

Abstract
Diabetic nephropathy (DN) is the leading cause of end-stage renal failure, and podocyte injury plays a major role in the development of DN. In this study, we investigated whether tacrolimus (FK506), an immunosuppressor, can attenuate podocyte injury in a type 2 diabetic mellitus (T2DM) rat model with DN. Transmission electron microcopy was used to morphologically evaluate renal injury. The urinary albumin (UAL), creatinine clearance rate (Ccr) and major biochemical parameters, including glucose, insulin, serum creatinine (Scr), urea nitrogen, total cholesterol (CHO) and triglyceride (TG), were examined 12 weeks after the administration of FK506. The expressions of the canonical transient receptor potential 6 (TRPC6), nuclear factor of activated T-cells (NFAT) and nephrin were detected by Western blotting and qPCR. In the rat model of DN, the expressions of TRPC6 and NFAT were significantly elevated compared with the normal rat group; however, the treatment with FK506 normalized the increased expression of TRPC6 and NFAT and attenuated podocyte ultrastructure injury. UAL, Ccr and the biochemical parameters were also improved by the use of FK506. In cell experiments, FK506 improved the decreased expression of nephrin and suppressed the elevated expression of both TRPC6 and NFAT caused by high glucose in accordance with TRPC6 blocker U73122. Our results demonstrated that FK506 could ameliorate podocyte injury in T2DM, which may be related to suppressed expressions of TRPC6 and NFAT.
Keywords: Diabetic nephropathy, podocyte, TRPC6, NFAT, tacrolimus
Introduction
Diabetic nephropathy (DN) is a major microvascular complication of diabetes and a leading cause of end stage renal disease (ESRD) [1]. DN is clinically characterized by proteinuria and pathologically by glomerular hypertrophy and glomerular basement membrane (GBM) thickening with foot process effacement [2]. The passage of albumin into Bowman’s space is overwhelmingly impeded by the size- and charge-selective glomerular filtration barrier, which consists of three interdependent layers: endothelial cells, the GBM, and podocytes. Porosities and its slit diaphragm structure are of vital importance to the maintenance of the filtration barrier. Studies of diabetic patients and animal models have revealed that the onset of albuminuria is most closely associated with podocytopathies involving foot process effacement, podocyte hypertrophy, detachment, apoptosis, and perhaps epithelial-to-mesenchymal transition (EMT); therefore, podocyte injury plays a crucial role in the initiation of DN [3-5]. Thus, research to identify the mechanism of podocyte injury is essential. Earlier studies have shown that the renin-angiotensin system (RAS), reactive oxygen species (ROS), peroxisome proliferator-activated receptors (PPAR), advanced glycation end-products, adiponectin, and microRNA are involved in the injury of podocytes [3]. Moreover, vascular endothelial growth factor (VEGF) [6], mechanical stress [7], the NOTCH pathway [8,9] and TGF-β [10] were also found to serve as important mediators of podocyte injuries. As reported by Brenner et al., ACE inhibition and angiotensin receptor blockers reduced the progression of clinical DKD [11]. Although early aggressive blood glucose control, improvement of blood lipid abnormalities and antihypertensive treatment, including blockade of the renin-angiotensin system, have improved prognosis, they have limited effects on reducing proteinuria, and additional treatments to slow progression are still lacking. Considering the severe consequences from ESRD, finding new targets and more effective therapies for DN is urgent.
Many drugs can exert a podocyte protective effect. In clinical settings, ARBs, ACEIs, renin inhibitors, aldosterone inhibitors, beta-blockers, calcium channel blockers (CCB) and diuretics are current therapeutic modalities for patients with DN. Among them, monotherapy with either an ACEI or ARB is currently recommended as first line therapy for diabetic nephropathy [1]. RAS activation, a major mediator of renal injury, can increase the local formation of angiotensin II (Ang II) in the kidneys and then cause many pathophysiological changes associated with DN [12,13]. According to the aforementioned studies, angiotensin-converting enzyme inhibitors (ACEIs) and/or AT1R blockers (ARBs) suppress the development and progression of DN in both type 1 and type 2 diabetic patients [14]. Zhou et al. recently reported that valsartan, an ARB, slowed the progression of diabetic nephropathy in db/db mice via a reduction in podocyte injury and renal oxidative stress and inflammation. Nijenhuis et al. found that Ang II contributed to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway [15]. Except for the above drugs, some immunosuppressor DN treatments have demonstrated efficacy in recent years. Wu et al. found that mycophenolate mofetil (MMF) treatment ameliorates early renal injury by inhibiting oxidative stress and the overexpression of intercellular adhesion molecule-1, monocyte chemotactic protein-1 and transforming growth factor-β1 in renal tissue in diabetic rats [16]. As reported by Gooch et al., calcineurin (CaN) play an important role in glomerular hypertrophy and extracellular matrix accumulation (ECM) in early diabetic nephropathy [17]. The CaN inhibitor cyclosporine A (CsA) had a preventive effect on glomerular hypertrophy and ECM accumulation in early diabetic rats [18]. However, the mechanism of this protective effect is still under debate. A recent study showed that CsA had a direct antiproteinuric effect on podocytes by inhibiting NFAT, as NFAT induces proteinuria and podocyte apoptosis. FK506 is a newer immunosuppressor and CaN inhibitor with few side effects and better tolerance, and it is widely used in post-transplantation. FK506 also showed rapid proteinuria remission in refractory IgAN patients, and the possible mechanism of FK506 in proteinuria remission might involve podocyte cytoskeleton stabilization by inhibiting CaN expression [19]. Additionally, a recent research study showed that FK506 could ameliorate renal injury in early experimental diabetic rats; the underlying mechanism may at least partly relate to the suppression of increased CaN in the renal tissue of diabetic rats [20]. Detailed information about the effects of FK506 on podocytes remains to be elucidated.
Transient receptor potential cation channel 6 (TRPC6) is an essential component of podocyte slit diaphragms. An increasing amount of research has demonstrated the relationship between TRPC6 and podocyte injuries. Mutations in the TRPC6 gene can cause both inherited glomerular disease such as FSGS [21]. Additionally, TRPC6 was also up-regulated in a subset of acquired human proteinuric kidney diseases, as well as in experimental models of acquired glomerular disease [22]. Furthermore, high glucose can induce apoptosis in podocytes by stimulating TRPC6 [23]. Previous evidence showed that angiotensin II (AngII) increased TPRC6 expression in non-renal cells, including mesenteric artery myocytes and ventricular myocytes [24,25]. The TRPC6 channel plays a crucial role in activating the nuclear factor of activated T cells (NFAT) induced by AngII, which participates in the progression of cardiac hypertrophy [26]. New evidence showed that podocyte TRPC6 expression is enhanced in hyperglycemic and proteinuric STZ-induced diabetic rats and that chronic enhanced TRPC6 expression could be an important factor in inducing persistent podocyte injury in the pathogenesis of proteinuria [27]. A recent study indicated that the activation of TRPC6 by AngII drives a positive feedback pathway in which AngII-induced, TRPC6-mediated Ca2+ influx stimulates NFAT expression, leading to enhanced TRPC6 expression and podocyte injuries [15]. The expression of TRPC6 and NFAT contributes to the initiation of podocyte injury.
In our study, we mainly focused on the effect of FK506 on podocytes in T2DM rats because of the pivotal role of podocyte injury in DN. We aimed to investigate whether FK506 could attenuate podocyte injury in T2DM rats, and if so, how FK506 realizes this effect and whether this protection is mediated by NFAT and TRPC6.
Materials and methods
Animals
Eight-week-old male Wistar SPF rats (weight 200 g, Institute of Drug Control of Qingdao, China) were housed in individual cages in a temperature-controlled room and had free access to food and water under a 12-h light/dark cycle. To establish the T2DM model, rats were fed with a high-calorie diet (10% animal fat, 20% cane sugar, 2.5% cholesterol, 1% cholate and 66.5% regular chow). After 8 weeks, the rats that fed on the high-calorie diet were intraperitoneally administered a low dose of streptozotocin (30 mg/kg, Sigma, St. Louis, MO, USA). And insulin sensitivity index (ISI) [ISI=22.5/[FBG (fasting blood glucose)×FINS (fasting serum insulin)], HOMA method] was used for assessing the T2DM model.
Then, the rats were divided into four groups: NC group (normal control, N=10), DM group (DM+NS, 0.9% normal saline, N=10), DTL group (DM+ low dose FK506, 0.5 mg/kg·d, N=10), and DTH group (DM+ high dose FK506, 1 mg/kg·d, N=10). Gastric gavage of FK506 (Astellas Ireland Co., Ltd.) and NS was conducted once a day for 12 weeks, and all rats were given a normal caloric diet. Then, the body weights (BWs) of the rats were examined, and urine samples were collected. Afterwards, the rats were sacrificed to collect blood samples and kidneys.
Cell culture
Conditionally immortalized mouse podocytes (MPC-5) were cultured as described previously [28]. Cells were cultured under growth-permissive conditions at 33°C with 5% CO2 in RPMI-1640 medium (Hyclone, USA) supplemented with 10% fetal bovine serum (Hyclone), 10 U/ml mouse recombinant interferon-gamma (IFN-γ; PeproTech USA) and 100 U/ml penicillin plus 100 mg/ml streptomycin (Sigma). To induce differentiation, podocytes were maintained in nonpermissive conditions at 37°C in the absence of IFN-γ for at least 2 weeks and then used for experiments. After serum starvation for 24 hours, differentiated podocytes were divided into five groups: NG group (normal glucose, 5.6 mmol/L), HG group (high glucose, 30 mmol/L), HM group (NG+ mannitol 25 mmol/L), FK group (HG+ FK506 5 µg/ml), and U73122 group (HG+TRP channel inhibitor, U73122 10 µmol/L). After 48 hours, the expressions of mRNA and TRPC6, NFAT2, and nephrin proteins were detected by real-time quantitative PCR and Western blot analysis.
Measurement of biochemical parameters
FBG was measured by tail vein blood with a glucometer (One Touch™Surestep™; Lifescan, Inc., Milpitas, CA, USA). The serum insulin level was determined using an enzyme-linked immunosorbent assay (ELISA) kit (Aquatic Diagnostic Ltd & Glasgow, Scotland). Serum creatinine (Scr), blood urea nitrogen (BUN), total cholesterol (TC), triglyceride (TG), aspartate transaminase (AST) and alanine transaminase (ALT) levels were determined using an automatic biochemistry analyzer. All of these biochemical parameters were examined at the 12th week after the administration of FK506.
Determination of urine albumin and creatinine concentrations
To determine the urine albumin (UAL) and creatinine (Ucr) concentrations, 24 h urine samples were collected before and 12 weeks after the administration of FK506. UAL excretion was measured using a turbidimetric immunoassay kit (Shibayagi Co., Ltd., Shibukawa, Japan). The Ucr level was evaluated using an automatic biochemistry analyzer (Hitachi Ltd., Tokyo, Japan). Creatinine clearance (Ccr) was then calculated (Ccr=UCr/Scr × V, V: ml/min, urine per minute).
Noninvasive blood pressure measurement
Blood pressure was measured using the tail-cuff method (LE5002, DSL, USA) at a resting, conscious condition in a climate-controlled room (23°C). Systolic blood pressure (SBP) was consecutively measured five times.
Transmission electron microscopy
The morphological characteristics of renal cortex sections (50 nm), including the thickness of GBM, and the condition of podocytes were observed by transmission electron microscopy. The renal specimens were examined and photographed using a JEM-1200 transmission electron microscope (Jeol Ltd., Tokyo, Japan). The detailed procedure of morphological evaluation was in accordance with previous studies [29,30].
Immunohistochemistry staining of TRPC6
Immunostaining was performed on 3-µm-thick sections after deparaffinization. Microwave antigen retrieval was performed in citrate buffer at pH 6.0 for 10 min prior to peroxide quenching with 3 % H2O2 in PBS for 10 min and pre-blocked with normal goat serum for 20 min. For primary antibody labeling, slides were incubated with antibodies that recognize goat anti-TRPC6 at a 1:500 dilution overnight at 4°C and incubated with biotinylated secondary antibodies (1:1,000) for 20 min. Streptavidin-peroxidase was applied, and the sections were rinsed (3 × 5 min) in PBS. Finally, the immunostaining was visualized using DAB and counterstained with hematoxylin. Semiquantitative immunohistochemical analysis was scored as follows: no staining (-), weak staining (+), moderate staining (++), and strong staining (+++). Twenty glomeruli were examined randomly per treatment group.
Western blotting
Cells and the homogenized renal cortex tissue were lysed in cold cell lysis buffer with protease and phosphatase inhibitors. The proteins were separated on 10% SDS-PAGE gels and subsequently transferred to nitrocellulose membranes. The primary antibodies for TRPC6 (rabbit anti-rat, Santa Cruz Biotechnology, Inc.), NFAT (rabbit anti-rat, Santa Cruz Biotechnology, Inc.) and nephrin (rabbit anti-rat, Santa Cruz Biotechnology, Inc.) were used at a ratio of 1:500-1:4000. Horseradish peroxidase-conjugated anti-immunoglobulin IgG (Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd., China) was used as a secondary antibody. The blots were detected using an enhanced chemiluminescence (ECL) system (Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd., China).
Quantitative polymerase chain reaction (qPCR) analysis
Frozen renal cortex tissues were homogenized, and total RNA was extracted with Trizol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA). The extracted RNA was measured by agarose gel electrophoresis for quality and by spectrometry for quantity. A reverse transcription reaction kit (Cat. no. DRR035A; TaKaRa Biotechnology Co. Ltd., Dalian, China) was used. For fluorescence qPCR, a 1,000 ng mRNA sample was added to the reverse transcription system. This sample was diluted with EASY dilution (TaKaRa Biotechnology Co. Ltd., Dalian, China) in a 10-fold series to generate a standard curve. The qPCR process was performed in an ABI Prism®7000 HT sequence detection system (cat. no. 11744-100; Applied Biosystems, Invitrogen Life Technologies). Primers for nephrin, NFAT and TRPC6 were designed and synthesized by Shanghai Sangon Biological Engineering Technology Co., Ltd. The primers used in the PCR process are presented in Tables 1, 2. The results are presented as the ratio of the target gene copy to the housekeeping gene GAPDH copy.
Table 1.
Primers used in the study (mouse)
| GenBank accession number | Primer | Sequence | Length (bp) |
|---|---|---|---|
| NM_001282086.1 | TRPC6 (F) | 5’-TCTCTGGTTTACGGCAGCAGA-3’ | 228 |
| TRPC6 (R) | 5’-GGAGCTTGGTGCCTTCAAATC-3’ | ||
| NM_001164109.1 | NFAT2 (F) | 5’-GGTGCCTTTTGCGAGCAGTA-3’ | 185 |
| NFAT2 (R) | 5’-TGAGCCCTGTGGTGAGACTTG-3’ | ||
| NM_019459.2 | Nephrin (F) | 5’-ACCTCCTGTCATTGATTGGCC-3’ | 147 |
| Nephrin (R) | 5’-CCCCAAGCTATGGACACTGGT-3’ | ||
| NM_001289726.1 | GAPDH (F) | 5’-CTCATGACCACAGTCCATGC-3’ | 201 |
| GAPDH (R) | 5’-CACATTGGGGGTAGGAACAC-3’ |
Table 2.
Primers used in the study (rat)
| GenBank accession number | Primer | Sequence | Length (bp) |
|---|---|---|---|
| NM_053559.1 | TRPC6 (F) | 5’-TACGGATTGTGGAGGCTATTCT-3’ | 98 |
| TRPC6 (R) | 5’-AAAGTCATCTTGCTGGAGTTCA-3’ | ||
| NM_001244933.1 | NFAT2 (F) | 5’-GAGGGAAGAAGATGGTGTTGTC-3’ | 125 |
| NFAT2 (R) | 5’-GCACAGGTCTCGGTCAGTTT-3’ | ||
| NM_022628.1 | Nephrin (F) | 5’-AAGTACGAATGGACCCCTATGAC-3’ | 176 |
| Nephrin (R) | 5’-CAGGGCTGTAGGAAACGGGTG-3’ | ||
| NM_017008.4 | GAPDH (F) | 5’-GGCACAGTCAAGGCTGAGAATG-3’ | 143 |
| GAPDH (R) | 5’-ATGGTGGTGAAGACGCCAGTA-3’ |
Statistics analysis
All data are presented as the means ± SEM. Statistically significant differences were assessed by analysis of variance (one-way ANOVA). All statistical analyses were performed using a commercially available statistical package (SPSS 17.0). A P value of <0.05 indicates statistical significance.
Results
Biochemical parameters
As shown in Table 3, FBG and ISI in the DM, DTL and DTH groups were significantly higher than that in the NC group. However, the results did not show significant alteration between the DM and FK506-treated groups. Moreover, the lipid metabolic parameters, including TG and TC levels, were significantly elevated in the DM group compared with the NC group. However, no significant differences in TG or TC levels were found between the untreated diabetic group and FK506-treated group. AST and ALT, which indicate hepatic function, did not change significantly in the FK506-treated group.
Table 3.
General data of the rats used in the study after 12 weeks of treatment (mean ± SEM)a
| Characteristics | NC (n=10) | DM (n=10) | DTL (n=10) | DTH (n=10) |
|---|---|---|---|---|
| ISI | 0.221±0.024 | 0.052±0.004* | 0.060±0.005* | 0.059±0.004* |
| FINS ng/ml | 18.57±1.01 | 22.09±1.75 | 20.16±1.57 | 20.49±1.61 |
| FBG mmol/L | 5.56±0.64 | 19.44±0.47* | 18.58±0.58* | 18.55±0.52* |
| TG mmol/L | 1.49±0.15 | 4.83±0.69* | 4.42±0.48* | 4.30±0.70* |
| TC mmol/L | 0.76±0.24 | 3.99±0.25* | 3.78±0.26* | 3.75±0.34* |
| AST U/L | 47.8±4.57 | 48.9±4.12 | 48.2±3.45 | 47.9±5.04 |
| ALT U/L | 42.9±3.54 | 44.1±3.60 | 43.2±3.29 | 43.1±3.14 |
ISI: insulin sensitivity index; FINS: Fasting serum insulin; FBG: Fasting blood glucose; TC: Total cholesterol; TG: Triglyceride; AST: aspartate aminotransferase; ALT: alanine aminotrans; NC: Control group; DM: Type 2 diabetes model group; DTL: valsartan group; DTH:
P<0.05 (compared with the NC group).
The urinary albumin, Ccr and SBP, KW/BW
The DM group exhibited significant increases in UAL, Ccr and SBP, and KW/BW compared with the NC group (P<0.05), and UAL, Ccr and KW/BW were suppressed by the 12 week FK506 treatment (P<0.05). However, no significant difference in SBP was found between the untreated diabetic group and FK506-treated diabetic group (Figure 1).
Figure 1.
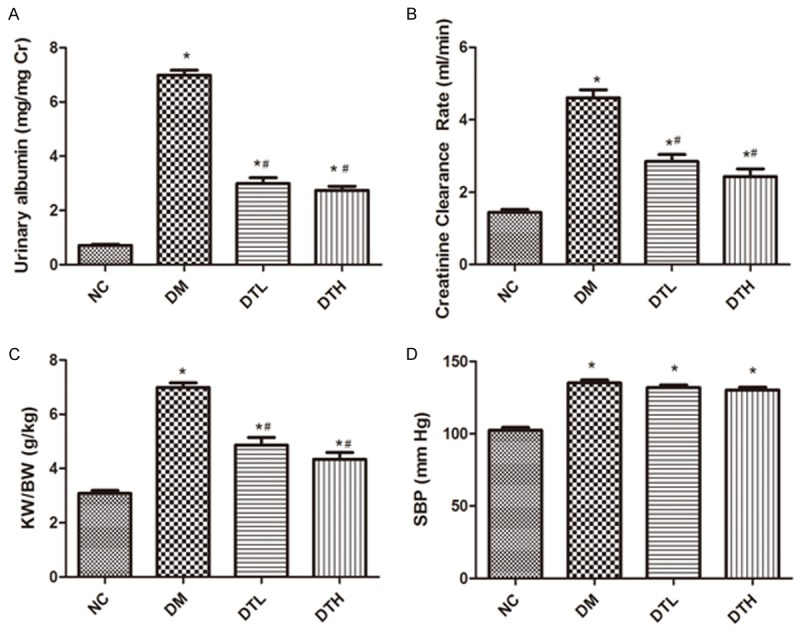
FK506 suppressed the elevated level of UAL, Ccr and KW/BW. The DM group showed a significant elevation in UAL (A), Ccr (B) and KW/BW (C) compared to the NC group, which can be improved by FK506. However, FK506 did not affect the increased SBP of DM rats (D).
Variation of podocyte ultrastructure
The transmission electron microscopy images revealed that the foot processes of neighboring podocytes were interdigitated, and the thicknesses of GBM were uniform in the NC group (Figure 2A). GBM thickness (731.30±76.16 versus 238.00±17.73; P<0.01), foot process fusion rate (75.00±3.30 versus 4.40±2.71; P<0.01) and foot process width (515.40±31.08 versus 261.10±31.96; P<0.01) increased in the DM group compared with the NC group (Figure 2B, 2E-G). However, treatment with low-dose FK506 suppressed the thickening of the GBM (321.40±32.30 versus 731.30±76.16; P<0.01) and reduced the foot process fusion rate (19.80±1.99 versus 75.00±3.30; P<0.01) and foot process width (318.17±29.49 versus 515.4±31.8; P<0.01) compared with the DM group. Compared with the DM group, after the high-dose FK506 treatment, the above indicators were also ameliorated, but there was no significant difference between the DTL group and DTH group (Figure 2C-G).
Figure 2.
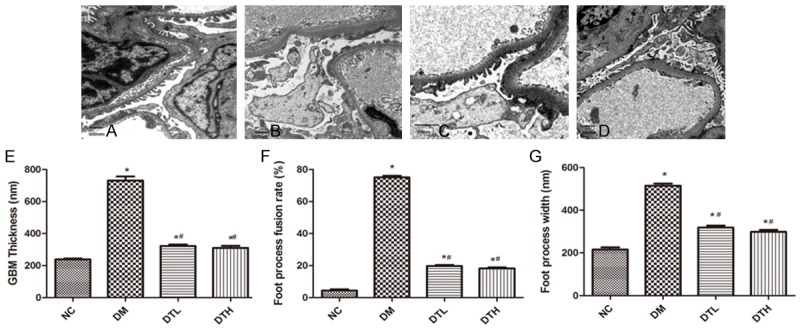
Effects of FK506 on ultrastructural variations of podocyte in DN rats. Four groups were included in this morphological observation (A. NC, B. DM, C. DTL, D. DTH). GBM thickness (E), foot process fusion rate (F) and foot process width (G) all increased in DM rats, and the FK506 treatment improved the impaired ultrastructure of podocytes in DM rats.
The distribution and expression of TRPC6 in diabetic rats
The immunohistochemistry in kidney tissues showed that the staining intensity expression level of TRPC6 was increased (as +++) in DM rats compared with NC rats (Figure 3A, 3B). With the FK506 treatment, TRPC6 expression decreased (++ to +) (Figure 3C, 3D). There were no significant changes in the TRPC6 expression level between the DTL and DTH groups.
Figure 3.
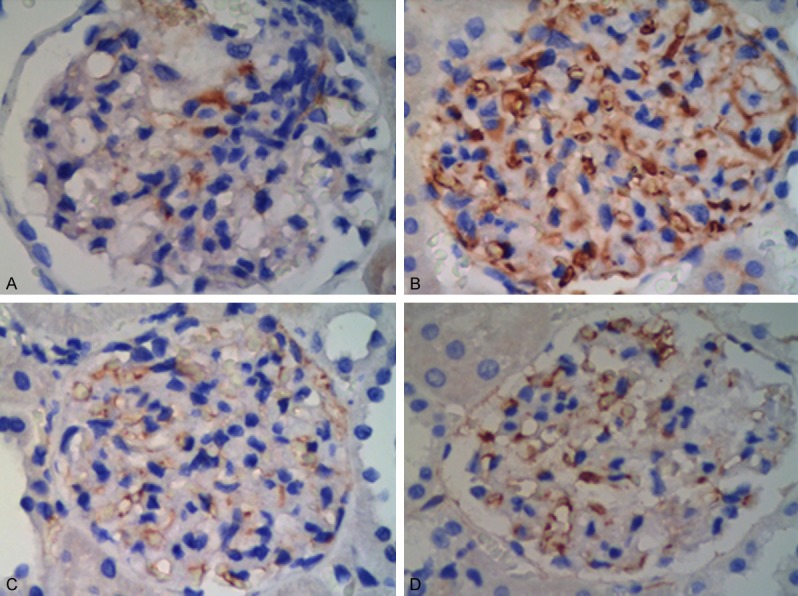
The distribution and expression of TRPC6 in DM rats (×400). In the DM group (B), TRPC6 showed an increased staining intensity (+++) in podocytes compared to the NC group (A). In the DTL (C) and DTH groups (D), FK506 had a lower staining intensity (as ++~+) than in the DM group.
Expression of nephrin, NFAT2 and TRPC6 at both protein level and mRNA level in animal experiment
In animal experiments, nephrin mRNA and proteins had lower expressions in the DM group compared with that in the NC group (Figure 4A, 4B), but the protein and mRNA expression of NFAT were significantly higher in the DM group than in the NC group (Figure 4C, 4D). After the 12-week FK506 treatment, the protein and mRNA expression of NFAT was significantly suppressed in both the DTL and DTH groups compared with the DM group (Figure 4C, 4D). The suppressed expression of nephrin in the DM group was improved by FK506 (Figure 4A, 4B). Similarly, the FK506 treatment also normalized the up-regulated expression of TRPC6 in the DM group (Figure 4E, 4F).
Figure 4.
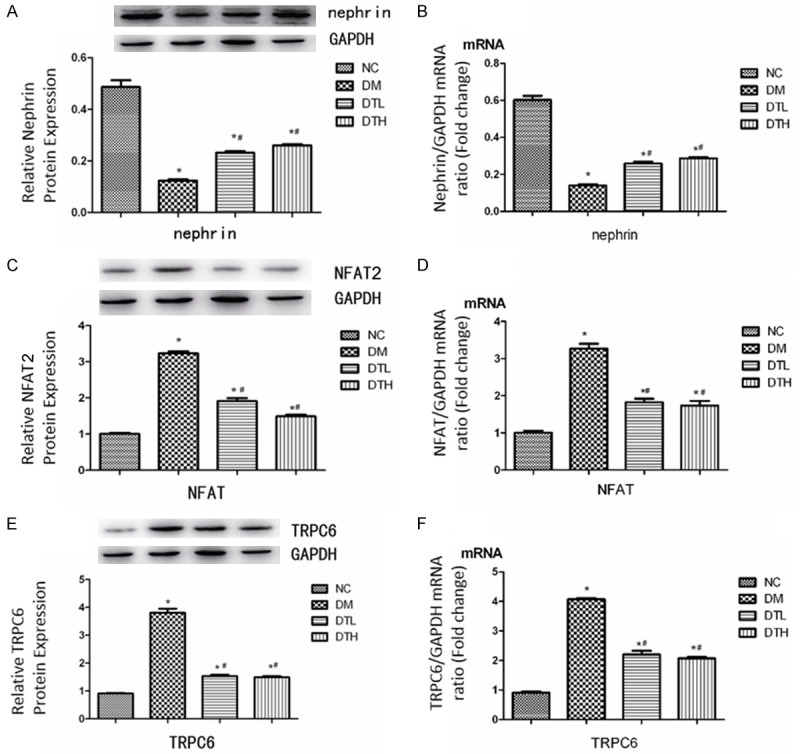
Effects of FK506 on protein and mRNA expressions of Nephrin (A, B), NFAT2 (C, D) and TRPC6 (E, F) in podocytes in vivo (N=10). Data are presented as the means ± S.E.M. *P<0.05 (compared with the NC group), #P<0.05 (compared with the DM group).
Expression of nephrin, NFAT2 and TRPC6 protein and mRNA levels in vitro
In the HG group, the expression of nephrin protein and mRNA was significantly diminished in HG group compared with the NG and HM groups, but this deficiency of nephrin in DM was remarkably improved by the FK506 treatment (Figure 5A, 5B). The expression of NFAT and TRPC6 were both elevated by high glucose intake and then suppressed by the use of FK506 (Figure 5C-F). In the U73122 group, the TRPC6 blocker U73122 clearly stimulates the expression of nephrin (Figure 5A, 5B) and decreases the expression of NFAT (Figure 5C, 5D) and TRPC6 (Figure 5E, 5F) compared to the HG group, which indicated that the decreased expression of nephrin induced by high glucose intake was mediated by TRPC6.
Figure 5.
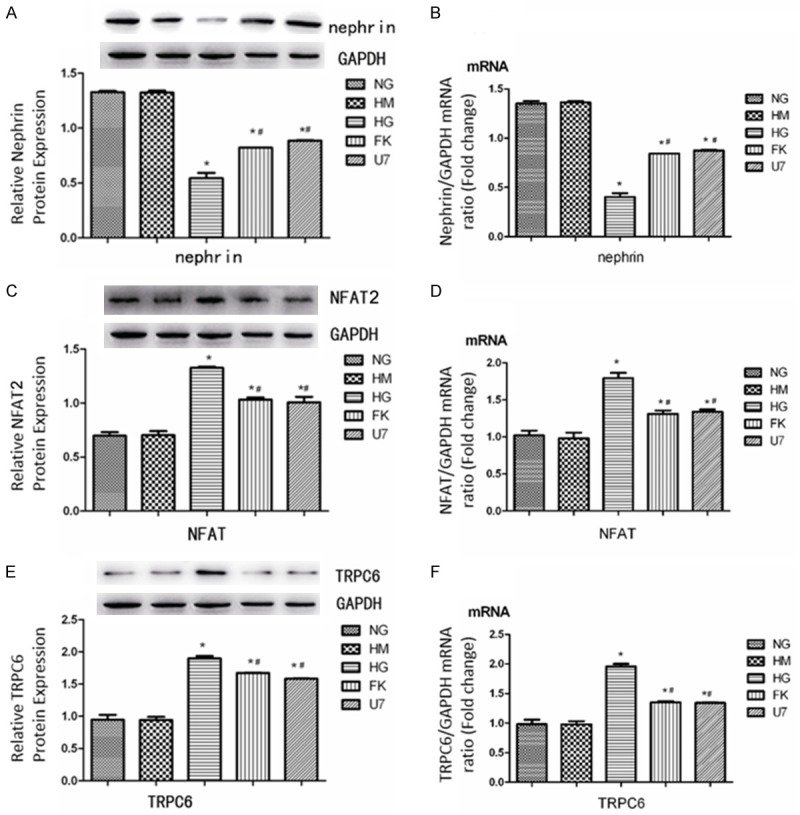
Effects of high glucose, U73122 and FK506 on protein and mRNA expressions of Nephrin (A, B), NFAT2 (C, D), and TRPC6 (E, F) in podocytes in vitro (N=10). Data were presented as means ± S.E.M. *P<0.05 (compared with NG group), #P<0.05 (compared with HG group).
Discussion
The increasing prevalence and incidence of DN has imposed global socio-economic stress on healthcare systems worldwide. According to the projections, the number of adult diabetic patients will be greater than 430 million in 2030 [31]. Therefore, it has become increasingly urgent to understand the detailed mechanisms underlying the pathogenesis of diabetic nephropathy and to find novel targets for the prevention and therapy of this disease. In this study, we found that the DM rats showed disrupted biochemical conditions, impaired podocyte ultrastructure and significant changes in some crucial markers, such as nephrin, NFAT and TRPC6, compared to normal control rats. These findings revealed that the DM model that we used successfully represents the condition of diabetic nephropathy. Our results also showed that FK506, a new immunosuppressor, decreased proteinuria, attenuated the impaired podocyte ultrastructure, improved renal function, elavated the expression of nephrin and decreased the expression of TRPC6 and NFAT in DM rats. These results indicate that FK506 exerted a podocyte protective function by mediating TRPC6 and NFAT.
It is currently widely recognized that microalbuminuria is an early clinically identifiable marker of DN, and microalbuminuria may develop into proteinuria and hyperfiltration, followed by a reduction in the glomerular filtration rate (GFR). It is thought that proteinuria is not simply an indicator of glomerular damage, nor just a predictor of DN progression, but affects disease processes [32]. We detected the 24 h UAL and Ccr before and 12 weeks after the administration of FK506 and found that treatment with FK506 decreased 24 h UAL and Ccr in T2DM rats. This result indicates a possible positive effect of FK506 on the early stage of DN. Additionally, FK506 treatment did not affect BUN, AST and ALT, which means that there could be no nephrotoxicity and hepatotoxicity in T2DM rats at both doses. We also found that FK506 did not aggravate the disturbance of lipid metabolism in DN. Moreover, although FK506 is toxic to beta cells in the pancreas and it can lead to increased blood glucose level, we did not find increased blood glucose after the 12-week treatment with both doses, particularly the low dose, in our experiment. This finding may indicate that the doses we used are safe. Above all, FK506 treatment in DN patients is an emerging clinical therapeutic strategy.
Research on the pathogenesis of albuminuria has demonstrated major achievements. Podocytopathy, involving foot process effacement, podocyte hypertrophy, detachment and apoptosis, plays a crucial role in the initiation and progression of albuminuria [33]. Nephrin was the first protein demonstrated to compose the slit diaphragm, and previous studies revealed the diminished expression and altered localization of nephrin in a nephropathy model of both type 1 and type 2 diabetes [34]. In our study, Western blotting and qPCR showed reduced expression of nephrin in T2DM, and the transmission electron microscopy clearly indicated the increased GBM thickness, foot process fusion rate and foot process width in T2DM rats, which was consistent with a previous report [35]. However, FK506 overexpressed nephrin and improved the ultrastructure of podocytes in T2DM rats. A cell experiment confirmed the effect of FK506 on nephrin with an identical result to a test in vitro. Based on these findings, we presume that FK506 may elevate the expression of nephrin and improve the damaged podocyte ultrastructure, as well as exert a protective effect on podocytes in DN. Therefore, FK506 is a potentially novel safe agent in patients with DN.
As reported, FK506 has a renoprotective function in various models. FK506 could improve renal function by decreasing CaN activity in both the cytoplasm and the nucleus of renal tubule cells in a rat model of ischemia-reperfusion injury [36] Liu et al. found that FK506 had a therapeutic effect on the progression of proteinuria and renal damage by downregulating TRPC6 and CaN in the rat model of adriamycin-induced nephropathy [35]. Moreover, in an early experimental diabetic rats model induced by streptozotocin, FK506 also ameliorated renal injury, and the underlying mechanism may be at least partly correlated with suppressing increased CaN [20]. In that study, the expression of CaN protein increased 2.4-fold in the kidney of diabetic rats, and FK506 treatment with 0.5 and 1.0 mg/kg reduced increased expression of CaN protein by 38.0% and 73.2%, respectively. Their results showed that the amelioration of renal injury induced by FK506 may be at least partly related to suppression of increased CaN in renal tissue in diabetic rats. However, the detailed underling mechanism remains to be elucidated. This study provides important information about podocyte protection induced by FK506.
Many studies have provided strong evidence that CaN, NFAT and TRPC6 are closely related to the initiation of proteinuria. In podocytes, TRPC6-mediated Ca2+ influx activates Ca2+-dependent CaN, which dephosphorylates NFAT. The activation of NFAT enhances TRPC6 transcription and is eventually detrimental to the glomerular filter, causing proteinuria [15]. It has been demonstrated that TRPC6-mediated Ca2+ influx stimulates NFAT-dependent TRPC6 expression, which is regarded as positive feedback [15,37]. FK506 is a well-known CaN inhibitor that inhibits CaN activity by binding to the FK binding protein (FKBP) domain [38]. According to a literature review, the renoprotection by FK506 may be at least partly related to the suppression of TRPC6 and NFAT. In our experiments, we used a different rat model of DN, which was closer to natural T2DM. There was a significant elevation of NFAT and TRPC6 expression in DN rats, and the 12 week FK506 treatment remarkably inhibited this elevation. Both animal experiments and cell experiments presented the same results. The use of a TRPC6 blocker and FK506 both inhibited the expression of NFAT and elevated the expression of nephrin in high glucose treated podocytes, which indicated that TRPC6 played a pivotal role in the initiation and progression of podocyte injury and that FK506 exerts its renoprotective effect by suppressing TRPC6.
Overall, we may reasonably conclude that FK506 attenuates podocyte injury in this DN rat model of T2DM. FK506 protects podocytes via a CaN/NFAT/TRPC6 pathway. FK506 may first inhibit CaN and then inhibit its substrate NFAT. Then, the down-regulated expression of NFAT leads to the suppression of transcription of NFAT-responsive genes such as TRPC6. Because the overexpression of TRPC6 is involved in podocyte injuries [27], FK506-induced suppressed expression of TRPC6 may be a crucial factor that results in the amelioration of podocyte injury and clinical symptoms.
Our experiments explored the effect of FK506 on diabetic nephropathy and conducted a preliminary investigation of the probable mechanism of this effect. The application of low-dose FK506 in DN patients is a promising treatment. However, more studies need to be conducted in the future. More samples and longer study periods are needed, and the efficacy and complications of FK506 in diabetic nephropathy and their detailed mechanisms need to be elucidated.
Acknowledgements
The authors thank Prof. Zhimin Wei from the Pathology Department of Qingdao University Affiliated Hospital, Prof. Junhui from the Pathology Department of Qilu Hospital of Shan Dong University and Prof. Jing Dong from the Physiology Department of Qingdao University for their helpful suggestions.
Disclosure of conflict of interest
None.
References
- 1.Van Buren PN, Toto R. Current update in the management of diabetic nephropathy. Curr Diabetes Rev. 2013;9:62–77. [PubMed] [Google Scholar]
- 2.Li JJ, Kwak SJ, Jung DS, Kim JJ, Yoo TH, Ryu DR, Han SH, Choi HY, Lee JE, Moon SJ. Podocyte biology in diabetic nephropathy. Kidney Int Suppl. 2007:S36–42. doi: 10.1038/sj.ki.5002384. [DOI] [PubMed] [Google Scholar]
- 3.Stitt-Cavanagh E, MacLeod L, Kennedy C. The podocyte in diabetic kidney disease. ScientificWorldJournal. 2009;9:1127–1139. doi: 10.1100/tsw.2009.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jefferson JA, Shankland SJ, Pichler RH. Proteinuria in diabetic kidney disease: a mechanistic viewpoint. Kidney Int. 2008;74:22–36. doi: 10.1038/ki.2008.128. [DOI] [PubMed] [Google Scholar]
- 5.Reddy GR, Kotlyarevska K, Ransom RF, Menon RK. The podocyte and diabetes mellitus: is the podocyte the key to the origins of diabetic nephropathy? Curr Opin Nephrol Hypertens. 2008;17:32–36. doi: 10.1097/MNH.0b013e3282f2904d. [DOI] [PubMed] [Google Scholar]
- 6.Chen S, Ziyadeh FN. Vascular endothelial growth factor and diabetic nephropathy. Curr Diab Rep. 2008;8:470–476. doi: 10.1007/s11892-008-0081-3. [DOI] [PubMed] [Google Scholar]
- 7.Lewko B, Stepinski J. Hyperglycemia and mechanical stress: targeting the renal podocyte. J Cell Physiol. 2009;221:288–295. doi: 10.1002/jcp.21856. [DOI] [PubMed] [Google Scholar]
- 8.Mertens PR, Raffetseder U, Rauen T. Notch receptors: a new target in glomerular diseases. Nephrol Dial Transplant. 2008;23:2743–2745. doi: 10.1093/ndt/gfn279. [DOI] [PubMed] [Google Scholar]
- 9.Lin CL, Wang FS, Hsu YC, Chen CN, Tseng MJ, Saleem MA, Chang PJ, Wang JY. Modulation of notch-1 signaling alleviates vascular endothelial growth factor-mediated diabetic nephropathy. Diabetes. 2010;59:1915–1925. doi: 10.2337/db09-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ziyadeh FN. Different roles for TGF-beta and VEGF in the pathogenesis of the cardinal features of diabetic nephropathy. Diabetes Res Clin Pract. 2008;82(Suppl 1):S38–41. doi: 10.1016/j.diabres.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 11.Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 12.Halimi JM, Asmar R, Ribstein J. Optimal nephroprotection: use, misuse and misconceptions about blockade of the renin-angiotensin system. Lessons from the ONTARGET and other recent trials. Diabetes Metab. 2009;35:425–430. doi: 10.1016/j.diabet.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 13.Kamiyama M, Zsombok A, Kobori H. Urinary angiotensinogen as a novel early biomarker of intrarenal renin-angiotensin system activation in experimental type 1 diabetes. J Pharmacol Sci. 2012;119:314–323. doi: 10.1254/jphs.12076fp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mehdi UF, Adams-Huet B, Raskin P, Vega GL, Toto RD. Addition of angiotensin receptor blockade or mineralocorticoid antagonism to maximal angiotensin-converting enzyme inhibition in diabetic nephropathy. J Am Soc Nephrol. 2009;20:2641–2650. doi: 10.1681/ASN.2009070737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nijenhuis T, Sloan AJ, Hoenderop JG, Flesche J, van Goor H, Kistler AD, Bakker M, Bindels RJ, de Boer RA, Moller CC. Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am J Pathol. 2011;179:1719–1732. doi: 10.1016/j.ajpath.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu YG, Lin H, Qi XM, Wu GZ, Qian H, Zhao M, Shen JJ, Lin ST. Prevention of early renal injury by mycophenolate mofetil and its mechanism in experimental diabetes. Int Immunopharmacol. 2006;6:445–453. doi: 10.1016/j.intimp.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 17.Gooch JL, Gorin Y, Zhang BX, Abboud HE. Involvement of calcineurin in transforming growth factor-beta-mediated regulation of extracellular matrix accumulation. J Biol Chem. 2004;279:15561–15570. doi: 10.1074/jbc.M308759200. [DOI] [PubMed] [Google Scholar]
- 18.Gooch JL, Barnes JL, Garcia S, Abboud HE. Calcineurin is activated in diabetes and is required for glomerular hypertrophy and ECM accumulation. Am J Physiol Renal Physiol. 2003;284:F144–154. doi: 10.1152/ajprenal.00158.2002. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Q, Shi SF, Zhu L, Lv JC, Liu LJ, Chen YQ, Zhang H, Wang HY. Tacrolimus improves the proteinuria remission in patients with refractory IgA nephropathy. Am J Nephrol. 2012;35:312–320. doi: 10.1159/000337175. [DOI] [PubMed] [Google Scholar]
- 20.Qi XM, Wu YG, Liang C, Zhang P, Dong J, Ren KJ, Zhang W, Fang F, Shen JJ. FK506 ameliorates renal injury in early experimental diabetic rats induced by streptozotocin. Int Immunopharmacol. 2011;11:1613–1619. doi: 10.1016/j.intimp.2011.05.023. [DOI] [PubMed] [Google Scholar]
- 21.Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 22.Moller CC, Wei C, Altintas MM, Li J, Greka A, Ohse T, Pippin JW, Rastaldi MP, Wawersik S, Schiavi S. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J Am Soc Nephrol. 2007;18:29–36. doi: 10.1681/ASN.2006091010. [DOI] [PubMed] [Google Scholar]
- 23.Liu BC, Song X, Lu XY, Li DT, Eaton DC, Shen BZ, Li XQ, Ma HP. High glucose induces podocyte apoptosis by stimulating TRPC6 via elevation of reactive oxygen species. Biochim Biophys Acta. 2013;1833:1434–1442. doi: 10.1016/j.bbamcr.2013.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saleh S, Albert A, Peppiatt C, Large W. Angiotensin II activates two cation conductances with distinct TRPC1 and TRPC6 channel properties in rabbit mesenteric artery myocytes. J Physiol. 2006;577:479–495. doi: 10.1113/jphysiol.2006.119305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuwahara K, Wang Y, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, Olson EN. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest. 2006;116:3114–3126. doi: 10.1172/JCI27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Onohara N, Nishida M, Inoue R, Kobayashi H, Sumimoto H, Sato Y, Mori Y, Nagao T, Kurose H. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J. 2006;25:5305–5316. doi: 10.1038/sj.emboj.7601417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sonneveld R, van der Vlag J, Baltissen MP, Verkaart SA, Wetzels JF, Berden JH, Hoenderop JG, Nijenhuis T. Glucose specifically regulates TRPC6 expression in the podocyte in an AngII-dependent manner. Am J Pathol. 2014;184:1715–1726. doi: 10.1016/j.ajpath.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 28.Graham S, Ding M, Sours-Brothers S, Yorio T, Ma JX, Ma R. Downregulation of TRPC6 protein expression by high glucose, a possible mechanism for the impaired Ca2+ signaling in glomerular mesangial cells in diabetes. Am J Physiol Renal Physiol. 2007;293:F1381–1390. doi: 10.1152/ajprenal.00185.2007. [DOI] [PubMed] [Google Scholar]
- 29.Dissard R, Klein J, Caubet C, Breuil B, Siwy J, Hoffman J, Sicard L, Ducasse L, Rascalou S, Payre B. Long term metabolic syndrome induced by a high fat high fructose diet leads to minimal renal injury in C57BL/6 mice. PLoS One. 2013;8:e76703. doi: 10.1371/journal.pone.0076703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu HF, Guo LQ, Huang YY, Chen K, Tao JL, Li SM, Chen XW. Thiazolidinedione attenuate proteinuria and glomerulosclerosis in Adriamycin-induced nephropathy rats via slit diaphragm protection. Nephrology. 2010;15:75–83. doi: 10.1111/j.1440-1797.2009.01146.x. [DOI] [PubMed] [Google Scholar]
- 31.Garcia-Garcia PM, Getino-Melian MA, Dominguez-Pimentel V, Navarro-Gonzalez JF. Inflammation in diabetic kidney disease. World J Diabetes. 2014;5:431–443. doi: 10.4239/wjd.v5.i4.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Remuzzi G, Benigni A, Remuzzi A. Mechanisms of progression and regression of renal lesions of chronic nephropathies and diabetes. J Clin Invest. 2006;116:288–296. doi: 10.1172/JCI27699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mundel P, Kriz W. Structure and function of podocytes: an update. Anat Embryol. 1995;192:385–397. doi: 10.1007/BF00240371. [DOI] [PubMed] [Google Scholar]
- 34.Doublier S, Salvidio G, Lupia E, Ruotsalainen V, Verzola D, Deferrari G, Camussi G. Nephrin expression is reduced in human diabetic nephropathy: evidence for a distinct role for glycated albumin and angiotensin II. Diabetes. 2003;52:1023–1030. doi: 10.2337/diabetes.52.4.1023. [DOI] [PubMed] [Google Scholar]
- 35.Liu Y, Ji Z. FK506 alleviates proteinuria in rats with adriamycin-induced nephropathy by down-regulating TRPC6 and CaN expression. J Nephrol. 2012;25:918–925. doi: 10.5301/jn.5000192. [DOI] [PubMed] [Google Scholar]
- 36.Lee SH, Choi J, Kim H, Lee DH, Roh GS, Kim HJ, Kang SS, Choi WS, Cho GJ. FK506 reduces calpain-regulated calcineurin activity in both the cytoplasm and the nucleus. Anat Cell Biol. 2014;47:91–100. doi: 10.5115/acb.2014.47.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, Chang JM, Choi HY, Campbell KN, Kim K. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008;14:931–938. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grigoriu S, Bond R, Cossio P, Chen JA, Ly N, Hummer G, Page R, Cyert MS, Peti W. The molecular mechanism of substrate engagement and immunosuppressant inhibition of calcineurin. PLoS Biol. 2013;11:e1001492. doi: 10.1371/journal.pbio.1001492. [DOI] [PMC free article] [PubMed] [Google Scholar]