

Abstract
The host response to pathogens through nuclear factor κB (NF-κB) is an essential defense mechanism for eukaryotic organisms. NF-κB-mediated host responses inhibit bone and other connective tissue synthesis and are thought to affect the transcription of matrix proteins through multiple indirect pathways. We demonstrate that inhibiting NF-κB in osteoblasts increases osteocalcin expression in vivo in mice with periodontal disease. Mutating NF-κB binding sites on osteocalcin (OC) or bone sialoprotein (Bsp) promoters rescues the negative impact of NF-κB on their transcription and that NF-κB can inhibit Wnt- and Bmp-induced OC and Bsp transcription, even when protein synthesis is inhibited, indicating a direct effect of NF-κB. This inhibition depends on p65-p50 NF-κB heterodimer formation and deacetylation by HDAC1 but is not affected by the noncanonical NF-κB pathway. Moreover, NF-κB reduces Runx2 and β-catenin binding to OC/Bsp promoters independently of their nuclear localization. Thus, inflammatory signals stimulate the direct interaction of NF-κB with response elements to inhibit binding of β-catenin and Runx2 binding to nearby consensus sites and reduce expression of matrix proteins. This direct mechanism provides a new explanation for the rapid decrease in new bone formation after inflammation-related NF-κB activation.
Keywords: BONE FORMATION, BMP, INFLAMMATION, MATRIX PROTEINS, OSTEOBLASTS, TNFA, NF-KB, WNT
Introduction
In response to inflammation, host defense mechanisms result in the secretion of inflammatory cytokines, leading to the activation of the nuclear factor κB (NF-κB) pathway.(1) NF-κB plays a central role in the cellular stress response and in inflammation by controlling the expression of a network of inducers and effectors that define responses to pathogens.(2) This pathway stands as the central regulator of inflammatory factors mediating crucial events in response to inflammation, leading to inhibition of matrix synthesis.(3) The canonical NF-κB pathway involves formation of homo- or heterodimers of p65 (RelA) and p50.(4) In the resting stage, NF-κB subunits exist as a cytoplasmic trimer that includes IkBβ that masks the nuclear localization signal of NF-κB.(5) Inflammatory signals activate IkB-kinase (IKK), which phosphorylates IkB and target it for degradation, releasing NF-κB and facilitating its nuclear localization and subsequent DNA binding.
Inflammatory mediators may negatively impact osteoblasts by inhibiting bone formation.(6) Several in vitro and in vivo studies have demonstrated that inflammatory cytokines such as TNFα inhibit matrix protein synthesis.(7,8) Inflammation inhibits bone formation in a number of disease states, including rheumatoid arthritis, and in failing dental and orthopedic implants.(9–12) Inflammatory mediators contribute to impaired bone formation linked to postmenopausal osteoporosis.(13,14) Inflammation inhibits production of bone matrix proteins by interfering with signals that stimulate bone production. One mechanism involves interfering with bone morphogenetic protein (Bmp) signaling by downregulation of Runx2, a transcription factor that stimulates and coordinates osteoblast activity.(15,16) Bmps are required for postnatal bone formation and stimulate expression of matrix proteins osteocalcin (OC) and bone sialoprotein (Bsp).(17–20) Loss of Bmp activity leads to osteopenia, bone fragility, and spontaneous fracture.(21,22)
The Wnt signaling pathway also stimulates production of bone.(23) Activation of Wnt signaling increases nuclear expression of β-catenin, which leads to increased expression of osteocalcin and Bsp.(23) Inflammation interferes with Wnt signaling by increasing expression of Wnt antagonists, Dkk1, or sclerostin (SOST).(24) Dkk1 or SOST prevent the Wnt ligand from binding to its receptor by competitive inhibition.(25,26) Both Dkk1 and SOST expression is stimulated by TNFα, which is mediated at the transcriptional level by NF-κB.(27) A third indirect mechanism through which NF-κB may inhibit expression of bone matrix proteins is by reducing expression of Fos-related antigen-1 (Fra1), which is a positive regulator of bone matrix protein transcription.(6)
In almost all cases, NF-κB is a positive regulator of transcription. More recent studies have also determined that NF-κB may negatively regulate gene transcription. Binding of noncanonical NF-κB subunits to the κB sites leads to repression of chemokines.(28,29) Activation of noncanonical NF-κB directly represses the expression of interferon-β at the promoter level.(30) In these cases, NF-κB exerts a negative effect through activation of the noncanonical pathway, involving the formation of heterodimers consisting of RelB-p52.
Although the impact of NF-κB on bone formation is profound, the only mechanisms known are indirect. We determined whether there were alternative mechanisms to the well-defined indirect effects of NF-κB on expression of bone matrix proteins. Mutating putative NF-κB binding sites on the promoter of OC and Bsp rescued the negative impact of TNFα, indicating that NF-κB can directly interfere with induction of these proteins. Moreover, NF-κB inhibited Bmp-induced Runx2 and Wnt-induced β-catenin binding to consensus response elements to reduce OC and Bsp transcription. This direct mechanism provides new insight into the rapid effect of inflammation on new bone formation, which is important in a number of diseases, including rheumatoid arthritis and periodontal disease.
Materials and Methods
Transgenic male and female mice (Col1α1.IKK-DN) were generated that express a dominant negative IKK under the control of a 2.3-kbp element of the collagen 1α1 promoter that restricts activation of NF-κB in osteoblast lineage cells.(31) Periodontitis was induced by inoculation of P. gingivalis and F. nucleatum and compared with mice inoculated with vehicle alone (2% methylcellulose) as described.(32) Mice were euthanized 6 weeks after oral inoculation. All animal procedures were approved by the Institutional Animal Care and Use Committee.
Preparation of histologic specimens
Specimens were fixed in 4% paraformaldehyde overnight at 4°C and decalcified in 10% EDTA for 3 to 4 weeks. Paraffin-embedded histologic sections were prepared for the region between the first and second and the second and third molars that included the teeth, gingiva, bone, and periodontal ligament as described.(33)
Detection of osteocalcin and NF-κB
Paraffin sections were subjected to antigen retrieval in citrate buffer at 95°C. Sections were incubated with antibody to osteocalcin (Takara, Mountain View, CA, USA) or NF-κB-p65 (Rockland, Gillbertsville, PA, USA). Antibody was localized with biotinylated secondary antibody and avidin-biotin horseradish peroxidase complex (Vector Laboratories, Burlingame, CA, USA). Antibodies were visualized using streptavidin Alexa-546 (Invitrogen, Carlsbad, CA, USA) and counterstained with DAPI. Tyramide signal amplification (PerkinElmer, Waltham, MA, USA) was used to enhance the signal. Fluorescent staining of cuboidal-shaped osteoblastic bone-lining cells, osteocytes in bone, or cells in the gingival connective tissue was observed under 400× magnification of images captured with a Nikon Eclipse 90i microscope (Nikon, Melville, NY, USA) and NIS Elements-AR software (Nikon). Osteocalcin matrix was assessed by immunofluorescence in 0.02 mm of bone adjacent to the edge and the mean fluorescence intensity measured.
Cell culture
MC3T3-E1 murine osteoblastic cells (ATCC, Rockville, MD, USA) were grown in α-MEM (Invitrogen) containing 10% FBS (Atlanta Biologicals, Atlanta, GA, USA) and 1% penicillin/streptomycin (Cellgro, Manassas, VA, USA) at 37°C in 5% CO2. Tumor necrosis factor α (TNFα) (1 ng/mL), bone morphogenetic protein 2 (Bmp2) (100 ng/mL), and Wnt3a (100 ng/mL) were all purchased from Peprotech (Rocky Hill, NJ, USA). Osteoblastic cells were isolated from calvarial fragments from 8-week-old mice. Parietal bone was cut, minced, and placed in digestion media containing collagenase (Sigma-Aldrich, St. Louis, MO, USA) in a 37°C shaker and subjected to multiple digestions. Osteoblasts were isolated by combining digestions 2 to 6(34) and cultured as above.
Expression of bone matrix proteins
For in vitro studies, total RNA was extracted and RT-PCR was carried out using fluorescent cDNA probes from Roche (Indianapolis, IN, USA) and primers from Integrated DNA Technologies (IDT, Coralville, IA, USA). MC3T3 cells were fixed in paraformaldehyde and incubated overnight at 4°C with primary antibody against osteocalcin (Takara) or antibody to bone sialoprotein (Bsp) generously provided by Dr Renny Franceschi (University of Michigan, School of Dentistry). Immunofluorescence was carried out as described above, and cells were examined at 200× magnification to measure mean fluorescence intensity. Total RNA was extracted from the periodontium of the molar teeth and mRNA levels of osteocalcin were assessed by real-time PCR as described.(35,36) Results were normalized to a housekeeping gene, ribosomal protein L32. The experiments were carried out with 6 to 7 animals per group.
Plasmids, siRNA, inhibitors, and transfection
Bsp and osteocalcin luciferase constructs with mouse-specific Bsp- and OC-promoter regions were kindly provided by Dr Renny Franceschi (University of Michigan, School of Dentistry) and Dr Chawnshang Chang (University of Rochester Medical Center).(37,38) pGL3 empty vector and pcDNA-IKK (wild-type) plasmid were obtained from Addgene (Cambridge, MA, USA). Small molecule inhibitors BAY-117082 (10 μM) and parthenolide (10 μM) and siRNA against p65, p50, IKK-beta and HDAC1 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Cells seeded in αMEM media containing 0.3% FBS were transfected with corresponding constructs using Lipofectamine 2000 (Invitrogen), followed by incubation with TNFα (10 ng/mL) and Bmp2 (100 ng/mL) for 48 hours. Luciferase reporter activity was measured using dual-luciferase reporter assay system (Promega, Madison, WI, USA) as described.(39) For mRNA assays, cells were lysed and mRNA was isolated using the RNeasy Mini kit (Qiagen, Valencia, CA, USA) and RTPCR was performed using SYBR Green (Applied Biosystems, Carlsbad, CA, USA) as described.(39)
In situ mutagenesis
Site-directed mutagenesis was carried out on Bsp and osteocalcin luciferase promoter constructs. Putative NF-κB binding sites on the promoters were identified using PROMO v. 8.3 software (Technical University of Catalonia, Barcelona, Spain). Mutants were generated using the QuikChange Lightning multisite-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA). The following primers were used to create site-directed mutagenesis on the Bsp promoter region for each of the three NF-κB binding sites: site 1 (gatatcccagtgtcgggttatttgagggcagggagg), site 2 (gtggatgggtaggtgggttaacaccctcatagaagc), and site 3 (caaagttagtttcctttgcaaacttaggaaatgttc). Similarly, primers used to create site-directed mutagenesis on the osteocalcin promoter region for each of the three NF-κB binding sites are site 1 (attgagactaagactggggtcacagctgcagaattgctca), site 2 (cccacaatgggctaggtccttccccaccaaccac), and site 3 (ttgacataaaactaaccagttcactcccccccaacacaca). MC3T3 cells and primary osteoblasts were cotransfected with corresponding constructs as described above and luciferase activity was measured.
Preparing mini-genes for Bsp promoter luciferase construct
Specific regions of Bsp promoter construct containing potential NF-κB binding sites (both wild-type and mutated ones) were amplified by PCR. The amplified region was cloned using restriction enzymes, Sac-I and XhoI (NEB, Ipswich, MA, USA). The cloned region of interest was ligated into pGL3-pCMV using T4 ligase (NEB). MC3T3 cells were cotransfected with these cloned mini-constructs and p65-p50 expression vectors using Lipofectamine 2000. The next day, transcriptional activity was measured by dual-luciferase assay (Promega).
Inhibition of protein synthesis
MC3T3 cells were stimulated with Bmp2 (100 ng/mL) for 48 hours. Some of the cells were preincubated with cycloheximide (10 μg/mL) (C7698; Sigma) for 1 hour, after which they were incubated with TNFα (10 ng/mL) (Peprotech, Rock Hill, NJ, USA) and cyloheximide for 6 hours. RNA was extracted using Quick-RNA MicroPrep Kit (Zymo Research, Irvine, CA, USA). The RNA was converted to cDNA using High Capacity RNA-to-cDNA Kit (Applied Biosystems), and RTPCR was performed using SYBR Green (Applied Biosystems) as described.(39)
Preparation of lysate
MC3T3 cells were transfected with siRNA and/or stimulated with TNFα (10 ng/mL) for 1 hour. Whole-cell and nuclear lysates were prepared using RIPA buffer and isolation kit, respectively (Thermo Scientific, Rockford, IL, USA). Immunoblotting was carried out as described.(39) For some of the experiments, MC3T3 cells were preincubated with TNFα (10 ng/mL) for 45 minutes, followed by incubation with Bmp2/Wnt3a (100 ng/mL) for 4 hours. Some of the cells were then incubated with cycloheximide (10 μg/mL) for 3 hours. Nuclear and cytosolic lysates were prepared following the manufacturer’s recommendations (Thermo Scientific), and immunoblotting was carried out as described earlier using antibodies against β-catenin or Runx2 (Cell Signaling Technology, Beverly, MA, USA) according to the manufacturer’s recommendations.(35)
Chromatin-immunoprecipitation
MC3T3 cells were transfected with siRNA and/or stimulated with TNFα (10 ng/mL) for 1 hour. ChIP assays were performed using ChIP-IT kit (Active Motif, Carlsbad, CA, USA).(39) Isolated DNA was subjected to real-time PCR using specific primers from IDT.
For some of the experimental studies, the cells were washed and incubated with Wnt3a (100 ng/mL) or Bmp2 (100 ng/mL) for 4 hours. About 45 minutes before stimulation, some of the cells were incubated with TNFα (10 ng/mL). After stimulation, protein synthesis was inhibited by incubating the cells with cycloheximide for 3 hours, after which ChIP assay was carried out. The chromatin was immunoprecipitated using antibodies for p65 (Active Motif), p50 (Cell Signaling Technology), HDAC1 (Active Motif), β-catenin (Cell Signaling Technology), Runx2 (Cell Signaling Technology), or RNAP-II (Millipore, Billerica, MA, USA) using the manufacturer’s recommendations.
Co-immunoprecipitation
MC3T3 cells were transfected with siRNA and/or stimulated with TNFα (10 ng/mL) for 2 hours. Cells were lysed, and co-immunoprecipitation was carried out using Pierce Co-Immunoprecipitation (Co-IP) Kit (Thermo Scientific) using antibodies against p65 and HDAC1 (Active Motif) following the manufacturer’s recommendations. The immune complex was resolved in 4% to 20% SDS-PAGE (Bio-Rad Laboratories, Hercules, CA, USA) and transferred onto PVDF membrane (Thermo Fisher). The membranes were incubated with primary antibodies against p65 (Active Motif), HDAC1 (Active Motif), phosphor-p65 (Thr505) (Santa Cruz Biotechnology), acetyl lysine (Cell Signaling Technology), β-actin (Sigma-Aldrich), or matched control IgG (Santa Cruz Biotechnology) according to the manufacturer’s recommendations.(39) Bands were detected using SuperSignal Chemiluminescent Substrate kit (Pierce, Thermo Scientific).
Statistical analysis
Statistical analyses were performed using SPSS software (SPSS, Chicago, IL, USA). One-way ANOVA was used to compare the differences between groups at a given time point and to compare differences from baseline values with the other time points. The significance level was set at p < 0.05. In vitro experiments were carried out three to four times with similar results. In vivo experiments were analyzed by a double-blind examiner with 6 to 7 specimens per group.
Results
NF-κB-mediated response inhibits bone and matrix protein synthesis
It has been suggested but never established that a significant component of periodontal bone loss is because of the effect of inflammation on inhibiting bone formation.(40) To investigate this issue, periodontitis was induced in transgenic Col1α1.IKK-DN mice (TG) that inhibits NF-κB activation in osteoblast lineage cells,(6,31) and expression of bone matrix proteins was examined. Oral infection stimulated a threefold increase in nuclear localization of the NF-κB subunit p65 in osteoblasts from infected WT mice but not in TG mice (p < 0.05) (Fig. 1A). mRNA expression of osteocalcin, a bone-specific matrix protein, was 72% higher in TG mice compared with WT mice after infection (Fig. 1B). The expression of osteocalcin at the protein level was twofold higher in TG mice (p < 0.05) (Fig. 1C). Thus, periodontal inflammation activates NF-κB in osteoblasts, and this activation suppresses expression of a bone matrix protein. In vitro experiments were carried out to establish that TNFα and IKK transfection, which directly activates NF-κB, inhibits OC and Bsp expression stimulated by Bmp2. TNFα reduced 75% to 85% of the stimulated Bsp and OC expression at the protein level with a similar inhibition caused by transfection with IKK in Bmp2-stimulated cells (p < 0.05) (Fig. 1D–I). A small molecule NF-κB blocker (BAY-117082) rescued the inhibitory effect of both TNFα and IKK transfection (p < 0.05) (Fig. 1D–I). Incubating the cells with TNFα resulted in a 51% to 63% decrease in osteocalcin and Bsp expression in Bmp2-unstimulated cells (Supplemental Fig. S1).
Fig. 1.
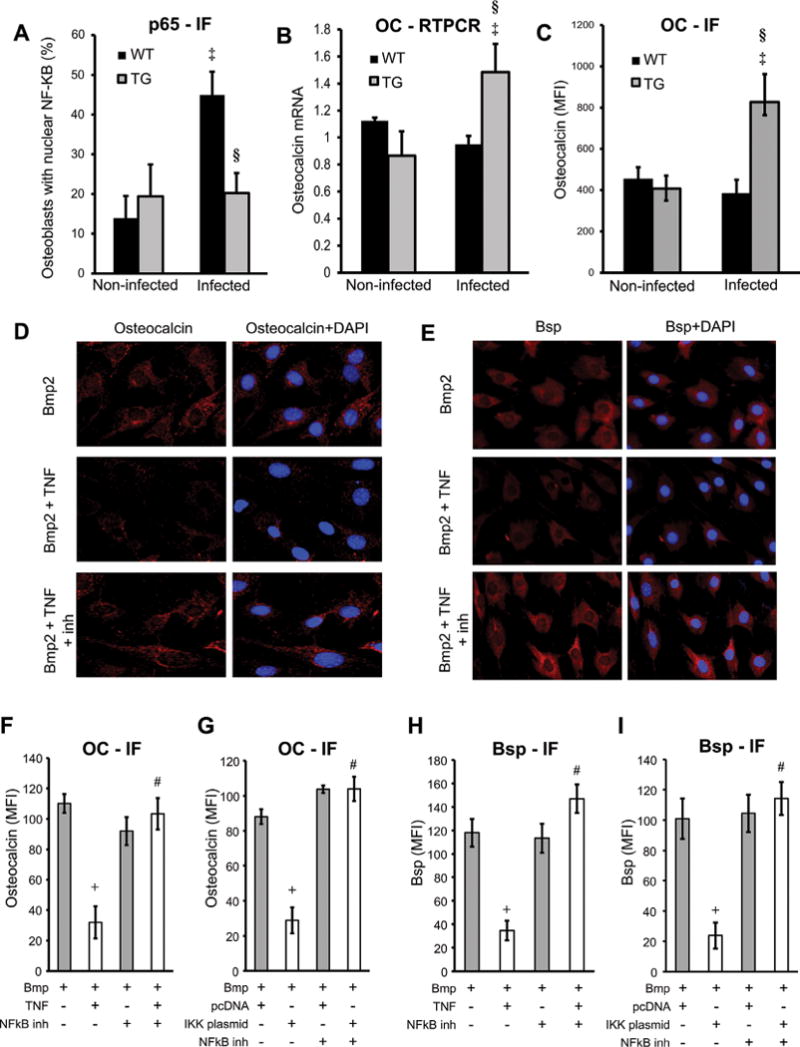
NF-κB-mediated response inhibits bone and matrix protein synthesis. Destructive periodontitis was initiated in IKK-DN transgenic mice (TG) or wild-type (WT) control mice by oral inoculation of the periodontal pathogens P. gingivalis plus F. nucleatum and compared with vehicle alone. Mice were euthanized 6 weeks after oral inoculation. Nuclear NF-κB was measured by immunofluorescence by colocalization of p65, a subunit of NF-κB and DAPI nuclear staining. (A) Number of NF-κB-positive cells was determined. Localization to bone-lining osteoblastic cells was determined in cells with a cuboidal appearance and with a nucleus that was not fusiform or parallel to the bone surface. (B) RNA was isolated from periodontal tissue, and osteocalcin mRNA levels were measured by real-time PCR. (C) OC expression was measured by immunofluorescence with antibody to OC and expressed as the mean fluorescence intensity. (D–I) MC3T3 cells were incubated with TNFα or transfected with IKK-overexpression vector compared with pcDNA (empty vector) alone. NF-κB was inhibited with the specific inhibitor BAY117082. Cells were stimulated with Bmp2 for 48 hours. Cells were stained for OC or Bsp (red) and counterstained with DAPI (blue). Immunofluorescence was analyzed and expressed as mean expression above baseline (MFI). ‡Significantly different in infected compared with matched noninfected group; §significantly different in infected TG compared with infected WT. Data shown are representative of three independent experiments. +Significant inhibition with TNFα or IKK transfection; #significantly different between cells incubated with no inhibitor.
Mutation of NF-κB binding sites on osteocalcin or Bsp promoters rescues the negative impact of TNFα or IKK transfection, indicating a direct effect of NF-κB
Three consensus NF-κB binding sites were identified on the promoter regions of osteocalcin and Bsp and were mutated in luciferase transcription reporter constructs (Supplemental Fig. S2). Bmp2 and Wnt stimulated a six- to eightfold increase in promoter activity, which was reduced by 85% to 95% by TNFα or IKK transfection (p < 0.05) (Fig. 2A–D). Mutation of the first or second putative NF-κB binding sites partially reversed the effect of TNFα by 47% to 58% (p < 0.05), while mutating the third putative response element had little impact (Fig. 2A). The effect of TNFα on osteocalcin transcription activity was completely reversed by mutating the first two sites simultaneously (p < 0.05) (Fig. 2A).
Fig. 2.
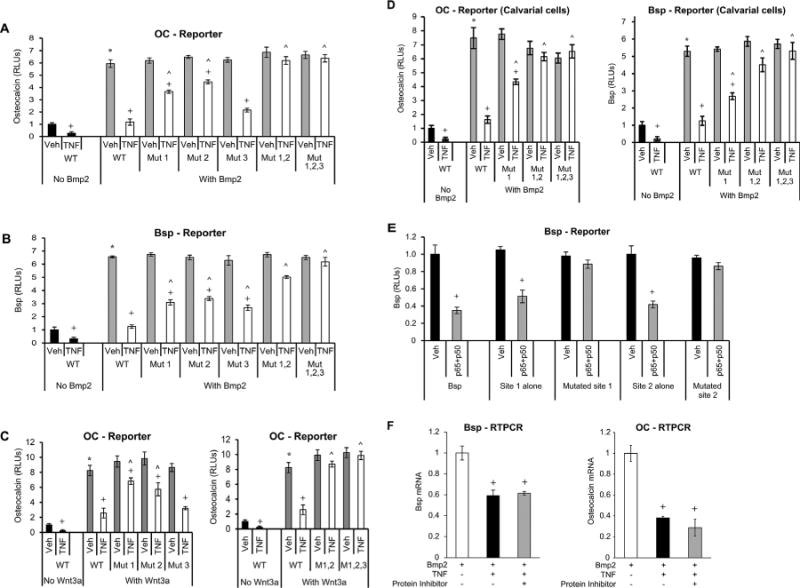
Mutation of NF-κB binding sites in osteocalcin or Bsp promoters rescues the negative effect of TNFα, indicating a direct effect of NF-κB. NF-κB binding sites were mutated as indicated (Mut1; Mut2; Mut3; Mut1,2; Mut1,2,3). Cells were transfected with wild-type and mutant constructs. After a 1-hour incubation with TNFα, cells were stimulated with Bmp2 or Wnt3a as indicated. (A–C) OC or Bsp promoter activity was analyzed by luciferase reporter construct. Luciferase activity was normalized by renilla control and expressed as relative luciferase units (RLUs). (D) Primary mouse osteoblastic cells were transfected with osteocalcin or Bsp reporter construct with or without mutated NF-κB binding sites. Transcriptional activity was measured using a luciferase assay and expressed as relative luciferase units (RLUs). (E) Cells were transfected with wild-type or mutated Bsp luciferase constructs containing all the NF-κB putative sites. Using subcloning, Bsp mini genes were prepared from wild-type and mutated promoter regions as described in Materials and Methods. NF-κB was activated by cotransfecting cells with p65- and p50-overexpressing plasmids. (F) Cells were stimulated with Bmp2 for 48 hours and then incubated with TNFα for 6 hours without or with cycloheximide. Bsp or OC mRNA levels were analyzed by RT-PCR. *Significantly different from Bmp2-stimulated control; +significant inhibition with TNFα; ˄significantly different between cells transfected with WT (wild-type construct) versus matched control (p < 0.05).
Experiments were also carried out with the Bsp promoter. Simply mutating any one of three putative NF-κB binding sites in the Bsp promoter had a partial effect (38% to 50%) on reversing TNFα-suppressed Bsp promoter activity (p < 0.05) (Fig. 2B). Mutating all three was necessary to completely reduce the inhibitory effect of TNFα (p < 0.05) (Fig. 2B). Mutation of the first or second putative NF-κB binding site in the osteocalcin promoter partially reversed the effect of TNFα inhibition in Wnt3a-stimulated cells by 25% to 40%, but mutating the third putative response element had less impact (p < 0.05) (Fig. 2C). The effect of TNFα on osteocalcin transcription activity in Wnt3a-stimulated cells was completely reversed by mutating the first two sites simultaneously (p < 0.05) (Fig. 2C). This suggests that NF-κB binding to consensus elements of bone matrix proteins plays a role in inhibiting their transcription activity whether induced by Wnt or Bmp2 stimulation.
The experiments were repeated using primary osteoblasts isolated from mouse calvariae. TNFα reduced the Bmp2-stimulated osteocalcin transcriptional activity by 92% to 95% (p < 0.05) (Fig 2D). Mutation of a single NF-κB response element partially reduced the negative impact of TNFα, and simultaneous mutation of the first two response elements completely abolished it (p < 0.05) (Fig. 2D). For Bsp, mutation of one site reduced the effect of TNFα by 33% in primary osteoblasts, two sites by almost 70%, and three sites completely reversed it (p < 0.05) (Fig. 2D).
To further investigate the impact of the first and second NF-κB binding sites, “Bsp-mini-genes,” each bearing a single NF-κB binding site, were cloned. Cotransfection of NF-κB subunits p65 and p50 with wild-type promoter constructs significantly reduced Bsp promoter activity for both NF-κB binding sites (p < 0.05) (Fig. 2E). Mutation of either site completely reversed the effect of p65/p50 transfection (p < 0.05) (Fig. 2E), further demonstrating the importance of a direct interaction of NF-κB to either of the first two binding sites in the Bsp promoter region. A direct mechanism is also supported by findings that TNFα diminishes the mRNA levels of Bsp and OC independently of new protein synthesis (Fig. 2F). Incubation with TNFα induced a 40% to 60% decrease in mRNA expression of both bone matrix proteins, which was not affected by incubation with cycloheximide (p < 0.05).
NF-κB p65-p60 heterodimer binds to Bsp and OC promoters
To analyze the binding of the p65-p50 heterodimer to the promoter region of osteocalcin or Bsp, chromatin immunoprecipitation (ChIP) studies were undertaken. TNFα stimulated a fourfold increase in binding of NF-κB p65 subunit to the promoter region of osteocalcin and Bsp (p < 0.05) (Fig. 3A, B), whereas there was no specific binding in an alternative region of the osteocalcin promoter lacking the NF-κB response elements (Fig. 3C). ChIP was also carried out using TNFα-stimulated cells, and the complex was immunoprecipitated with the NF-κB p50 subunit. TNFα stimulation resulted in a fourfold increase in binding to the OC and Bsp promoter (p < 0.05) (Fig. 3A, B), further demonstrating the binding of the p65-p50 heterodimer to the promoter region. To further analyze the association between p65 and the promoter region, ChIP was carried out in TNFα-stimulated cells upon p65 knockdown using siRNA. Knockdown of p65 substantially reduced the binding of p65 to the promoter region (p < 0.05) (Fig. 3D).
Fig. 3.
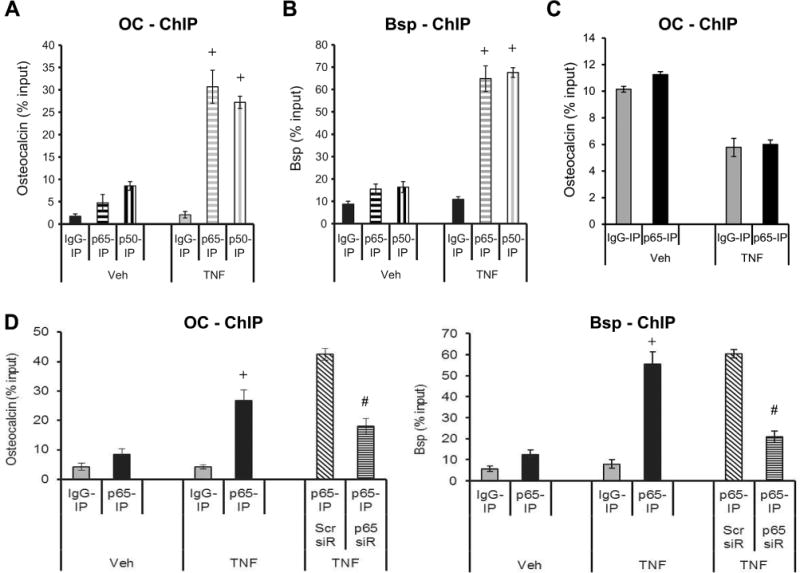
NF-κB p65-p50 heterodimer binds to Bsp and osteocalcin promoters. Cells were stimulated with TNFα and compared with vehicle. After incubation, cells were fixed, DNA isolated, and subjected to chromatin immunoprecipitation using antibodies for p65 and p50 or matched IgG control. (A, B) ChIP was performed using antibodies for p65, p50 or control IgG. RT-PCR was performed using primers to Bsp or osteocalcin promoter that contained NF-κB response elements or (C) to a region further upstream of the promoter region. (D) Cells were incubated with TNFα after knockdown with p65 siRNA or scrambled siRNA and chromatin was immunoprecipitated with antibody to p65 or control IgG. RT-PCR was performed using primers to OC or Bsp promoter that contained NF-κB response elements. Data shown are representative of three independent experiments. +Significantly different with TNFα versus matched control; #significantly different between cells transfected with scrambled siRNA versus p65 siRNA (p < 0.05).
NF-κB activation decreases Bmp2-stimulated Runx2 and Wnt-stimulated β-catenin binding to osteocalcin or Bsp promoters
Chromatin immunoprecipitation was carried out to assess interaction of Runx2 and β-catenin binding to Bsp and OC promoters. Stimulation with Bmp2 resulted in a six- to sevenfold increase in Runx2 binding to the promoter region of OC and Bsp (p < 0.05) (Fig. 4A). When NF-κB was activated using TNFα, this increase was reduced by 53% to 56% (p < 0.05) (Fig. 4A). Similarly, Wnt stimulation resulted in a seven- to ninefold increase in β-catenin binding to the promoter region, and a 45% to 70% decrease in binding was observed upon TNFα stimulation (p < 0.05) (Fig. 4B). To rule out the possibility that reduced β-catenin or Runx2 binding to OC or Bsp promoters was because of reduced β-catenin or Runx2 nuclear localization, immunoblots were carried out. TNFα did not reduce β-catenin levels in the nucleus or cytoplasm (Fig. 4C). Similarly, TNFα did not alter nuclear or cytoplasmic levels of Runx2 (Fig. 4D). These results demonstrate that the effect of TNFα on β-catenin or Runx2-DNA interactions was not because of changes in nuclear localization or expression. Results demonstrate that TNFα reduces RNAP-II binding to bone sialoprotein and osteocalcin promoters (Fig. 4E). Inhibition of NF-κB using small molecule inhibitor, diminishes the inhibitory effect of TNFα on the binding of RNAP-II to the promoter region of bone matrix proteins by 67–91% (Fig. 4E).
Fig. 4.
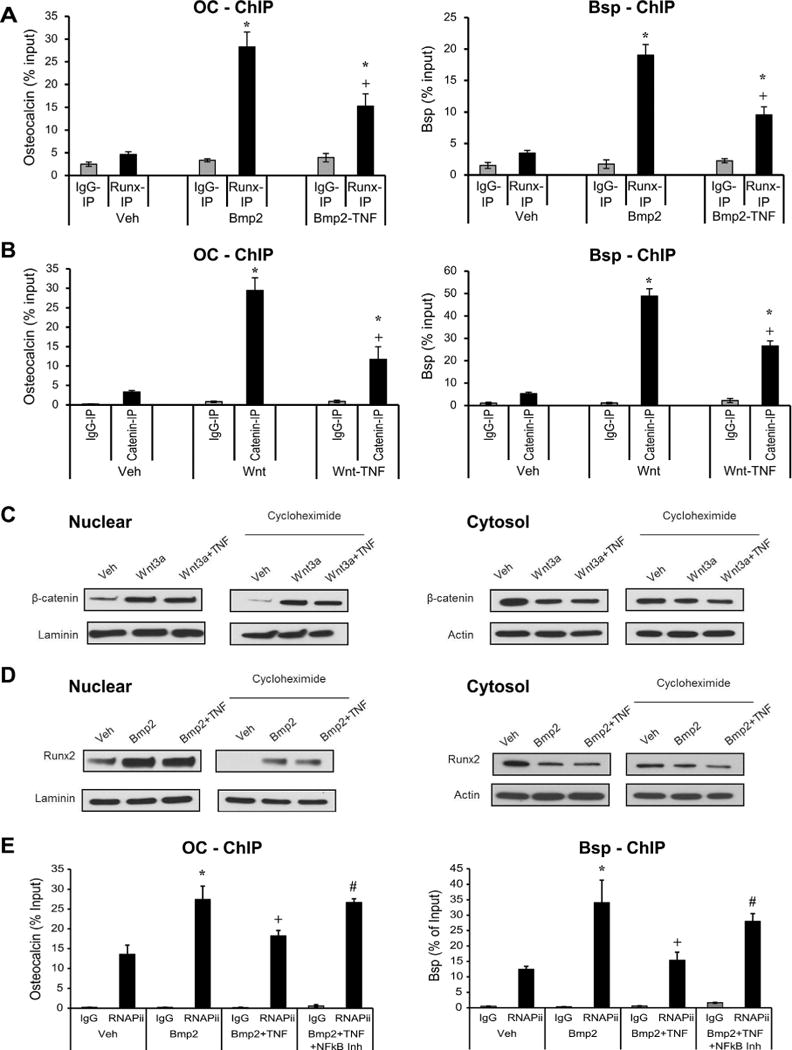
NF-κB activation decreases Bmp2-stimulated Runx2 and Wnt-stimulated β-catenin binding to osteocalcin or Bsp promoters. Cells were incubated with Bmp2 or Wnt3a for 4 hours with or without a 45-minute pre-incubation with TNFα. Some of the cells were incubated with NF-κB small molecule inhibitor (BAY-117082). Cells were fixed, DNA isolated, and subjected to ChIP using antibodies for Runx2, β-catenin, RNAP-II or control IgG. (A, B) RT-PCR was performed using primers to Bsp or OC promoter that contained NF-κB response elements. (C, D) Immunoblotting with antibody to beta-catenin, Runx2, actin or laminin of nuclear or cytosolic lysates of cells incubated with Wnt or Bmp for 4 hours, followed by a 3-hour cycloheximide incubation. Before stimulation, some of the cells were incubated with TNFα for 45 minutes. (E) ChIP assay was performed with antibody to RNAP-II or control IgG. RT-PCR was performed using primers to Bsp or OC promoter that contained binding regions of RNAP-II. *Significantly different from No-Bmp2/Wnt3a-stimulated control; +significant inhibition with TNFα; #significantly different between cells incubated with no inhibitor (p < 0.05).
The canonical NF-κB suppresses matrix protein transcription
Experiments were performed to determine whether canonical or noncanonical NF-κB pathways were involved in suppressing Bsp or OC transcription activity. Knockdown of either p65 or p50 diminished the suppressive effect of NF-κB activation to a similar extent as IKKβ knockdown on OC or Bsp mRNA expression (p < 0.05) (Fig. 5A). Transfection by IKK inhibited the Bmp2-stimulated transcriptional activity of OC/Bsp by more than 90% (p < 0.05) (Fig. 5B). Wnt3a stimulated a sixfold increase in osteocalcin transcription activity, and this increase was inhibited by 80% by incubation with TNFα (p < 0.05) (Fig. 5C). The inhibitory effect of TNFα was rescued by knockdown of the canonical NF-κB subunits, p50 and p65 (p < 0.05) but not by the noncanonical (RelB) subunit (Fig. 5C). An alternative approach to examine the effect of the canonical NF-κB subunits was taken by cotransfection of osteocalcin reporter construct and expression vectors for p50, p65, and a p65 mutant containing an alteration of Ser276 to alanine (p65S276A) (Fig. 5D). Transfection of either p65 or p50 alone did not have a significant effect in decreasing OC transcriptional activity (Fig. 5D). Cotransfection of both p65 and p50 was necessary to inhibit osteocalcin transcriptional activity, which it did by 90% (p < 0.05) (Fig. 5D). The degree of inhibition owing to cotransfection of p65/p50 was similar to that of TNFα (p < 0.05) (Fig. 5D). Knockdown of RelB, the noncanonical NF-κB subunit, had no effect on inhibiting osteocalcin and Bsp promoter activity compared with scrambled siRNA (Fig. 5E). In contrast, knockdown of p65 subunit rescued the inhibitory effect of TNFα (p < 0.05) (Fig. 5E). p65 and RelB siRNA resulted in substantial knockdown of p65 and RelB expression (Fig. 5F). This suggests that the noncanonical NF-κB does not play a role in the repressive effect of bone matrix genes.
Fig. 5.
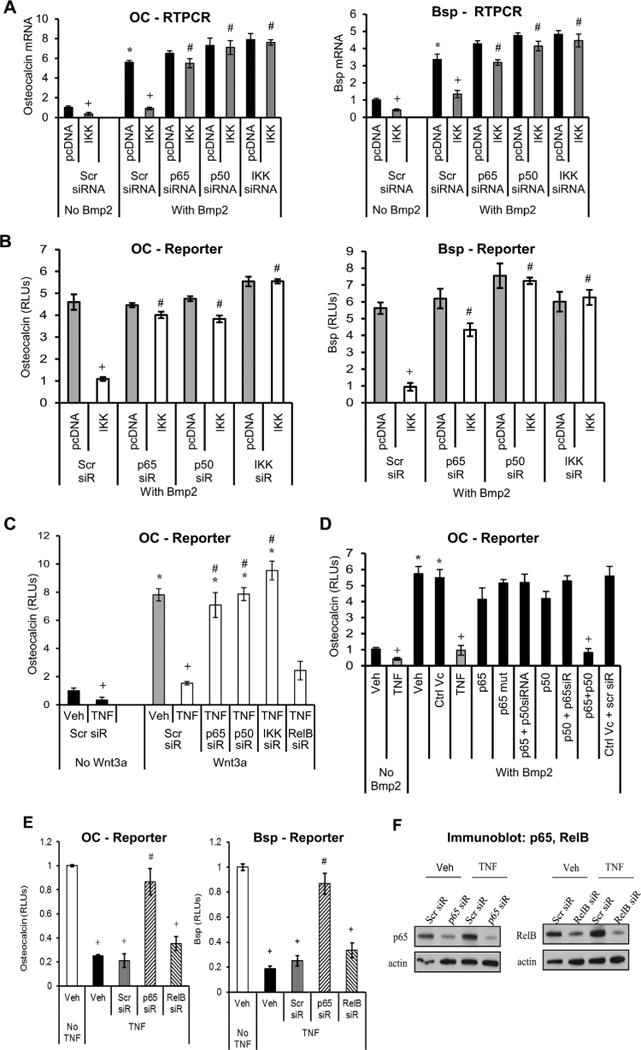
The canonical NF-κB suppresses matrix protein transcription. Cells were stimulated with TNFα or transfected with IKK-, p65-, p50-overexpression vector compared with pcDNA (empty vector) alone. Some of the cells were also transfected with siRNA specific for IKK, the NF-κB subunits p65, p50, RelB, or scrambled siRNA (Scr). Cells were stimulated with Bmp2 or Wnt3a for 48 hours. (A) Osteocalcin or Bsp mRNA levels were measured by real-time PCR. (B–E) Osteocalcin or Bsp promoter activity was analyzed by luciferase reporter construct in cells incubated with Bmp2 or Wnt. (F) Immunoblot analysis of whole-cell lysate probed for p65 and RelB. Data shown are representative of three independent experiments. +Significant inhibition with TNFα or IKK transfection versus matched control; #significantly different between cells transfected with scrambled siRNA versus targeted siRNA (p < 0.05).
HDAC1 is needed for NF-κB suppression of Bsp and OC
Because acetylation of NF-κB regulates its activity, we carried out co-immunoprecipitation (Co-IP) and ChIP assays to determine whether HDAC1, which deacetylates NF-κB, played a role in its suppressive effect. Immunoprecipitation with antibody to p65 followed by immunoblot analysis demonstrated an association between p65 and p50, as well as an association between p65 and HDAC1 (Fig. 6A). Similarly, immunoprecipitation with antibody against HDAC1 followed by immunoblot showed an association between HDAC1 and p65 (Fig. 6A). Interestingly, mutual detection of HDAC1, p65, and p50 occurred to a much greater extent in TNFα-stimulated cells, indicating that formation of these associations was stimulated by TNFα (Fig. 6A). In addition, TNFα stimulation increased phosphorylation of p65 at Thr505 (Fig. 6A). Immunoprecipitation/immunoblot experiments also demonstrate that HDAC1 was important in deacetylating the NF-κB p65 subunit because its level of acetylation is significantly increased with knockdown of HDAC1 by siRNA (Fig. 6B). ChIP assays demonstrated that TNFα stimulated a five- to sevenfold increase in HDAC1 binding to the promoter region of OC and Bsp (p < 0.05) (Fig. 6C). HDAC1 siRNA resulted in substantial knockdown of HDAC1 (Fig. 6D) and reduced p65 binding to the OC or Bsp promoter by more than 50% (p < 0.05) (Fig. 6E). Thus, HDAC1 was needed for optimal interaction of NF-κB with Bsp or OC promoters. A potential mechanism by which HDAC1 promotes NF-κB-OC or NF-κB-Bsp interactions is by increasing nuclear localization. TNFα dramatically increased Ser276-phosphorylation of p65 and knockdown or inhibition of HDAC1 inhibited it, indicating that HDAC1 modulated NF-κB activation (Fig. 6F). TNFα also stimulated nuclear localization of both p65 and p50 (Fig. 6F), and inhibition or knockdown of HDAC1 decreased nuclear localization as shown by immunoblot (Fig. 6F) and by colocalization with DAPI in immunofluorescence experiments (Fig. 6G).
Fig. 6.
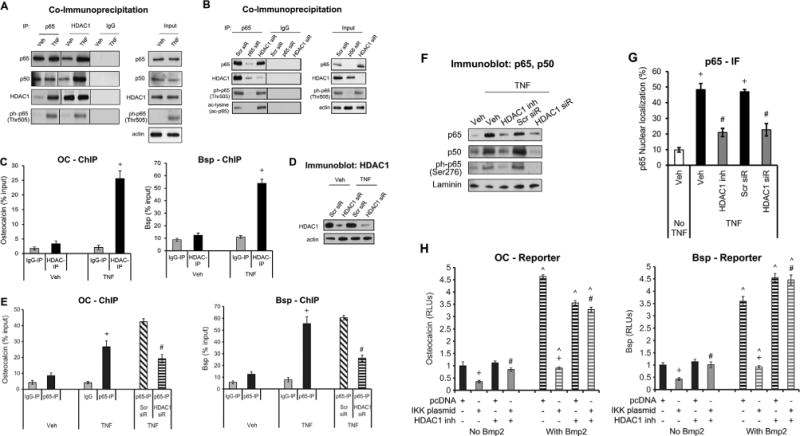
HDAC1 interacts with NF-κB to regulate BSP and OC transcription activity. Cells were stimulated with TNFα alone with or without pre-incubation with HDAC1-specific inhibitor (parthenolide)(48) or transfection with siRNA specific for HDAC1 or scrambled (Scr). (A, B) Cell lysates were incubated with antibody to p65 or control IgG. Immunoprecipitated proteins or input cell lysate was then examined by immunoblot analysis with antibody to p65, p50, HDAC1, ph-p65 (Thr505), or acetylated lysine. (C, E) Cells were fixed, DNA isolated, and subjected to chromatin immunoprecipitation using antibodies for HDAC1, p65, or matched IgG control. RT-PCR was performed using primers to Bsp or OC promoter that contained NF-κB response elements. (D) Immunoblot analysis of whole-cell lysate probed for HDAC1. (F) Immunoblot analysis of nuclear lysate in cells incubated with HDAC1 inhibitor or transfected with HDAC1-siRNA were probed for p65, p50, or ph-p65. (G) Nuclear localization of NF-κB was measured by immunofluorescence with an antibody specific for p65 and counterstained with DAPI. (H) Cells were pre-incubated with the HDAC1 inhibitor (parthenolide), transfected with IKK or pcDNA (empty vector), and transfected with a Bsp or osteocalcin reporter vector with or without Bmp2 stimulation. Luciferase activity was normalized by renilla control and expressed as relative luciferase units (RLUs). Data shown are representative of three independent experiments. +Significantly different between TNFα versus matched control; #significantly different between cells transfected with scrambled siRNA versus HDAC1 siRNA or between cells incubated with no inhibitor; ˄significantly different between cells with Bmp2 stimulation (p < 0.05).
Consistent with reduced activation of NF-κB, inhibition of HDAC1 reduced the suppressive effects of NF-κB on bone matrix protein expression. A decrease in OC- and Bsp-luciferase activity was observed upon transfection with IKK-overexpression plasmid in the presence or absence of Bmp2 stimulation (Fig. 6H). An HDAC1 inhibitor rescued the inhibitory effect of IKK transfection on OC and Bsp transcriptional activity (p < 0.05) (Fig. 6H). The inhibitor also rescued the suppressive effect of TNFα on OC or Bsp mRNA levels (p < 0.05) (Fig. 7A). Similarly, HDAC1 inhibitor reduced the inhibitory effect of TNFα on OC or Bsp protein expression (p < 0.05) (Fig. 7B–D). Incubation of cells with TNFα resulted in a 50% decrease in OC or Bsp protein expression in Bmp2-unstimulated cells (p < 0.05) (Fig. 7B–D). Thus, HDAC1 deacetylation of NF-κB is needed for NF-κB activation, nuclear localization, binding to Bsp and OC promoter elements, and inhibition of Bsp and OC transcription activity and expression.
Fig. 7.
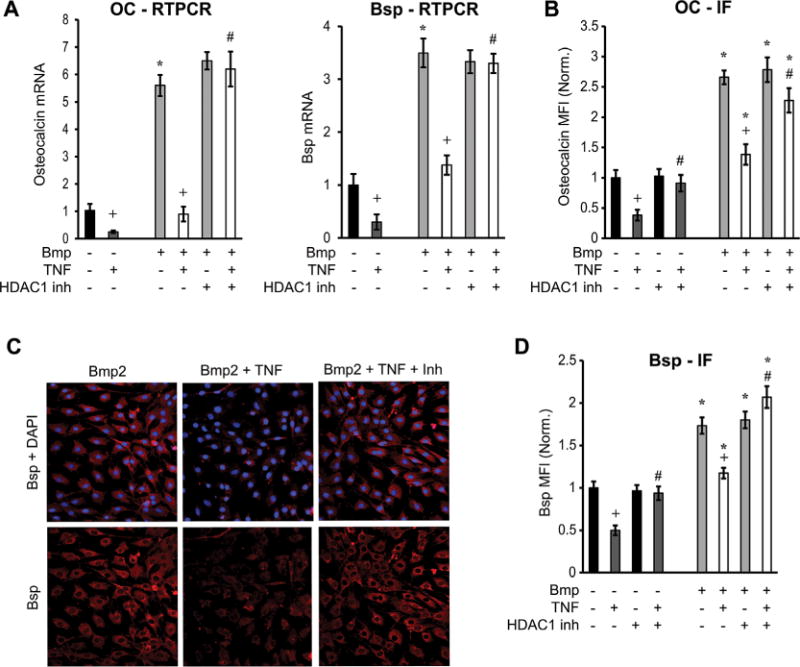
HDAC1 plays a role in decreased mRNA and protein expression of matrix proteins. Cells were stimulated with TNFα with or without pre-incubation with HDAC1-specific inhibitor (parthenolide). Some of the cells were stimulated with Bmp2. (A) OC or Bsp mRNA was measured by real-time PCR in cells incubated with TNFα and parthenolide, with or without Bmp2 stimulation. (B–D) Protein was measured by immunofluorescence using specific antibody for osteocalcin or Bsp (red) and counterstained with DAPI. Protein expression was measured by immunofluorescence and expressed as mean fluorescence intensity (MFI). Values were normalized to unstimulated controls. Data are expressed as level above baseline. Data shown are representative of three independent experiments. *Significantly different from No-Bmp2-stimulated control; +significant inhibition with TNFα; #significantly different between cells with no inhibitor (p < 0.05).
Discussion
Chronic inflammation has a debilitating effect on matrix proteins, which contributes to significant morbidity by interfering with fracture healing and contributing to formation of osteolytic lesions in rheumatoid arthritis, osteoporosis, periodontal disease, and other conditions.(4,41,42) We present findings here that demonstrate a novel mechanism by which inflammation directly affects the repression of matrix proteins through NF-κB. NF-κB inhibits expression of matrix proteins by interacting with the promoter region of these genes. We also demonstrate for the first time that periodontal infection stimulates NF-κB activation in osteoblasts in vivo and that the effect is to reduce osteocalcin expression, which is part of the reparative process. In vitro studies also showed that NF-κB activation by IKK transfection or TNFα stimulation decreased both Wnt- and Bmp-stimulated bone matrix protein expression. That this process required direct interaction with NF-κB response elements in the Bsp and OC promoters was shown by site-directed mutagenesis. Mutation of these consensus NF-κB response elements rescued the inhibitory effect of TNFα on Wnt- and Bmp-stimulated Bsp and OC transcription. The mechanism is likely to involve the direct effect of NF-κB on transcription factors that stimulate OC and Bsp expression. Incubation with TNFα decreased Wnt-stimulated β-catenin binding to OC and Bsp promoters and also diminished the Bmp2-induced Runx2 binding to these promoters. In addition, we demonstrate that the capacity of NF-κB to inhibit OC or Bsp synthesis is dependent upon NF-κB forming a complex with HDAC1 and deacetylation, which facilitates NF-κB nuclear localization and subsequent binding to Bsp and OC promoters. In summary, we propose that the p65-p50 complex binds to the promoter region of Bsp or OC and prevents the binding of the positive regulators (β-catenin or Runx2) to initiate transcription of bone matrix proteins. These studies provide new insight into mechanisms through which inflammation affects matrix proteins and the potential importance of targeting NF-κB during inflammatory conditions when bone formation is critical.
The inhibitory effect of NF-κB on Bsp and OC expression largely occurred at the transcriptional level. TNFα reduced the Bmp2-stimulated transcription of OC by 90%. That this inhibitory effect was directly related to the classical NF-κB pathway was shown by the use of a NF-κB inhibitor (BAY117082), which completely blocked the inhibitory effect of TNFα or IKK transfection. Moreover, knockdown of the NF-κB canonical subunits, p65 and p50, but not the noncanonical NF-κB subunit RelB, rescued the inhibitory effect of both IKK transfection and TNFα stimulation. In addition, cotransfection of p65 and p50 simultaneously inhibited the Bmp-stimulated transcription compared with either alone, consistent with the canonical heterodimer mediating the suppressive effect.
Promoter analyses of OC and Bsp identified three putative NF-κB binding sites on OC and Bsp promoters. Singly mutating the first two sites partially reversed the effect of TNFα inhibition on Bmp2- and Wnt-stimulated OC transcriptional activity, whereas the third site had no effect. Double mutation of the first two sites completely blocked the inhibitory effect of TNFα or IKK transfection. For the Bsp promoter, singly mutating each individual site partially reversed the inhibitory effect of TNFα. Mutating all three sites on Bsp was needed to completely reverse TNFα inhibition. This suggests that NF-κB binding to consensus elements of bone matrix proteins played an essential role in inhibiting OC and Bsp transcriptional activity in MC3T3 cells whether induced by Bmp or Wnt. That the first two OC NF-κB response elements and all three OC NF-κB response elements had inhibitory activity was confirmed by creating mini Bsp promoters containing a single consensus element. Transfection of p65 and p50 NF-κB subunits resulted in a 50% to 60% decrease in transcriptional activity for each NF-κB response element individually, whereas mutating the site reversed the inhibitory effect of NF-κB activation.
At baseline, osteocalcin mRNA levels reflect basal bone remodeling. Basal osteocalcin mRNA levels were not affected by blocking NF-κB activation in transgenic mice, consistent with previous reports that it does not affect physiologic bone remodeling in adult mice.(6) Infection induced resorption and coupled bone formation, which was blocked in wild-type mice through a NF-κB-mediated mechanism. Inhibition of NF-κB activation did not affect basal bone remodeling but did block the increase stimulated by infection-induced coupled new bone formation. In addition, periodontal bone loss was blocked in transgenic Col1α1.IKK-DN mice (data not shown).
A critical finding from our studies is that TNFα prevented two essential matrix protein-promoting transcription factors, β-catenin and Runx2, from binding to promoters of OC and Bsp. Bsp and OC promoters contain consensus binding sites for β-catenin that are 2 to 57 nucleotides from NF-κB response elements. Moreover, consensus binding sites for Runx2 are 15 to 160 nucleotides from NF-κB response elements. Bmp2-stimulated Runx2 binding to the promoter region of both OC and Bsp was substantially reduced when NF-κB was activated by TNFα. Similarly, TNFα reduced Wnt-stimulated β-catenin binding to the Bsp and OC promoters region. In addition, the capacity of TNFα to inhibit Runx2 or β-catenin binding occurred within six hours, which is sooner than the known indirect mechanisms through which TNFα has previously been shown to affect bone matrix proteins. This was confirmed by demonstrating that the effect on Bmp2-induced Runx2 binding was not the result of changes in Runx2 expression or Runx2 nuclear localization. This is consistent with the idea that NF-κB directly reduces their binding to Bsp or OC promoters. A direct effect is also supported by a significant reduction in Bsp and osteocalcin mRNA levels induced by TNFα even in the presence of a protein synthesis inhibitor. Thus, TNFα inhibits β-catenin and Runx2 from interacting with the Bsp and OC promoters and inhibits their transcriptional activity. Results demonstrate that TNFα reduces the binding of RNAP-II to Bsp and osteocalcin promoters. Inhibiting NF-κB blunted the effect of TNFα on RNAP-II binding to the promoter region of bone matrix proteins, thereby reinforcing the mechanistic link. This is the first evidence that NF-κB can directly repress the binding of transcription factors to response elements of matrix proteins to inhibit expression.
HDAC1 is a histone deacetylase that regulates gene expression through histone-dependent and histone-independent mechanisms.(43) We have observed a direct correlation between phosphorylation of p65 at Thr505 and recruitment of HDAC1. This is in line with other studies that suggest that phosphorylation of p65 at Thr505 increases HDAC1 recruitment.(44) We demonstrated that HDAC1 formed complexes with p65-p50 and was functionally important in deacetylating NF-κB. It was needed for NF-κB activation as measured by increased p65 phosphorylation and nuclear localization. Inhibition of HDAC1 substantially reduced binding of NF-κB (p65) to both the OC and Bsp promoters and reversed the inhibitory effect of TNFα on the transcriptional activity and expression of OC and Bsp. Thus, HDAC1 interaction with NF-κB was needed for the repression of matrix protein expression. A previous report has shown that(45) NF-κB interacts with HDAC1 and promotes NF-κB nuclear localization most likely through deacetylation.
In summary, NF-κB plays a central role in decreased expression of matrix proteins caused by inflammatory conditions, ultimately resulting in decreased bone formation. We demonstrate a novel mechanism through which both Wnt-stimulated and Bmp-stimulated expression of matrix proteins is inhibited upon activation of NF-κB. This inhibition involves the direct interaction of NF-κB with response elements in the promoter regions of bone matrix proteins and the inhibition of β-catenin and Runx2 binding to nearby consensus sites. Although NF-κB is typically thought of as a positive regulator for inflammatory cytokines,(46) it has recently been shown to inhibit transcription of other genes.(47) Contrary to most reports, we found that the inhibitory effect was mediated by the canonical NF-κB heterodimer p65-p50 rather than the more common NF-κB p50-p50 homodimer. Lastly, the capacity of NF-κB to repress Bsp and osteocalcin expression was dependent upon TNFα-stimulated HDAC1-NF-κB interactions that led to NF-κB deacetylation and nuclear localization.
Supplementary Material
Acknowledgments
We thank Dr Chawnshang Chang (University of Rochester) for OC reporter plasmid. We thank Dr Renny Franceschi (University of Michigan) for generously providing the antibody and reporter construct for bone sialoprotein. We thank Dr Rob Riccardi for his technical assistance and for generously donating the p65-, p50-, p65(S275A)-overexpressing plasmids. We also thank Jyothsna Meda for assistance in microscopy. This work was supported by grants DE-017732 and AR-060055 from the National Institutes of Health.
Authors’ roles: RT planned the experimental design, conducted experiments, and wrote the manuscript. JL and TC conducted experiments. SP and WX conducted in vivo experiments. DR and MM analyzed data. HG gave technical suggestions. BY and CW generated and provided the transgenic mice. DG conceived the project, designed experiments, interpreted the data and wrote the manuscript.
Footnotes
Additional Supporting Information may be found in the online version of this article.
Disclosures
All authors state that they have no conflicts of interests.
References
- 1.Rahman MM, McFadden G. Modulation of tumor necrosis factor by microbial pathogens. PLoS Pathog. 2006;2(2):e4. doi: 10.1371/journal.ppat.0020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vitiello M, Galdiero M, Finamore E, Galdiero S, Galdiero M. NF-kappaB as a potential therapeutic target in microbial diseases. Mol Biosyst. 2012;8(4):1108–20. doi: 10.1039/c2mb05335g. [DOI] [PubMed] [Google Scholar]
- 3.Marcu KB, Otero M, Olivotto E, Borzi RM, Goldring MB. NF-kappaB signaling: multiple angles to target OA. Curr Drug Targets. 2010;11(5):599–613. doi: 10.2174/138945010791011938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyce BF, Yao Z, Xing L. Functions of nuclear factor kappaB in bone. Ann NY Acad Sci. 2010;1192:367–75. doi: 10.1111/j.1749-6632.2009.05315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Novack DV. Role of NF-kappaB in the skeleton. Cell Res. 2011;21(1):169–82. doi: 10.1038/cr.2010.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang J, Wang Z, Tang E, et al. Inhibition of osteoblastic bone formation by nuclear factor-kappaB. Nat Med. 2009;15(6):682–9. doi: 10.1038/nm.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamazaki M, Fukushima H, Shin M, et al. Tumor necrosis factor alpha represses bone morphogenetic protein (BMP) signaling by interfering with the DNA binding of Smads through the activation of NF-kappaB. J Biol Chem. 2009;284(51):35987–95. doi: 10.1074/jbc.M109.070540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou FH, Foster BK, Zhou XF, Cowin AJ, Xian CJ. TNF-alpha mediates p38 MAP kinase activation and negatively regulates bone formation at the injured growth plate in rats. J Bone Miner Res. 2006;21(7):1075–88. doi: 10.1359/jbmr.060410. [DOI] [PubMed] [Google Scholar]
- 9.Pathak JL, Bravenboer N, Verschueren P, et al. Inflammatory factors in the circulation of patients with active rheumatoid arthritis stimulate osteoclastogenesis via endogenous cytokine production by osteoblasts. Osteoporos Int. 2014;25(10):2453–63. doi: 10.1007/s00198-014-2779-1. [DOI] [PubMed] [Google Scholar]
- 10.Corrado A, Neve A, Maruotti N, Cantatore FP. Bone effects of biologic drugs in rheumatoid arthritis. Clin Dev Immunol. 2013;2013:945945. doi: 10.1155/2013/945945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matuszewska A, Szechinski J. Evaluation of selected bone metabolism markers in rheumatoid arthritis patients. Adv Clin Exp Med. 2013;22(2):193–202. [PubMed] [Google Scholar]
- 12.Stanford CM. Surface modification of biomedical and dental implants and the processes of inflammation, wound healing and bone formation. Int J Mol Sci. 2010;11(1):354–69. doi: 10.3390/ijms11010354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bridgeman MB, Pathak R. Denosumab for the reduction of bone loss in postmenopausal osteoporosis: a review. Clin Ther. 2011;33(11):1547–59. doi: 10.1016/j.clinthera.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 14.Nabipour I, Larijani B, Vahdat K, et al. Relationships among serum receptor of nuclear factor-kappaB ligand, osteoprotegerin, high-sensitivity C-reactive protein, and bone mineral density in postmenopausal women: osteoimmunity versus osteoinflammatory. Menopause. 2009;16(5):950–5. doi: 10.1097/gme.0b013e3181a181b8. [DOI] [PubMed] [Google Scholar]
- 15.Greenblatt MB, Shim JH, Glimcher LH. Mitogen-activated protein kinase pathways in osteoblasts. Annu Rev Cell Dev Biol. 2013;29:63–79. doi: 10.1146/annurev-cellbio-101512-122347. [DOI] [PubMed] [Google Scholar]
- 16.Long F, Ornitz DM. Development of the endochondral skeleton. Cold Spring Harb Perspect Biol. 2013;5(1):a008334. doi: 10.1101/cshperspect.a008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee KS, Kim HJ, Li QL, et al. Runx2 is a common target of transforming growth factor beta1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Mol Cell Biol. 2000;20(23):8783–92. doi: 10.1128/mcb.20.23.8783-8792.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89(5):747–54. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 19.Little RD, Carulli JP, Del Mastro RG, et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet. 2002;70(1):11–9. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Centrella M, Christakos S, McCarthy TL. Skeletal hormones and the C/EBP and Runx transcription factors: interactions that integrate and redefine gene expression. Gene. 2004;342(1):13–24. doi: 10.1016/j.gene.2004.06.036. [DOI] [PubMed] [Google Scholar]
- 21.Wu XB, Li Y, Schneider A, et al. Impaired osteoblastic differentiation, reduced bone formation, and severe osteoporosis in noggin-overexpressing mice. J Clin Invest. 2003;112(6):924–34. doi: 10.1172/JCI15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devlin RD, Du Z, Pereira RC, et al. Skeletal overexpression of noggin results in osteopenia and reduced bone formation. Endocrinology. 2003;144(5):1972–8. doi: 10.1210/en.2002-220918. [DOI] [PubMed] [Google Scholar]
- 23.Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19(2):179–92. doi: 10.1038/nm.3074. [DOI] [PubMed] [Google Scholar]
- 24.Lories R. The balance of tissue repair and remodeling in chronic arthritis. Nat Rev Rheumatol. 2011;7(12):700–7. doi: 10.1038/nrrheum.2011.156. [DOI] [PubMed] [Google Scholar]
- 25.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 26.Pearse RN. Wnt antagonism in multiple myeloma: a potential cause of uncoupled bone remodeling. Clin Cancer Res. 2006;12(20 Pt 2):6274s–8s. doi: 10.1158/1078-0432.CCR-06-0648. [DOI] [PubMed] [Google Scholar]
- 27.Heiland GR, Zwerina K, Baum W, et al. Neutralisation of Dkk-1 protects from systemic bone loss during inflammation and reduces sclerostin expression. Ann Rheum Dis. 2010;69(12):2152–9. doi: 10.1136/ard.2010.132852. [DOI] [PubMed] [Google Scholar]
- 28.Marienfeld R, May MJ, Berberich I, Serfling E, Ghosh S, Neumann M. RelB forms transcriptionally inactive complexes with RelA/p65. J Biol Chem. 2003;278(22):19852–60. doi: 10.1074/jbc.M301945200. [DOI] [PubMed] [Google Scholar]
- 29.Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell. 2002;9(3):625–36. doi: 10.1016/s1097-2765(02)00477-x. [DOI] [PubMed] [Google Scholar]
- 30.Jin J, Hu H, Li HS, et al. Noncanonical NF-kappaB pathway controls the production of type I interferons in antiviral innate immunity. Immunity. 2014;40(3):342–54. doi: 10.1016/j.immuni.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braut A, Kollar EJ, Mina M. Analysis of the odontogenic and osteogenic potentials of dental pulp in vivo using a Col1α1-2.3-GFP transgene. Int J Dev Biol. 2003;47(4):281–92. [PubMed] [Google Scholar]
- 32.Liu R, Desta T, Raptis M, Darveau RP, Graves DT. Gingivalis and E. coli lipopolysaccharides exhibit different systemic but similar local induction of inflammatory markers. J Periodontol. 2008;79(7):1241–7. doi: 10.1902/jop.2008.070575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pacios S, Andriankaja O, Kang J, et al. Bacterial infection increases periodontal bone loss in diabetic rats through enhanced apoptosis. Am J Pathol. 2013;183(6):1928–35. doi: 10.1016/j.ajpath.2013.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Marijanovic I, Kronenberg MS, et al. Expression and function of Dlx genes in the osteoblast lineage. Dev Biol. 2008;316(2):458–70. doi: 10.1016/j.ydbio.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ponugoti B, Xu F, Zhang C, Tian C, Pacios S, Graves DT. FOXO1 promotes wound healing through the up-regulation of TGF-beta1 and prevention of oxidative stress. J Cell Biol. 2013;203(2):327–43. doi: 10.1083/jcb.201305074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pacios S, Kang J, Galicia J, et al. Diabetes aggravates periodontitis by limiting repair through enhanced inflammation. FASEB J. 2012;26(4):1423–30. doi: 10.1096/fj.11-196279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benson MD, Bargeon JL, Xiao G, et al. Identification of a homeodomain binding element in the bone sialoprotein gene promoter that is required for its osteoblast-selective expression. J Biol Chem. 2000;275(18):13907–17. doi: 10.1074/jbc.275.18.13907. [DOI] [PubMed] [Google Scholar]
- 38.Lin SJ, Ho HC, Lee YF, et al. Reduced osteoblast activity in the mice lacking TR4 nuclear receptor leads to osteoporosis. Reprod Biol Endocrinol. 2012;10:43. doi: 10.1186/1477-7827-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ponugoti B, Xu F, Zhang C, Tian C, Pacios S, Graves DT. FOXO1 promotes wound healing through the up-regulation of TGF-beta1 and prevention of oxidative stress. J Cell Biol. 2013;203(2):327–43. doi: 10.1083/jcb.201305074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Graves DT, Li J, Cochran DL. Inflammation and uncoupling as mechanisms of periodontal bone loss. J Dent Res. 2011;90(2):143–53. doi: 10.1177/0022034510385236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilbert LC, Chen H, Lu X, Nanes MS. Chronic low dose tumor necrosis factor-alpha (TNF) suppresses early bone accrual in young mice by inhibiting osteoblasts without affecting osteoclasts. Bone. 2013;56(1):174–83. doi: 10.1016/j.bone.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 42.Graves DT, Li J, Cochran DL. Inflammation and uncoupling as mechanisms of periodontal bone loss. J Dent Res. 2011;90(2):143–53. doi: 10.1177/0022034510385236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGee-Lawrence ME, Westendorf JJ. Histone deacetylases in skeletal development and bone mass maintenance. Gene. 2011;474(1–2):1–11. doi: 10.1016/j.gene.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-kappaB in the heart: to be or not to NF-kappaB. Circ Res. 2011;108(9):1122–32. doi: 10.1161/CIRCRESAHA.110.226928. [DOI] [PubMed] [Google Scholar]
- 45.Rocha S, Garrett MD, Campbell KJ, Schumm K, Perkins ND. Regulation of NF-kappaB and p53 through activation of ATR and Chk1 by the ARF tumour suppressor. EMBO J. 2005;24(6):1157–69. doi: 10.1038/sj.emboj.7600608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mantovani A. Molecular pathways linking inflammation and cancer. Curr Mol Med. 2010;10(4):369–73. doi: 10.2174/156652410791316968. [DOI] [PubMed] [Google Scholar]
- 47.Hayden MS, Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26(3):203–34. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gopal YN, Arora TS, Van Dyke MW. Parthenolide specifically depletes histone deacetylase 1 protein and induces cell death through ataxia telangiectasia mutated. Chem Biol. 2007;14(7):813–23. doi: 10.1016/j.chembiol.2007.06.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.