

Abstract
Synapsin I is a neuronal phosphoprotein contained in the synaptic vesicles of mammalian central and peripheral nervous systems. It regulates both neurotransmitter release and synaptic formation. Variations in synapsin I expression in the brain have been reported to cause brain malfunction. In severe malaria, neurological complications, such as convulsion, delirium and coma, suggest abnormalities in the release of neurotransmitters. This study evaluated synapsin I expression in cerebral malaria (CM). An immunohistochemical method was used to study the semi-quantitative and qualitative expression of synapsin I in the brain of CM patients (10 cases) who died with Plasmodium falciparum, compared with non-cerebral malaria (NCM) (4 cases), and control brain tissues (5). Synapsin I was expressed in the gray matter of the cerebral cortex and the molecular layer of the cerebellum, as a diffusely dense precipitate pattern in the neuropil, with no immunoreactivity in the neurons, neuronal dendrites, glial cells, endothelial cells, and Purkinje cells. The findings were similarly demonstrated in CM, NCM, and control brain tissues. However, in the granular layer of the cerebellum, a significant increase in synapsin I expression was observed in the granule cells, and the glomerular synaptic complex, from the CM group, compared with the NCM, and control brain tissues (all P < 0.05). Parasitemia showed a positive correlation with synapsin I expression in the granule cells (on admission: Spearman’s ρ = 0.600, P = 0.023) (before death: Spearman’s ρ = 0.678, P = 0.008), and glomerular synaptic complex (before death: Spearman’s ρ = 0.571, P = 0.033). It was hypothesized that CM causes pre-synaptic excitation and eventually activation of synapsin I, leading to increased neurotransmitter release. Synapsin I inhibitor should be investigated further as a target for a therapeutic intervention to alleviate neurological symptoms in severe malaria.
Keywords: Malaria, Plasmodium falciparum, cerebral malaria, synapsin I, immunohistochemistry
Introduction
Cerebral malaria (CM) is one of the major fatal complications of Plasmodium falciparum infection. The condition is associated with a mortality rate of 15-20% in severe malaria cases [1]. According to the World Health Organization (WHO), CM is a clinical syndrome defined as a potentially reversible diffuse encephalopathy, characterized mainly by coma (unconsciousness state and inability to localize a painful stimulus), and the presence of asexual forms of P. falciparum parasites in the peripheral blood smears, in the absence of other causes of encephalopathy [2]. CM can cause variations in clinical presentation, including cognitive, behavioral and motor dysfunctions, seizures and coma [3]. The release of chemical mediators, immunological responses, and the process of cytoadhesion/sequestration are the main pathologic hypotheses for severe malaria, particularly CM. A dominant hypothesis of CM is the sequestration of parasitized red blood cells (PRBCs) to the cerebral microvascular endothelium, causing multifocal abnormalities in cerebral blood flow, resulting in hypoxemia, acidosis, hypoglycemia, and other metabolic derangements affecting brain function [4,5]. The underlying pathogenic mechanism of CM is still incompletely understood. Previous study suggests that during P. falciparum malaria infection, the axons are vulnerable to damage. Axonal transport may be disrupted within the nerve fibers, and may lead to neurological dysfunction in CM [1]. Among the neuronal phosphoproteins, synapsin I plays an important role in the regulation of neurotransmitter release [6-8]. A recently study found a significant increase in glutamate level (the major excitatory neurotransmitter in the mammalian central nervous system) in the cerebral cortex of mice infected with P. berghei ANKA [3].
The physiologic neuro-regulating process in the pre-synaptic terminal is multifactorial, and occurs rapidly. Synapsin proteins may be involved in the process of acute infection with neurological symptoms, such as in severe malaria. This study investigated the expression of synapsin I in post-mortem CM brain tissues, compared with non-cerebral malaria (NCM), and normal brain samples.
Materials and methods
Specimen collection
Brain specimens from fatal P. falciparum malaria patients and control brain samples were obtained from the Department of Tropical Pathology, Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand. All tissues were embedded in paraffin blocks. The specimens were divided into 3 groups--cerebral malaria (CM), non-cerebral malaria (NCM), and control. CM diagnosis was based on clinical presentation in the patient’s chart and autopsy findings of cytoadhesion and sequestration in the blood vessels of the brain tissue. Patients were grouped into NCM when no cerebral involvement was documented in both clinical and autopsy reports. Control brain tissues were collected from patients who died from accidents with no brain injury. Histologically, control brain tissues showed normal neurons and supporting glial cells, with no sign of infection or hemorrhage. The use of left-over brain specimens was approved by the Ethics Committee of the Faculty of Tropical Medicine, Mahidol University (MUTM 2011-046-1).
Immunohistochemical study of synapsin I in brain tissues
The paraffin-embedded specimens were re-embedded with new paraffin medium, and sectioned with microtome at 4 µm thickness for hematoxylin and eosin (H&E) staining and immunohistochemical study. The primary antibody, synapsin I rabbit monoclonal antibody, detects endogenous levels of total synapsin proteins, which covered human synapsin-Ia and synapsin-Ib (Cell Signaling Technology, Inc., USA). The brain-tissue sections were placed on an adhesive slide coated with poly-L-lysine (Sigma, St. Louis, CA), and deparaffinized with a series of xylene, then re-hydrated with graded alcohol. In the antigen retrieval technique, sections were treated with 0.1 M sodium citrate buffer (pH 6.0), and heated in a microwave (Samsung MW71B) at high power (800 W) for 20 mins, modified from previous study [9]. Then, endogenous peroxidase activity was blocked by treating sections with 3% hydrogen peroxide (H2O2) in distilled water (dH2O) for 30 mins at 37°C, and rinsed in running tap-water for 10 mins. Then, the sections were incubated with goat serum for 30 mins at room temperature to block non-specific staining, and washed twice with phosphate buffered saline (PBS). The sections were then incubated with primary antibody, synapsin I rabbit monoclonal antibody (Cell Signaling Technology, Inc., USA) (1:200 dilution) overnight at 4°C. The following day, the brain sections were washed with PBS, and overlaid with secondary antibody, biotinylated goat anti-rabbit IgG (Vector Laboratories, Inc., USA), and incubated for 30 mins at room temperature. After washing with PBS, the sections were incubated with avidin-biotin peroxidase complex (ABC) conjugated with horseradish peroxidase (HRP) (Vector Laboratories, Inc., USA) or alkaline phosphatase (AP) (Vector Laboratories, Inc., USA) for 30 mins. The enzymatic reaction was visualized by adding 3,3’-diaminobenzidine (DAB) (Vector Laboratories, Inc., USA) or Vector® Red substrate kit (Vector Laboratories, Inc., USA), resulting in the formation of a brown (DAB) or red (Vector® Red) color at the antigen sites. Mouse brain and eyeball (retina) were used as the positive control, to validate the specificity of the primary antibody.
Evaluation of immunohistochemical staining
Positive cells showed characteristic granular brown or bright-red stained synapsin I in the cytoplasm, nucleus, or both, or formed into complexes called glomerular synaptic complex in the granular layer of the cerebellum. The number of immunopositive cells and staining intensity for synapsin I were evaluated in the granular layer of the cerebellum. Each slide was examined at 1,000 × magnification in 10 random fields. The positive cells per field were counted and the percentage was calculated in relation to the total number of cells; the averages of these were taken. Staining intensity was graded on a scale ranging from 0 to 3 (0 = no staining, 1 = weak positive staining, 2 = moderate positive staining, and 3 = strong positive staining). The total score (TS) was calculated by taking the product of the percentage of immunopositive cells (P) and intensity (I) [Total score (TS) = percentage of positive cells (P) × intensity (I); maximum = 300], according to previous studies [10-13]. Immunoexpression in the glomeruli was quantified based on the number of immunopositive areas in the granular layer of the cerebellum per high power field (400 ×).
Statistical analysis
Demographic data were described using descriptive statistics. The test for normality was used for all quantitative data. The normality of distribution was tested with Kolmogorov-Smirnov test. Data were presented as mean ± standard error of mean (SEM). Statistical significance was determined by Mann Whitney U test to compare the percentage of immunopositive cells and staining intensity for synapsin I in the brain between groups, including CM, NCM, and control groups. Statistical analysis was performed using the Statistical Package for the Social Science (SPSS) version 11.0 software (SPSS, Chicago, IL). The level of significance was set at p-value ≤ 0.05.
Results
Summary of clinical data
Fourteen cases of patients who died from P. falciparum malaria were included in the study. Brain specimens consisted of 10 cases of CM and 4 cases of NCM. Normal brain tissues (5 cases) were used as control. A clinical summary of the malaria cases is shown in Table 1. Other complications in CM included anemia (4/10, 40%), jaundice (8/10, 80%), pneumonia 5/10, (50%), shock (2/10, 20%), pulmonary edema (4/10, 40%), disseminated intravascular coagulation (DIC) (4/10, 40%), acute kidney injury (AKI) (6/10, 60%), hypoglycemia (2/10, 20%), and acidosis (2/10, 20%). Among the NCM, the causes of death were respiratory distress syndrome with pulmonary edema (1), septicemia (2), and various organ failures (1).
Table 1.
Clinical data of malaria patients
| Cerebral malaria (n = 10) | Non-cerebral malaria (n = 4) | |
|---|---|---|
| Male: Female | 7:3 | 3:1 |
| Age (P = 0.539) | 31.7 ± 3.38 | 28.75 ± 5.91 |
| Hemoglobin (g/dl) (P = 0.304) | 11.82 ± 0.49 | 12.58 ± 0.33 |
| White blood cell count (P = 0.304) | 11,992.40 ± 1115.37 | 14,988.50 ± 2,332.76 |
| Parasite count (on admission) (parasites/µl) (P = 0.002) | 601,668.77 ± 68,153.77 | 63,215.25 ± 16,748.60 |
| Last parasite count (parasites/µl) (P = 0.002) | 54,721.80 ± 11,707.06 | 0 |
Histopathological evaluation of brain tissues
Brain tissues from CM cases generally showed congestion, minute petechial hemorrhages, hemosiderin pigment deposits, parasitized red blood cells in the blood vessels, and a degree of sequestration. The brain tissues from NCM cases showed unremarkable findings, similar to the normal brain. Representative images of changes in the brain tissue in CM are illustrated in Figure 1.
Figure 1.
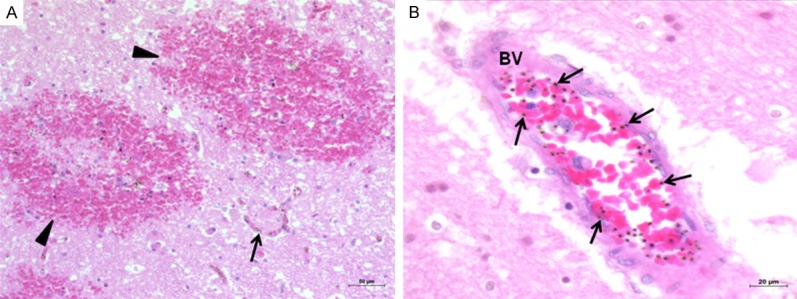
Histopathologic changes of the brain in CM. Representative images showing petechial hemorrhages (arrowheads) in the cerebral cortex (A) (200 ×), and sequestration of PRBCs (arrows) in the cerebral blood vessels (BV) (B) (400 ×). Hematoxylin and eosin stain.
Cerebrum
The neuropil of the grey matter showed similar immunoexpression of synapsin I in CM, NCM, and control brain tissues. Generalized uniform intense staining in the neuropil of the gray matter was demonstrated. Quantification of immunoexpression was not possible due to diffused immunostaining of synapsin I protein in the gray matter. Synapsin I expression was absent in the neurons, dendrites of neurons, glial cells, and endothelial cells in both white and gray matters (Figure 2).
Figure 2.
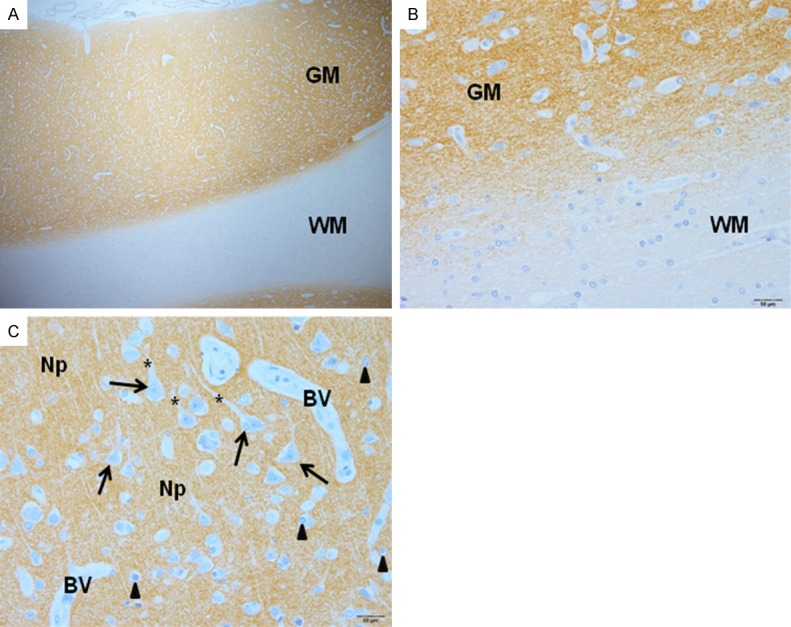
Synapsin I expression in the cerebral cortex, illustrating strong immunostaining in the gray matter (GM), and no staining in the white matter (WM) (A) (400 ×). At higher magnification (B), the junction between gray and white matter is shown. In the pyramidal cell layer (C), neuropil (Np) shows dispersed, strong immunostaining for synapsin I, while immunostaining is absent from the neurons (arrows), dendrites (asterisks), glial cells (arrowheads), and blood vessels (BV) (avidin-biotin peroxidase complex technique).
Cerebellum
The cerebellum consists of the cerebellar cortex (gray matter) and an inner, white matter. The cerebellar cortex can be distinguished into 3 layers--molecular, Purkinje cell, and granular layers. For the molecular and Purkinje cell layers, the expression of synapsin I was similar in CM, NCM, and control brain tissues. In the molecular layer, intense precipitate was scattered diffusely in the neuropil (Figure 3). There was no synapsin I expression in the body of neurons, dendrites of neurons, glial cells, or endothelial cells. This pattern was similar to the findings in the cerebral cortex. The Purkinje cells and associated dendrites in the Purkinje cell layer also showed no immunoreactivity to synapsin I (Figure 3). The granular layer consists of granule cells, Golgi type II cells, and small, irregular dispersed, faint staining, clear spaces called the glomeruli [14]. Within the granule cells, many fine punctate precipitates of synapsin I were observed in the nuclei and cytoplasm. The glomerular synaptic complex demonstrated clusters of immunoreactive areas (Figures 3C and 4). The small granule cells in the granular layer of the cerebellum showed differential synapsin I expression in CM, NCM, and control brain tissues. Synapsin I expression was distinct in the nucleus and cytoplasm of the granule cells. CM cases showed significantly increase in mean percentage immunopositive cells (27.85 ± 2.93) and total score (TS) (72.68 ± 8.94) of synapsin I in the granule cells of the cerebellar cortex, compared with NCM cases (4.99 ± 1.84, TS = 9.46 ± 3.98, P = 0.002) and control brains (6.08 ± 2.61, TS = 11.18 ± 5.41, P = 0.001), (NCM vs. control brain: P = 0.905) (Figure 5A). The spaces between the granule cells contain the glomerular synaptic complex. Although the intensity of synapsin I expression was similar in all 3 groups, the density of immunoreactive glomeruli per high power field was significantly increased in CM (80.44 ± 5.57), compared with NCM (16.45 ± 2.64, P = 0.002) and control groups (14.62 ± 1.84, P = 0.001), (NCM vs. control brain: P = 0.73) (Figure 5B).
Figure 3.
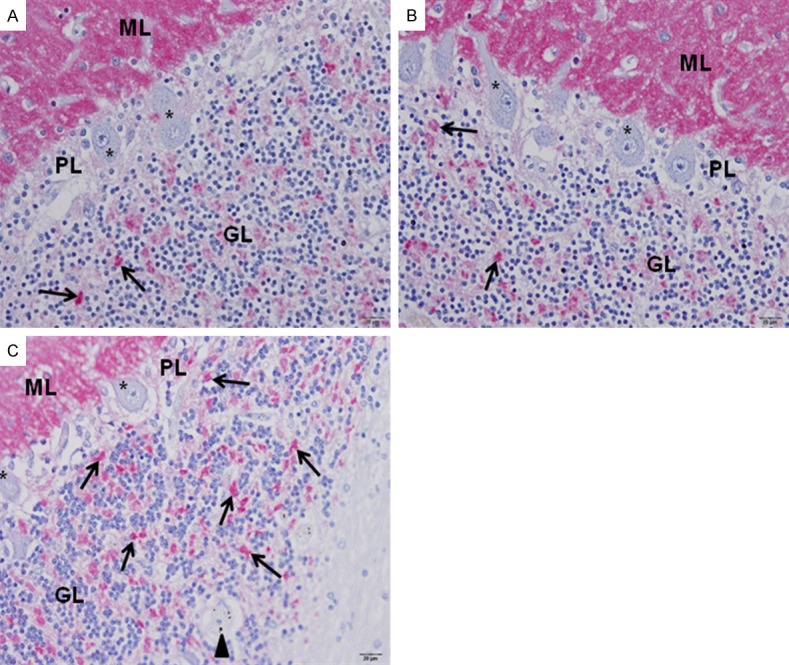
Synapsin I expression in the gray matter of the cerebellar cortex. Similar immunoexpression in the molecular layer (ML), and Purkinje cell layer (PL) was demonstrated in the control (A), NCM (B), and CM (C) brain tissues. Strong and diffused immunostaining was seen in the ML, while Purkinje cells failed to express synapsin I protein (asterisks). The granule cells in the granular layer (GL) shows increased in immuno-positive cells in CM (C), compared to NCM (B) and control brain (A) tissues. Increased synapsin I expression was seen in glomerular synaptic complex (arrows) in CM (C). Parasitized red blood cells (arrowhead) are seen in a blood vessel (C). All images are 400 × magnification. Bar = 20 µm. (avidin-biotin alkaline phosphatase complex technique).
Figure 4.
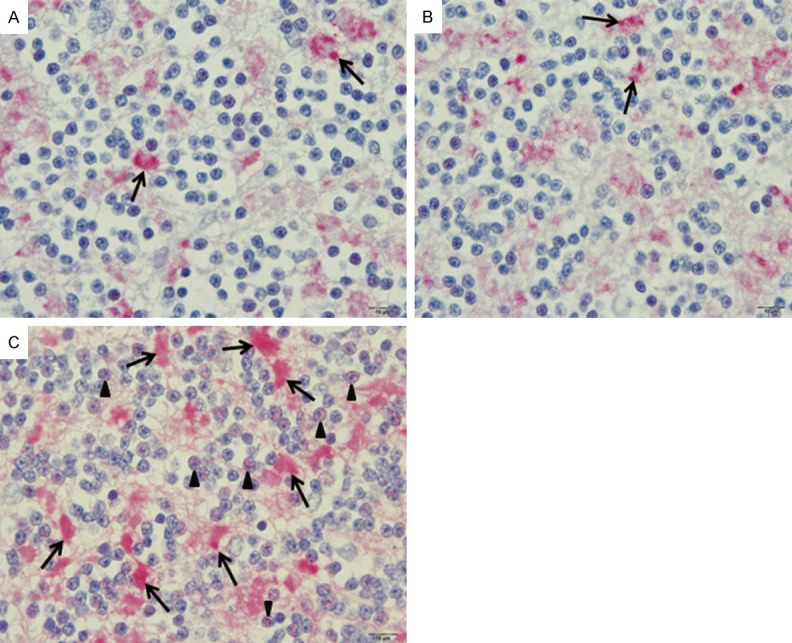
Synapsin I expression in the granular layer of the cerebellar cortex. The granular cells in the control (A) and NCM (B) groups show few immunopositive cells compared to CM (C) brain tissues (arrowheads). Increased synapsin I expression in the glomeruli synaptic complex (arrows) was demonstrated in the CM (C). All images are 1,000 × magnification, Bar = 10 µm (avidin-biotin alkaline phosphatase complex technique).
Figure 5.
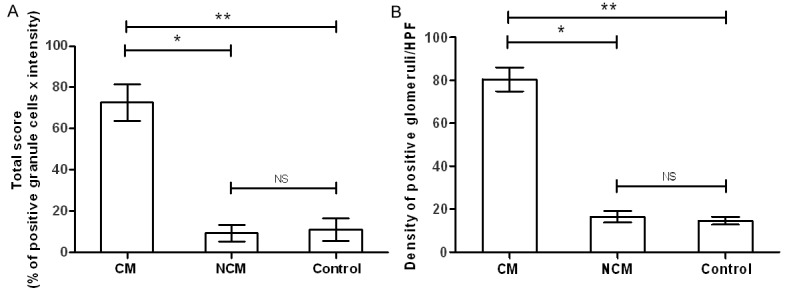
Comparative findings of synapsin I expression in the cerebellum, showing a significant increase in immune-reactivity against synapsin I antibody for granule cells (A) and glomerular synaptic complex (B) in CM group. (*-P < 0.001, **-P < 0.002, NS- not significant).
Correlations between parasitemia and synapsin I expression
Parasitemia was positively correlated with synapsin I expression in the granule cells (On admission: Spearman’s ρ = 0.600, P = 0.023) (before death: Spearman’s ρ = 0.678, P = 0.008), and glomerular synaptic complex (before death: Spearman’s ρ = 0.571, P = 0.033) (Figure 6).
Figure 6.
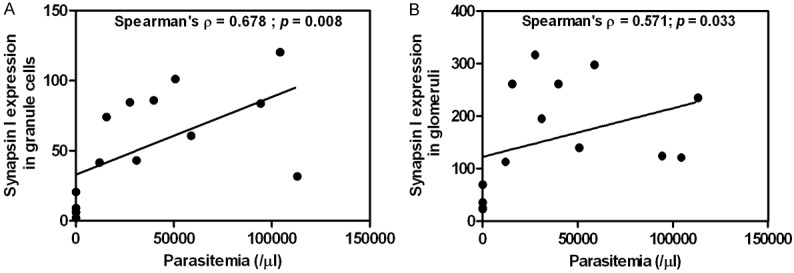
Correlations between parasitemia and synapsin I expression in the granule cells (A), and glomerular synaptic complex (B) of the cerebellum.
Discussion
Synapsin I is a neuronal phosphoprotein bound to the synaptic vesicles, which regulates the exocytosis of the synaptic vesicles and neurotransmitter release, neurite elongation, and synaptogenesis [15]. Synapsin I immunoreactivity has been used as an indirect indicator of synapses in the central nervous system [15-18]. The present work used immunohistochemical methods to demonstrate the expression of synapsin I in terms of distribution and intensity in the cerebrum and cerebellum of P. falciparum malaria patients. Synapsin I and II have been demonstrated to be widely distributed in the central nervous system, including the olfactory bulb, cerebral cortex, hippocampus, basal ganglia, thalamus, hypothalamus, cerebellum, pons-medulla, spinal cord and posterior pituitary gland [19]. For the cerebrum, the present study found that the immunostaining pattern was similar to previous reports, where a diffused precipitate was observed in the neuropil, but neurons and dendrites showed no or minimal immunoreactivity [15,18]. Synapsin I expression was found in the gray matter (neuropil) of the cerebral cortex, the molecular layer and the granular layer of the cerebellar cortex. Moreover, synapsin I was observed in the nucleus and cytoplasm of granule cells, and in the glomerular synaptic complex of the cerebellum. Glomeruli are specialized structures where mossy fibers and Golgi cells are connected together synaptically [20]. In the CM group, the glomerular synaptic complex expressed an increase in synapsin I protein compared with NCM and normal brain tissues, which may indicate that CM can induce synapsin I activation.
Synapsin I expression has been associated with ischemia. A study in the hippocampus of Mongolian gerbils after transient forebrain ischemia showed a strong increase in synapsin I immunoreactivity in the hilus of dentate gyrus and in the mossy fiber layer of the hippocampus, suggesting a role for synapsin I in plastic adaptation of the hippocampus following injury [15]. In CM, cytoadhesion and sequestration of parasitized red blood cells are important mechanical processes that lead to transient obstruction and possibly brain ischemia. Increased synapsin I expression in CM could stimulate the activation of pre-synaptic vesicles and modulate neurotransmitter release. A previous study has shown an association between synapsin I and brain spectrin in neurotransmitter release [21]. In an animal model, vesicular glutamate transporter (VGLUT) 1, VGLUT 2, and the vesicular GABA transporter (VGAT), were decreased in mouse forebrains devoid of synapsin I and II [22]. It would be useful to determine the neurotransmitter level and its correlation with synapsin I activation in cerebral malaria. Synapsin I has not been studied in the infectious process with neurological involvement. So far, research on synapsin I has been linked to neurodegenerative diseases, such as Alzheimer’s disease [23]. An impairment in synapsin I has also been documented in epileptic seizures in human [24] and animal models [25-28], in behavioral alteration [29], learning deficit [30], and Huntington’s disease [31]. Moreover, synapsin protein has been identified as a novel drug target for the treatment of refractory hypertension, a common side effect of cyclosporine A (CsA), the immunosuppressive drug used in transplant patients and as a treatment for autoimmune diseases [32].
A murine model study of CM found an increase in glutamate levels in the cerebral cortex and cerebrospinal fluid, suggesting a role for glutamate in the central nervous system dysfunction found in CM [3]. In addition, the metabolites of the kynurenine pathway, particularly quinolinic acid, has been linked to convulsion in CM [33], and has been detected at a high level in the cerebrospinal fluid of Kenyan children with CM [34]. It can be speculated that synapsin I is involved in neurotransmitter release in CM. The findings illustrate a functional correlation between synapsin I level and clinical severity. Further investigations of synapsin I inhibition might eventually help alleviate CM symptoms.
Acknowledgements
This work was supported by the Office of the Higher Education Commission and Mahidol University under the National Research Universities Initiative, Thailand. Printing of the manuscript was partially supported by the Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand. The authors would like to thank Dr. Mario Riganti for the use of left-over specimen.
Disclosure of conflict of interest
None.
References
- 1.Medana IM, Day NP, Hien TT, Mai NT, Bethell D, Phu NH, Farrar J, Esiri MM, White NJ, Turner GD. Axonal injury in cerebral malaria. Am J Pathol. 2002;160:655–666. doi: 10.1016/S0002-9440(10)64885-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Severe falciparum malaria. World Health Organization, Communicable Diseases Cluster. Trans R Soc Trop Med Hyg. 2000;94(Suppl 1):S1–90. [PubMed] [Google Scholar]
- 3.Miranda AS, Vieira LB, Lacerda-Queiroz N, Souza AH, Rodrigues DH, Vilela MC, Gomez MV, Machado FS, Rachid MA, Teixeira AL. Increased levels of glutamate in the central nervous system are associated with behavioral symptoms in experimental malaria. Braz J Med Biol Res. 2010;43:1173–1177. doi: 10.1590/s0100-879x2010007500130. [DOI] [PubMed] [Google Scholar]
- 4.Idro R, Jenkins NE, Newton CR. Pathogenesis, clinical features, and neurological outcome of cerebral malaria. Lancet Neurol. 2005;4:827–840. doi: 10.1016/S1474-4422(05)70247-7. [DOI] [PubMed] [Google Scholar]
- 5.Medana IM, Chaudhri G, Chan-Ling T, Hunt NH. Central nervous system in cerebral malaria: ‘Innocent bystander’ or active participant in the induction of immunopathology? Immunol Cell Biol. 2001;79:101–120. doi: 10.1046/j.1440-1711.2001.00995.x. [DOI] [PubMed] [Google Scholar]
- 6.Bennett AF, Hayes NV, Baines AJ. Site specificity in the interactions of synapsin 1 with tubulin. Biochem J. 1991;276:793–799. doi: 10.1042/bj2760793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirokawa N, Sobue K, Kanda K, Harada A, Yorifuji H. The cytoskeletal architecture of the presynaptic terminal and molecular structure of synapsin 1. J Cell Biol. 1989;108:111–126. doi: 10.1083/jcb.108.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bahler M, Greengard P. Synapsin I bundles F-actin in a phosphorylation-dependent manner. Nature. 1987;326:704–707. doi: 10.1038/326704a0. [DOI] [PubMed] [Google Scholar]
- 9.Wu LM, Han H, Wang QN, Hou HL, Tong H, Yan XB, Zhou JN. Mifepristone repairs region-dependent alteration of synapsin I in hippocampus in rat model of depression. Neuropsychopharmacology. 2007;32:2500–2510. doi: 10.1038/sj.npp.1301386. [DOI] [PubMed] [Google Scholar]
- 10.Konstantinidou AE, Givalos N, Gakiopoulou H, Korkolopoulou P, Kotsiakis X, Boviatsis E, Agrogiannis G, Mahera H, Patsouris E. Caspase-3 immunohistochemical expression is a marker of apoptosis, increased grade and early recurrence in intracranial meningiomas. Apoptosis. 2007;12:695–705. doi: 10.1007/s10495-006-0001-4. [DOI] [PubMed] [Google Scholar]
- 11.Charafe-Jauffret E, Tarpin C, Bardou VJ, Bertucci F, Ginestier C, Braud AC, Puig B, Geneix J, Hassoun J, Birnbaum D, Jacquemier J, Viens P. Immunophenotypic analysis of inflammatory breast cancers: identification of an ‘inflammatory signature’. J Pathol. 2004;202:265–273. doi: 10.1002/path.1515. [DOI] [PubMed] [Google Scholar]
- 12.Punsawad C, Maneerat Y, Chaisri U, Nantavisai K, Viriyavejakul P. Nuclear factor kappa B modulates apoptosis in the brain endothelial cells and intravascular leukocytes of fatal cerebral malaria. Malar J. 2013;12:260. doi: 10.1186/1475-2875-12-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Viriyavejakul P, Khachonsaksumet V, Punsawad C. Liver changes in severe Plasmodium falciparum malaria: histopathology, apoptosis and nuclear factor kappa B expression. Malar J. 2014;13:106. doi: 10.1186/1475-2875-13-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eroschenko VP. diFiore’s atlas of histology with functional correlations. 11th edition. Baltimore. MD: Lippincott Williams & Wilkins; 2008. [Google Scholar]
- 15.Marti E, Ferrer I, Blasi J. Transient increase of synapsin-I immunoreactivity in the mossy fiber layer of the hippocampus after transient forebrain ischemia in the mongolian gerbil. Brain Res. 1999;824:153–160. doi: 10.1016/s0006-8993(99)01158-0. [DOI] [PubMed] [Google Scholar]
- 16.Masliah E, Terry RD, Alford M, DeTeresa R. Quantitative immunohistochemistry of synaptophysin in human neocortex: an alternative method to estimate density of presynaptic terminals in paraffin sections. J Histochem Cytochem. 1990;38:837–844. doi: 10.1177/38.6.2110586. [DOI] [PubMed] [Google Scholar]
- 17.Stone LM, Browning MD, Finger TE. Differential distribution of the synapsins in the rat olfactory bulb. J Neurosci. 1994;14:301–309. doi: 10.1523/JNEUROSCI.14-01-00301.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitagawa K, Matsumoto M, Sobue K, Tagaya M, Okabe T, Niinobe M, Ohtsuki T, Handa N, Kimura K, Mikoshiba K, Kamada T. The synapsin I brain distribution in ischemia. Neuroscience. 1992;46:287–299. doi: 10.1016/0306-4522(92)90051-3. [DOI] [PubMed] [Google Scholar]
- 19.Walaas SI, Browning MD, Greengard P. Synapsin Ia, synapsin Ib, protein IIIa, and protein IIIb, four related synaptic vesicle-associated phosphoproteins, share regional and cellular localization in rat brain. J Neurochem. 1988;51:1214–1220. doi: 10.1111/j.1471-4159.1988.tb03089.x. [DOI] [PubMed] [Google Scholar]
- 20.Manto M, De Zeeuw C. Diversity and Complexity of Roles of Granule Cells in the Cerebellar Cortex. Editorial. Cerebellum. 2012;11:1–4. doi: 10.1007/s12311-012-0365-7. [DOI] [PubMed] [Google Scholar]
- 21.Zimmer WE, Zhao Y, Sikorski AF, Critz SD, Sangerman J, Elferink LA, Xu XS, Goodman SR. The domain of brain beta-spectrin responsible for synaptic vesicle association is essential for synaptic transmission. Brain Res. 2000;881:18–27. doi: 10.1016/s0006-8993(00)02796-7. [DOI] [PubMed] [Google Scholar]
- 22.Bogen IL, Boulland JL, Mariussen E, Wright MS, Fonnum F, Kao HT, Walaas SI. Absence of synapsin I and II is accompanied by decreases in vesicular transport of specific neurotransmitters. J Neurochem. 2006;96:1458–1466. doi: 10.1111/j.1471-4159.2005.03636.x. [DOI] [PubMed] [Google Scholar]
- 23.Parks KM, Sugar JE, Haroutunian V, Bierer L, Perl D, Wallace WC. Reduced in vitro phosphorylation of synapsin I (site 1) in Alzheimer’s disease postmortem tissues. Brain Res Mol Brain Res. 1991;9:125–134. doi: 10.1016/0169-328x(91)90137-m. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen DK, Rouleau I, Senechal G, Ansaldo AI, Gravel M, Benfenati F, Cossette P. X-linked focal epilepsy with reflex bathing seizures: Characterization of a distinct epileptic syndrome. Epilepsia. 2015;56:1098–1108. doi: 10.1111/epi.13042. [DOI] [PubMed] [Google Scholar]
- 25.Etholm L, Bahonjic E, Heggelund P. Sensitive and critical periods in the development of handling induced seizures in mice lacking synapsins: differences between synapsin I and synapsin II knockouts. Exp Neurol. 2013;247:59–65. doi: 10.1016/j.expneurol.2013.03.025. [DOI] [PubMed] [Google Scholar]
- 26.Suemaru S, Sato K, Morimoto K, Yamada N, Sato T, Kuroda S. Increment of synapsin I immunoreactivity in the hippocampus of the rat kindling model of epilepsy. Neuroreport. 2000;11:1319–1322. doi: 10.1097/00001756-200004270-00034. [DOI] [PubMed] [Google Scholar]
- 27.Morimoto K, Sato K, Sato S, Suemaru S, Sato T, Yamada N, Hayabara T. Increases in mRNA levels for synapsin I but not synapsin II in the hippocampus of the rat kindling model of epilepsy. Seizure. 1998;7:229–235. doi: 10.1016/s1059-1311(98)80041-1. [DOI] [PubMed] [Google Scholar]
- 28.Li L, Chin LS, Shupliakov O, Brodin L, Sihra TS, Hvalby O, Jensen V, Zheng D, McNamara JO, Greengard P, et al. Impairment of synaptic vesicle clustering and of synaptic transmission, and increased seizure propensity, in synapsin I-deficient mice. Proc Natl Acad Sci U S A. 1995;92:9235–9239. doi: 10.1073/pnas.92.20.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silva AJ, Rosahl TW, Chapman PF, Marowitz Z, Friedman E, Frankland PW, Cestari V, Cioffi D, Sudhof TC, Bourtchuladze R. Impaired learning in mice with abnormal short-lived plasticity. Curr Biol. 1996;6:1509–1518. doi: 10.1016/s0960-9822(96)00756-7. [DOI] [PubMed] [Google Scholar]
- 30.Cesca F, Baldelli P, Valtorta F, Benfenati F. The synapsins: key actors of synapse function and plasticity. Prog Neurobiol. 2010;91:313–348. doi: 10.1016/j.pneurobio.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 31.Lievens JC, Woodman B, Mahal A, Bates GP. Abnormal phosphorylation of synapsin I predicts a neuronal transmission impairment in the R6/2 Huntington’s disease transgenic mice. Mol Cell Neurosci. 2002;20:638–648. doi: 10.1006/mcne.2002.1152. [DOI] [PubMed] [Google Scholar]
- 32.Zhang W, Li JL, Hosaka M, Janz R, Shelton JM, Albright GM, Richardson JA, Sudhof TC, Victor RG. Cyclosporine A-induced hypertension involves synapsin in renal sensory nerve endings. Proc Natl Acad Sci U S A. 2000;97:9765–9770. doi: 10.1073/pnas.170160397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Idro R, Marsh K, John CC, Newton CR. Cerebral malaria: mechanisms of brain injury and strategies for improved neurocognitive outcome. Pediatr Res. 2010;68:267–274. doi: 10.1203/PDR.0b013e3181eee738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobbie M, Crawley J, Waruiru C, Marsh K, Surtees R. Cerebrospinal fluid studies in children with cerebral malaria: an excitotoxic mechanism? Am J Trop Med Hyg. 2000;62:284–290. doi: 10.4269/ajtmh.2000.62.284. [DOI] [PubMed] [Google Scholar]