

Abstract
Tuberculosis and nontuberculous mycobacterial infections constitute a high burden of pulmonary disease in humans, resulting in over 1.5 million deaths per year. Building on the premise that genetic factors influence the instance, progression, and defense of infectious disease, we undertook a systems biology approach to investigate relationships among genetic factors that may play a role in increased susceptibility or control of mycobacterial infections. We combined literature and database mining with network analysis and pathway enrichment analysis to examine genes, pathways, and networks, involved in the human response to Mycobacterium tuberculosis and nontuberculous mycobacterial infections. This approach allowed us to examine functional relationships among reported genes, and to identify novel genes and enriched pathways that may play a role in mycobacterial susceptibility or control. Our findings suggest that the primary pathways and genes influencing mycobacterial infection control involve an interplay between innate and adaptive immune proteins and pathways. Signaling pathways involved in autoimmune disease were significantly enriched as revealed in our networks. Mycobacterial disease susceptibility networks were also examined within the context of gene-chemical relationships, in order to identify putative drugs and nutrients with potential beneficial immunomodulatory or anti-mycobacterial effects.
Introduction
Tuberculosis (TB), an airborne infectious disease caused by the bacterium Mycobacterium tuberculosis (MTB), is an ongoing global health crisis [1,2] resulting in over 9 million illnesses and 1.5 million deaths each year [3]. In contrast, nontuberculous mycobacterial (NTM) disease, caused by phylogenetically related environmental mycobacteria [4], has emerged as an increasingly prevalent infectious disease, particularly over the last two decades [5–8].
Although exposure to TB is common in certain regions of the world, a relatively small proportion of exposed people progress to develop active pulmonary disease. As an example, one third of the world’s population is latently infected with MTB, but only 10% of those individuals will ever progress to become ill with active TB. Similarly, many individuals come into contact with NTM through soil or municipal water sources [4], but few develop pulmonary NTM disease. Certain clinical conditions, including immunodeficiencies and individuals with compromised lungs, increase susceptibility, but most TB and NTM disease occur in otherwise healthy people [3,9]. We hypothesized that a systems biology approach would help reveal critical human pathways involved in mycobacterial susceptibility, and help elucidate why some individuals progress to active disease while most do not.
Accumulating evidence suggests that host genetic factors influence the susceptibility to MTB and NTM infection. Research studies utilizing twin design [10,11], linkage analysis [12,13], candidate gene association [14–17], genome-wide association analysis [18–21], and fine mapping studies [22] have implicated numerous human genetic markers as contributing factors to the susceptibility of MTB infection. Although fewer studies have examined the human genetic contribution to NTM susceptibility, familial clustering of pulmonary NTM [23] and candidate gene association studies have implicated certain genetic factors [24–26] and support the hypothesis of a genetic predisposition to NTM in some individuals.
In this study, we examine genes critical to the human response to TB and NTM infection, as well as highlight enriched biological pathways and networks that may play a critical role in mycobacterial susceptibility. We explore whether there is any commonality between TB and NTM susceptibility genes and the functional implications of these shared genes and pathways. Shared susceptibility genes or pathways may suggest related mechanisms for the response or control of TB and NTM disease. Furthermore, we examined the resulting networks in order to identify drugs and nutrients with potential immunomodulatory or anti-mycobacterial effects.
Materials and Methods
Data sources & gene selection
We identified genes associated with TB and NTM utilizing three publically available databases: the Online Mendelian Inheritance in Man (OMIM) [27, 28] database, the Comparative Toxicogenomics Database (CTD) [29], and the Human Genome Epidemiology encyclopedia (HuGE Navigator) [30]. The Online Mendelian Inheritance in Man (OMIM) database [27] is considered to be the best curated resource of genotype-phenotype relationships [28]. The Comparative Toxicogenomics Database (CTD) [29] curates relationships between chemicals, genes and human diseases, and is unique because it integrates chemical and gene/protein-disease relationships with the goal of understanding the effects of environmental chemicals on human health. In CTD, disease-gene associations are reported as curated or inferred. We selected only curated associations due to a higher confidence than inferred associations. Lastly, we used the Human Genome Epidemiology encyclopedia (HuGE Navigator) [30] which mines the scientific literature on human gene-disease associations and maintains a comprehensive database of population-based epidemiologic studies of human genes [31]. We selected these databases because of their unique approach, breadth, and depth to cataloguing human disease-gene associations.
TB key word search
For searches of OMIM, we used the key words: “Mycobacterium tuberculosis, susceptibility to” which resulted in 11 genes associated with Mycobacterium tuberculosis susceptibility or protection.
For searches of CTD, we used the key words “Mycobacterium tuberculosis, susceptibility to infection by”, which resulted in 15 genes associated with Mycobacterium tuberculosis susceptibility.
For searches of the HuGE Navigator, we searched using key words “mycobacterium infections”. Tuberculosis was defined by disease phenotypes, such as, “Tuberculosis, Gastrointestinal”, “Tuberculosis, Pleural”, or “Tuberculosis, Pulmonary”. We therefore chose to use “Tuberculosis, Pulmonary” as our search term, since our focus for TB and NTM disease in this study is related to lung disease. We excluded genes that have been implicated in hepatotoxicity and other adverse reactions, rather than susceptibility to infection. None of these excluded genes were later identified in the network analyses. In the HuGE database, we found 154 genes that were associated with pulmonary tuberculosis. We further refined this list and selected only genes with at least 3 references. This restricted our list to 42 genes that were associated with pulmonary tuberculosis. Three of the excluded genes were identified from other databases and thus included in the overall gene list.
NTM key word search
For searches of OMIM, we searched using the key words “mycobacterium infection, nontuberculous, familial”. Three separate phenotypes, “Atypical mycobacteriosis, familial”, Atypical mycobacteriosis, familial, x-linked”, “Atypical mycobacteriosis, familial, x-linked 2”, were associated with 7 genes.
For searches of CTD, we searched using key words “mycobacterium infections, nontuberculous”, identifying 6 genes.
For searches of the HuGE Navigator, we searched using key words “mycobacterium infections”, and then selected “Mycobacterium avium-intracellulare infection” identifying 10 genes and “Mycobacterium Infections, Atypical” identifying 10 genes.
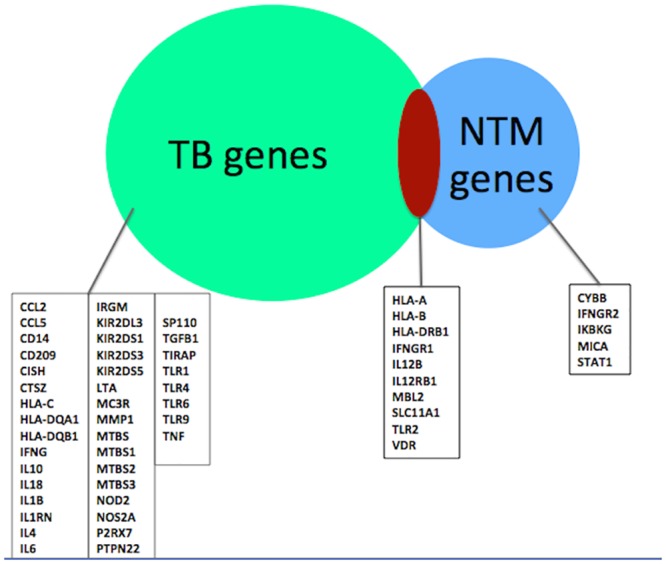
From each of the three databases, we compiled lists of genes that were associated with susceptibility to TB and NTM infection. All subsequent analyses included 50 TB-associated genes and 15 NTM-associated genes (Fig 1). In the overall TB list, ten genes were identified in more than 1 database, and seven genes in the overall NTM list were identified in more than 1 database.
Fig 1. Database-derived TB and NTM-associated genes.
Pathway and network analysis
We utilized the Ingenuity Pathway Analysis (IPA), a web-based software application (Ingenuity Systems, www.ingenuity.com), to investigate gene-gene relationships within the context of networks and pathway enrichment. IPA uses a database of human, mouse, and rat genes/proteins, as well as other biological and chemical targets of interest, to find interactions between genes, proteins, related networks, functions/diseases, and canonical pathways. IPA uses a manually curated database, mined from over 300 top journals using the full text, from over 3,600 journals using abstracts, as well as interaction data from third party databases such as GNF, IntAct, BIND, DIP, MINT, MIPS, BIOGRID, COGNIA, GNF, GO, Entrez Gene, OMIM, RefSeq, ClinVar, COSMIC, GWAS Database, HMDB, clinicaltrials.gov, TarBase, TargetScan, miRecords, DrugBank, HSDB, CCRIS, (www.ingenuity.com), [32]. IPA’s data analysis allows for understanding the significance of genes or gene products of interest within a larger biological system.
We generated TB and NTM-associated gene networks with the IPA Core Analysis function using 50 TB focus genes and 15 NTM focus genes (Fig 1). Focus genes are genes that were identified in our initial OMIM, CTD, and HuGE database queries. Focus genes represented in networks must interact with at least one other gene in the database. Interactions between genes are represented as nodes connected by edges during network generation. Non-focus genes are genes that were not in our initial database derived search, but are connected in the resulting networks. We also refer to these as “network-suggested genes”. We trimmed the resulting gene networks such that nodes with four or fewer edges were removed to facilitate visualization of the most highly connected nodes and networks.
Gene-chemical network
Our list of input genes for the gene-chemical network analysis was obtained by combining genes from the union of networks depicted in Figs 2 & 3. We defined our threshold for gene inclusion as the top 50% of all connections between genes. This corresponded to 9 or more edges per node in the union network 1, and 5 or more edges per node in the union network 2. The complete list of input genes included: BCR (complex), CCL2, CCL5, IKBKG, IL10, IL12 (complex), IL1B, IL1RN, IL4, IL6, Immunoglobulin, MMP1, NFkB (complex), TLR1, TLR2, TLR 4, TLR 9, TNF, Vegf. The final list of genes corresponds to only those that mapped in CTD (this excluded: BCR, IL12, Immunoglobulin, and TNF). The chemical-gene network was constructed using data from CTD [29] and DrugBank [33]. Chemicals listed in CTD were excluded if they did not have a synonymous drug name in DrugBank. Gene-gene interactions and gene-chemical interactions were extracted from CTD based on our gene list. In this network analysis, gene-gene interactions represent a variety of interaction or relational types, including genes encoding proteins that physically interact or participate in sequential steps of a biochemical pathway. This analysis builds upon our previous work on disease-gene-chemical networks [34].
Fig 2. Union of TB and NTM gene sets: network 1.
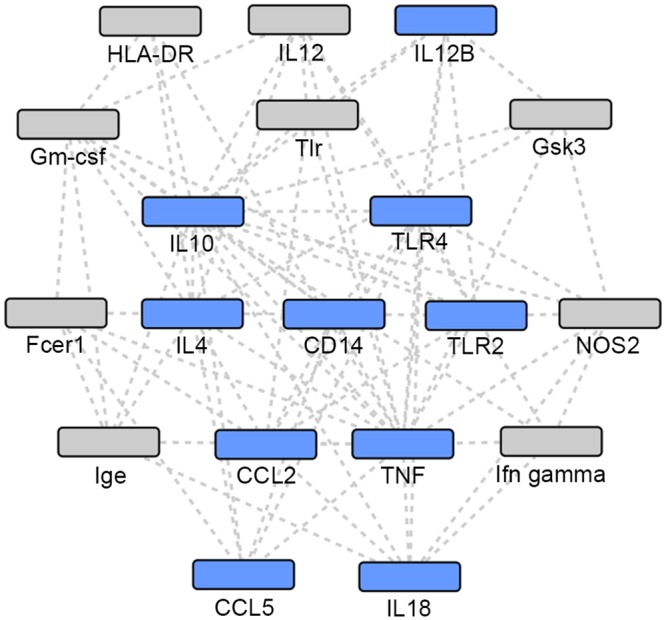
Grey: Network identified genes; Blue: TB or NTM focus genes.
Fig 3. Union of TB and NTM gene sets: network 2.
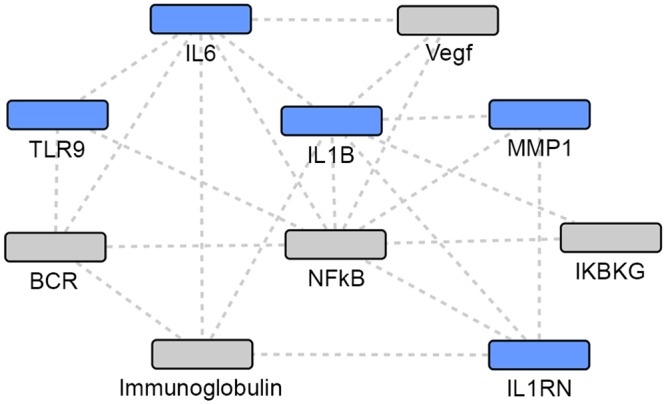
Grey: Network identified genes; Blue: TB or NTM focus genes.
Chemical exclusion criteria were based upon literature evaluation in that any intervention deemed potentially harmful to a patient would not be considered in these networks. Therefore, we excluded: known toxins (ie. alcohols and arsenic), drugs with obvious negative side effects (ie. anticonvulsants, warfarin, opiods/opiates, mood stabilizers, narcotics), drugs with known negative side effects (ie. NSAIDs, immune suppressing steroids/hormones), non-concise chemicals that are generally seen as harmful (ie. dust, CTD definition: “Earth or other dry matter in fine, dry particles”), anti-retrovirals due to frequent HIV-TB co-infection, their use in HIV treatment and concern for development of drug resistance, as well as antibiotics that are effective against the bacteria but detrimental to the patient. The remaining chemicals included: statins/fibrates, select group of antibiotics, and vitamins. Macrolides and Gemfibrozil were grouped together because of their potential to positively alter the human immunological response to infection as well as both being pharmaceutical drugs. Gene networks and gene-chemical networks were recreated using Cytoscape [35].
Results and Discussion
Many studies using conventional genetic methods have identified single genes/loci influencing the susceptibility to TB or NTM infection and control of disease [12–22,24–26]. Despite these reports, little has been done to examine these reported loci within the context of biological networks. Since the progression to active TB and NTM disease are influenced by multiple genetic factors, genes identified through single gene approaches, in and of themselves, may provide an incomplete etiological story [36,37]. As a result, we undertook a comprehensive network and pathway analysis of human genes associated with TB and NTM infection susceptibility. Using this approach we identify new candidate genes, as well as investigate the overlap of genes and pathways involved in both TB and NTM susceptibility and control.
Based on our database search, we identified 50 TB-associated focus genes and 15 NTM-associated focus genes (Fig 1). We generated 10 TB-associated networks, 1 NTM-associated network, and 1 network using the intersection of TB- and NTM-associated focus genes. Five networks were generated using the union of TB- and NTM-associated genes (Tables 1–4). In the following text, we discuss the most statistically significant networks from each gene set.
Table 1. Network associated with TB.
| Molecules in Network | Score* | Focus Molecules | Top Diseases and Functions |
|---|---|---|---|
| BCR (complex), caspase, CISH, Fcger3, HLA-A, HLA-B, HLA-C, HLA-DQB1, HLA-DRB1, IFN Beta, IFNG, lga, Ikb, IL12 (family), IL12RB1, IL1B, IL1RN, Immunoglobulin, Interferon alpha, KIR, KIR2DL1/KIR2DL3, KIR2DS4, LTA, MBL2, MMP1, NFkB (complex), P2RX7, PTPN22, Ras, SAA, TCR, TIRAP, TLR1, TLR9, Vegf | 42 | 20 | Cell-To-Cell Signaling and Interaction, Immunological Disease, Connective Tissue Disorders |
Bold: TB focus genes.
*Score: -log(Fisher’s Exact p-value)
Table 4. Networks associated with the union of TB and NTM genes.
| Molecules in Network | Score* | Focus Molecules | Top Diseases and Functions |
|---|---|---|---|
| CCL2, CCL5 CD14, CTSZ, CYBB, Fcer1, Fcgr2, Gm-csf, Gsk3, HLA-DQ, HLA-DR, lfn gamma, IFNGR1, IFNGR2, lge, IgG1, IL4, IL10, IL18, IL23, IL12 (complex), IL12B, lymphotoxin-alpha1-beta2, MHC Class II (complex), NOD2, NOS2, Nr1h, SCAVENGER receptor CLASS A, Sod, Tlr, TLR2, TLR4, TLR6, TNF, U1 snRNP | 33 | 17 | Cellular Function and Maintenance, Hematological System Development and Function, Infectious Disease |
| 26s Proteasome, BCR (complex), caspase, CD209, Cdk, Collagen type 1, Eotaxin, Fcgr3, HLA-DQB1, Hsp27, Iga, Ikb, IKBKG, IL1, IL6, IL12 (family), IL1B, IL1RN, Immunoglobulin, JK, LTA, MBL2, MICA, MMP1, N-cor, NFkB(complex), P2RX7, PTPN22, Ras homolog, SAA, TIRAP, TLR1, TLR9, VDR, Vegf | 33 | 16 | Cell-To-Cell Signaling and Interaction, Hematological System Development and Function, Immunological Disease |
Bold: TB and NTM focus genes.
*Score: -log(Fisher’s Exact p-value)
Gene Network Analyses
Using the IPA enrichment algorithm, five networks were generated using the union of TB and NTM genes. The most statistically significant network (p = 10−33) included 17 focus genes implicated in either infection (Table 4, Fig 2). The second network (p = 10−30) included 16 focus genes in a 35 gene network (Table 4, Fig 3).
The top TB network was highly significant (p = 10−42) and included 20 focus genes implicated in susceptibility to Mycobacteria tuberculosis infection in a 35 gene network (Table 1, Fig 4).
Fig 4. TB network.
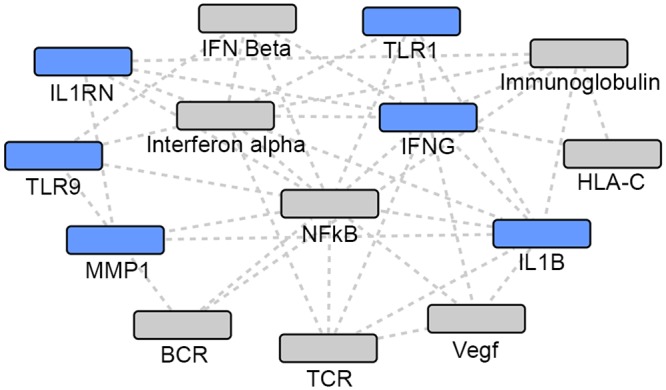
Grey: Network identified genes; Blue: TB focus genes.
The top NTM network was also highly significant (p = 10−36). This 35 gene network included 14 focus genes, which have been previously implicated in susceptibility to NTM infection (Table 2, Fig 5).
Table 2. Network associated with NTM.
| Molecules in Network | Score* | Focus Molecules | Top Diseases and Functions |
|---|---|---|---|
| CLEC11A, CYBB, DUOX2, ERK1/2, FXN, GRM1, Gsk3, HLA-A, HLA-B, HLA-DRB1, lfn gamma, IFNGR1, IFNGR2, IKBKG, IL12B, IL12RB1, IL36A, Immunoglobulin, IRAK, MBL2, MICA, NEU1, NFkB (complex), NLRP2, P38 MAPK, PASK, SH3GLB2, STAT1, SUMO4, TCR, TFG, TLR2, TLR10, USP21, VDR | 36 | 14 | Cancer, Cell Death and Survival, Cellular Compromise |
Bold: NTM focus genes.
*Score: -log(Fisher’s Exact p-value)
Fig 5. NTM network.
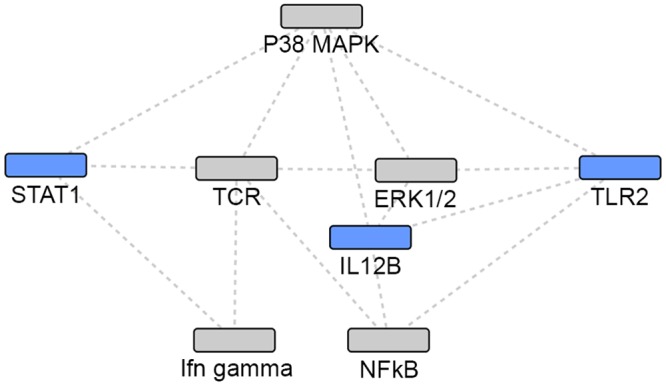
Grey: Network identified genes; Blue: NTM focus genes.
Using the intersection of TB- and NTM-associated genes, one network was generated (p = 10−22). From the 10 focus genes common to both infections, 9 genes were implicated in this 35 gene network (Table 3, Fig 6).
Table 3. Network associated with the intersection of TB and NTM genes.
| Molecules in Network | Score* | Focus Molecules | Top Diseases and Functions |
|---|---|---|---|
| CCL8, CCL26, CD6, CLEC11A, ERK1/2, FXN, HLA-A, HLA-B, HLA-DRB1, IFNGR1, IL25, IL10RA, IL12B, IL12RB1, IL36A, Immunoglobulin, IRAK, KIR, LILRB1, MAP3K10, MBL2, MED28, NEU1, NFkB (complex), NOD1, P2RY6, P38 MAPK, PTPN12, SH3GLB2, SUMO4, TLR2, TLR10, TNIP3, UNC93B1, VDR | 22 | 9 | Inflammatory Response, Hematological System Development and Function, Tissue Morphology |
Bold: TB and NTM focus genes.
*Score: -log(Fisher’s Exact p-value)
Fig 6. Intersection of TB and NTM gene sets.
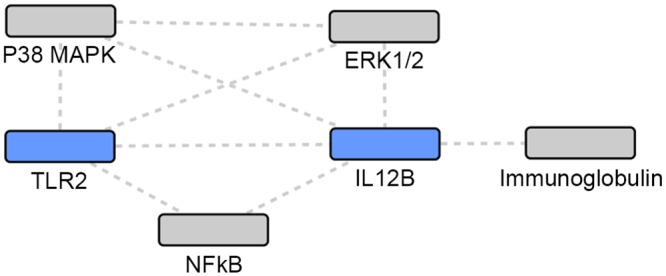
Grey: Network identified genes; Blue: TB and NTM focus genes.
Discussion of network-central genes
Networks using the union of TB and NTM gene sets
The two most statistically significant networks demonstrated that NTM-associated focus genes were the most peripheral, while the TB-associated focus genes and the focus genes common to both TB and NTM infection were the more central and connected nodes in the network. When comparing the two networks (Figs 2 and 3), different genes appear to be most central and interconnected. In network 1 (Fig 2), the most central and highly connected genes are the TB- associated focus genes (blue), IL4, IL10, TNF, CCL2, CCL5, and TLR4 as well as the focus genes common to both TB and NTM (blue) IL12B, and TLR2. While in network 2 (Fig 3), different TB- associated focus genes (blue), IL6 and IL1B, are the most central and highly connected, while genes common to both TB and NTM infection are not central or highly connected nodes. Among the genes putatively associated with TB or NTM identified via network analysis, Gm-csf (Fig 2) and NFkB complex (Fig 3) are the most central and highly connected nodes. Overall, NTM-associated genes appear the most peripheral, while the TB-associated focus genes seem to play a more central role in these networks.
Focus genes
TLR2: TLR2 stands out as being one of the most central and highly interconnected nodes in all the networks. Toll Like Receptors (TLRs) can recognize molecular patterns of MTB and initiate signaling pathways to activate the innate and adaptive immune responses [38]. The major receptors for MTB and those most critical in the recognition process are TLR-1, -2, and -6. Because of the TLR’s essential role in MTB recognition, many studies have examined associations of TLR polymorphisms and TB, however results have been inconsistent. In a recent meta-analysis [38], authors reported a positive association between a TLR2 polymorphism and TB risk, especially among Asians and Europeans. Yim et al. demonstrated that a GT repeat microsatellite polymorphism in intron II of TLR2 contributes to the development of NTM lung disease, especially for M. avium-intracellulare complex (MAC) infection [39], although another study from Korea did not find an association between TLR2 and NTM susceptibility [40]. Supported by previous studies, our network analyses highlight the important role that TLR2 may play in the immune response to both TB and NTM infection.
IL12B: The IL12B gene encodes the p40 subunit of IL-12 and IL-23 cytokines, which play important roles in bridging the innate and adaptive immune systems. Interleukin 12 (IL-12) is a proinflammatory cytokine and acts as a key regulator in determining the T helper 1 or 2 immune response [41]. IL12B polymorphisms have been associated with increased susceptibility to TB [42], as well as to Mycobacterium leprae [43], but findings have been inconsistent [44,45]. IL12B has also been implicated in the development of autoimmune disease. A recent meta-analysis demonstrated a significant association between IL12B polymorphisms and risk of psoriasis [46]. IL12B has also been implicated [47] as a susceptibility gene for leprosy and inflammatory bowel disease. This may imply a shared genetic susceptibility to inflammation and infectious disease.
IFNG: Interferon-gamma (IFNG) binds to its own receptor made of transmembrane proteins, IFNGR1 and IFNGR2, to induce antimicrobial mechanisms and upregulates antigen processing and presentation pathways [48]. Consequently, IFNG is central to innate and adaptive cell-mediated immunity against intracellular pathogens and thus crucial to controlling MTB replication. Studies have shown that low production of IFNG has been associated with active TB [49–51]. A recent meta-analysis reported a statistically significant protective association of an IFNG polymorphism and pulmonary/extrapulmonary TB in different ethnicities [52]. Polymorphisms in this gene may be good genetic markers for TB resistance.
Network-suggested genes
NFkB complex: Among the putatively associated genes identified via network analysis, the nuclear factor-kB (NFkB) complex stands out as one of the most highly central and interconnected nodes in all our networks. The NFkB family of transcription factors plays a key role in the regulation of genes involved in immune and inflammatory responses [53,54]. A wide range of stimuli, including viral and bacterial products, can lead to the activation of NFkB. It regulates gene expression in many different cell types during development in response to injury and infection, thus it is often referred to as a central mediator of the immune response [53–55]. Research has demonstrated that activation of NFkB enhances immunity against certain microbial pathogens [56,57], however some NFkB pathways can be exploited to promote pathogen survival [56]. A recent study demonstrated that inhibiting NFkB reduced MTB survival in human macrophages [58], emphasizing the complex role of NFkB in MTB infection. While the human immune response against NTM infection is not well understood, many studies have shown that the role of the NFkB is a crucial step in the regulation of genes involved in the killing of NTM. One study [59] focused on the role of NFkB in the innate immune response of macrophages. The authors reported that M. smegmatis, a nonpathogenic mycobacteria, induces NFkB activation and is killed by macrophages, while M. avium, a pathogenic mycobacteria, represses NFkB activation and survives within macrophages. Additional cell, mouse and bovine studies [60–63] have also examined the influence of NFkB on the response to infection. These studies demonstrated that NFkB is involved in the initiation of a proinflammatory cytokine response in the macrophage and is rapidly activated by the interaction of host cell and bacterium [60,63]. However, the mechanisms and sequence of events underlying NFkB activation and cytokine response to NTM infection remains unclear.
ERK1/2 & p38 MAPK: In response to invading pathogens, innate immune cells, such as macrophages/monocytes and dendritic cells, use pattern recognition receptors (PRRs) to recognize bacteria, which in turn, leads to the activation of signaling pathways, such as the mitogen-activated protein (MAP) kinase pathway [64–66]. The NTM gene network (Fig 5) and the network using the intersection of TB and NTM gene sets (Fig 6) identify ERK1/2 and p38 MAPK, both members of the MAP kinases, as highly central and interconnected nodes. Depending on the stimulus, activation of ERK1/2 together with p38 can have contrasting functions. A greater amount of ERK activity relative to p38 can promote cell proliferation and survival, whereas a greater amount of p38 activity relative to ERK can trigger cell death and apoptosis [67,68]. In an in vitro study using primary human monocytes, authors reported that Mycobacterium avium-intracellulare (MAI) infection activates both ERK and p38. While ERK appears to regulate pathogenic MAI replication in human monocytes, p38 influences cytokine release more than ERK does [69].
VEGF: Vascular endothelial growth factor (VEGF) is a key regulator of normal angiogenesis. It promotes endothelial cell survival, growth and migration, in addition to being implicated in pathological angiogenesis associated with tumor growth [70]. VEGF has been also studied as a prognostic biomarker for infectious diseases but studies have yielded inconsistent results. In sepsis infection, some studies found higher VEGF levels in survivors compared with non-survivors, while other studies found the opposite [71,72]. Some studies examining dengue infection have reported significantly higher VEGF levels in patients with dengue hemorrhagic fever compared with dengue fever [73,74], other studies have reported a lack of association between VEGF and illness severity [75]. Similarly, several TB studies have reported elevated plasma VEGF levels in active TB [76–78]. And in a separate TB study, authors found that plasma VEGF concentrations were significantly reduced upon TB treatment and could potentially represent a surrogate marker to monitor sputum culture conversion [79].
Pathway Enrichment Analysis
Canonical Pathway Analysis using IPA
For TB, the top two canonical pathways most significantly associated with this gene set were “altered T cell and B cell signaling in rheumatoid arthritis” and “communication between innate and adaptive immune cells” (Table 5). For NTM, the top two pathways most significantly associated with this gene set were “type 1 diabetes mellitus signaling” and “T Helper cell differentiation” (Table 6). Examining the intersection of the TB and NTM gene sets, pathway analysis revealed that the top two canonical pathways were “communication between Innate and Adaptive Immune Cells” and “type 1 diabetes mellitus signaling” (Table 7). Examining the union of TB and NTM gene sets, pathway analysis revealed that the top two canonical pathways were “altered T cell and B cell signaling in rheumatoid arthritis” and “communication between innate and adaptive immune cells” (Table 8).
Table 5. Top canonical pathways associated with TB gene set.
| Pathways | p-value | Ratio* | Molecules in pathway |
|---|---|---|---|
| Altered T Cell and B Cell Signaling in Rheumatoid Arthritis | 1.14E-33 | 19/81 (0.235) | HLA-DQA1, HLA-DQB1, HLA-DRB1, IFNG, IL4, IL6, IL10, IL18, IL12B, IL1B, IL1RN, LTA, TGFB1, TLR1, TLR2, TLR4, TLR6, TLR9, TNF |
| Communication between Innate and Adaptive Immune Cells | 1.48E-33 | 19/82 (0.232) | CCL5, HLA-A, HLA-B, HLA-C, HLA-DRB1, IFNG, IL4, IL6, IL10, IL18, IL12B, IL1B, IL1RN, TLR1, TLR2, TLR4, TLR6, TLR9, TNF |
*Ratio: number of genes from our dataset that map to a canonical pathway divided by the total number of genes in that pathway.
Table 6. Top canonical pathways associated with NTM gene set.
| Pathways | p-value | Ratio* | Molecules in pathway |
|---|---|---|---|
| Type 1 Diabetes Mellitus Signaling | 9.08E-15 | 8/106 (0.075) | HLA-A, HLA-B, HLA-DRB1, IFNGR1, IFNGR2, IKBKG, IL12B, STAT1 |
| T Helper Cell Differentiation | 1.27E-11 | 6/67 (0.09) | HLA-DRB1, IFNGR1, IFNGR2, IL12B, IL12RB1, STAT1 |
*Ratio: number of genes from our dataset that map to a canonical pathway divided by the total number of genes in that pathway.
Table 7. Top canonical pathways associated with the intersection of TB and NTM gene sets.
| Pathways | p-value | Ratio* | Molecules in pathway |
|---|---|---|---|
| Communication between Innate and Adaptive Immune Cells | 5.11E-10 | 5/82 (0.047) | HLA-A, HLA-B, HLA-DRB1, IFNGR1, IL12B, |
| Type 1 Diabetes Mellitus Signaling | 1.89E-09 | 5/106 (0.047) | HLA-A, HLA-B, HLA-DRB1, IFNGR1, IL12B, |
*Ratio: number of genes from our dataset that map to a canonical pathway divided by the total number of genes in that pathway.
Table 8. Top canonical pathways associated with the union of TB and NTM gene sets.
| Pathways | p-value | Ratio* | Molecules in pathway |
|---|---|---|---|
| Altered T Cell and B Cell Signaling in Rheumatoid Arthritis | 6.25E-33 | 19/81 (0.235) | HLA-DQA1, HLA-DQB1, HLA-DRB1, IFNG, IL4, IL6, IL10, IL18, IL12B, IL1B, IL1RN, LTA, TGFB1, TLR1, TLR2, TLR4, TLR6, TLR9, TNF |
| Communication between Innate and Adaptive Immune Cells | 8.13E-33 | 19/82 (0.232) | CCL5, HLA-A, HLA-B, HLA-C, HLA-DRB1, IFNG, IL4, IL6, IL10, IL18, IL12B, IL1B, IL1RN, TLR1, TLR2, TLR4, TLR6, TLR9, TNF |
*Ratio: number of genes from our dataset that map to a canonical pathway divided by the total number of genes in that pathway.
Signaling pathways for autoimmune disease and communication or differentiation of immune cells are overrepresented in our pathway enrichment analyses. This may indicate similar susceptibility or pathogenic mechanisms for TB and NTM infections.
The top canonical pathway for the TB gene set and the union of TB and NTM gene sets includes “altered T cell and B cell signaling in rheumatoid arthritis” (RA). Our analysis has identified a signaling pathway specific to RA, which has been confirmed in the literature. The relationship between mycobacterial infection and autoimmune disease, in particular, Inflammatory Bowel Disease (IBD) and RA, has been reported previously [80–84]. Studies have shown that persons with RA are at increased risk for TB and NTM disease, independent of immunosuppressive medications used in RA treatment [85,86]. The link between specific genes, for example SLC11A1 (NRAMP1) and susceptibility to autoimmune disease and infectious disease has been widely explored. Shaw et al (1996) demonstrated genetic linkage of NRAMP1 and RA. Searle & Blackwell (1999) found that high expression of NRAMP1 allele 3 contributes to autoimmune susceptibility, specifically to RA [80]. This association was also found in diabetes patients with a first or second degree relative with RA [87]. Conversely, low expression of NRAMP1 allele 2 was found to contribute to infectious disease, specifically tuberculosis [88,89]. Interestingly, Mobley (2004) demonstrated similarity in the epidemiology of RA and the epidemiology of tuberculosis deaths from 2 centuries ago [90]. The author suggested the possibility that genetic factors influencing tuberculosis survival may now be influencing susceptibility to the development of RA. Other genetic associations have reported between mycobacterial infection and chronic inflammation. IL12B, for example, has been identified as a susceptibility gene for leprosy, psoriasis and IBD [46,47]. Similarly, the IRGM autophagy gene, a strong mediator of inflammation, has also been associated with an increased risk to leprosy [91], Crohn’s disease and IBD [92,93].
“Type 1 diabetes mellitus signaling” was also ranked among the top canonical pathways for both the NTM gene set and the intersection of TB and NTM gene sets. Diabetes mellitus (DM) has been associated with increased risk of progression to TB, increased TB severity and with poor TB treatment outcomes [94,95]. While some studies hypothesize that DM impairs the immune responses necessary to control mycobacterial replication [96], the exact mechanisms by which DM increases TB risk have not yet been elucidated.
Network-identified immune-modulating chemicals
Pro-immune altering chemicals remain an understudied and underutilized area of disease treatment. Within our gene-chemical network, there were three macrolides (Azithromycin, Clarithromycin, and Roxithromycin) and a fibrate (Gemfibrozil) that are known to have immune altering effects (Fig 7). Although macrolides play a central role in the treatment of NTM infection [97,98], they are not commonly used to treat TB. There is conflicting evidence regarding the implications of potential macrolide induced alterations in macrophage activation [99–101] and immune cell composition [102] during the course of infection in humans. In mice models, it has been hypothesized that azithromycin may contribute to NTM infection by decreasing autophagy [103], despite evidence showing azithromycin successfully treats NTM infections in mice with chronic pulmonary infection [104]. In humans with COPD, azithromycin increases phagocytosis [105,106], which suggests a beneficial effect on macrophages-mediated degradation of microorganisms. It has been shown that TB can inhibit phagosome maturation by using mannose receptors to mediate transport to phagosomes with limited fusion capabilities [107], the same receptors that are greatly increased by azithromycin treatment [106]. However, given that mycobacteria often reside in inactivated macrophages [108] and that azithromycin promotes altered activation of macrophages [99], this altered immune response may be beneficial to treating TB [34], even though direct macrolide effects are inconsistently seen in vitro [109–114]. Gemfibrozil represents a novel drug for both NTM and TB treatment. This drug inhibited 27 strains of MTB grown in macrophages by decreasing their ability to acquire fatty acids from macrophages [115], leading to novel mechanisms for aiding the host in combating active TB.
Fig 7. Network-identified immune-modulating chemicals.
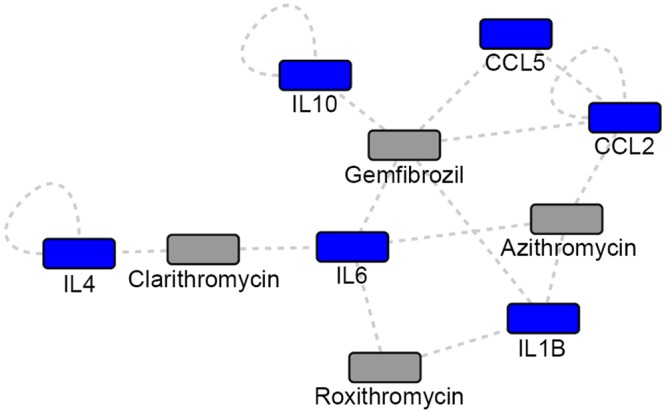
Grey: Network identified chemicals; Blue: Network identified genes.
Network-identified nutrients
Nutrients play an important role in both basic immune health and pathogen virulence. There is a correlation between malnutrition and active TB; malnutrition increases the likelihood of progressing to TB and TB causes a decrease in available nutrients [116]. There are guidelines for providing nutrient supplementation in malnourished individuals and pregnant women; however, there are no guidelines for additional supplementation in individuals receiving adequate food intake [116]. The overall chemical-gene network contains a variety of vitamins (A, B, C, D, E, K, lycopene), suggesting the importance of micronutrients in both immune regulation and mycobacterial infection (Fig 8). While there is clear support for nutrient supplementation in malnourished individuals, particularly in treating malaria [117], there is some evidence that nutrient modulation in healthy individuals may improve response to infection. Vitamin A mediates anti-microbial activity, which may be beneficial in MTB treatment [118]. MTB is highly sensitive to vitamin C induced Fenton reaction [119]. This reaction generates hydroxyl radicals, chemicals that promote eradication of actively growing MTB [120]. Vitamin D supplementation improved treatment response among patients with specific vitamin D receptor mutations who were infected with MTB [121]. Vitamin D deficiency has also been associated with susceptibility to NTM infection [122]. However, based on the evidence from randomized controlled trials, vitamin D supplementation has not shown benefit in the general population of TB patients [121,123]. Further, a Cochrane Review concludes that “there is currently no reliable evidence that routinely supplementing at or above recommended daily amounts has clinical benefits” [124].
Fig 8. Network-identified nutrients.
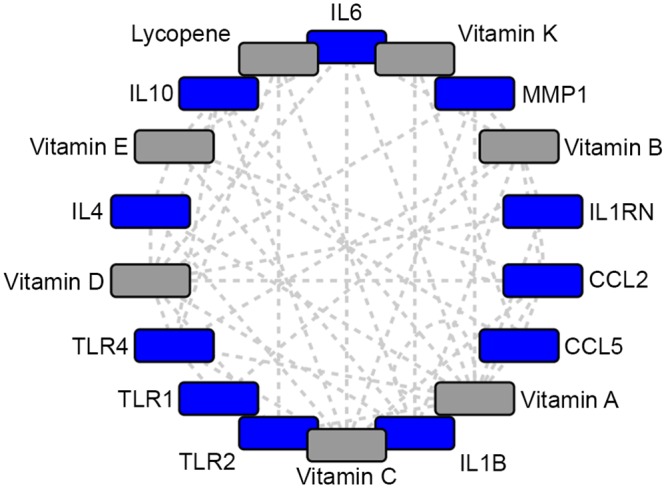
Grey: Network identified vitamins; Blue: Network identified genes.
Just as nutrients can help augment host response, nutrients also represent key chemicals for regulating the pathogen. As iron is a necessary molecule for NTM and TB survival and virulence [125], the human host is the main source of iron acquisition during infection [125,126]. The heme acquisition system offers a possible target for drugs [126] by limiting mycobacterial nutrient uptake and preserving these essential nutrients for the host. The folate pathway represents another potential target for therapeutics against mycobacterium [127]. Folate cofactors are an essential carbon donor used for amino acid and DNA synthesis [127] and inhibition can possibly disrupt bacterial growth and replication.
Conclusion
Our findings suggest that the genetic contribution to MTB and NTM infection operates through similar genes and pathways, providing insight into the underlying pathogenesis and human immune response to mycobacterial disease. Genes involved in bridging the innate and adaptive immune responses are central to both TB and NTM infection susceptibility and control. Genes in these processes are essential to host protection, thus providing an important basis for further research. TB and NTM disease gene sets were also overrepresented in autoimmune signaling pathways. This implies that overlapping physiological mechanisms and pathways influence susceptibility to mycobacterial infection and the development of autoimmune disease, suggesting another avenue for future research.
We identified drugs and nutrients via network analysis that interact with genes of interest, and suggest potential therapeutics with immune modulating effects. Our network findings suggest that three well-known macrolides and a fibrate may target our genes of interest and may boost human immune response to infection. While research examining the effectiveness of these treatments on mycobacterial disease is inconclusive, our findings suggest that further research may be warranted to expand the repertoire of treatment options for mycobacterial disease. Lastly, we identified nutrients that target our genes of interest, which may improve response to infection even in healthy individuals. If vitamins A, B, C, D, E, K, and lycopene positively impact response to infection, then nutrient supplementation may be a simple intervention to reduce the incidence and prevalence of mycobacterial disease worldwide.
Although literature dependent network analyses are often limited by an incomplete knowledge base, by relying on what has been published in the literature, network inferences can suggest important pathways involved in disease and lead to novel hypotheses. In our study, for example, our TB input gene list was larger compared with the NTM gene list, as a result of TB being a more extensively studied disease. The fact that a gene is associated with risk of TB but not NTM may either be due to a true functional difference, or alternatively, may result as a byproduct of a smaller pool of published NTM literature. Nonetheless, our network analysis examines two important categories of mycobacterial disease, TB and NTM, and the resulting networks provide a visually intuitive and statistically sound methodology for data interpretation and examination. The resulting network and pathway analysis offers a powerful and complementary approach to other methods, to help identify underlying mechanisms and pathways involved in complex diseases where multiple genes and gene products interact.
In summary, this analysis explores the connectivity between TB and NTM-associated genes, susceptibility to infection, and possible therapeutics. It also provides a foundation for further examination of these target genes among infected and uninfected individuals.
Acknowledgments
We thank Dr. Nicholas Walter and Dr. Lynn Petukhova for thorough reviews and feedback.
Data Availability
All relevant data are within the paper.
Funding Statement
MS acknowledges funding from the Boettcher Foundation Webb Waring Award. EML and BJG acknowledge support from a NLM Institutional Training Grant, NIH 5T15LM009451.
References
- 1.Donoghue HD, Spigelman M, Greenblatt CL, Lev-Maor G, Bar-Gal GK, Matheson C, et al. Tuberculosis: from prehistory to Robert Koch, as revealed by ancient DNA. Lancet Infect Dis. 2004. September;4(9):584–92. [DOI] [PubMed] [Google Scholar]
- 2.Zink AR, Grabner W, Reischl U, Wolf H, Nerlich AG. Molecular study on human tuberculosis in three geographically distinct and time delineated populations from ancient Egypt. Epidemiol Infect 2003. March;130(2):239–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization. Global tuberculosis report 2014. World Health Organization; 2014. [Google Scholar]
- 4.Falkinham JO. Surrounded by mycobacteria: nontuberculous mycobacteria in the human environment. J Appl Microbiol. 2009. August;107(2):356–67. 10.1111/j.1365-2672.2009.04161.x [DOI] [PubMed] [Google Scholar]
- 5.Kennedy TP, Weber DJ. Nontuberculous mycobacteria. An underappreciated cause of geriatric lung disease. Am J Respir Crit Care Med. 1994. June;149(6):1654–8 [DOI] [PubMed] [Google Scholar]
- 6.Adjemian J, Olivier KN, Seitz AE, Holland SM, Prevots DR. Prevalence of Nontuberculous Mycobacterial Lung Disease in U.S. Medicare Beneficiaries. Am J Respir Crit Care Med. 2012. April 15;185(8):881–6. 10.1164/rccm.201111-2016OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prevots DR, Shaw PA, Strickland D, Jackson LA, Raebel MA, Blosky MA, et al. Nontuberculous mycobacterial lung disease prevalence at four integrated health care delivery systems. Am J Respir Crit Care Med 2010. October 1;182(7):970–6. 10.1164/rccm.201002-0310OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kendall B, Winthrop K. Update on the Epidemiology of Pulmonary Nontuberculous Mycobacterial Infections. Semin Respir Crit Care Med. 2013. March 4;34(01):087–94. [DOI] [PubMed] [Google Scholar]
- 9.Wagner D, Young LS. Nontuberculous Mycobacterial Infections: A Clinical Review. Infection. 2004. October;32(5):257–70. [DOI] [PubMed] [Google Scholar]
- 10.Simonds B. Twin research in tuberculosis. Eugen Rev. 1957. April;49(1):25–32. [PMC free article] [PubMed] [Google Scholar]
- 11.Jepson A, Fowler A, Banya W, Singh M, Bennett S, Whittle H, et al. Genetic Regulation of Acquired Immune Responses to Antigens of Mycobacterium tuberculosis: a Study of Twins in West Africa. Infection and Immunity. 2001. June 1;69(6):3989–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bellamy R, Beyers N, McAdam KP, Ruwende C, Gie R, Samaai P, et al. Genetic susceptibility to tuberculosis in Africans: A genome-wide scan. Proceedings of the National Academy of Sciences. 2000. June 20;97(14):8005–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cooke GS, Campbell SJ, Bennett S, Lienhardt C, McAdam KP, Sirugo G, et al. Mapping of a Novel Susceptibility Locus Suggests a Role for MC3R and CTSZ in Human Tuberculosis. Am J Respir Crit Care Med. 2008. July 15;178(2):203–7. 10.1164/rccm.200710-1554OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoal EG, Lewis L-A, Jamieson SE, Tanzer F, Rossouw M, Victor T, et al. SLC11A1 (NRAMP1) but not SLC11A2 (NRAMP2) polymorphisms are associated with susceptibility to tuberculosis in a high-incidence community in South Africa. Int J Tuberc Lung Dis. 2004. December;8(12):1464–71 [PubMed] [Google Scholar]
- 15.Möller M, Nebel A, Valentonyte R, van Helden PD, Schreiber S, Hoal EG. Investigation of chromosome 17 candidate genes in susceptibility to TB in a South African population. Tuberculosis. 2009. March;89(2):189–94. 10.1016/j.tube.2008.10.001 [DOI] [PubMed] [Google Scholar]
- 16.Rossouw M, Nel HJ, Cooke GS, van Helden PD, Hoal EG. Association between tuberculosis and a polymorphic NFkappaB binding site in the interferon gamma gene. Lancet. 2003. May 31;361(9372):1871–2. [DOI] [PubMed] [Google Scholar]
- 17.Pan H, Dai Y, Tang S, Wang J. Polymorphisms of NOD2 and the risk of tuberculosis: a validation study in the Chinese population. Int J Immunogenet. 2012. June;39(3):233–40. 10.1111/j.1744-313X.2011.01079.x [DOI] [PubMed] [Google Scholar]
- 18.Thye T, Vannberg FO, Wong SH, Owusu-Dabo E, Osei I, Gyapong J, et al. Genome-wide association analyses identifies a susceptibility locus for tuberculosis on chromosome 18q11.2. Nat Genet. 2010. September;42(9):739–41. 10.1038/ng.639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thye T, Owusu-Dabo E, Vannberg FO, van Crevel R, Curtis J, Sahiratmadja E, et al. Common variants at 11p13 are associated with susceptibility to tuberculosis. Nat Genet. 2012. March;44(3):257–9. 10.1038/ng.1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Png E, Alisjahbana B, Sahiratmadja E, Marzuki S, Nelwan R, Balabanova Y, et al. A genome wide association study of pulmonary tuberculosis susceptibility in Indonesians. BMC Medical Genetics. 2012. January 13;13:5 10.1186/1471-2350-13-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahasirimongkol S, Yanai H, Mushiroda T, Promphittayarat W, Wattanapokayakit S, Phromjai J, et al. Genome-wide association studies of tuberculosis in Asians identify distinct at-risk locus for young tuberculosis. J Hum Genet. 2012. June;57(6):363–7. 10.1038/jhg.2012.35 [DOI] [PubMed] [Google Scholar]
- 22.Cervino AC, Lakiss S, Sow O, Bellamy R, Beyers N, Hoal-van Helden E, et al. Fine mapping of a putative tuberculosis-susceptibility locus on chromosome 15q11-13 in African families. Hum Mol Genet. 2002. July 1;11(14):1599–603. [DOI] [PubMed] [Google Scholar]
- 23.Colombo RE, Hill SC, Claypool RJ, Holland SM, Olivier KN. Familial clustering of pulmonary nontuberculous mycobacterial disease. Chest. 2010. March;137(3):629–34. 10.1378/chest.09-1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Um S-W, Ki C-S, Kwon OJ, Koh W-J. HLA Antigens and Nontuberculous Mycobacterial Lung Disease in Korean Patients. Lung. 2009. February 14;187(2):136–40. 10.1007/s00408-009-9136-8 [DOI] [PubMed] [Google Scholar]
- 25.Koh W-J, Kwon OJ, Kim EJ, Lee KS, Ki C-S, Kim JW. NRAMP1 Gene Polymorphism and Susceptibility to Nontuberculous Mycobacterial Lung Diseases. Chest. 2005. July 1;128(1):94–101. [DOI] [PubMed] [Google Scholar]
- 26.Hwang JH, Kim EJ, Koh W-J, Kim SY, Lee S-H, Suh GY, et al. Polymorphisms of interferon-gamma and interferon-gamma receptor 1 genes and non-tuberculous mycobacterial lung diseases. Tuberculosis. 2007. March;87(2):166–7. [DOI] [PubMed] [Google Scholar]
- 27.Hamosh A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Research. 2004. December 17;33(Database issue):D514–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Research. 2015. January;43: D789–D798. 10.1093/nar/gku1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davis AP, Murphy CG, Johnson R, Lay JM, Lennon-Hopkins K, Saraceni-Richards C, et al. The Comparative Toxicogenomics Database: update 2013. Nucleic Acids Research. 2013. January;41(D1):D1104–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu W, Gwinn M, Clyne M, Yesupriya A, Khoury MJ. A navigator for human genome epidemiology. Nat Genet. 2008. February;40(2):124–5. 10.1038/ng0208-124 [DOI] [PubMed] [Google Scholar]
- 31.Yesupriya A, Evangelou E, Kavvoura FK, Patsopoulos NA, Clyne M, Walsh MC, et al. Reporting of human genome epidemiology (HuGE) association studies: an empirical assessment. BMC Med Res Methodol. 2008. May 20;8:31 10.1186/1471-2288-8-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomas S, Bonchev D. A survey of current software for network analysis in molecular biology. Hum Genomics. 2010. June;4(5):353–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014. January;42(Database issue): D1091–7. 10.1093/nar/gkt1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia B, Datta G, Cosgrove GP, Strong M. Network and matrix analysis of the respiratory disease interactome. BMC Syst Biol. 2014;8(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smoot ME, Ono K, Ruscheinski J, Wang P- L, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011. February 1;27(3):431–2. 10.1093/bioinformatics/btq675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Wit E, van der Merwe L, van Helden PD, Hoal EG. Gene-gene interaction between tuberculosis candidate genes in a South African population. Mamm Genome. 2010. August 27;22(1–2):100–10. 10.1007/s00335-010-9280-8 [DOI] [PubMed] [Google Scholar]
- 37.Möller M, Hoal EG. Current findings, challenges and novel approaches in human genetic susceptibility to tuberculosis. Tuberculosis. 2010. March;90(2):71–83 10.1016/j.tube.2010.02.002 [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y, Jiang T, Yang X, Xue Y, Wang C, Liu J, et al. Toll-like receptor -1, -2, and -6 polymorphisms and pulmonary tuberculosis susceptibility: a systematic review and meta-analysis. PLoS ONE. 2013;8(5):e63357 10.1371/journal.pone.0063357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yim J-J, Kim HJ, Kwon OJ, Koh W-J. Association between microsatellite polymorphisms in intron II of the human Toll-like receptor 2 gene and nontuberculous mycobacterial lung disease in a Korean population. Hum Immunol. 2008. September;69(9):572–6. 10.1016/j.humimm.2008.06.003 [DOI] [PubMed] [Google Scholar]
- 40.Ryu YJ, Kim EJ, Koh W-J, Kim H, Kwon OJ, Chang JH. Toll-like receptor 2 polymorphisms and nontuberculous mycobacterial lung diseases. Clin Vaccine Immunol. 2006. July;13(7):818–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hall MA, McGlinn E, Coakley G, Fisher SA, Boki K, Middleton D, et al. Genetic polymorphism of IL-12 p40 gene in immune-mediated disease. Genes Immun. 2000. February;1(3):219–24. [DOI] [PubMed] [Google Scholar]
- 42.Tso HW, Lau YL, Tam CM, Wong HS, Chiang AK. Associations between IL12B polymorphisms and tuberculosis in the Hong Kong Chinese population. The Journal of infectious diseases. 2004. September 1;190(5):913–9. [DOI] [PubMed] [Google Scholar]
- 43.Ali S, Srivastava AK, Chopra R, Aggarwal S, Garg VK, Bhattacharya SN, et al. IL12B SNPs and copy number variation in IL23R gene associated with susceptibility to leprosy. Journal of Medical Genetics. 2013. January;50(1):34–42. 10.1136/jmedgenet-2012-101214 [DOI] [PubMed] [Google Scholar]
- 44.Morahan G, Kaur G, Singh M, Rapthap CC, Kumar N, Katoch K, et al. Association of variants in the IL12B gene with leprosy and tuberculosis. Tissue Antigens. 2007. April;69 Suppl 1:234–6. [DOI] [PubMed] [Google Scholar]
- 45.Ma X, Reich RA, Gonzalez O, Pan X, Fothergill AK, Starke JR, et al. No evidence for association between the polymorphism in the 3' untranslated region of interleukin-12B and human susceptibility to tuberculosis. The Journal of Infectious Diseases. 2003. October 15;188(8):1116–8. [DOI] [PubMed] [Google Scholar]
- 46.Zhu K-J, Zhu C-Y, Shi G, Fan Y-M. Meta-analysis of IL12B polymorphisms (rs3212227, rs6887695) with psoriasis and psoriatic arthritis. Rheumatol Int. 2013. July;33(7):1785–90. 10.1007/s00296-012-2637-4 [DOI] [PubMed] [Google Scholar]
- 47.Liu H, Irwanto A, Tian H, Fu X, Yu Y, Yu G, et al. Identification of IL18RAP/IL18R1 and IL12B as leprosy risk genes demonstrates shared pathogenesis between inflammation and infectious diseases. American Journal of Human Genetics. 2012. November 2;91(5):935–41. 10.1016/j.ajhg.2012.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manry J, Laval G, Patin E, Fornarino S, Tichit M, Bouchier C, et al. Evolutionary genetics evidence of an essential, nonredundant role of the IFN-γ pathway in protective immunity. Hum Mutat. 2011. June;32(6):633–42. 10.1002/humu.21484 [DOI] [PubMed] [Google Scholar]
- 49.Zhang M, Lin Y, Iyer DV, Gong J, Abrams JS, Barnes PF. T-cell cytokine responses in human infection with Mycobacterium tuberculosis. Infect Immun. 1995. August;63(8)3231–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vallinoto ACR, Graça ES, Araújo MS, Azevedo VN, Cayres-Vallinoto I, Machado LF, et al. IFNG +874T/A polymorphism and cytokine plasma levels are associated with susceptibility to Mycobacterium tuberculosis infection and clinical manifestation of tuberculosis. Hum Immunol. 2010. July;71(7):692–6. 10.1016/j.humimm.2010.03.008 [DOI] [PubMed] [Google Scholar]
- 51.Onwubalili JK, Scott GM, Robinson JA. Deficient immune interferon production in tuberculosis. Clin Exp Immunol. 1985. February;59(2):405–13. [PMC free article] [PubMed] [Google Scholar]
- 52.Pacheco AG, Cardoso CC, Moraes MO. IFNG +874T/A, IL10 -1082G/A and TNF -308G/A polymorphisms in association with tuberculosis susceptibility: a meta-analysis study. Hum Genet. 2008. June;123(5):477–84. 10.1007/s00439-008-0497-5 [DOI] [PubMed] [Google Scholar]
- 53.Li X. NFκB-dependent signaling pathways. Experimental Hematology. 2002. April;30(4):285–96. [DOI] [PubMed] [Google Scholar]
- 54.Sha WC. Regulation of immune responses by NF-kappa B/Rel transcription factor. J Exp Med. 1998. January 19;187(2):143–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bowie A, O’Neill LA. Oxidative stress and nuclear factor-κB activation. Biochemical Pharmacology. 2000. January;59(1):13–23. [DOI] [PubMed] [Google Scholar]
- 56.Tato CM, Hunter CA. Host-Pathogen Interactions: Subversion and Utilization of the NF- B Pathway during Infection. Infection and Immunity. 2002. July 1;70(7):3311–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yamada H, Mizuno S, Reza-Gholizadeh M, Sugawara I. Relative Importance of NF-κB p50 in Mycobacterial Infection. Infect Immun. 2001. November;69(11):7100–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bai X, Feldman NE, Chmura K, Ovrutsky AR, Su W- L, Griffin L, et al. Inhibition of nuclear factor-kappa B activation decreases survival of Mycobacterium tuberculosis in human macrophages. PLoS ONE. 2013;8(4):e61925 10.1371/journal.pone.0061925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gutierrez MG, Mishra BB, Jordao L, Elliott E, Anes E, Griffiths G, et al. NF- B Activation Controls Phagolysosome Fusion-Mediated Killing of Mycobacteria by Macrophages. J Immunol. 2008. August 15;181(4)2651–63. [DOI] [PubMed] [Google Scholar]
- 60.Giri DK, Mehta RT, Kansal RG, Aggarwal BB. Mycobacterium avium-intracellulare complex activates nuclear transcription factor-kappaB in different cell types through reactive oxygen intermediates. J Immunol. 1998. November 1;161(9): 4834–4841. [PubMed] [Google Scholar]
- 61.Kim K-H, Kim T-S, Lee JG, Park J-K, Yang M, Kim JM, et al. Characterization of Proinflammatory Responses and Innate Signaling Activation in Macrophages Infected with Mycobacterium scrofulaceum. Immune Netw. 2014. December;14(6):307–320. 10.4110/in.2014.14.6.307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weiss DJ, Souza CD, Evanson OA. Effects of nuclear factor-kappaB on regulation of cytokine expression and apoptosis in bovine monocytes exposed to Mycobacterium avium subsp paratuberculosis. Am J Vet Res. 2008. June;69(6): 804–810. 10.2460/ajvr.69.6.804 [DOI] [PubMed] [Google Scholar]
- 63.Ghassemi M, Andersen BR, Roebuck KA, Rabbi MF, Plate JM, Novak RM. Mycobacterium avium Complex Activates Nuclear Factor κB via Induction of Inflammatory Cytokines. Cellular Immunology. 1999. February 1;191(2)117–23. [DOI] [PubMed] [Google Scholar]
- 64.Kawai T, Akira S. Signaling to NF-κB by Toll-like receptors. Trends in Molecular Medicine. 2007. November;13(11):460–9. [DOI] [PubMed] [Google Scholar]
- 65.Kawai T, Akira S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity. 2011. May;34(5):637–50. 10.1016/j.immuni.2011.05.006 [DOI] [PubMed] [Google Scholar]
- 66.Schorey JS, Cooper AM. Macrophage signalling upon mycobacterial infection: the MAP kinases lead the way. Cell Microbiol. 2003. March;5(3):133–42. [DOI] [PubMed] [Google Scholar]
- 67.Xia Z, Dickens M, Raingeaud JL, Davis RJ, Greenberg ME. Opposing Effects of ERK and JNK-p38 MAP Kinases on Apoptosis. Science. 1995. November 24;270(5240):1326–31. [DOI] [PubMed] [Google Scholar]
- 68.Pearson G, Robinson F, Beers Gibson T, Xu B-E, Karandikar M, Berman K, et al. Mitogen-Activated Protein (MAP) Kinase Pathways: Regulation and Physiological Functions. Endocrine Reviews. 2001. April;22(2):153–83. [DOI] [PubMed] [Google Scholar]
- 69.Shiratsuchi H, Ellner JJ, Basson MD. Extracellular-regulated kinase activation regulates replication of Mycobacterium avium intracellularly in primary human monocytes. Cell Tissue Res. 2008. May;332(2):237–44. 10.1007/s00441-008-0594-8 [DOI] [PubMed] [Google Scholar]
- 70.Ferrara N, Gerber H-P, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003. June;9(6):669–76. [DOI] [PubMed] [Google Scholar]
- 71.van der Flier M, van Leeuwen HJ, van Kessel KP, Kimpen JL, Hoepelman AI, Geelen SP. Plasma Vascular Endothelial Growth Factor in Severe Sepsis. Shock. 2005. January 1;23(1):35 [DOI] [PubMed] [Google Scholar]
- 72.Karlsson S, Pettilä V, Tenhunen J, Lund V, Hovilehto S, Ruokonen E, et al. Vascular endothelial growth factor in severe sepsis and septic shock. Anesth Analg. 2008. June;106(6):1820–6. 10.1213/ane.0b013e31816a643f [DOI] [PubMed] [Google Scholar]
- 73.Srikiatkhachorn A, Ajariyakhajorn C, Endy TP, Kalayanarooj S, Libraty DH, Green S, et al. (2007) Virus-induced decline in soluble vascular endothelial growth receptor 2 is associated with plasma leakage in dengue hemorrhagic Fever. J Virol. 2007 February;81(4):1592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tseng C-S, Lo H-W, Teng H-C, Lo W-C, Ker C-G. Elevated levels of plasma VEGF in patients with dengue hemorrhagic fever. FEMS Immunol Med Microbiol. 2005. January 1;43(1):99–102. [DOI] [PubMed] [Google Scholar]
- 75.Sathupan P, Khongphattanayothin A, Srisai J, Srikaew K, Poovorawan Y. The role of vascular endothelial growth factor leading to vascular leakage in children with dengue virus infection. Ann Trop Paediatr. 2007. September;27(3):179–84. [DOI] [PubMed] [Google Scholar]
- 76.Matsuyama W, Hashiguchi T, Matsumuro K, Iwami F, Hirotsu Y, Kawabata M, et al. Increased serum level of vascular endothelial growth factor in pulmonary tuberculosis. Am J Respir Crit Care Med. 2000. September;162(3 Pt 1):1120–2. [DOI] [PubMed] [Google Scholar]
- 77.Abe Y, Nakamura M, Oshika Y, Hatanaka H, Tokunaga T, Ohkubo Y, et al. Serum levels of vascular endothelial growth factor and cavity formation in active pulmonary tuberculosis. Respiration. 2001;68(5):496–500. [DOI] [PubMed] [Google Scholar]
- 78.Alatas F, Alatas O, Metintas M, Özarslan A, Erginel S, Yildirim H. Vascular Endothelial Growth Factor Levels in Active Pulmonary Tuberculosis. Chest. 2004. June 1;125(6):2156–9. [DOI] [PubMed] [Google Scholar]
- 79.Riou C, Perez Peixoto B, Roberts L, Ronacher K, Walzl G, Manca C, et al. Effect of standard tuberculosis treatment on plasma cytokine levels in patients with active pulmonary tuberculosis. PLoS ONE. 2012;7(5):e36886 10.1371/journal.pone.0036886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shaw MA, Clayton D, Atkinson SE, Williams H, Miller N, Sibthorpe D, et al. Linkage of rheumatoid arthritis to the candidate gene NRAMP1 on 2q35. Journal of Medical Genetics. 1996;33:672–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Searle S, Blackwell JM. Evidence for a functional repeat polymorphism in the promoter of the human NRAMP1 gene that correlates with autoimmune versus infectious disease susceptibility. Journal of Medical Genetics. 1999;36:295–299. [PMC free article] [PubMed] [Google Scholar]
- 82.Blackwell JM, Searle S, Mohamed H, White JK. Divalent cation transport and susceptibility to infectious and autoimmune disease: continuation of the Ity/Lsh/Bcg/Nramp1/Slc11a1 gene story. Immunol Lett. 2003. January 22;85(2):197–203. [DOI] [PubMed] [Google Scholar]
- 83.Ates O, Dalyan L, Müsellim B, Hatemi G, Türker H, Ongen G, et al. NRAMP1 (SLC11A1) gene polymorphisms that correlate with autoimmune versus infectious disease susceptibility in tuberculosis and rheumatoid arthritis. Int J Immunogenet. 2009. February;36(1):15–9. 10.1111/j.1744-313X.2008.00814.x [DOI] [PubMed] [Google Scholar]
- 84.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012. November 1;491(7422):119–24. 10.1038/nature11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Winthrop KL, Iseman M. Bedfellows: mycobacteria and rheumatoid arthritis in the era of biologic therapy. Nat Rev Rheumatol. 2013. June 25;9(9):524–31. 10.1038/nrrheum.2013.82 [DOI] [PubMed] [Google Scholar]
- 86.Yeh J-J, Wang Y-C, Sung F-C, Kao C-H. Rheumatoid arthritis increases the risk of nontuberculosis mycobacterial disease and active pulmonary tuberculosis. PLoS ONE. 2014;9(10):e110922 10.1371/journal.pone.0110922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Esposito L, Hill NJ, Pritchard LE, Cucca F. Genetic analysis of chromosome 2 in type 1 diabetes: analysis of putative loci IDDM7, IDDM12, and IDDM13 and candidate genes NRAMP1 and IA-2 and the interleukin-1 gene cluster. IMDIAB Group. Diabetes. 1998. November;47(11):1797–9. [DOI] [PubMed] [Google Scholar]
- 88.Bellamy R, Ruwende C, Corrah T, McAdam KP, Whittle HC, Hill AV. Variations in the NRAMP1 gene and susceptibility to tuberculosis in West Africans. N Engl J Med. 1998. March 5;338(10):640–4. [DOI] [PubMed] [Google Scholar]
- 89.Blackwell JM, Black GF, Peacock CS, Miller EN, Sibthorpe D, Gnananandha D, et al. Immunogenetics of leishmanial and mycobacterial infections: the Belem Family Study. Philos Trans R Soc Lond B Biol Sci. 1997. September 29;352(1359):1331–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mobley JL. Is rheumatoid arthritis a consequence of natural selection for enhanced tuberculosis resistance? Med Hypotheses. 2004;62(5):839–43. [DOI] [PubMed] [Google Scholar]
- 91.Yang D, Chen J, Zhang L, Cha Z, Han S, Shi W, et al. Mycobacterium leprae upregulates IRGM expression in monocytes and monocyte-derived macrophages. Inflammation. 2014. August;37(4):1028–34. 10.1007/s10753-014-9825-1 [DOI] [PubMed] [Google Scholar]
- 92.Zhang H, Massey D, Tremelling M, Parkes M. Genetics of inflammatory bowel disease: clues to pathogenesis. Br Med Bull. 2008;87:17–30. 10.1093/bmb/ldn031 [DOI] [PubMed] [Google Scholar]
- 93.Lu XC, Tao Y, Wu C, Zhao PL, Li K, Zheng JY, et al. Association between variants of the autophagy related gene—IRGM and susceptibility to Crohn's disease and ulcerative colitis: a meta-analysis. PLoS ONE. 2013. November 13;8(11):e80602 10.1371/journal.pone.0080602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Baker MA, Harries AD, Jeon CY, Hart JE, Kapur A, Lonnroth K, et al. The impact of diabetes on tuberculosis treatment outcomes: A systematic review. BMC Med. 2011;9(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dooley KE, Tang T, Golub JE, Dorman SE, Cronin W. Impact of diabetes mellitus on treatment outcomes of patients with active tuberculosis. Am J Trop Med Hyg. 2009. April;80(4):634–9. [PMC free article] [PubMed] [Google Scholar]
- 96.Dooley KE, Chaisson RE. Tuberculosis and diabetes mellitus: convergence of two epidemics. Lancet Infect Dis. 2009. December;9(12):737–46. 10.1016/S1473-3099(09)70282-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Griffith DE, Brown-Elliott BA, Langsjoen B, Zhang Y, Pan X, Girard W, et al. Clinical and Molecular Analysis of Macrolide Resistance in Mycobacterium avium Complex Lung Disease. Am J Respir Crit Care Med. 2006. October 15;174(8):928–34. [DOI] [PubMed] [Google Scholar]
- 98.Griffith DE, Brown BA, Girard WM, Griffith BE, Couch LA, Wallace RJ Jr. Azithromycin-Containing Regimens for Treatment of Mycobacterium avium Complex Lung Disease. Clin Infect Dis. 2001. June 1;32(11):1547–53. [DOI] [PubMed] [Google Scholar]
- 99.Murphy BS, Sundareshan V, Cory TJ, Hayes D, Anstead MI, Feola DJ. Azithromycin alters macrophage phenotype. Journal of Antimicrobial Chemotherapy. 2008. February 4;61(3):554–60. 10.1093/jac/dkn007 [DOI] [PubMed] [Google Scholar]
- 100.Xu G, Fujita J, Negayama K, Ohnishi T, Miyawaki H, Hojo S, Takigawa K, et al. Effect of clarithromycin on macrophage functions. Kansenshogaku Zasshi. 1995. August 1;69(8):864–72. [DOI] [PubMed] [Google Scholar]
- 101.Kumar V, Harjai K, Chhibber S. Effect of Clarithromycin on Lung Inflammation and Alveolar Macrophage Function in Klebsiella pneumoniaeB5055-Induced Acute Lung Infection in BALB/c mice. Journal of Chemotherapy. 2008. October;20(5):609–14. [DOI] [PubMed] [Google Scholar]
- 102.Feola DJ, Garvy BA, Cory TJ, Birket SE, Hoy H, Hayes D Jr, et al. Azithromycin Alters Macrophage Phenotype and Pulmonary Compartmentalization during Lung Infection with Pseudomonas. Antimicrobial Agents and Chemotherapy. 2010. May 17;54(6):2437–47. 10.1128/AAC.01424-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Renna M, Schaffner C, Brown K, Shang S, Tamayo MH, Hegyi K, et al. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J Clin Invest. 2011. September;121(9):3554–63. 10.1172/JCI46095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.De Groote MA, Johnson L, Podell B, Brooks E, Basaraba R, Gonzalez-Juarrero M. GM-CSF knockout mice for preclinical testing of agents with antimicrobial activity against Mycobacterium abscessus. Journal of Antimicrobial Chemotherapy. 2014. March 17;69(4):1057–64. 10.1093/jac/dkt451 [DOI] [PubMed] [Google Scholar]
- 105.Hodge S, Hodge G, Jersmann H, Matthews G, Ahern J, Holmes M, et al. Azithromycin improves macrophage phagocytic function and expression of mannose receptor in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008. July 15;178(2):139–48. 10.1164/rccm.200711-1666OC [DOI] [PubMed] [Google Scholar]
- 106.Hodge S, Reynolds PN. Low-dose azithromycin improves phagocytosis of bacteria by both alveolar and monocyte-derived macrophagesin chronic obstructive pulmonary disease subjects. Respirology. 2012. July;17(5):802–7. 10.1111/j.1440-1843.2012.02135.x [DOI] [PubMed] [Google Scholar]
- 107.Kang PB. The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. Journal of Experimental Medicine. 2005. October 3;202(7):987–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Leemans JC, Thepen T, Weijer S, Florquin S, van Rooijen N, van de Winkel JG, et al. Macrophages Play a Dual Role during Pulmonary Tuberculosis in Mice. J Infect Dis. 2005. January 1;191(1):65–74. [DOI] [PubMed] [Google Scholar]
- 109.Watt B, Rayner A, Harris G. Comparative activity of azithromycin against clinical isolates of mycobacteria. J Antimicrob Chemother. 1996. September;38(3):539–42. [DOI] [PubMed] [Google Scholar]
- 110.Prammananan T, Arjratanakool W, Chaiprasert A, Tingtoy N, Leechawengwong M, Asawapokee N, et al. Second-line drug susceptibilities of Thai multidrug-resistant Mycobacterium tuberculosis isolates. Int J Tuberc Lung Dis. 2005. February;9(2)216–9. [PubMed] [Google Scholar]
- 111.van Halsema C, Humphreys S, Bonington A. Extensively drug-resistant tuberculosis: early access to bedaquiline for a UK patient. European Respiratory Journal. 2013. December 31;43(1):292–4. 10.1183/09031936.00128613 [DOI] [PubMed] [Google Scholar]
- 112.Luna-Herrera J, Reddy VM, Daneluzzi D, Gangadharam PR. Antituberculosis activity of clarithromycin. Antimicrobial Agents and Chemotherapy. 1995. December 1;39(12):2692–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bolhuis MS, van Altena R, van Soolingen D, de Lange WC, Uges DR, van der Werf TS, et al. Clarithromycin increases linezolid exposure in multidrug-resistant tuberculosis patients. European Respiratory Journal. 2013. December;42(6):1614–21. 10.1183/09031936.00001913 [DOI] [PubMed] [Google Scholar]
- 114.Bosne-David S, Barros V, Verde SC, Portugal C, David HL. Intrinsic resistance of Mycobacterium tuberculosis to clarithromycin is effectively reversed by subinhibitory concentrations of cell wall inhibitors. J Antimicrob Chemother. 2000. September;46(3):391–5. [DOI] [PubMed] [Google Scholar]
- 115.Reich-Slotky R, Kabbash CA, Della-Latta P, Blanchard JS, Feinmark SJ, Freeman S, et al. Gemfibrozil Inhibits Legionella pneumophila and Mycobacterium tuberculosis Enoyl Coenzyme A Reductases and Blocks Intracellular Growth of These Bacteria in Macrophages. Journal of Bacteriology. 2009. July 27;191(16):5262–71. 10.1128/JB.00175-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Guideline: Nutritional Care and Support for Patients with Tuberculosis. Geneva: World Health Organization; 2013. [PubMed] [Google Scholar]
- 117.Shankar AH. Nutritional modulation of malaria morbidity and mortality. The Journal of Infectious Diseases. 2000. September;182 Suppl 1:S37–53. [DOI] [PubMed] [Google Scholar]
- 118.Wheelwright M, Kim EW, Inkeles MS, De Leon A, Pellegrini M, Krutzik SR, et al. All-trans retinoic acid-triggered antimicrobial activity against Mycobacterium tuberculosis is dependent on NPC2. J Immunol. 2014. March 1;192(5):2280–90. 10.4049/jimmunol.1301686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vilchèze C, Hartman T, Weinrick B, Jacobs WR. Mycobacterium tuberculosis is extraordinarily sensitive to killing by a vitamin C-induced Fenton reaction. Nat Comms. 2013;4:1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Grant SS, Kaufmann BB, Chand NS, Haseley N, Hung DT. Eradication of bacterial persisters with antibiotic-generated hydroxyl radicals. Proc Natl Acad Sci USA. 2012. July 24;109(30):12147–52. 10.1073/pnas.1203735109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Martineau AR, Timms PM, Bothamley GH, Hanifa Y, Islam K, Claxton AP, et al. High-dose vitamin D(3) during intensive-phase antimicrobial treatment of pulmonary tuberculosis: a double-blind randomised controlled trial. Lancet. 2011. January 15;377(9761):242–50. 10.1016/S0140-6736(10)61889-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jeon K, Kim S- Y, Jeong B- H, Chang B, Shin SJ, Koh WJ. Severe vitamin D deficiency is associated with non-tuberculous mycobacterial lung disease: a case-control study. Respirology. 2013. August;18(6):983–8. 10.1111/resp.12109 [DOI] [PubMed] [Google Scholar]
- 123.Wejse C, Gomes VF, Rabna P, Gustafson P, Aaby P, Lisse IM, et al. Vitamin D as supplementary treatment for tuberculosis: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2009. May 1;179(9):843–50. 10.1164/rccm.200804-567OC [DOI] [PubMed] [Google Scholar]
- 124.Sinclair D, Abba K, Grobler L. Nutritional supplements for people being treated for active tuberculosis. Cochrane Database Syst Rev. 2011. November 9;(11):CD006086 10.1002/14651858.CD006086.pub3 [DOI] [PubMed] [Google Scholar]
- 125.Ratledge C. Iron, mycobacteria and tuberculosis. Tuberculosis. 2004;84(1–2):110–30. [DOI] [PubMed] [Google Scholar]
- 126.Tullius MV, Harmston CA, Owens CP, Chim N, Morse RP, McMath LM, et al. Discovery and characterization of a unique mycobacterial heme acquisition system. Proceedings of the National Academy of Sciences. 2011. March 22;108(12):5051–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nixon MR, Saionz KW, Koo M-S, Szymonifka MJ, Jung H, Roberts JP, et al. Folate Pathway Disruption Leads to Critical Disruption of Methionine Derivatives in Mycobacterium tuberculosis. Chemistry & Biology. 2014. July;21(7):819–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.