

Abstract
Spindle cell/sclerosing rhabdomyosarcoma is a rare skeletal-muscle tumor with distinctive clinicopathologic characteristics. 10 cases (6 cases of spindle cell rhabdomyosarcoma and 4 cases of scleroisng rhabdomyosarcoma) were composed of 6 males and 4 females aging from 5 months to 57 years, with median age 33 years, most of who represented a painless solid mass. Histologically, the tumors were composed of fascicles of spindle cells or primitive round cells embed in sclerotic matrix with presence of rhabdomyoblasts in varying proportion. Immunohistochemically, the tumor cells expressed MyoD1 (10/10), Desmin (10/10), myogenin (6/10), AE1/AE3 (2/10), EMA (2/10), but were negative for SMA, caldesmon, S-100. All of the patients underwent a complete surgical resection without or with chemotherapy (2/10) or radiotherapy (1/10). During the follow-up period (1 to 24 months), 1 patient was succumbed, and 2 cases showed in situ recurrence with 1 of them adopting metastasis. Our cases further demonstrate there do present some clincopathologic relations between spindle cells rhabdomyosarcoma and sclerosing rhabdomyosarcoma, but the latter seems to have a better prognosis. Exact grading and staging contribute to predict the outcome.
Keywords: Spindle rhabdomyosarcoma, sclerosing rhabdomyosarcoma, pathology, immunohistochemistry, differential diagnosis, prognosis
Introduction
Rhabdomyosarcoma (RMS) is most frequently malignant soft tissue tumor developing in the childhood and adolescence with skeletal muscle differentiation. Traditionally, it falls into three main groups: embryonal, alveolar and pleomorphic. However, both spindle and sclerosing subset were reported subsequently [1-3]. Spindle RMS was originally defined as a variant of embryonal RMS predominantly affecting children with a favorable prognosis compared to other categories of RMS, but adult patients were also appreciated subsequently, however, with evidences from sporadic cases showing no prognostic advantage [1,2,4]. Sclerosing RMS was recently recognized and also can develop in either pediatric or adult population [3,5]. In light of existence of morphologic overlap and clinical similarities, both of the two entities shared a common designation as “spindle cell/sclerosing rhabdomyosarcoma” in newly WHO classification [6]. As an uncommon subtype of RMS, spindle cell/sclerosing RMS was only described by limited literature, most of which were reported by case studies. To further understand the clinicopathologic features, we herein retrospectively reviewed 10 cases from our institution emphasizing the distinctive morphology, immunochemistry and follow-up.
Materials and methods
Cases diagnosed as Spindle cell or sclerosing RMS from 2010 to 2014 in the first affiliated hospital of Zhengzhou university were selected and reviewed. Complete clinicopathologic and follow-up data were obtained through the records or inquiring by phone. Grading and staging were performed by FNCLCC and AJCC assessment system [7,8]. All of the surgical specimens were fixed in 4% formalin, embedded routinely in paraffin and stained with hematoxylin and eosin. Immuohistochemical studies were performed using commercial antibodies in the Ventana BenchMark XT instrument (Ventana System, Tucson AZ). The antibodies included desmin, myogenin, MyoD1, SMA, caldesmon, AE1/AE3, EMA, S-100 and Ki-67 (all above from Ventana, prediluted).
Results
Clinical findings and follow-up
The main clinical and follow-up data were summarized in Table 1. Cases were composed of 6 cases of spindle cell RMS and 4 cases of scleroisng RMS containing 6 males and 4 females aging from 5 months to 57 years (median, 33 years; mean, 34 years). The tumor involving the anatomic locations including upper arm, abdominal wall, glottis, orbit, nasopharynx, thigh, hand and groin, most of which affecting trunks or extremities presented with a solid painless mass with a relative demarcation in physical examination. But the bulky lesions or local nerve compression can result in hoarseness (case 3), protopsis (case 4), nasal obstruction (case 5), mobility limitation (case 6, 10), peripheral vessels compression (case 8), and painful feelings (case 6, 9). All of patients denied familial heredity diseases and underwent a complete tumor resection with or without additional adjuvant chemo- (case 1, 7) or radio- (case 3) therapy. Generally, the spindle variant in our series was more likely to have a high grading and staging compared to sclerosing variant and accordingly, 1 patient was succumbed and 2 patients suffered from in situ recurrence, one of who was clinically manifested evidences of metastasis during the follow-up period. Another 5 cases of sclerosing RMS in our group had relatively low grading and staging and therefore behaved a good prognosis in our group.
Table 1.
Clinical Features and Follow-up Data
| Case no. | Diagnosis (RMS) | Age/sex | site | Size (cm × cm × cm) | Presentation | Treatment | Gradea | Stageb | Follow-up (mo) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Spindle cell | 42 y/M | Left upper arm | 5.2×3.2×2.5 | Painless solid mass with poor mobility and related demarcation | Surgery and chemotherapy | 3 | III | DOD (24) |
| 2 | Spindle cell | 30 y/M | Left lower quadrant of abdomen wall | 14.0×9.8×9.0 | Painless and cystic-solid mass with demarcation and peritoneum involved focally | Surgery | 3 | III | In situ Rec (10) |
| 3 | Spindle cell | 49 y/M | Next to left glottis in larynx | 2.4×1.8×1.5 | Hoarseness; solid mass with focal calcification and cervical lymphadenopathy | Surgery and radiotherapy | 1 | I B | AWOD (5) |
| 4 | Spindle cell | 5 mo/F | Left orbit | 2.8×2.7×0.6 | Left eye swelling, expanded palpebral fissure and protopsis | Surgery | 2 | II A | AWD (5) |
| 5 | Spindle cell | 28 y/M | nasopharynx | 5.0×5.0×1.0 | Nasal obstruction, rhinorrhoe and occasional headache; solid mass involved sphenoid sinus | Surgery | 3 | III | AWD (1) |
| 6 | Spindle cell | 52/M | Left thigh | 21×9.0×8.5 | Solid mass with heavy tenderness; mobility limitation | Surgery | 2 | IV | In situ Rec and metastasis to stomach (12) |
| 7 | sclerosing | 24/F | Back of right hand | 3.4×2.5×1.5 | Solid and demarcated mass gradually increase during 10 years and repeatedly recurred | Surgery | 1 | I A | NED (1) |
| 8 | sclerosing | 18/M | Right groin | 16.5×9.0×9.0 | Painless solid mass with compressed peripheral vessels | Surgery and chemotherapy | 1 | I B | AWOD (6) |
| 9 | sclerosing | 36/F | Left pars buccalis | 4.8×3.5×2 | Solid increasing mass with ulcer formation and pain feelings | Surgery | 1 | I A | AWOD (8) |
| 10 | sclerosing | 57/F | Right parotid gland | 2.3×1.8×1.5 | Solid painless mass with mouth-opening limitation | Surgery | 1 | I A | AWOD (13) |
using FNCLCC grading system;
sing AJCC anatomic staging system;
AWD, alive with diseases; AWOD, alive without evidence of disease; DOD, die of disease; mo, months; NA, not available; NED, no evidence of disease; Rec, recurrence.
Pathological findings
Grossly, the pinkish-grey or grayish solid tumors ranging from 2.3 cm to 21 cm in maximum diameter, tended to be well circumscribed with a firm, gray-white to tan cut surface, some of which possessed a gritty texture, necrosis or cystic degeneration. Histologically, in 6 cases (case 1 to case 6), the tumor predominantly consisted of sheets or multiple nodules of spindle cells arranging in whorls or fascicles infiltrating normal muscles or adipose tissues, usually, in pushing type that stimulated leiomyosarcoma (Figure 1). Perivascular accentuation, significant necrosis (Figure 2), inflammatory infiltration and hemorrhage also can be appreciated. Areas of case 1 imparted an evident herringbone growth pattern mimicking adult fibrosarcoma (Figure 3). The spindle tumor cells had elongated and fusiform nuclei, small nucleoli and abundant eosinophilic cytoplasm with varying nuclear atypia, mitotic activity and pleomorphism. In the remaining cases (case 7 to case 10), undifferentiated small round cells were mainly interpreted as cords, strands (Figure 4), alveolar or packet pattern (Figure 5) embed in intensely hyalinized matrix with a little eosionphilic or pale cytoplasm, inconspicuous nuclei and nucleoli with or without mitotic figures. Necrosis was relatively uncommon compared with the former 6 cases. More or less, transition zones between spindle cell and sclerosing areas (Figure 6) and presence of rhabdomyoblasts (Figure 7) can be found when performing an attentively observation. The immunohistochemical profile was summarized in Table 2. Most of tumor cells were typically positive for MyoD1 (10/10) (Figure 8), Desmin (10/10) (Figure 9) and focally positive for myogenin (6/10) (Figure 10), but totally negative for SMA, caldesmon, S-100. Only two cases focally expressed AE1/AE3 and EMA. Proliferative index Ki67 varied from 15% to 80%.
Figure 1.
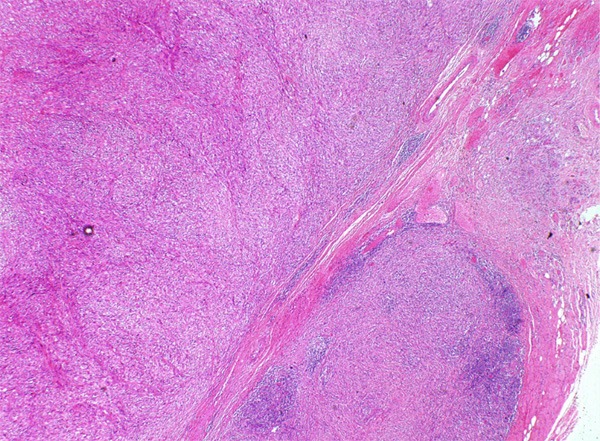
Multiple nodules of spindle cells infiltrating normal tissues in pushing type.
Figure 2.
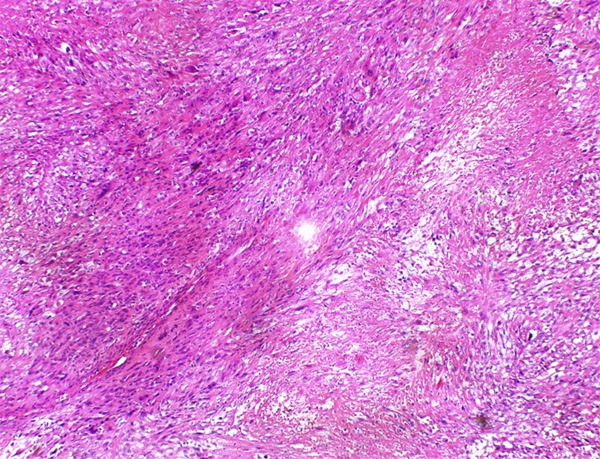
Intersecting fascicles of spindle tumor cells with significant necrosis.
Figure 3.
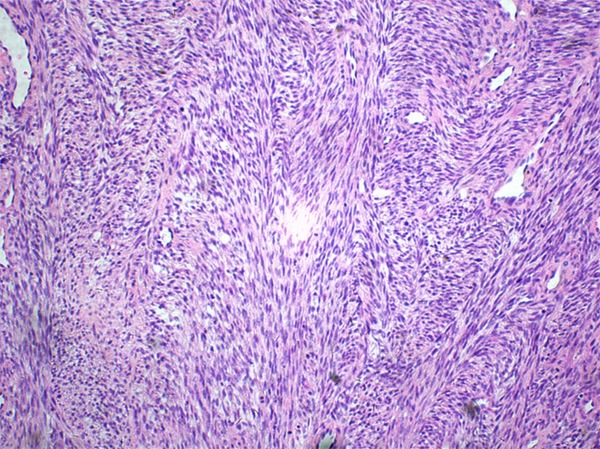
Herringbone growth pattern mimicking adult fibrosarcoma.
Figure 4.
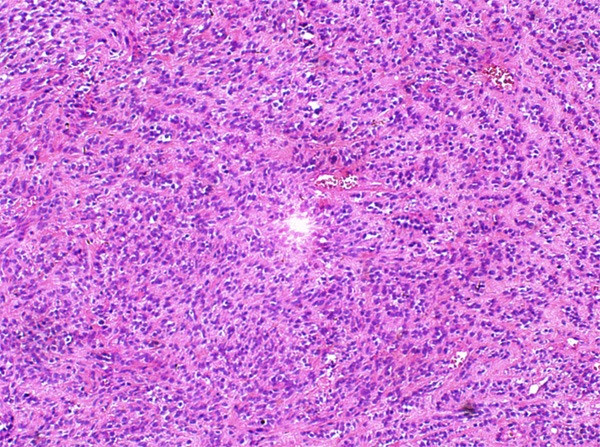
Primitive-like small round cells arranging in cords or strands with intensely hyalinized matrix.
Figure 5.
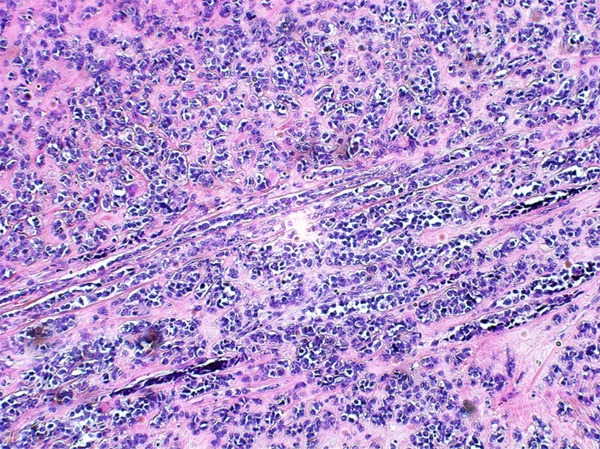
Small alveolar or packet growth pattern embed in sclerotic stroma.
Figure 6.
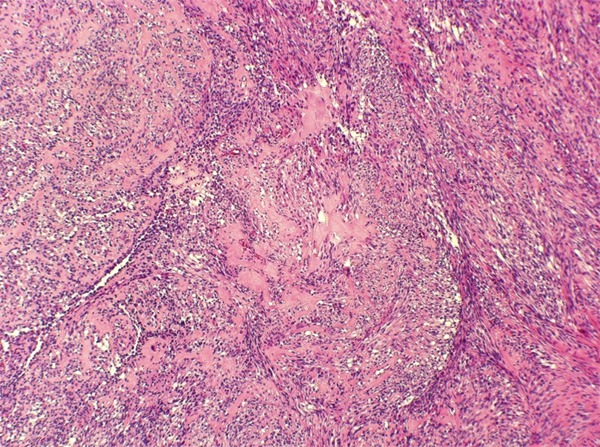
Transition zone between spindle cell and sclerosing area.
Figure 7.
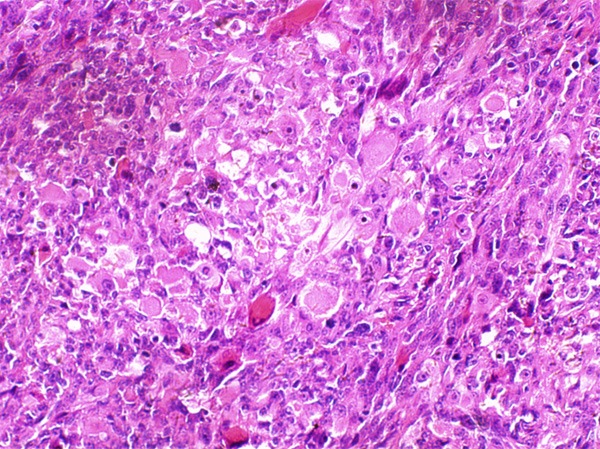
Focal presence of rhabdomyobalsts.
Table 2.
Immunohistochemical profiles of spindle cell/sclerosing RMS
| Case no. | Desmin | Myogenin | MyoD1 | SMA | Caldesmon | AE1/AE3 | EMA | S-100 | Ki-67 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | ++ | - | +++ | - | - | + | ++ | - | 80% |
| 2 | ++ | + | ++ | - | - | - | - | - | 70% |
| 3 | +++ | + | ++ | - | - | - | - | - | 15% |
| 4 | + | - | ++ | - | - | - | - | - | 30% |
| 5 | +++ | + | +++ | - | - | ++ | + | - | 60% |
| 6 | +++ | - | +++ | - | - | - | - | - | 60% |
| 7 | +++ | ++ | +++ | - | - | - | - | - | 50% |
| 8 | + | ++ | +++ | - | - | - | - | - | 50% |
| 9 | +++ | ++ | +++ | - | - | - | - | - | 70% |
| 10 | +++ | - | ++ | - | - | - | - | - | 30% |
-, negative; +, tumor cell expressed less than 10%; ++, tumor cells expressed 10%-50%; +++, tumor cells expressed above 50%.
Figure 8.
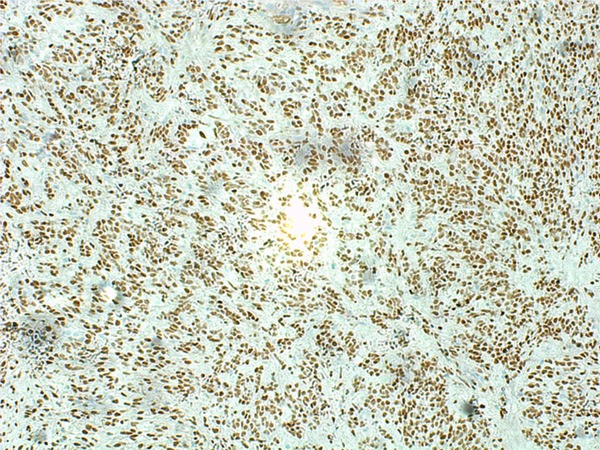
MyoD1 was diffusely expressed.
Figure 9.
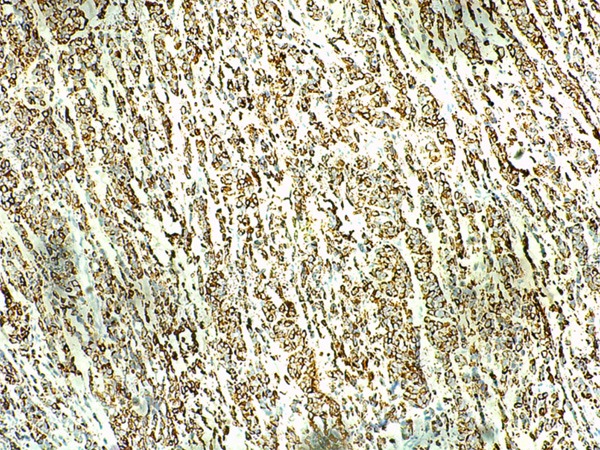
Diffuse immunoreactivity for Desmin.
Figure 10.
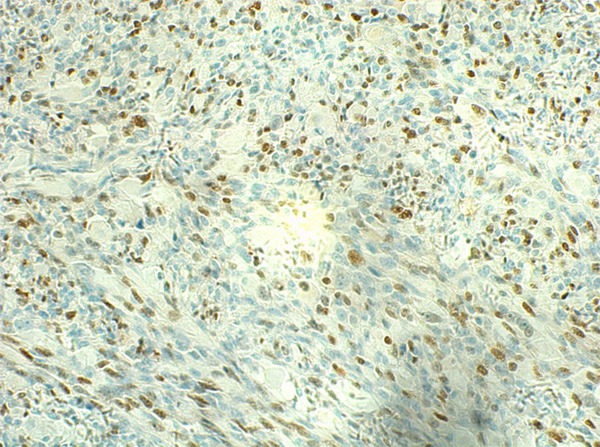
Myogenin was focally positive.
Discussion
Spindle cell RMS first reported by Cavazzana et al, mainly arise in paratesticular region and followed by the head and neck in pediatric population and has a better prognosis with lower lymph node metastasis and favorable 5-year survival compared to other subtypes of RMS [1,9]. Cases of adults affected were subsequently founded, but head and neck region and extremities are the most frequent anatomic locations [4,10,11]. The prognosis seems to be aggressive, however, still better than other adult RMS, such as polymorphic variant [12]. A mass with or without pain is the most common complaint, which usually demonstrates a firm and fine demarcation from surrounding tissues in gross examination [1]. Histologically, spindle cell RMS is typically composed of long fascicles of relatively uniform spindle cells arranged in intersecting or herring bone pattern mimicking leiomysarcoma or fibrosarcoma. The majority of tumor cells have pale and eosinophilic indistinct cytoplasm with small and long nuclei with vesicular chromatin and small nucleoli. Significant mitotic activity usually can be seen. Sclerosing RMS, originally reported by Mentzel et al, also can rise in pediatrics or adults, but paratesticular region is rarely implicated [2,3]. Microscopically, the prominent hyaline matrix separate the undifferentiated round or oval tumor cells into cords, nests, or small alveolar patterns, frequently reminiscent of sclerosing epithelioid fibrosarcoma, chondrosarcoma or even angiosarcoma [2,13].
However, cases with synchronous presentation of areas of spindle cells or small primitive-like cells embed in abundant collagen stroma can be founded, suggesting their morphologic overlaps and therefore these two entities have shared the same designation, namely, “spindle cell/sclerosing RMS” [6]. Although scattered rhabdomyoblasts with eccentric nuclei and significant ample eosinophilic cytoplasm might suggest there existed relevance between spindle cell/sclerosing RMS and embryonal RMS, clear genetic data link have been not yet founded [13-15]. By immunohistochemistry, tumor cells usually show strong and diffuse positivity for desmin and MyoD1, and a variable extent of nuclear reactivity for myogenin from focal to diffuse pattern, with or without focal expression of cytokeratin [5,16].
Other RMS subtypes have different pathologic features from spindle cell/sclerosing RMS. Although sclerosing RMS may have a pseudovascular or acinar growth pattern, the prominently hyaline stroma, smaller alveolar spaces and focal presence of fascicles of spindle cells are different from alveolar RMS. In addition, the latter tends to consistently express both MyoD1 and myogenin and was therefore distinct from the priority of MyoD1 expression in spindle cell/sclerosing RMS [17]. As we discussed above, the real relationship between embryonal RMS and spindle cell/sclerosing RMS is still controversial, but ubiquitous strap- or radpole-like rhabdomyoblasts with abundant eosinophilic cytoplasm in a myxoid matrix and scattered nuclear expression of MyoD1 and myogenin are far more likely to support embryonal RMS [18]. Accurate diagnosis also needs to rule out several entities with spindle and sclerotic morphology, including sarcomatoid carcinoma, spindle cell melanoma, malignant peripheral nerve sheath tumor with or without rhabdomyosarcoma differentiation, leiomyosarcoma, fibrosarcoma, sclerosing epithelioid fibrosarcoma, osteosarcoma and so on [2,16,19]. However, deficiency of relationship with nerve fibers, “marble-like” growth pattern with myxoid matrix, better differentiated epithelial areas, lace- or filigree-like neoplastic bone, and broad-spectrum cytokeratins (focal positivity permissible), CK5/6, P63, S-100, HMB45, CEA, SMA, caldesmon expression, and presence of typical immunophenotype feature of MyoD1 and myogenin can easily tell them apart.
Although the consensus of optimal treatment for spindle cell/sclerosing RMS has been not reached, the mainstay therapeutic method should also, similar to most soft tissue tumors, be surgery and adjuvant chemotherapy or radiotherapy can be added [19]. 2 cases and 1 case in our group received a chemotherapy and radiotherapy after tumor resection, but whether the adjuvant treatments worked was unclear. Lesions developing in some anatomic locations are hard to be eliminated may have a recurrent risk, such as case 4 and 5 in our series which need a close supervision. Generally, spindle cell/sclerosing RMS has a relatively favorable outcome in pediatric group, but worse prognosis in adults with a higher propensity of recurrence and metastasis [6,9,19]. In our limited cases, influence of prognosis derived from age seems insignificant but more relate to the grading and staging. In addition, our group of spindle subtype presented with more common necrosis, mitotic images and rhabdomyoblasts with a comparatively higher grading and staging in clinic and therefore more frequent recurrence, metastasis and even mortality. However, that situation still needs ongoing follow-up in our cases and more prognostic data from other republications.
In conclusion, spindle cell/sclerosing RMS is a rare skeletal-muscle tumor with distinct morphology, immunohistochemistry and relatively favorable prognosis. An accurate diagnosis based on the well-known recognition and an exact clinical grading and staging contribute to predict the outcome of this entity.
Acknowledgements
This work was financially supported by the first affiliated hospital of Zhengzhou University.
Disclosure of conflict of interest
None.
References
- 1.Cavazzana AO, Schmidt D, Ninfo V, Harms D, Tollot M, Carli M, Treuner J, Betto R, Salviati G. Spindle cell rhabdomyosarcoma. A prognostically favorable variant of rhabdomyosarcoma. Am J Surg Pathol. 1992;16:229–235. doi: 10.1097/00000478-199203000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Folpe AL, McKenney JK, Bridge JA, Weiss SW. Sclerosing rhabdomyosarcoma in adults: report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. Am J Surg Pathol. 2002;26:1175–1183. doi: 10.1097/00000478-200209000-00008. [DOI] [PubMed] [Google Scholar]
- 3.Mentzel T, Katenkamp D. Sclerosing, pseudovascular rhabdomyosarcoma in adults. Clinicopathological and immunohistochemical analysis of three cases. Virchows Arch. 2000;436:305–311. doi: 10.1007/s004280050451. [DOI] [PubMed] [Google Scholar]
- 4.Fernando Val-Bernal J, Fernandez N, Gomez-Roman JJ. Spindle cell rhabdomyosarcoma in adults. A case report and literature review. Pathol Res Pract. 2000;196:67–72. doi: 10.1016/s0344-0338(00)80024-2. [DOI] [PubMed] [Google Scholar]
- 5.Rekhi B, Singhvi T. Histopathological, immunohistochemical and molecular cytogenetic analysis of 21 spindle cell/sclerosing rhabdomyosarcomas. APMIS. 2014;122:1144–1152. doi: 10.1111/apm.12272. [DOI] [PubMed] [Google Scholar]
- 6.Nascimento AF, Barr FG. In: Spindle cell/sclerosing rhabdomyosarcoma. Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, editors. Lyon: IARC; 2013. pp. 134–135. [Google Scholar]
- 7.Trojani M, Contesso G, Coindre JM, Rouesse J, Bui NB, de Mascarel A, Goussot JF, David M, Bonichon F, Lagarde C. Soft-tissue sarcomas of adults; study of pathological prognostic variables and definition of a histopathological grading system. Int J Cancer. 1984;33:37–42. doi: 10.1002/ijc.2910330108. [DOI] [PubMed] [Google Scholar]
- 8.Edge SB, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A. AJCC cancer staging mannual. 7th edition. New York: Springer; 2010. [Google Scholar]
- 9.Leuschner I, Newton WA Jr, Schmidt D, Sachs N, Asmar L, Hamoudi A, Harms D, Maurer HM. Spindle cell variants of embryonal rhabdomyosarcoma in the paratesticular region. A report of the Intergroup Rhabdomyosarcoma Study. Am J Surg Pathol. 1993;17:221–230. doi: 10.1097/00000478-199303000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Rubin BP, Hasserjian RP, Singer S, Janecka I, Fletcher JA, Fletcher CD. Spindle cell rhabdomyosarcoma (so-called) in adults: report of two cases with emphasis on differential diagnosis. Am J Surg Pathol. 1998;22:459–464. doi: 10.1097/00000478-199804000-00011. [DOI] [PubMed] [Google Scholar]
- 11.Nascimento AF, Fletcher CD. Spindle cell rhabdomyosarcoma in adults. Am J Surg Pathol. 2005;29:1106–1113. [PubMed] [Google Scholar]
- 12.Stock N, Chibon F, Binh MB, Terrier P, Michels JJ, Valo I, Robin YM, Guillou L, Ranchere-Vince D, Decouvelaere AV, Collin F, Birtwisle-Peyrottes I, Gregoire F, Aurias A, Coindre JM. Adulttype rhabdomyosarcoma: analysis of 57 cases with clinicopathologic description, identification of 3 morphologic patterns and prognosis. Am J Surg Pathol. 2009;33:1850–1859. doi: 10.1097/PAS.0b013e3181be6209. [DOI] [PubMed] [Google Scholar]
- 13.Croes R, Debiec-Rychter M, Cokelaere K, De Vos R, Hagemeijer A, Sciot R. Adult sclerosing rhabdomyosarcoma: cytogenetic link with embryonal rhabdomyosarcoma. Virchows Arch. 2005;446:64–67. doi: 10.1007/s00428-004-1131-0. [DOI] [PubMed] [Google Scholar]
- 14.Chiles MC, Parham DM, Qualman SJ, Teot LA, Bridge JA, Ullrich F, Barr FG, Meyer WH. Sclerosing rhabdomyosarcomas in children and adolescents: a clinicopathologic review of 13 cases from the Intergroup Rhabdomyosarcoma Study Group and Children’s Oncology Group. Pediatr Dev Pathol. 2004;7:583–594. doi: 10.1007/s10024-004-5058-x. [DOI] [PubMed] [Google Scholar]
- 15.Bouron-Dal Soglio D, Rougemont AL, Absi R, Barrette S, Montpetit A, Fetni R, Fournet JC. SNP genotyping of a sclerosing rhabdomyosarcoma: Reveals highly aneuploid profile and a specific MDM2/HMGA2 amplification. Hum Pathol. 2009;40:1347–1352. doi: 10.1016/j.humpath.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 16.Carroll SJ, Nodit L. Spindle cell rhabdomyosarcoma: A brief diagnostic review and differential diagnosis. Arch Pathol Lab Med. 2013;137:1155–1158. doi: 10.5858/arpa.2012-0465-RS. [DOI] [PubMed] [Google Scholar]
- 17.Sebire NJ, Malone M. Myogenin and MyoD1 expression in paediatric rhabdomyosarcomas. J Clin Pathol. 2003;56:412–416. doi: 10.1136/jcp.56.6.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parham DM, Ellison DA. Rhabdomyosarcomas in adults and children: an update. Arch Pathol Lab Med. 2006;130:1454–1465. doi: 10.5858/2006-130-1454-RIAACA. [DOI] [PubMed] [Google Scholar]
- 19.Yasui N, Yoshida A, Kawamoto H, Yonemori K, Hosono A, Kawai A. Clinicopathologic analysis of spindle cell/sclerosing rhabdomyosarcoma. Pediatr Blood Cancer. 2015;62:1011–1016. doi: 10.1002/pbc.25367. [DOI] [PubMed] [Google Scholar]