, Bag6, Bai1, Bai2, Bdkrb2, Carm1, Ccdc88c, Ccnd1, Cdc42ep1, Celsr2, Col1a1, Cpz, Csnk1g2, Cspg4, Ctnnb1, Cyr61, Dchsl, Ddr1, Dhx58, Dlk2, Dock1, Dusp3, Dvl1, Ece1, Epha8, Erbb3, Esrra, Fat4, Fgfr4, Fkbp1a, Fkbp8, Foxs1, Fuz, Gadd45b, Gdf1, Gli2, Gna11, Gpr142, Gpr162, Gpr3, Gprc5a, Grid2ip, Grik3, Grik5, Grina, Grk6, Hap1. Hdac4, Hipk4, Ifitm3, Irs1, Irs2, Itga11, Itpr3, Jag2, Kif26a, Klf16, Lama5, Lck, Lingo1, Lphn1, Lynx1, Map3k14, Mapk11, Mapt, Mchr1, Men1, Mknk2, Myo9b, Nacc2, Notch1, Notch3, Npas1, Npas2, Nr1h2, Nr1h3, Olfr222, Olfr279, Pacsin1, Padi2, Pi4kb, Pkd1, Pkn3, Plekhg3, Plxnb1, Plxnd1, Ppp5c, Prdm16, Prex1, Prrx2, Pth1r, Ptprf, Ptpru, Rab3d, Rasgef1a, Rasgrp2, Ret, Rhof, Rhot2, Rxra, Rxrb, Sez6, Sgsm1, Slc27a1, Slc9a1, Sphk2, Spns2, Sstr2, Stk10, Tab1, Tas1r3, Tbc1d24, Tbc1d9b, Tbx6, Tead3, Ticam1, Tmem109, Tmem145, Tnxb, Trim62, Tsc2, Ulk4, Unc13a, Unc5cl, Vasn, Vegfa, Wnt5b, Wnt7a, Wnt7b, Wnt9a, Wwox |
| cell projection organization | 0.00313 | Atg7, Ccnd1, Celsr2, Ddr1, Nr1h2, Nr1h3, Vegfa |
| regulation of developmental process | 0.00418 | Fgfr4, Nr1h2, Nr1h3 |
P-values were obtained using the Fischer test.
Known stress related genes from the GeneCards database are highlighted in bold
Table 2.
Neuronal enriched gene ontology of biological processes on Hypo DhMR-associated genes
| Gene Ontology Term | * P-value | Significant GO genes |
|---|---|---|
| cell adhesion | 0.00017 | Adam23, Alcam, Braf, Cadm2, Col11a1, Col19a1, Dlg1, Efna5, Efnb2, Fat4, Itgb3, Macf1, Nrcam, Nrxn1, Pcdh8, Ppfibp1, Ppp1r12a, Ptprk, Srgap2, Tesk2, Thbs2, Tnr |
| biological adhesion | 0.00018 | Adam23, Alcam, Braf, Cadm2, Col11a1, Col19a1, Dlg1, Efna5, Efnb2, Fat4, Itgb3, Macf1, Nrcam, Nrxn1, Pcdh8, Ppfibp1, Ppp1r12a, Ptprk, Srgap2, Tesk2, Thbs2, Tnr |
| cell communication | 0.00023 | Ahr, Arhgap32, Arhgef3, Arpp19, Atg4c, Atrn, Bai3, Btnpr1a, Braf, Chrm2, Cnih3, Cnot2, Dlg1, Dusp16, Efna5, Efnb2, Fat4, Gpr85, Gria1, Hey1, Insr, Itgb3, Kcnh1, Klf9, Lepr, Lphn2, Macf1, Mc2r, Mllt3, Murc, Notch2, Nrcam, Nrxn1, Pcdh8, Pde4d, Pde7a, Pde7b, Pdgfc, Ppfibp1, Ppp2r5c, Prkd1, Psd3, Ptprk, Qk, Ralgapa2, Ralgps1, Rap1gds1. Rfx3, Rgs2, Ror1, Scn2a1, Sncaip, Snx13, Sostdc1, Sox7, Srgap2. Ss18, Tas2r134, Tbck, Tbx3, Thbs2, Tnr, Tom1l1, Tox3, Unc13c,Wrn |
| regulation of GTPase activity | 0.00144 | Arhgap32. EfnaS. Murc, Psd3, Ralgapa2, Ralgps1, Rap1gds1, Rgs2, Snx13, Srgap2, Tbck |
| regulation of Ras protein signal transduction | 0.00173 | Arhgef3, Arpp19, Efna5, Murc, Notch2, Psd3, Ralgapa2, Ralgps1, Rap1gds1, Srgap2, Tbck |
| DNA replication | 0.00197 | Cdc6, Insr, Nfia, Orc3, Orc5, Pdgfc, Rbms1, Wrn |
| transmission of nerve impulse | 0.0022 | Atrn, Dlg1, Gria1, Nrcam, Qk, Scn2a1, Tnr |
| Ras protein signal transduction | 0.00225 | Arhgap32. Arhgef3, Arpp19, Efna5, Murc, Notch2, Psd3, Ralgapa2, Ralgps1, Rap1gds1, Srgap2, Tbck |
| regulation of small GTPase mediated signal transduction | 0.00251 | Arhgef3, Arpp19, Efna5, Murc, Notch2, Psd3, Ralgapa2, Ralgps1, Rap1gds1, Srgap2, Tbck |
| anatomical structure morphogenesis | 0.00301 | Ahr, Alcam, Bai3, Bmpr1a, Braf, Cnot2, Col11a1, Dlg1, Efna5, Efnb2, Fal4, Hey1, Insr, Itgb3, Lepr, Macf1, MIII3, Murc, Notch2, Nrcam, Pcdh8, Pdgfc, Prkd1, Qk, Rfx3, Sostdc1, Sox7, Srgap2, Ss18, Tbccdl, Tbx3, Thbs2, Tnr, Zfpm2, Zswim6 |
P-values were obtained using the Fischer test.
Known stress related genes from the GeneCards database are highlighted in bold
Identification of Potentially Functional DhMRs
To gain insight into the potential mechanism(s) for stress-related DhMRs, we used RNA sequencing (RNAseq) to profile gene expression in the hippocampal tissue of the same mice surveyed for 5hmC (see Methods). Comparison of transcript levels in experimental and control animals reveals 415 genes that were altered by an acute stress (P < 0.05; Dataset S6; Methods). Many of these genes were previously shown to be differentially expressed in response to acute stress, and are associated with recent neural activity, including Arc, cfos, Fkbp5, Irs2, Nfkbia, Sgk1 (Dataset S6)7,46–48. Examination of the gene ontologies of these 415 differentially expressed genes found a significant enrichment of neuronal ontological terms, including sensory perception of chemical stimulus and defense response (permutation P-value = 0.01; Dataset S7; Methods). DhMRs associated with differentially expressed genes represent candidate functional DhMRs that they may have a direct role in gene regulation. An overlay of the DhMR data with the differentially expressed genes revealed eleven potentially functional DhMRs in genes with documented roles in stress and/or stress-induced psychiatric related disorders, including Irs2, Gadd45b, and Klf15 (Fig. 3; Dataset S7)46,49,50. Notably, these data also revealed functional DhMRs in genes that have an uncharacterized role in stress but have been implicated in mental illness, such as Ulk4 (Fig. 3)51. Together, these data suggest that 5hmC is correlated to altered gene expression in response to acute stress.
Fig. 3.
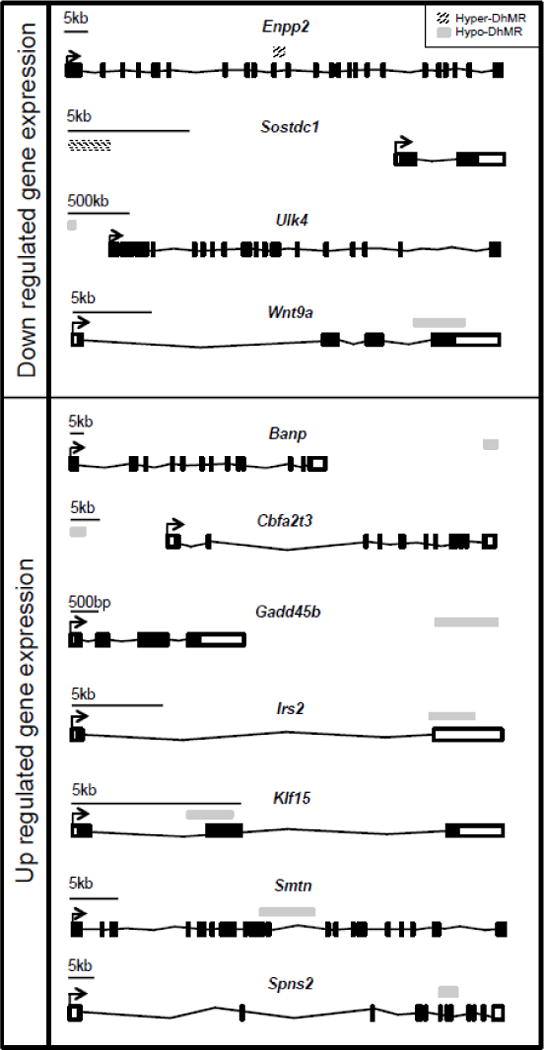
Potentially functional DhMR-associated genes. Shown are the eleven DhMR-associated genes that are either down regulated (top panel) or up regulated (bottom panel) following exposure to acute stress. The name of each gene is shown above the gene schematic, which depicts the relative location of each transcription start site (broken arrow), exon (white and black boxes), intron (black line connecting exons), and DhMR (stripped (hyper) or grey (hypo) box above each gene). A relative scale bar is shown for each gene.
Syndromic forms of mental illness can involve the disruption of transcription factor function52,53. Thus we investigated whether the stress-related DhMRs contained enrichments of known transcription factor binding sequences, compared to the total 5hmC peaks, using the DREME suite software package (Discriminative Regular Expression Motif Elicitation)40. This analysis found five sequence motifs significantly associated with the stress-related DhMRs (Fig. 4). Collectively these sequence motifs preferentially bind to ten transcription factors, many of which have links to stress-related behaviors and disorders, such as Tcf3, Gmeb2, and Olig1 (see Discussion)54–56. Subsequent examination of the sequence data for each DhMR residing in the acute stress-related differentially expressed genes revealed that all candidate functional DhMRs contained at least one of the enriched transcription factor binding sequence motifs (Fig. S3). These findings suggest that 5hmC may regulate the expression of neuronal genes following exposure to acute stress by modulating the binding or function of transcription factors.
Fig. 4.
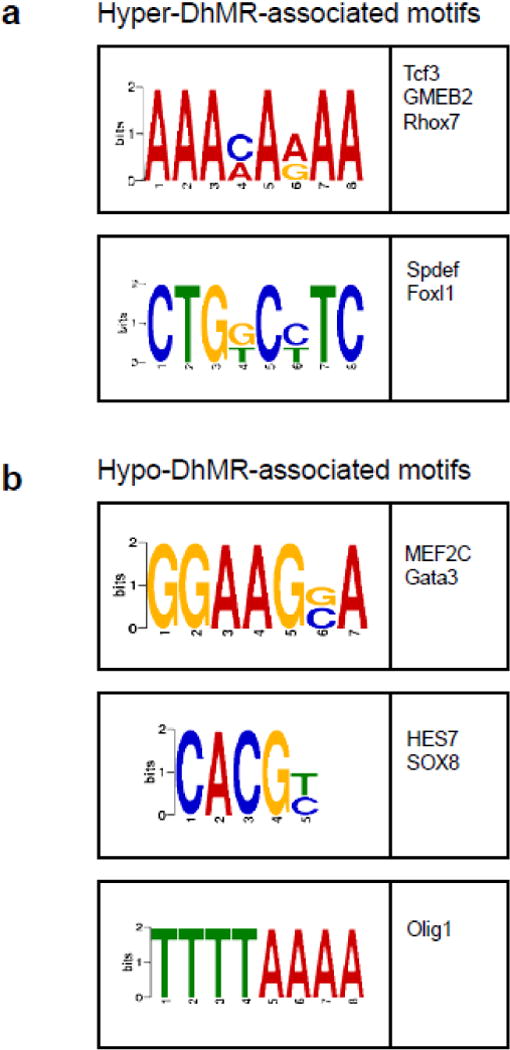
Characterization of potential role(s) for DhMRs in gene expression. (a–b) Identification of DhMR-associated sequence motifs and their putative transcription factors. The DREME suite was used to predict the sequence motifs in hyper-(a) and hypo-(b) DhMRs. Motifs with E-value <10−3 were considered to be significant. Putative binding factors were predicted using SpaMo directly from the DREME suite software package and are listed in the tables shown with their putative transcription factors.
Discussion
Here we report the identification of genome-wide DhMRs in DNA from the hippocampus of male mice exposed to acute stress followed by a one-hour recovery. Importantly, these DhMRs significantly reside in known stress-related genes, which serves to validate a role for 5hmC in response to stress and also reveals potentially novel stress-related genes. Together, the DhMRs identified here represent a framework that will facilitate the future study of the complex interactions between the genes involved in stress and stress recovery. Moreover, a clearer understanding of the molecular mechanisms regulating the expression of these genes may provide potentially modifiable substrates that ultimately could be targeted to prevent the onset of some forms of psychiatric-related disorders.
The DhMRs revealed a role for 5hmC in known and potentially novel genes in the stress pathway, which suggests that 5hmC is marking biologically relevant plasticity genes in response to acute stress. For example, the DhMR-associated genes include neurotransmission genes (e.g., glutamate receptors, which have been shown to be a core feature of mental illnesses57 (Grik3, Grik5, Grina, Gria1, and Grip1), and potassium channels, which have been shown to induce anxiety-like behaviors in mice58 (Kcnc3, Kcnd3, Kcnh1, Kcnj16, Kcnk1, Kcnq1, and Kcnv2)) and genes in the Notch pathway, which regulate the development of the central nervous system (Notch1, Notch2, Notch3 and Jag2)54, the latter of which is consistent with our earlier study that identified functional 5mC (Jag1) in relation to the development of anxiety and depressive disorders17. In addition, four members of the Kruppel-like factor (KLF) family of transcription factors contain DhMRs (e.g., Klf3, Klf9, Klf15, and Klf16). These transcription factors are required for late phase neuronal maturation in developing dentate gyrus during adult hippocampal neurogenesis59. While murine studies find associations with Klf-9 and anxiety, human studies link Klf-11 with chronic stress and depressive disorders59,60. A common function of many of the DhMR-associated genes involves their role in general cell growth processes, including cell adhesion (e.g., cadherin and protocadherin gene families) and cellular developmental processes (Bcl11a, Bcl11b, and Gdf1). Together, these data demonstrate that a remarkable number of known stress-related genes are associated with changes in 5hmC abundance and implicate a role for genes not known to respond to acute stress.
A descriptive analysis of the hydroxymethylation events found in the hippocampus revealed that the majority of the hydroxymethylation is located in the genic structures (i.e., within the gene body), which is consistent with previous reports of genomic distribution of 5hmC31. However, nearly 20% of the total hydroxymethylation was found in the intergenic region. Despite this somewhat diverse distribution of total 5hmC, the vast majority of DhMRs are found in the genic structures. Notably, nearly all the genomic regions investigated (9 of 11) contained significant shifts in their 5hmC distribution following acute stress. For example the significant enrichment in the intragenic region and depletion in the intergenic region suggests that acute stress redistributes 5hmC into gene bodies. Enrichment in exons and intron/exon boundary regions and depletion in intronic regions may further suggest that 5hmC is moving towards areas associated with gene expression, potentially leading to transcriptional modifications. Finally, we also found that the region just downstream of the gene (TES1.5K) was enriched for DhMRs. This finding may indicate that 5hmC changes in these regions contribute to direct and/or indirect molecular mechanism(s) that include regulatory sites for transcription factors such as those we found associated with the DhMRs.
Comparison of methylome and transcriptome data found genes with potentially functional DhMRs, which extends previous studies that lacked the identity of specific molecular mechanism(s) that could be regulating stress-related expression changes. We were encouraged that ~20% (11/55) of the differentially expressed genes contain DhMRs. Although we only identified eleven candidate functional DhMRs, it is tempting to speculate about the relationships between 5hmC abundance and gene expression that are beginning to emerge. For example, the majority of candidate functional DhMRs are hyper-DhMRs (9/11) that are associated with an increase in gene expression (7/11). In addition, if we only consider the candidate functional DhMRs that overlap an intron/exon boundary (N = 6), we found that 5 of the 6 are associated with an increase in gene expression, perhaps further suggesting that 5hmC facilitates transcriptional modifications. On the other hand, DhMRs that are not correlated to changes in gene expression may reveal that these methylation levels mediate the action of a noncoding, but functional, RNA found in the genome. Noncoding RNAs are highly expressed in the brain and are involved in neurodevelopment and neuroplasticity-related functions; their dysregulation has been described in numerous neurological diseases61. Hence, future studies investigating stress related 5hmC levels need to consider the role of 5hmC in non-coding RNA expression. Interestingly, when we removed the FDR correction from the DhMR data (i.e., raw P < 0.05) we found a hyper-DhMR in the glucocorticoid receptor gene (Nr3c1), which is consistent with our previous finding28, and functional DhMRs in additional well-known stress related genes, including Fkbp5 and Sgk147,48. However, because the role of hydroxymethylation in gene regulation is not fully characterized, these data warrant a deeper investigation of the role of 5hmC on individual gene expression changes caused by a novel short stress. It will be important for this future study to also seek to characterize the DNA methylation and expression profiles from distinct hippocampal subregions and cell types.
Several of the transcription factors whose binding sites are enriched among the DhMRs have known roles in neurogenesis and neurological activities, suggesting that 5hmC may play a role in transcription factor binding in response to acute stress. For example, transcription factor 3 (Tcf3) is a neurodevelopmental transcription factor that directly enhances the expression of Hes1, which regulates the development of the central nervous system through Notch signaling54. The transcription factor glucocorticoid modulatory element binding protein 2 (Gmeb2) has intrinsic transactivation activity and can modify the induction properties of glucocorticoid receptors55. Moreover, its binding and subsequent induction properties were shown to be sensitive to DNA methylation in numerous human promoters62. DNA methylation-sensitive gene induction properties are consistent with our finding that acute stress-related candidate functional DhMRs contain Gmeb2 sequence motifs. Thus, since Enpp2, Sostdc1, Ulk4, Smtn, and Spns2 each contain Gmeb2 binding sites, it is of high interest to functionally test the relationship between 5hmC and Gmeb2 binding in the expression of these genes. Finally, oligodendrocyte transcription factor 1 (Olig1) function is required for oligodendrocyte differentiation in the mouse brain and has become an important focus in psychiatric disorders, including major depressive disorder, bipolar disorder, and schizophrenia56,63. Notably, none of the genes that encode these transcription factors contain DhMRs, suggesting that 5hmC does not disrupt their expression following exposure to acute stress. Clearly, further studies are needed to definitively determine the role of DhMRs within transcription factor binding sites in response to stress.
Previous studies have identified differential distributions of 5hmC associated with neuronal development and the pathogenesis of neurological and neurodegenerative diseases21–27. Whereas these changes represent molecular consequences over a lifespan, here we show that in a relatively short amount of time (i.e., 90 minutes) genome-wide changes in 5hmC can occur, indicating that 5hmC is more dynamic that was previously appreciated. Moreover, we found that 20% of the changes in 5hmC following acute stress are correlated with altered transcript levels, supporting an environmentally-sensitive functional role for 5hmC. While there is still much to learn about 5hmC and its contributions to stress response, these results can serve as a benchmark for future comparison of trauma type and duration and it will be important to relate these findings to chronic stress paradigms and to behavioral phenotypes. In addition, it is of great interest to examine the response of the female epigenome to stress and compare it to that of the male response, which might provide insight into the gender differences of psychiatric-related disorders. These findings support the emergence of 5hmC in environmentally-induced psychopathy, which is important, because at this time there is a substantial need to identify and characterize neuromolecular substrates contributing to psychiatric disorders that ultimately could be targeted to prevent individuals from developing full-blown anxiety and depressive disorders.
Supplementary Material
Acknowledgments
The authors would like to thank Kim Sorens, the WISPIC animal facility, and the UW biotechnology center. This work was supported in part by the University of Wisconsin-Madison department of Psychiatry (RSA), the University of Wisconsin Neuroscience training grant T32-GM007507 (SL), NARSAD Young Investigator Grant from the Brain & Behavioral Research Foundation #22669 (LP). All experiments in this study were approved by the Institutional Animal Care and Use Committee at the University of Wisconsin (UW IACUC protocol #M02529).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no competing financial interests.
References
- 1.McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiological reviews. 2007;87:873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- 2.Conrad CD. Chronic stress-induced hippocampal vulnerability: the glucocorticoid vulnerability hypothesis. Reviews in the neurosciences. 2008;19:395–411. doi: 10.1515/revneuro.2008.19.6.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Y, et al. Modulation of dendritic differentiation by corticotropin-releasing factor in the developing hippocampus. Proc Natl Acad Sci U S A. 2004;101:15782–15787. doi: 10.1073/pnas.0403975101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yan XX, Toth Z, Schultz L, Ribak CE, Baram TZ. Corticotropin-releasing hormone (CRH)-containing neurons in the immature rat hippocampal formation: light and electron microscopic features and colocalization with glutamate decarboxylase and parvalbumin. Hippocampus. 1998;8:231–243. doi: 10.1002/(SICI)1098-1063(1998)8:3<231::AID-HIPO6>3.0.CO;2-M. doi:10.1002/(SICI)1098-1063(1998)8:3<231∷AID-HIPO6>3.0.CO;2-M. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Regev L, Baram TZ. Corticotropin releasing factor in neuroplasticity. Frontiers in neuroendocrinology. 2014;35:171–179. doi: 10.1016/j.yfrne.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klengel T, Pape J, Binder EB, Mehta D. The role of DNA methylation in stress-related psychiatric disorders. Neuropharmacology. 2014;80:115–132. doi: 10.1016/j.neuropharm.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 7.Gray JD, Rubin TG, Hunter RG, McEwen BS. Hippocampal gene expression changes underlying stress sensitization and recovery. Molecular psychiatry. 2014;19:1171–1178. doi: 10.1038/mp.2013.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hunter RG, McCarthy KJ, Milne TA, Pfaff DW, McEwen BS. Regulation of hippocampal H3 histone methylation by acute and chronic stress. Proc Natl Acad Sci U S A. 2009;106:20912–20917. doi: 10.1073/pnas.0911143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 10.Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- 11.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. doi: 10.1038/nature05918. doi:nature05918 [pii]10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 12.Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–919. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
- 13.Grayson DR, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci U S A. 2005;102:9341–9346. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abdolmaleky HM, et al. Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Hum Mol Genet. 2006;15:3132–3145. doi: 10.1093/hmg/ddl253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuratomi G, et al. Aberrant DNA methylation associated with bipolar disorder identified from discordant monozygotic twins. Molecular psychiatry. 2008;13:429–441. doi: 10.1038/sj.mp.4002001. [DOI] [PubMed] [Google Scholar]
- 16.Poulter MO, et al. GABAA receptor promoter hypermethylation in suicide brain: implications for the involvement of epigenetic processes. Biological psychiatry. 2008;64:645–652. doi: 10.1016/j.biopsych.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 17.Alisch RS, et al. Differentially methylated plasticity genes in the amygdala of young primates are linked to anxious temperament, an at risk phenotype for anxiety and depressive disorders. J Neurosci. 2014;34:15548–15556. doi: 10.1523/jneurosci.3338-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pidsley R, et al. Methylomic profiling of human brain tissue supports a neurodevelopmental origin for schizophrenia. Genome biology. 2014;15:483. doi: 10.1186/s13059-014-0483-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao B, Jin P. Cytosine modifications in neurodevelopment and diseases. Cell Mol Life Sci. 2014;71:405–418. doi: 10.1007/s00018-013-1433-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khare T, et al. 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nature structural & molecular biology. 2012;19:1037–1043. doi: 10.1038/nsmb.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Al-Mahdawi S, Virmouni SA, Pook MA. The emerging role of 5-hydroxymethylcytosine in neurodegenerative diseases. Frontiers in neuroscience. 2014;8:397. doi: 10.3389/fnins.2014.00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–1430. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhubi A, et al. Increased binding of MeCP2 to the GAD1 and RELN promoters may be mediated by an enrichment of 5-hmC in autism spectrum disorder (ASD) cerebellum. Translational psychiatry. 2014;4:e349. doi: 10.1038/tp.2013.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Villar-Menendez I, et al. Increased 5-methylcytosine and decreased 5-hydroxymethylcytosine levels are associated with reduced striatal A2AR levels in Huntington’s disease. Neuromolecular medicine. 2013;15:295–309. doi: 10.1007/s12017-013-8219-0. [DOI] [PubMed] [Google Scholar]
- 25.Wang F, et al. Genome-wide loss of 5-hmC is a novel epigenetic feature of Huntington’s disease. Hum Mol Genet. 2013;22:3641–3653. doi: 10.1093/hmg/ddt214. [DOI] [PubMed] [Google Scholar]
- 26.Chouliaras L, et al. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiology of aging. 2013;34:2091–2099. doi: 10.1016/j.neurobiolaging.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bradley-Whitman MA, Lovell MA. Epigenetic changes in the progression of Alzheimer’s disease. Mech Ageing Dev. 2013;134:486–495. doi: 10.1016/j.mad.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li S, et al. Hippocampal increase of 5-hmC in the glucocorticoid receptor gene following acute stress. Behavioural brain research. 2015;286:236–240. doi: 10.1016/j.bbr.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X, et al. Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc Natl Acad Sci U S A. 2014;111:7120–7125. doi: 10.1073/pnas.1318906111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song CX, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nature biotechnology. 2011;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szulwach KE, et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nature neuroscience. 2011;14:1607–1616. doi: 10.1038/nn.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawrence M, et al. Software for computing and annotating genomic ranges. PLoS computational biology. 2013;9:e1003118. doi: 10.1371/journal.pcbi.1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.topGO. topGO: Enrichment analysis for Gene Ontology. 2010 [Google Scholar]
- 35.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi Y, Sha G, Sun X. Genome-wide study of NAGNAG alternative splicing in Arabidopsis. Planta. 2014;239:127–138. doi: 10.1007/s00425-013-1965-2. [DOI] [PubMed] [Google Scholar]
- 38.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trapnell C, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nature biotechnology. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bailey TL. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011;27:1653–1659. doi: 10.1093/bioinformatics/btr261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cope JL, et al. Differential contribution of CBP:CREB binding to corticotropin-releasing hormone expression in the infant and adult hypothalamus. Stress. 2014;17:39–50. doi: 10.3109/10253890.2013.806907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldberg LB, Aujla PK, Raetzman LT. Persistent expression of activated Notch inhibits corticotrope and melanotrope differentiation and results in dysfunction of the HPA axis. Dev Biol. 2011;358:23–32. doi: 10.1016/j.ydbio.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anacker C, et al. Glucocorticoid-related molecular signaling pathways regulating hippocampal neurogenesis. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2013;38:872–883. doi: 10.1038/npp.2012.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gray AL, Hyde TM, Deep-Soboslay A, Kleinman JE, Sodhi MS. Sex differences in glutamate receptor gene expression in major depression and suicide. Molecular psychiatry. 2015;20:1139. doi: 10.1038/mp.2015.114. [DOI] [PubMed] [Google Scholar]
- 45.Grayton HM, Missler M, Collier DA, Fernandes C. Altered social behaviours in neurexin 1alpha knockout mice resemble core symptoms in neurodevelopmental disorders. PLoS One. 2013;8:e67114. doi: 10.1371/journal.pone.0067114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fox AS, et al. Central amygdala nucleus (Ce) gene expression linked to increased trait-like Ce metabolism and anxious temperament in young primates. Proc Natl Acad Sci U S A. 2012;109:18108–18113. doi: 10.1073/pnas.1206723109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klengel T, et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nature neuroscience. 2013;16:33–41. doi: 10.1038/nn.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anacker C, et al. Role for the kinase SGK1 in stress, depression, and glucocorticoid effects on hippocampal neurogenesis. Proc Natl Acad Sci U S A. 2013;110:8708–8713. doi: 10.1073/pnas.1300886110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sato H, et al. Large-scale analysis of glucocorticoid target genes in rat hypothalamus. Journal of neurochemistry. 2008;106:805–814. doi: 10.1111/j.1471-4159.2008.05489.x. [DOI] [PubMed] [Google Scholar]
- 50.Kigar SL, Chang L, Auger AP. Gadd45b is an epigenetic regulator of juvenile social behavior and alters local pro-inflammatory cytokine production in the rodent amygdala. Brain, behavior, and immunity. 2015;46:60–69. doi: 10.1016/j.bbi.2015.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lang B, et al. Recurrent deletions of ULK4 in schizophrenia: a gene crucial for neuritogenesis and neuronal motility. Journal of cell science. 2014;127:630–640. doi: 10.1242/jcs.137604. [DOI] [PubMed] [Google Scholar]
- 52.Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat Rev Genet. 2006;7:415–426. doi: 10.1038/nrg1878. [DOI] [PubMed] [Google Scholar]
- 53.Gharani N, Benayed R, Mancuso V, Brzustowicz LM, Millonig JH. Association of the homeobox transcription factor, ENGRAILED 2, 3, with autism spectrum disorder. Molecular psychiatry. 2004;9:474–484. doi: 10.1038/sj.mp.4001498. [DOI] [PubMed] [Google Scholar]
- 54.Ikawa T, Kawamoto H, Goldrath AW, Murre C. E proteins and Notch signaling cooperate to promote T cell lineage specification and commitment. The Journal of experimental medicine. 2006;203:1329–1342. doi: 10.1084/jem.20060268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen J, He Y, Simons SS., Jr Structure/activity relationships for GMEB-2: the second member of the glucocorticoid modulatory element-binding complex. Biochemistry. 2004;43:245–255. doi: 10.1021/bi035311b. [DOI] [PubMed] [Google Scholar]
- 56.Dai J, Bercury KK, Ahrendsen JT, Macklin WB. Olig1 function is required for oligodendrocyte differentiation in the mouse brain. J Neurosci. 2015;35:4386–4402. doi: 10.1523/JNEUROSCI.4962-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nature reviews Neuroscience. 2012;13:22–37. doi: 10.1038/nrn3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wallace DL, et al. CREB regulation of nucleus accumbens excitability mediates social isolation-induced behavioral deficits. Nature neuroscience. 2009;12:200–209. doi: 10.1038/nn.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scobie KN, et al. Kruppel-like factor 9 is necessary for late-phase neuronal maturation in the developing dentate gyrus and during adult hippocampal neurogenesis. J Neurosci. 2009;29:9875–9887. doi: 10.1523/JNEUROSCI.2260-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Duncan J, Johnson S, Ou XM. Monoamine oxidases in major depressive disorder and alcoholism. Drug discoveries & therapeutics. 2012;6:112–122. [PubMed] [Google Scholar]
- 61.Qureshi IA, Mehler MF. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nature reviews Neuroscience. 2012;13:528–541. doi: 10.1038/nrn3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Burnett E, Christensen J, Tattersall P. A consensus DNA recognition motif for two KDWK transcription factors identifies flexible-length, CpG-methylation sensitive cognate binding sites in the majority of human promoters. J Mol Biol. 2001;314:1029–1039. doi: 10.1006/jmbi.2000.5198. [DOI] [PubMed] [Google Scholar]
- 63.Mosebach J, et al. Increased nuclear Olig1-expression in the pregenual anterior cingulate white matter of patients with major depression: a regenerative attempt to compensate oligodendrocyte loss? Journal of psychiatric research. 2013;47:1069–1079. doi: 10.1016/j.jpsychires.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 64.Geifman N, Monsonego A, Rubin E. The Neural/Immune Gene Ontology: clipping the Gene Ontology for neurological and immunological systems. BMC Bioinformatics. 2010;11:458. doi: 10.1186/1471-2105-11-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.