

Abstract
Grainyhead-like genes are part of a highly conserved gene family that play a number of roles in ectoderm development and maintenance in mammals. Here we identify a novel allele of Grhl2, cleft-face 3 (clft3), in a mouse line recovered from an ENU mutagenesis screen for organogenesis defects. Homozygous clft3 mutants have a number of phenotypes in common with other alleles of Grhl2. We note a significant effect of genetic background on the clft3 phenotype. One of these is a reduction in size of the telencephalon where we find abnormal patterns of neural progenitor mitosis and apoptosis in mutant brains. Interestingly, Grhl2 is not expressed in the developing forebrain, suggesting this is a survival factor for neural progenitors exerting a paracrine effect on the neural tissue from the overlying ectoderm where Grhl2 is highly expressed.
Keywords: ENU, mutagenesis, forebrain, development, mouse, cloning
Introduction
Embryological development requires the cooperative patterning of cell types from multiple germ layers, which undergo complex morphogenetic movements to create the final body plan. We have been using a forward genetics approach to both identify novel regulators of embryonic organogenesis and to ascertain previously uncharacterized roles for known genes in these processes (Ha et al., 2015; Herron et al., 2002; Stottmann and Beier, 2014; Stottmann and Beier, 2010). This has proven to be an efficient tool for gene and allele discovery. One important outcome from these screens is the recovery of an allelic series of a gene of interest, which may collectively give significant insight into the role of that gene in development that is not evident from a simple null allele.
The grainyhead gene was identified in the classic Drosophila larval patterning screen (Nusslein-Volhard et al., 1984) and has since been implicated in a number of functions such as epidermal barrier maturation, wound repair, tracheal tube size control and CNS development (Almeida and Bray, 2005; Baumgardt et al., 2009; Bray and Kafatos, 1991; Cenci and Gould, 2005; Hemphala et al., 2003; Mace et al., 2005; Maurange et al., 2008; Wang and Samakovlis, 2012). Mammalian genomes have three Grh genes (Wilanowski et al., 2002), now called grainyhead-like 1 (Grhl1), Grhl2, Grhl3. Consistent with the data from Drosophila mutants, the mammalian grainyhead gene family has been implicated in developmental process involving the ectoderm. Human mutations have been found in patients with ectodermal dysplasia (GRHL2: (Petrof et al., 2014), hearing loss (GRHL2; (Peters et al., 2002; Van Laer et al., 2008) and Van der Woude Syndrome (GRHL3, (Peyrard-Janvid et al., 2014). Surprisingly, the development of the CNS has not been studied to date in mammalian Grh homologs beyond the role of these genes in neural tube closure.
Here we describe our identification of an ENU mutation in the mouse in the Grhl2 gene, which recapitulates many of the previously described phenotypes in other Grhl2 alleles. Interestingly, our mutation results in a reduction in size of the telencephalon. This phenotype is dependent on the genetic background of the allele and has not previously been reported for mutations in Grhl2. We note that expression of Grhl2 has repeatedly been demonstrated to be confined to the surface ectoderm, suggesting we have identified a novel, non-tissue autonomous role for this gene.
RESULTS
Cleft-face 3 mutants have multiple defects in head development
We originally recovered the cleft-face3 (clft3) mutation as part of an ENU mutagenesis screen to identify genes important for development of the mammalian forebrain (Stottmann et al., 2011). Clft3 mutants were initially identified by their significant craniofacial defects at late embryonic stages. Mutants had shorter snouts than wild-type mice (Fig. 1B,C) and some had a complete failure of tissue fusion in the anterior craniofacial tissues, leading to a cleft face (Fig. 1D,E). Further analysis identified a number of other phenotypes. Clft3 mutant embryos are often smaller than littermate controls and exhibit edema or appear pale, suggesting a cardiovascular defect (Fig. 1B). We also note incompletely penetrant exencephaly and apparent hemangiomas (Fig. 1B,E) Histological analysis revealed these are, in fact, neuronal overgrowths (Fig. 1H). We also note a hypoplastic telencephalon in clft3 mutants with defects in dorsal midline structure formation (Fig. 1G).
Figure 1. Clft3 phenotypes.

(A,B) Wild-type (A) and clft3 (B) mutant embryo showing the reduced size of the mutant, exencephaly and edema. (C–E) Cleft face 3 mutants have a variety of anterior phenotypes including shortened snouts (C), failure of anterior midline fusion (D) and growths on top of the head (E). Histological analysis of mutants compared to wild-type (F) indicates a smaller telencephalon with loss of dorsal midline structures (G) and neuronal overgrowths (H; coronal sections at the approximate level of the eye and teeth). All paired images are shown at the same magnification.
In order to study the development of these phenotypes, we performed a phenotypic analysis at multiple stages of embryonic development. The clft3 phenotype is first evident at E10.5 when a significant proportion of clft3 mutants are either dead or significantly smaller than littermates (Fig. 2B,D). At E11.5 and E12.5 we began to notice defects in the forebrain, including an obviously smaller telencephalon in relation to the rest of the embryo, in clft3 mutant embryos (Fig. 2D,F). Even in embryos that are growth retarded, we often note a disproportionate decrease in size of the telencephalon (E11.5). We also noted some embryos with obvious signs of neuronal overgrowths at late organogenesis stages (Fig. 2H).
Figure 2. Developmental analysis of Clft3 phenotypes.
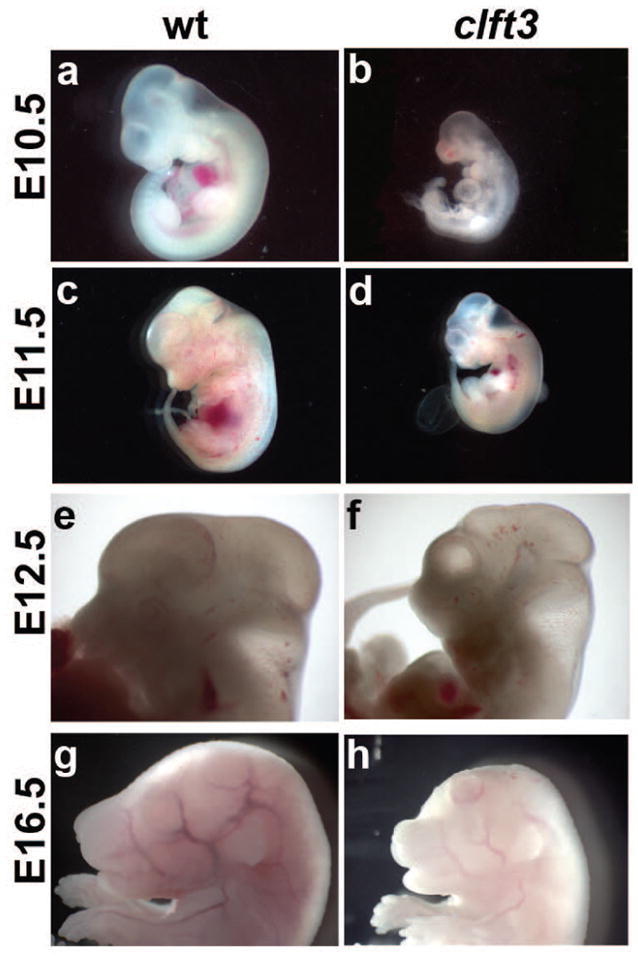
A comparison of wild-type (A,C,E,G) and cflt3 mutants (B,D,F,H) at E10.5 (A,B), E11.5 (C,D), E12.5 (E,F) and E16.5 (G,H). Mutants are often significantly smaller (B,D) than wild-type littermates or already dead. Surviving mutants at E11.5 and E12.5 (D,F) show a forebrain which is disproportionately reduced in size. At E16.5 (F), the initial stages of neuronal overgrowth can be seen. All paired images are shown at the same magnification.
The clft3 mutation is in the Grainyhead-like 2 gene
We took a positional cloning approach to identify the causal mutation in clft3 mutants. An initial SNP scan of four mutant genomes identified a 72.4 Mb region on chromosome 15 as the only region of shared homozygosity for A/J SNPs among all four embryos tested (proximal end of the chromosome to SNP rs13482643, Fig. 3A). Further mapping with microsatellite markers and SNPs with multiple affected embryos narrowed the region to a 19.5 Mb interval (D15Mit11 at 31.9Mb to D15Mit228 at 51.4Mb). A literature review on the 60 genes in this minimal interval focusing on gene expression patterns and known loss of function phenotypes suggested grainyhead-like 2 as a candidate gene for the clft3 mutation. Sequencing of the Grhl2 locus from two clft3 mutants from different litters identified an A-to-G coding change in the eleventh exon of the grhl2 locus (c.A1451G; p.D484G, genomic position chr3:37,336,311 GRCm38, mm10; Fig. 3B). The identity of this amino acid sequence in this region of the protein is well conserved among vertebrates and is in the DNA binding domain of this CP2 family transcription factor (Fig. 3C,D). Computational analysis suggests the missense mutation is highly likely to be damaging (Polyphen score of 1.00: “probably damaging: on a scale of 0.00–1.00). Other homologous point mutations from human syndromes cluster in the DNA binding domain as well: Y398H and I428L in ectodermal dysplasia (Petrof et al., 2014).
Figure 3. Cloning of the clft3 mutation.

(A) A whole-genome SNP scan originally identified a region on chromosome 15 containing the clft3 mutation from the proximal end to SNP rs13482643 (red = homozygous for A/J SNP, blue = homozygous for FVB/NJ SNP, yellow = heterozygous). (B) Sanger sequencing of the Grhl2 locus identified an A>G missense mutation in clft3 mutants. (C) The ENU-induced mutation changes the coding of a well-conserved amino acid from aspartic acid to glycine (red text). Amino acid sequence from all three mouse Grhl proteins and Grhl2 sequence from four other vertebrates are indicated (- indicates same residue as above sequence). (D) A graphical representation of the protein domains of Grhl2 shows the clft3 mutation is in the DNA binding domain (blue) of the Grhl2 protein. The location of point mutations in human ectodermal dysplasia patients (e.d.) are also indicated.
The clft3 phenotype has segregated with this mutation for at least twelve generations. Both a custom RFLP and a custom Taqman genotyping assay for the clft3 mutation have continued to show precise concordance between the clft3 phenotype (or carrier status in adults) and the c.A1451G variant in the Grhl2 gene. While there are some subtle differences between the clft3 allele and previously published alleles of Grhl2, the similarity in phenotypes in conjunction with mapping and sequencing data lead us to conclude that clft3 is most likely a mutation in Grhl2.
Clft3 mutants show a background dependent phenotype including early embryonic lethality
The embryonic lethality of homozygous mutants we noted at E10.5 became increasingly penetrant through development (Table 1). In aggregate, mutants are recovered from heterozygous intercrosses at a 19.1% frequency from E9.5–E12.5 and 16.8% frequency from E13.5–E18.5 (both ratios are statistically significantly decreased from the expected 25% as determined by a chi squared analysis). In addition, a significant proportion of the mutants we recovered are either obviously dead or growth retarded and noticeably smaller than their littermates (Table 2). From E10.5–E12.5, 45.7% of mutants are growth-retarded when compared to littermates and 13.6% are clearly dead or dying. From E12.5–E18.5, 48.0% of mutants are dead and 12.0% are growth retarded. Heterozygotes survive to weaning in appropriate Mendelian ratios, regardless of genetic background (data not shown).
Table 1.
A proportion of Grhlclft3 homozygous mutants do not survive to birth.#
| Age(E) | Total Embryos | Mutants Recovered | Percent |
|---|---|---|---|
| 8.5 | 26 | 2 | 7.7 |
| 9.5 | 37 | 9 | 24.3 |
| 10.5 | 138 | 28 | 20.3 |
| 11.5 | 158 | 33 | 20.9 |
| 12.5 | 180 | 31 | 17.2* |
| (E9.5–12.5) | 539 | 103 | 19.1** |
| 13.5 | 71 | 9 | 12.7* |
| 14.5 | 99 | 16 | 16.2 |
| 16.5 | 43 | 12 | 27.9 |
| 17.5 | 19 | 3 | 15.8 |
| 18.5 | 72 | 11 | 15.3* |
| (E13.5–E18.5) | 304 | 51 | 16.8** |
| total | 843 | 154 | 18.3** |
Data collected are from all crosses, irrespective of genetic background. Chi-square analyses were performed to indicate mutants are obtained at less than Mendelian ratios (*:p≤0.05; **:p≤0.01).
Table 2.
A proportion of Grhlclft3 homozygous mutants are dead or growth retarded.
| Age(E) | # Mutants Dead (%) | Small (%) | |
|---|---|---|---|
| 10.5 | 22 | 3 (13.6) | 6 (27.2) |
| 11.5 | 33 | 5 (15.1) | 16 (48.5) |
| 12.5 | 26 | 3 (11.5) | 15 (57.6) |
| (E10.5–12.5) | 81 | 11(13.6) | 37 (45.7) |
| 13.5 | 8 | 2 (25.0) | 3 (37.5) |
| 14.5 | 16 | 8 (50.0) | 1 (6.3) |
| 16.5 | 12 | 5 (41.7) | 1 (8.3) |
| 17.5 | 3 | 3 (100.0 | 0 |
| 18.5 | 11 | 6 (54.5) | 1 (9.1) |
| (E13.5–E18.5) | 50 | 24 (48.0) | 6 (12.0) |
During the course of our embryonic analysis and outcross breeding to positionally clone the causal mutation in clft3 mutants, we noted that the phenotype seemed to be getting progressively more severe and the penetrance of some phenotypes was changing. (Note the data presented above and in tables 1 and 2 are from all experiments combined.) We originally identified clft3 mutants in the third generation of a recessive screen (Stottmann et al., 2011) in which the G3 mutant embryos would have a significant contribution of the genome from the mutagenized A/J strain (approximately 3/8) and the rest from the FVB/NJ strain (Fig. 4). Details of the mutagenesis strategy can be found elsewhere (Stottmann and Beier, 2010; Stottmann et al., 2011) but, in brief, we mutagenized a population of inbred A/J males (G0, Fig. 4). These were mated with FVB/NJ inbred females to create G1 males which are genetically 50% from the A/J strain, and 50% FVB/NJ. In order to homozygose any potential ENU mutations, we again first crossed to FVB/NJ females to create the G2 females. These are (as an average) 25% A/J and 75% FVB/NJ, with respect to genetic background. Crossing the G2 females to the G1 males allows recovery of embryos with recessive mutations. These G3 embryos are approximately 37.5% A/J and 62.5% FVB/NJ. However, as we continue to outcross the carrier animals as part of our cloning strategy, the genetic background will therefore become increasingly the FVBN/J strain (Fig. 4).
Figure 4. Breeding strategy for the clft3 mutation.
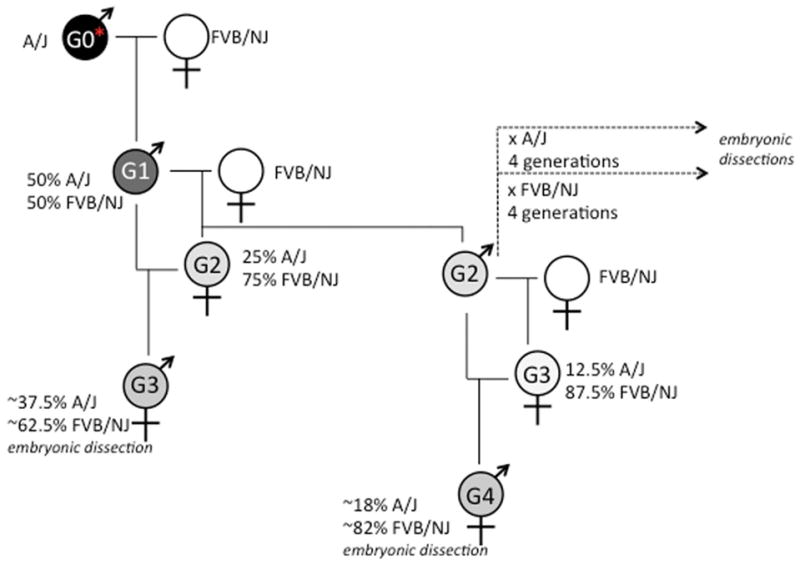
Details of the breeding are found in the text. The relative shading of gray is to represent the approximate percentage of the genome that comes from the A/J strain (100% = black) as compared to the FVB/NJ strain (100% white). G=generation.
In crosses involving the G1 founder, 4/5 mutants had the cleft face phenotype. In the next generation of crosses sired by a G2 male, one 1/195 mutants had a cleft face. Crosses of the G2 male to G2 and G3 females were combined in this analysis and the single mutant with the cleft face came from a G2 intercross in which the mutant genome would be approximately 25% derived from the A/J strain, and 75% from the FVB/NJ strain. Subsequently, we created two “substrains” of the clft3 mutation by outcrossing to A/J and FVBN/J respectively (Fig. 4). While scoring phenotypes in this analysis, several were found to be significantly affected by genetic background and earlier lethality on the FVBN/J background became evident. We again scored for specific phenotypes after crossing to each strain for at least 4 generations (Table 3). In crosses from FVB/NJ mice at the N4 generation or greater, we note 22/30 (73.3%) have growth retardation, 10/30 (33.3%) have an obviously cleft face, 1/30 (3.0%) had exencephaly and 11/30 (36.7%) have a noticeably smaller telencephalon. In embryos from the A/J “substrain,” we note 11/17 (64.7%) have a smaller body, 6/17 (35.3%) have a cleft face, 4/17 (23.5%) have exencephaly and none had the obviously smaller telencephalon. This supported our earlier findings that strain background appears to have an effect on penetrance and expressivity of the clft3 mutation in mouse. We were unable to maintain the mutation on the A/J background with any efficiency. This is likely due to a known reduced fecundity of that strain (Silver, 1995) and/or a maternal effect as has been previously noted (Pyrgaki et al., 2011). Any further embryologic data presented here are from the mouse colony enriched for the FVB background.
Table 3.
Genetic background affects Grhlclft3 phenotype*
| FVB/NJ (%) | A/J (%) | |
|---|---|---|
|
|
||
| Small Body | 22 (73.3) | 11 (64.7) |
| Cleft Face | 10 (33.3) | 6 (35.3) |
| Exencephaly | 1 (3.0) | 4 (23.5) |
| Reduced Telencephalon | 11 (36.7) | 0 |
|
| ||
| Total Sample | 30 | 17 |
Phenotypes tallied in a representative sample of embryos from outcrosses of the clft3 allele.
Wild-type and clft3 mutant Grhl2 proteins bind DNA with identical specificity
The clft3 mutation is a missense mutation in the DNA binding domain of Grhl2 suggesting it might alter the DNA binding motif for the protein. We tested this hypothesis by performing protein-binding microarray (PBM) experiments for the wild-type (WT) and clft3 mutant Grhl2 protein (expression of the DNA binding domain as well as flanking sequence). PBM experiments, using the universal PBM design (Berger and Bulyk, 2009; Berger et al., 2006) allow a comprehensive and unbiased assessment of protein-DNA to all possible 8-base pair sequences. PBM experiments performed for the WT and clft3 mutants showed highly similar DNA binding across the full-range of tested DNA sequences (data not shown), and resulted in identical DNA binding motifs (Fig. 5) that closely resemble the previously reported Grhl2 binding motif (e.g., (Walentin et al., 2015). These results demonstrate that the clft3 mutant does not change the DNA binding specificity of the mutant protein. However, GRHL2 is known to bind as a dimer and the clft3 mutation may affect dimer interactions.
Figure 5. PBM-determined DNA-binding Motifs for Grhl2 and Clft3.
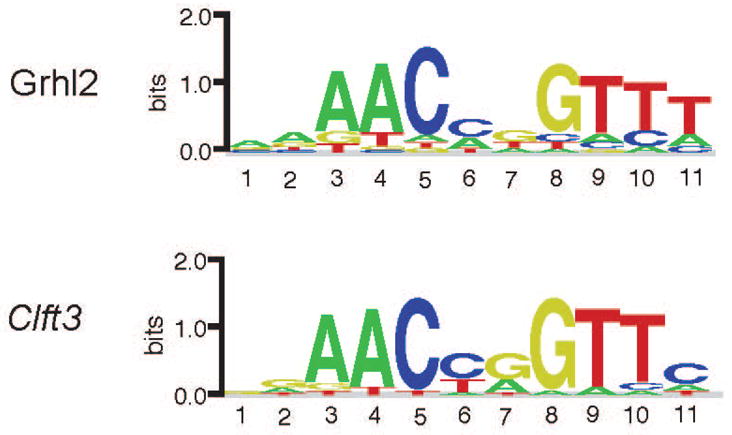
DNA-binding motif logos were determined from the PBM binding data determined for Grhl2 and Clft3. Motifs were determined using the Seed-and-Wobble algorithm (see Methods & Materials).
Proliferation and cell death in the cflt3 brains
We pursued a molecular analysis to explain the reduction in size of the telencephalon in the clft3 mutants at E10.5–E12.5 by measuring cell proliferation and cell death with immunohistochemistry for phospho-histone H3 and cleaved caspase 3, respectively. At E10.5, we see no change in the number of pHH3 mitotic cells in the region immediately adjacent to the ventricle (“ventricular”) between mutant and wild-type embryos (Fig. 6A–C). However, we do note a four-fold increase in cells immunoreactive for pHH3 which are not confined to the ventricular region of the neuroepithelium in mutants. These are a small proportion of all dividing cells but the difference in mutants in this subset is striking (“cortex” in Fig. 6C). Combining all mitotic cells we see a 76% increase in pHH3-positive cells in clft3 mutants at E10.5. At E11.5, this pattern continues as a true ventricular zone (VZ) emerges and we observe a slight decrease (11%) in VZ pHH3 positive cells in mutants as compared to wt. Again, we see a dramatic increase (244%) in dividing cells in cortical areas outside the VZ. Altogether, these comprise a 53% increase in total cortical mitotic activity (Fig. 6D–F). At E12.5, the VZ mitotic activity is now significantly reduced as compared to wild-type (Fig. 6G–I). We again see an increase in pHH3-positive cells away from the VZ (333% increase) and, in total, a 47% increase in mitotic activity.
Figure 6. Molecular analysis of forebrain development in clft3 mutants.

Proliferation analysis with immunohistochemistry for phosphorylated histone H3 (A–I). Representative results are shown for wild-type (A,D,G) and cflt3 mutants (B,E,H) at E10.5 (A,B), E11.5 (D,E) and E12.5 (G,H). Quantification for each stage is shown in C,F,I. Cells were separately counted in the first cell width immediately adjacent to the ventricle (“ventricular”) and the remaining neuroepithelium (“cortical”). Totals of all cells counted are also shown. In each case, the number relative to the wild-type average is shown. A similar analysis was performed for apoptosis with immunohistochemistry for cleaved-caspase 3 (J–N). Images are shown for E10.5 (J,K) and E11.5 (L,M) from wild-type (J,K) and clft3 mutant (K,M). All sections are in the coronal plane from the telencephalic neuroepithelium. Cartoon in (O) shows approximate location of sections in a stylized E11/12 embryo. *: p≤0.05, **: p≤0.01.
The overall increase in cell proliferation is not consistent with a dramatic reduction in brain size in the clft3 mutants, so we sought to determine what happened to these cells. We first analyzed levels of programmed cell death with immunohistochemistry for cleaved caspase 3. We see a 22-fold increase at E10.5, an 8-fold increase at E11.5 and a 2.4-fold increase at E12.5. We thus conclude that the clft3 mutants have the disproportionately small brain largely because of the significantly increased cell death. The proliferation dynamics (increases in numbers of mitotic cells away from the VZ) indicate other dysregulation but these are more than adequately masked by the dramatic increase in cell death suggesting the primary defect leading to the hypoplastic telencephalon is a reduction in cell survival.
DISCUSSION
Here we report a novel ENU-induced allele of Grhl2, which results in growth retardation and embryonic lethality with phenotypes in the head including a background dependent reduction in size of the telencephalon. Our data suggest this reduction in size is due to a significant increase in cell death in mutants indicating Grhl2 is normally acting as an important survival factor during neurogenesis.
The phenotype of clft3 is consistent with that found for previous genetic ablations of Grhl2. The first reported ablation of Grhl2 was through ES cell mediated homologous recombination (Grhl2tm1.1Jane, (Rifat et al., 2010). The homozygous null embryos were characterized on a C57BL/6 genetic background and had a fully penetrant anterior craniofacial fusion phenotype. Two independent gene traps were subsequently generated (Grhl2Gt(E115B04)Wrst, Grhl2lacz1 and Grhl2Gt(RRU622)Byg, Grhl2lacZ4) and maintained on a mixed 129/B6 background (Werth et al., 2010). These homozygous null mutants were embryonic lethal after E11.5 with anterior spina bifida, exencephaly, split face malformation and growth retardation after the 22 somite stage. Failed closure of the posterior neural tube closure was also noted with lumbosacral spina bifida and curled tail. A third gene trap allele was generated (Grhl2GT(AC0205)Wtsi, Grhl2GT) which also had an incompletely penetrant cranial neural tube defects (on a B6 genetic background) and lethality prior to E9.5 (Brouns et al., 2011). An independent ENU induced mutation has been identified (Grhl2m1Nisw) with similar phenotypes to our clft3 allele, including very limited survival past E9.5. Finally, a conditional allele has been generated which also resulted in death by E11.5 when Grhl2 was deleted throughout the embryo (Grhl2flox, (Walentin et al., 2015). Given the similarity between our phenotype and those reported for other alleles, and the predicted damaging nature of amino acid substitution, we are confident we have identified the correct mutation.
Differing phenotypes resulting from loss of Grhl2 on different genetic backgrounds as we report here has been previously noted, although A/J has not been one of the strains reported to date. A/J is not a commonly used strain for maintaining alleles but is able to withstand larger doses of ENU (Justice et al., 2000)., so is used often in our mutagenesis strategy. Brouns et al (Brouns et al., 2011) noted that crossing the Grhl2GT allele from a mixed 129/B6 background to BALB/c background prolonged the survival of the homozygous mutant embryos past E9.5 and also resulted in a spina bifida phenotype in 88% of mutants. These mice also showed exencephaly and craniofacial defects. Pyrgaki and colleagues (Pyrgaki et al., 2011) observed that moving the Grhl2m1Nisw allele from the 129 to C3H background allowed some embryos to survive to E18.5; these still had fully penetrant exencephaly and anterior clefting phenotypes. We note that none of the reported alleles have yet been crossed onto either the A/J or FVB/NJ genetic backgrounds that we used in our study. We used these genetic backgrounds because of their utility in ENU mutagenesis and mapping. Some aspects of our phenotype seem to be distinct from alleles reported so far; specifically, the reduction in telencephalon size. We cannot be certain whether this phenotype is then particular to our mutation or the genetic background(s) the alleles are maintained on. However, given the predicted severity of the mutation and the overall similarity between reported phenotypes and ours, we suspect our allele is essentially a null allele, or at least a severe hypomorph, and the differences in phenotypes are indeed due to genetic background effects. It would be intriguing to cross the previously reported alleles onto A/J or FVB/NJ to see if the neural phenotypes we see in our experiments begin to emerge (or to cross clft3 onto backgrounds similar to the studies described above). Alternatively, a tissue specific ablation could be pursued with the conditional allele (Walentin et al., 2015) to circumvent the early lethality and further understand the role of Grhl2 in survival of the forebrain tissue.
We find the primary molecular explanation for the decreased forebrain is the massive increase in cell death in the homozygous mutants. Grhl2 has been liked to the control of cell proliferation in a number of different contexts. Morpholino knockdown of grhl1b in fish leads to increased apoptosis in the CNS (Dworkin et al., 2012). Grh also regulates neuroblast proliferation in Drosophila (Brody and Odenwald, 2000; Cenci and Gould, 2005; Maurange et al., 2008). Interestingly, human ectodermal dysplasia syndrome patients with GRHL2 mutations had increased Ki67 expression in the skin (Petrof et al., 2014). One potential molecular explanation for these changes in proliferation is the finding in multiple systems that Grhl2 acts within the Fgf8 signaling pathway. In Drosophila, branchless/FGF upregulates grh activity post-transcriptionally and this is thought to be due to FGF-induced phosphorylation of grh, most likely by ERK2 (Hemphala et al., 2003). Grh is also downstream of erk signaling in Drosophila wound healing. In zebrafish, subphenotypic concentrations of grhl2b and fgf8 morpholino knockdown lead to MHB patterning defects (Dworkin et al., 2012). These links to FGF signaling are consistent with the finding FGF is required for neuronal survival in the mouse forebrain (Paek et al., 2009).
The most interesting aspect of this forebrain phenotype is that the reduced telencephalic size in clft3 mutants is apparently regulated by Grhl2 in a non-autonomous fashion. There is robust data on the expression patterns of Grhl2 from both of the gene trap alleles as well as the RNA in situ hydridization. All current evidence shows Grhl2 is expressed in the surface ectoderm, including the non-neural ectoderm during early stages of neural tube development (Auden et al., 2006; Brouns et al., 2011; Pyrgaki et al., 2011; Werth et al., 2010). None of the available data show any evidence of Grlh2 expression in the forebrain. Again, the use of the conditional allele of Grhl2 recently created would be an interesting approach to define the spatiotemporal requirement for Grhl2 in regulating the survival of the forebrain tissue (Walentin et al., 2015).
In conclusion, our findings from the cloning of the clft3 mouse mutant continue to implicate Grhl2 in a number of different roles in embryonic development. The non-tissue autonomous effect on brain size and the sensitivity of the phenotype to genetic background suggest that Grhl2 acts within a robust network to guide development of the embryonic ectoderm.
METHODS
Mouse husbandry
Animals were initially maintained as a mixed A/J, FVB stock. The allele was isolated in an ENU mutagenesis experiment in which A/J males were mutagenized and outcrossed to FVB females (Stottmann et al., 2011). Routine genotyping was performed via a RFLP assay wherein the clft3 mutation creates a Bstn1 restriction enzyme recognition sequence (F primer: TAAGATGAGGCCGGTAGCTG; R primer: GAGGGTGTGAGAGCAGGAGT ). We also used a custom TaqMan Sample2SNP assay (Assay ID:AHOJB7P, Life Technologies). All animals were maintained in accordance with Brigham and Women’s Hospital and Cincinnati Children’s Hospital Medical Center IACUC guidelines. Matings were monitored and noon of the day of copulation plug was determined to be E0.5. Embryos were collected via Cesarean section after the pregnant dams were sedated and euthanized. This line is available to the research community.
Genetic mapping
The initial mapping of the clft3 mutant has been described (Stottmann et al., 2011). Briefly, an initial genome scan was done with multiple mutant embryos using a 768 marker whole genome SNP panel to identify a region of shared A/J homozygosity among mutants, similar to a method described previously (Moran et al., 2006). Microsatellite mapping using standard protocol further localized the mutation as descrived. After identification of the candidate region, exon-directed sequencing revealed the clft3 mutation in Grhl2.
Histology and immunohistochemistry
Samples for histological analysis were fixed in Bouins fixative, prepared using a Leica TP1020 automated tissue processor, sectioned at 14 μm and stained using established protocols. Paired images presented are of equal magnification. Cryo-sections were used for immunohistochemistry. Antigen retrieval was performed with an Antigen Unmasking Solution (Vector Laboratories) in a microwave. Sections were blocked with 5% Normal Goat Serum/PBST and primary antibodies were incubated overnight. Primary antibodies used in this study were anti-Phospho-Histone H3 (pHH3, SIGMA, 1:500) and cleaved caspase-3 (Cell Signaling, 1:300, ). Sections were rinsed with PBST and incubated with an Alexa-Fluor 488 Goat anti-Rabbit secondary antibody (Molecular Probes, 1:500) for one hour at room temperature. Sections were stained with DAPI to visualize cell nuclei and slides were mounted in ProLong AntiFade (Invitrogen) and sealed. Microscopy was performed on a Zeiss AxioImager and cell count analysis was completed using IMARIS 7.5.1 software.
Quantification of immunohistochemistry
Quantification of mitotic and apoptotic cells was performed by counting fields of cells parallel to the ventricular zone with Imaris 7.5.1. Cells immunoreactive for pHH3 or cleaved caspase 3 were counted as a proportion of all cells in the defined field (DAPI-positive). All statistical analyses were performed in Excel.
Protein samples and protein-binding microarray experiments
Residues 196–625 of Grhl2 open reading frame (cDNA primers GGGGACAAGTTTGTACAAAAAAGCAGGCTCCTAGCCAGCCACAGCTCCTAT and GGGGACCACTTTGTACAAGAAAGCTGGGTCTATCAGATCTCCATCAGCGTGAT) was cloned into the bacterial expression vector pDEST15 to make GST-fusion protein. Site-directed mutagenesis was performed on the Grhl2 pDEST15 vector to recapitulate the clft3 mutation. Proteins (WT and mutant) were expressed in E. coli BL21 (DE3) cells at 37°C for 2 hours, and purified by GST affinity column. PBM experiments were performed using a custom-designed, universal ‘all-10mer’ microarray (Agilent Technologies Inc., AMADID #016060, 4x44K array format (Zhu et al., 2009) described previously (Berger et al., 2006). PBM experiments were performed as described previously (Siggers et al., 2014).
Acknowledgments
This work was supported by the National Institutes of Health [grant numbers HD36404, MH081187 to D.B., HG0003985 to M.L.B. and NS085023 to R.S.] and laboratory start-up funds from the Cincinnati Children’s Hospital Research Foundation (R.S.).
References
- Almeida MS, Bray SJ. Regulation of post-embryonic neuroblasts by Drosophila Grainyhead. Mech Dev. 2005;122:1282–1293. doi: 10.1016/j.mod.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Auden A, Caddy J, Wilanowski T, Ting SB, Cunningham JM, Jane SM. Spatial and temporal expression of the Grainyhead-like transcription factor family during murine development. Gene Expr Patterns. 2006;6:964–970. doi: 10.1016/j.modgep.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Baumgardt M, Karlsson D, Terriente J, Diaz-Benjumea FJ, Thor S. Neuronal subtype specification within a lineage by opposing temporal feed-forward loops. Cell. 2009;139:969–982. doi: 10.1016/j.cell.2009.10.032. [DOI] [PubMed] [Google Scholar]
- Berger MF, Bulyk ML. Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat Protoc. 2009;4:393–411. doi: 10.1038/nprot.2008.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Philippakis AA, Qureshi AM, He FS, Estep PW, 3rd, Bulyk ML. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat Biotechnol. 2006;24:1429–1435. doi: 10.1038/nbt1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray SJ, Kafatos FC. Developmental function of Elf-1: an essential transcription factor during embryogenesis in Drosophila. Genes Dev. 1991;5:1672–1683. doi: 10.1101/gad.5.9.1672. [DOI] [PubMed] [Google Scholar]
- Brody T, Odenwald WF. Programmed transformations in neuroblast gene expression during Drosophila CNS lineage development. Dev Biol. 2000;226:34–44. doi: 10.1006/dbio.2000.9829. [DOI] [PubMed] [Google Scholar]
- Brouns MR, De Castro SC, Terwindt-Rouwenhorst EA, Massa V, Hekking JW, Hirst CS, Savery D, Munts C, Partridge D, Lamers W, Kohler E, van Straaten HW, Copp AJ, Greene ND. Over-expression of Grhl2 causes spina bifida in the Axial defects mutant mouse. Hum Mol Genet. 2011;20:1536–1546. doi: 10.1093/hmg/ddr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci C, Gould AP. Drosophila Grainyhead specifies late programmes of neural proliferation by regulating the mitotic activity and Hox-dependent apoptosis of neuroblasts. Development. 2005;132:3835–3845. doi: 10.1242/dev.01932. [DOI] [PubMed] [Google Scholar]
- Dworkin S, Darido C, Georgy SR, Wilanowski T, Srivastava S, Ellett F, Pase L, Han Y, Meng A, Heath JK, Lieschke GJ, Jane SM. Midbrain-hindbrain boundary patterning and morphogenesis are regulated by diverse grainy head-like 2-dependent pathways. Development. 2012;139:525–536. doi: 10.1242/dev.066522. [DOI] [PubMed] [Google Scholar]
- Ha S, Stottmann RW, Furley AJ, Beier DR. A forward genetic screen in mice identifies mutants with abnormal cortical patterning. Cereb Cortex. 2015;25:167–179. doi: 10.1093/cercor/bht209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemphala J, Uv A, Cantera R, Bray S, Samakovlis C. Grainy head controls apical membrane growth and tube elongation in response to Branchless/FGF signalling. Development. 2003;130:249–258. doi: 10.1242/dev.00218. [DOI] [PubMed] [Google Scholar]
- Herron BJ, Lu W, Rao C, Liu S, Peters H, Bronson RT, Justice MJ, McDonald JD, Beier DR. Efficient generation and mapping of recessive developmental mutations using ENU mutagenesis. Nat Genet. 2002;30:185–189. doi: 10.1038/ng812. [DOI] [PubMed] [Google Scholar]
- Justice MJ, Carpenter DA, Favor J, Neuhauser-Klaus A, Hrabe de Angelis M, Soewarto D, Moser A, Cordes S, Miller D, Chapman V, Weber JS, Rinchik EM, Hunsicker PR, Russell WL, Bode VC. Effects of ENU dosage on mouse strains. Mamm Genome. 2000;11:484–488. doi: 10.1007/s003350010094. [DOI] [PubMed] [Google Scholar]
- Mace KA, Pearson JC, McGinnis W. An epidermal barrier wound repair pathway in Drosophila is mediated by grainy head. Science. 2005;308:381–385. doi: 10.1126/science.1107573. [DOI] [PubMed] [Google Scholar]
- Maurange C, Cheng L, Gould AP. Temporal transcription factors and their targets schedule the end of neural proliferation in Drosophila. Cell. 2008;133:891–902. doi: 10.1016/j.cell.2008.03.034. [DOI] [PubMed] [Google Scholar]
- Moran JL, Bolton AD, Tran PV, Brown A, Dwyer ND, Manning DK, Bjork BC, Li C, Montgomery K, Siepka SM, Vitaterna MH, Takahashi JS, Wiltshire T, Kwiatkowski DJ, Kucherlapati R, Beier DR. Utilization of a whole genome SNP panel for efficient genetic mapping in the mouse. Genome Res. 2006;16:436–440. doi: 10.1101/gr.4563306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusslein-Volhard C, Wieschaus E, Kluding H. Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster. I. Zygotic loci on the second chromosome. Wilhelm Roux’s Arch Dev Biol. 1984;193:267–282. doi: 10.1007/BF00848156. [DOI] [PubMed] [Google Scholar]
- Paek H, Gutin G, Hebert JM. FGF signaling is strictly required to maintain early telencephalic precursor cell survival. Development. 2009;136:2457–2465. doi: 10.1242/dev.032656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters LM, Anderson DW, Griffith AJ, Grundfast KM, San Agustin TB, Madeo AC, Friedman TB, Morell RJ. Mutation of a transcription factor, TFCP2L3, causes progressive autosomal dominant hearing loss, DFNA28. Hum Mol Genet. 2002;11:2877–2885. doi: 10.1093/hmg/11.23.2877. [DOI] [PubMed] [Google Scholar]
- Petrof G, Nanda A, Howden J, Takeichi T, McMillan JR, Aristodemou S, Ozoemena L, Liu L, South AP, Pourreyron C, Dafou D, Proudfoot LE, Al-Ajmi H, Akiyama M, McLean WH, Simpson MA, Parsons M, McGrath JA. Mutations in GRHL2 result in an autosomal-recessive ectodermal Dysplasia syndrome. Am J Hum Genet. 2014;95:308–314. doi: 10.1016/j.ajhg.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrard-Janvid M, Leslie EJ, Kousa YA, Smith TL, Dunnwald M, Magnusson M, Lentz BA, Unneberg P, Fransson I, Koillinen HK, Rautio J, Pegelow M, Karsten A, Basel-Vanagaite L, Gordon W, Andersen B, Svensson T, Murray JC, Cornell RA, Kere J, Schutte BC. Dominant mutations in GRHL3 cause Van der Woude Syndrome and disrupt oral periderm development. Am J Hum Genet. 2014;94:23–32. doi: 10.1016/j.ajhg.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyrgaki C, Liu A, Niswander L. Grainyhead-like 2 regulates neural tube closure and adhesion molecule expression during neural fold fusion. Dev Biol. 2011;353:38–49. doi: 10.1016/j.ydbio.2011.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifat Y, Parekh V, Wilanowski T, Hislop NR, Auden A, Ting SB, Cunningham JM, Jane SM. Regional neural tube closure defined by the Grainy head-like transcription factors. Dev Biol. 2010;345:237–245. doi: 10.1016/j.ydbio.2010.07.017. [DOI] [PubMed] [Google Scholar]
- Siggers T, Reddy J, Barron B, Bulyk ML. Diversification of transcription factor paralogs via noncanonical modularity in C2H2 zinc finger DNA binding. Mol Cell. 2014;55:640–648. doi: 10.1016/j.molcel.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver LM. Mouse genetics : concepts and applications. New York: Oxford University Press; 1995. p. xiii.p. 362. [Google Scholar]
- Stottmann R, Beier D. ENU Mutagenesis in the Mouse. Curr Protoc Hum Genet. 2014;82:15 14 11–15 14 10. doi: 10.1002/0471142905.hg1504s82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stottmann RW, Beier DR. Using ENU mutagenesis for phenotype-driven analysis of the mouse. Methods Enzymol. 2010;477:329–348. doi: 10.1016/S0076-6879(10)77017-8. [DOI] [PubMed] [Google Scholar]
- Stottmann RW, Moran JL, Turbe-Doan A, Driver E, Kelley M, Beier DR. Focusing forward genetics: a tripartite ENU screen for neurodevelopmental mutations in the mouse. Genetics. 2011;188:615–624. doi: 10.1534/genetics.111.126862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laer L, Van Eyken E, Fransen E, Huyghe JR, Topsakal V, Hendrickx JJ, Hannula S, Maki-Torkko E, Jensen M, Demeester K, Baur M, Bonaconsa A, Mazzoli M, Espeso A, Verbruggen K, Huyghe J, Huygen P, Kunst S, Manninen M, Konings A, Diaz-Lacava AN, Steffens M, Wienker TF, Pyykko I, Cremers CW, Kremer H, Dhooge I, Stephens D, Orzan E, Pfister M, Bille M, Parving A, Sorri M, Van de Heyning PH, Van Camp G. The grainyhead like 2 gene (GRHL2), alias TFCP2L3, is associated with age-related hearing impairment. Hum Mol Genet. 2008;17:159–169. doi: 10.1093/hmg/ddm292. [DOI] [PubMed] [Google Scholar]
- Walentin K, Hinze C, Werth M, Haase N, Varma S, Morell R, Aue A, Potschke E, Warburton D, Qiu A, Barasch J, Purfurst B, Dieterich C, Popova E, Bader M, Dechend R, Staff AC, Yurtdas ZY, Kilic E, Schmidt-Ott KM. A Grhl2-dependent gene network controls trophoblast branching morphogenesis. Development. 2015;142:1125–1136. doi: 10.1242/dev.113829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Samakovlis C. Grainy head and its target genes in epithelial morphogenesis and wound healing. Curr Top Dev Biol. 2012;98:35–63. doi: 10.1016/B978-0-12-386499-4.00002-1. [DOI] [PubMed] [Google Scholar]
- Werth M, Walentin K, Aue A, Schonheit J, Wuebken A, Pode-Shakked N, Vilianovitch L, Erdmann B, Dekel B, Bader M, Barasch J, Rosenbauer F, Luft FC, Schmidt-Ott KM. The transcription factor grainyhead-like 2 regulates the molecular composition of the epithelial apical junctional complex. Development. 2010;137:3835–3845. doi: 10.1242/dev.055483. [DOI] [PubMed] [Google Scholar]
- Wilanowski T, Tuckfield A, Cerruti L, O’Connell S, Saint R, Parekh V, Tao J, Cunningham JM, Jane SM. A highly conserved novel family of mammalian developmental transcription factors related to Drosophila grainyhead. Mech Dev. 2002;114:37–50. doi: 10.1016/s0925-4773(02)00046-1. [DOI] [PubMed] [Google Scholar]
- Zhu C, Byers KJ, McCord RP, Shi Z, Berger MF, Newburger DE, Saulrieta K, Smith Z, Shah MV, Radhakrishnan M, Philippakis AA, Hu Y, De Masi F, Pacek M, Rolfs A, Murthy T, Labaer J, Bulyk ML. High-resolution DNA-binding specificity analysis of yeast transcription factors. Genome Res. 2009;19:556–566. doi: 10.1101/gr.090233.108. [DOI] [PMC free article] [PubMed] [Google Scholar]