

Abstract
Rheumatic heart disease (RHD) makes a heavy burden in human lives and economy. The proteomic analysis of acute rheumatic heart disease (ARHD) can provide precious data to study RHD at the early stages, but no one has looked into. So based on our early research we applied the method of continuous GAS stimulation on Lewis rats to duplicate the animal model of ARHD. And the mitral valves of rats in control group (n=10) and ARHD group (n=10) were selected for proteomic analysis of ARHD with the iTRAQ labeling based 2D LC-ESI-MS/MS quantitative technology. We identified 3931 proteins in valve tissue out of which we obtained 395 differentially expressed proteins containing 176 up-regulated proteins and 119 down-regulated proteins. Changes in levels of GAPDH (6.793 times higher than the control group) and CD9 (2.63 times higher than the control group) were confirmed by Western blot or immunohistochemistry. The differentially expressed proteins such as GAPDH, CD9, myosin, collagen and RAC1 may be potential biomarkers for ARHD. Moreover, the mitral valve protein profile shed light on further understanding and investigating ARHD.
Keywords: Acute rheumatic heart disease, ITRAQ quantitative proteomics, mitral valve tissue, animal model, mitral valve protein profile
Introduction
The number of people with rheumatic heart disease (RHD) is 15.6-19.6 million worldwide [1]. The incidence, morbidity and mortality of RHD remain high among human population and make a heavy burden in their lives and economy [2-4]. The mitral valve dysfunction is the most common valve lesion in patients with RHD, particularly in the early stages [5,6].
To date, there are mostly two hypotheses about the development of RHD. Tandon et al. proposed the collagen-mediated autoimmunity in cardiac valve [7]; Another study indicated molecular mimicry between streptococcus and the heart, which may be closely related with the former hypothesis [8,9]. But the exact molecular pathway is still not completely elucidated.
Changes of protein profiles in RHD have been extensively detected in humans such as, PDIA3, HSPA5, vimentin and HSP60 in valve tissue of patients with chronic RHD [10,11]. Ago et al. [12] also have detected the altered plasma protein profiles. However, they were based on the chronic RHD in patients who were not diagnosed and treated at the initial stage of a disease. Further investigations with suitable animal model of RHD are required to study acute rheumatic heart disease (ARHD) that is defined as the early inflammatory injury in myocardium or valve without the formation of fibrosis and calcification. Although the animal model mainly on Lewis rats have been widely researched [13-15], the rate of success in animal model of RHD was not very high. In our previous study, we compared three immune methods of RHD and successfully found an optimal animal model that can overcome the defect and we can effortlessly obtain the mitral valve of ARHD.
In this study we used the Group A-hemolytic streptococci (GAS) with continuous stimulation on Lewis rats to duplicate the RHD model. We initially studied the proteomic analysis of valve tissue in Lewis rats of ARHD with iTRAQ coupled 2D LC-ESI-MS/MS quantitative technology. Ultimately, we aimed to identify putative protein biomarker candidates for ARHD, which can be useful for its molecular diagnosis and monitoring, and to provide the altered mitral valve protein profile for further understanding and investigating the ARHD.
Materials and methods
Animals and materials
Thirty-six 8-week-old female Lewis rats (150-180 g) provided by Vital River Laboratory Animal Technology Co. Ltd (Beijing, China) were used to create the RHD model. They were housed in the animal experiment center of Guangxi medical university, maintained under SPF conditions and 12 h light/dark cycle at 23±2°C, with free access to water and diet. All experimental protocols were carried out according to the ethical guidelines about the Care and Use of Laboratory Animals. Unless indicated, all reagents were purchased from Sigma Aldrich (St. Louis, MO, USA).
Bacterial culture, inactivation and antigen preparation
GAS were cultivated for 24 h, collected and washed by normal saline. Then inactivated in 10 ml of 40 g/l paraformaldehyde solution for 6 h, washed and suspended in sterile normal saline, adjusted concentration to 1×1011 CFU/ml at 600 nm absorbance. Finally emulsified in Complete Freund’s Adjuvant (CFA, Sigma, USA) at a 1:1 ratio (v/v) to compound the antigen I.
Induction of animal model of rheumatic heart disease
Rats were randomly assigned to two groups: RHD group and control group respectively had 18 rats. Rats in RHD group were immunized with antigen I at the foot pad firstly. After one week, antigen I was injected in abdominal subcutaneous mass once a week for 4 weeks, then given inactivated GAS until to 24 weeks. Rats in control group were immunized with the same protocol outlined as treatment groups but without GAS. At 12 weeks 24 rats were sacrificed (each group comprised of 12) and at 24 weeks 12 were sacrificed (each group comprised of 6).
Sample collection
Hematoxylin-eosin (HE) staining and Masson staining were used to observe the pathological changes in hearts. The anterior mitral valve of every rats stored with the myocardium were immersed in 10% neutral buffered formalin for 8 hours, then cut four-micrometer sections and stained. The posterior mitral valve was excised without myocardium and preserved at -80°C until analyzed.
Mitral valve tissue protein preparation
Rats sacrificed after 12 weeks of antigenic induction was called the ARHD group. The mitral valves of rats sacrificed at 12 weeks in ARHD (n=10) and control (n=10) group were selected for proteomic analysis. In ARHD group, 10 valves were chosen from the rat succeeded in developing the rheumatic valvulitis and carditis. Total valve proteins of ten rats in each group were pooled at the same amount. The valves were desalted and concentrated. The supernatant was taken for determination of protein concentration by the Bradford method with Bradford reagents (Thermo Fischer Scientific, USA).
iTRAQ labeling
Total protein (100 μg) was taken from each sample solution, digested with Trypsin Gold (Promega, Madison, WI, USA). Then peptides were dried by vacuum centrifugation, reconstituted in 0.5 M TEAB and labeled with iTRAQ reagents according to the manufacture’s protocol (Applied Biosystems). Samples were labeled with the iTRAQ tags as follows: 121 for the peptides of control group, 117 for RHD group.
LC-ESI-MS/MS analysis
A LC-20AB HPLC Pump system (Shimadzu, Kyoto, Japan) was used to perform the separation with strong cation exchange chromatography. Two iTRAQ labeled peptide mixtures were loaded onto a column (Ф5 μm, Phenomenex) in a gradient of buffer A or buffer B to be eluted. The eluted peptides were collected and pooled into 20 fractions, desalted with a Strata XC18 column. The fractionated peptides re-suspended in buffer A were loaded on a LC-20AD nanoHPLC (Shimadzu, Kyoto, Japan) by the autosampler onto a 2 cm C18 trap column. Then the peptides were eluted onto a 10 cm analytical C18 column (inner diameter 75 μm) packed in-house. The MS Data acquisition was performed with a Triple TOF 5600 System (AB SCIEX, Concord, ON) fitted with a Nanospray III source (AB SCIEX, Concord, ON) and a pulled quartz tip as the emitter (New Objectives, Woburn, MA).
Database search
The MS/MS data were searched for the species of Rattus using Mascot search engine (Matrix Science, London, UK; version 2.3.02) with the Uniprot website. For protein identification, we selected search parameters as follows: fragmented mass tolerance, 0.1 Da; peptide mass tolerance, 0.05 Da; max missed cleavages, 1; enzyme: trypsin. Only peptides with significance scores (≥20) at 99% confidence interval by a Mascot probability analysis showing greater than “identity” were considered as identified and every identified protein involved at least one unique peptide. The weighed and normalized quantitative protein ratios were subjected for analysis and the ratios with P-values < 0.05, fold changes of > 1.2 were counted as significant. Protein annotations were performed with the Blast2GO program and then applied the kEGG database to classify these identified proteins.
Western blotting and immunohistochemical analysis
The rest of protein samples were used for verifying the up-regulated expression of GAPDH by Western blotting. Immunohistochemistry analysis was used for qualitative analysis of CD9. 10% sodium dodecyl polyacrylamide gel electrophoresis and immunoblotting were performed according to the standard protocol with equal amounts of proteins (20 μg). The antibodies used for testing are as follows: mouse anti-β-ACTIN (1:5000), anti-GAPDH (1:2000) (Vazyme Biotech Co. Ltd) and the goat anti-mouse IgG (1:10000, licor, USA) was used as the secondary antibody. β-actin was used as a normalization control. Blots were scanned with LAS4000 (Fujifilm, Tokyo, Japan) and the data were analyzed with Image J software. Immunohistochemistry was performed with rabbit antibodies for CD9 (1:25) overnight at 4°C, and then sections were incubated with a secondary antibody (MaxVision HRP-Polymer IHC kit, Fuzhou Maixin Biotech. Co. Ltd) for 15 min at room temperature. The DAB substrate system was used for color development.
Results
Growth state observation of Lewis rats
Redness and swelling appeared at the foot joints of all rats after the first immunization and disappeared after 2 weeks. But in treatment group it persisted in other foot joins following successive immunizations, without causing any abnormalities.
Pathological examination of myocardium and valve
HE staining showed that none of the rats in control group had myocarditis or valvulitis (Figure 1A). In RHD group at 12 weeks: only two rats were not presented with valvulitis or myocarditis; Diffuse inflammatory cells infiltration and accumulated Anitschkow cells were found in myocardium and valve (Figure 1B-D). Fibrosis of valves appeared in five of six rats in RHD group at 24 weeks (Figure 1E, 1F). The results of Masson staining of valve in RHD group at 24 weeks were just similar to the HE staining which is an indication that more collagen fibers accumulated at valves in RHD group than in control group (Figure 1G, 1H).
Figure 1.
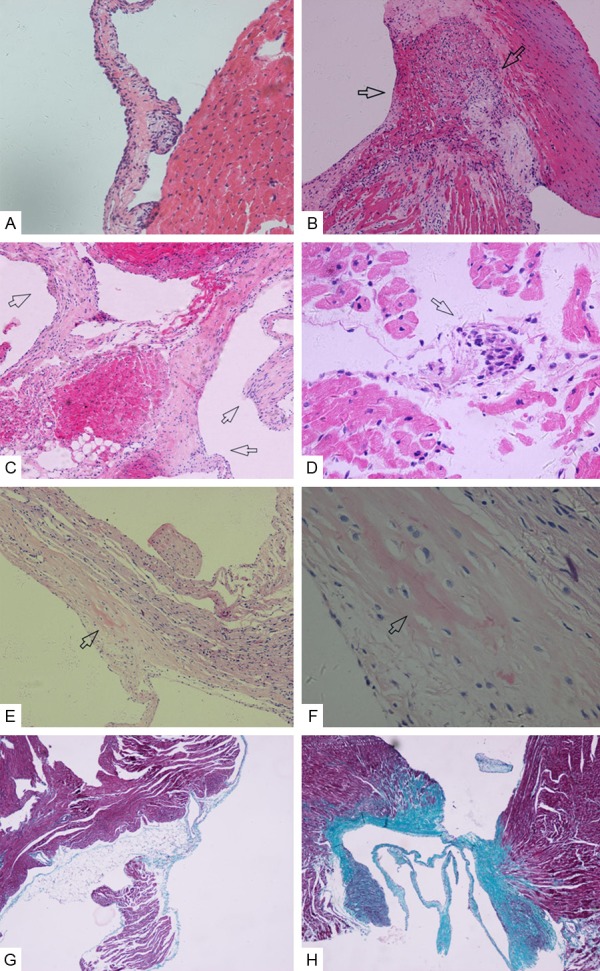
Hematoxylin and Eosin (H&E) staining and Masson staining of myocardial and valvular tissue. A. Normal myocardial and valve tissue in control group. Magnifications, ×200; B. Diffuse inflammatory cells infiltration in myocardium (arrows). Magnification, ×100; C. Diffuse inflammatory cells infiltration in valve (arrows). Magnification, ×100; D. Anitschkow cells (arrows). Magnification, ×400; E, F. Valve fibrosis (arrow) from 24 weeks of rats. Magnification, ×100 and ×400; G. Masson staining of valve in control group at 24 weeks. Magnification, ×40; H. Masson staining of valve in RHD group at 24 weeks. Magnification, ×40.
Differential expression of proteins between the ARHD group and control group
Using LC-ESI MS/MS, we identified 3931 proteins in the mitral valve of Lewis rat. Compared with the control group, a total of 295 proteins showed significant differences in ARHD group with 95% confidence. Since the fold change of 1.2 (fold change is the ratio of intensity of protein expression in ARHD to control group valve tissue) was considered as a threshold to minimize biological and technical errors, of which 176 proteins were found to be up-regulated and 119 proteins were down-regulated. The fold change (> 2.0) of proteins selected in our study described in Tables 1 and 2. The two closely related protein GAPDH and CD9 antigen are validated by western blotting or immunohistochemistry in order to check the technical reliability of iTRAQ result. The immunohistochemistry staining results showed significant difference of CD9 expression on mitral valve between ARHD and control group. Expression of CD9 was almost nil in control group (Figure 2A) and was increased in cytoplasmic membrane and cytoplasm in ARHD group (Figure 2B). The western blotting indicated increased GAPDH and the gray scale in ARHD group is 4.5 of that of the control group (Figure 3). The ontology analysis of the identified proteins and their correlation with molecular functions showed that, they were involved in binding (48.78%) and catalytic activity (27.49%) (Figure 4). The pathway analysis based on the KEGG database about focal adhesion pathway associated with binding was shown in Figure 5.
Table 1.
Up-regulated tissue protein in RHD group compared to control group
| Accession number | Protein name | Coverage (%) | RHD group_117: control group_121 | p-Value |
|---|---|---|---|---|
| Q4KM66 | LOC500183 protein | 42.7 | 2.388 | < 0.05 |
| M0R660 | Glyceraldehyde-3-phosphate dehydrogenase | 47.7 | 6.793 | < 0.05 |
| M0R7B4 | Protein LOC684828 | 17.8 | 2.357 | < 0.05 |
| P58775-2 | Isoform 2 of Tropomyosin beta chain | 51.1 | 2.864 | < 0.05 |
| M0R6R6 | Uncharacterized protein | 13.9 | 2.903 | < 0.05 |
| Q5PPG2 | Legumain | 7.6 | 2.02 | < 0.05 |
| D3ZP98 | Histocompatibility 13 | 2.7 | 2.111 | < 0.05 |
| P47875 | Cysteine and glycine-rich protein 1 | 36.8 | 2.3 | < 0.05 |
| P60203 | Myelin proteolipid protein | 7.6 | 2.972 | < 0.05 |
| Q62636 | Ras-related protein Rap-1b | 50 | 2.155 | < 0.05 |
| B0BMS8 | Myl9 protein | 33.7 | 2.795 | < 0.05 |
| P62912 | 60S ribosomal protein L32 | 15.6 | 2.409 | < 0.05 |
| P40241 | CD9 antigen | 15 | 2.63 | < 0.05 |
| P20767 | Ig lambda-2 chain C region | 41.3 | 2.155 | < 0.05 |
| Q6RUV5 | Ras-related C3 botulinum toxin substrate 1 (RAC1) | 11.5 | 2.826 | < 0.05 |
| P62836 | Ras-related protein Rap-1A (RAP1) | 35.3 | 2.31 | < 0.05 |
| F1LUS1 | Uncharacterized protein (Fragment) | 17 | 2.261 | < 0.05 |
| Q91V33 | KH domain-containing, RNA-binding, signal transduction-associated protein 1 | 6.5 | 2.536 | < 0.05 |
| B0K040 | Sh3bgr protein (Fragment) | 17.5 | 2.221 | < 0.05 |
| G3V6S3 | Calumenin | 32.1 | 2.001 | < 0.05 |
Table 2.
Down-regulated tissue protein in RHD group compared to control group
| Accession number | Protein name | Coverage (%) | RHD group_117: control group_121 | p-Value |
|---|---|---|---|---|
| G3V721 | WW domain binding protein 2, isoform CRA_b | 13 | 0.478 | < 0.05 |
| Q4FZZ3 | Glutathione S-transferase | 13.5 | 0.376 | < 0.05 |
| F1LM78 | Protein LOC100365958 | 9.8 | 0.334 | < 0.05 |
| F1LQQ1 | Malic enzyme (Fragment) | 12.2 | 0.471 | < 0.05 |
| D4A1W8 | Microsomal triglyceride transfer protein | 3.5 | 0.388 | < 0.05 |
| M0RDH0 | Glycine N-methyltransferase | 18.8 | 0.164 | < 0.05 |
| F1LR47 | Uncharacterized protein | 4.8 | 0.231 | < 0.05 |
| G3V6C2 | Protein Hgd | 16.6 | 0.211 | < 0.05 |
| Q68G44 | 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 2 (Mitochondrial) | 27 | 0.478 | < 0.05 |
| D3ZSF3 | Uncharacterized protein | 17.8 | 0.392 | < 0.05 |
| Q5M897 | Otc protein | 20.3 | 0.264 | < 0.05 |
| M0RDC5 | Acyl-CoA-binding protein (Fragment) | 51.2 | 0.443 | < 0.05 |
| P09034 | Argininosuccinate synthase | 28.6 | 0.28 | < 0.05 |
| P19112 | Fructose-1,6-bisphosphatase 1 | 8.3 | 0.442 | < 0.05 |
| P32755 | 4-hydroxyphenylpyruvate dioxygenase | 14 | 0.248 | < 0.05 |
| F1LS48 | Acetyl-CoA acetyltransferase, cytosolic | 2.8 | 0.205 | < 0.05 |
| P21213 | Histidine ammonia-lyase | 4.9 | 0.152 | < 0.05 |
| P11510 | Cytochrome P450 2C12, female-specific | 3.7 | 0.183 | < 0.05 |
| P80432 | Cytochrome c oxidase subunit 7C, mitochondrial | 17.5 | 0.207 | < 0.05 |
| P0CC09 | Histone H2A type 2-A | 35.4 | 0.208 | < 0.05 |
| Q71US7 | Cytochrome P450 2E1 (Fragment) | 5.5 | 0.216 | < 0.05 |
| P0C169 | Histone H2A type 1-C | 35.4 | 0.098 | < 0.05 |
| D3ZJ08 | Histone H3 | 43.4 | 0.138 | < 0.05 |
| P09811 | Glycogen phosphorylase, liver form | 10.4 | 0.439 | < 0.05 |
| D3ZDW1 | Protein Uroc1 | 6.1 | 0.347 | < 0.05 |
| P12928-2 | Isoform L-type of Pyruvate kinase PKLR | 6.1 | 0.327 | < 0.05 |
| P22734-2 | Isoform 2 of Catechol O-methyltransferase | 36.7 | 0.47 | < 0.05 |
| G3V885 | Myosin-6 | 62.1 | 0.106 | < 0.05 |
| P19468 | Glutamate--cysteine ligase catalytic subunit | 4.6 | 0.463 | < 0.05 |
Figure 2.
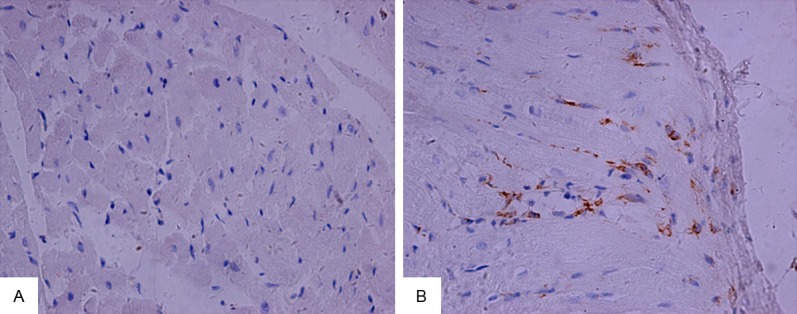
Validation of CD9 using immunohistochemical staining. A. CD9 was almost nil expressed in control group; B. CD9 overexpressed in cytoplasmic membrane and cytoplasm in ARHD group. magnification ×400.
Figure 3.
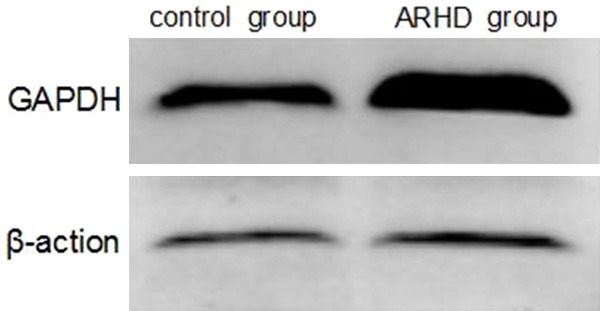
Validation of GAPDH on pooled lysates by Western blotting. GAPDH was up-regulated in ARHD groups and the gray scale in ARHD group is 4.5 of that of the control group, which is consistent with the iTRAQ result.
Figure 4.
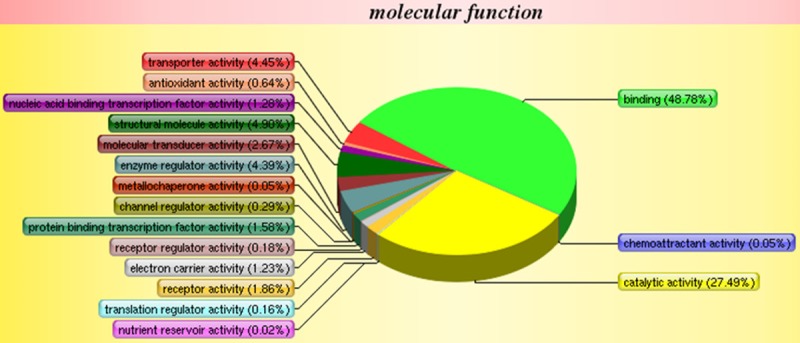
Gene ontology for molecular functions of the identified 3931 proteins between the valve of ARHD and control group. The function about binding accounts for the highest ratio of 48.78 percent.
Figure 5.

Focal adhesion pathway analysis based on the KEGG database. Ras-related C3 botulinum toxin substrate 1(RAC1), Ras-related protein Rap-1A, Chondroadherin, Myl9 protein, Protein Mylk and Collagen I increased in ARHD group, Talin increased in control group.
Discussion
In the past few years, proteomics was widely applied in cardiovascular disease research to make progress in the diagnostic biomarkers and mechanisms of disease [16,17]. However, the preceding proteomic analysis about RHD was restricted by the technique of 2-DE [10-12]. Moreover, it is worthy to highlight that the previous researches about RHD were either involved with chronic valve tissue or sera from RHD patients [10,12,18], which cannot be more intuitive to illuminate the early lesions of mitral valve. So the mitral valves of ARHD rats were selected for proteomic analysis. We obtained 295 different proteins, among them the significantly related protein such as GAPDH and CD9 antigen were confirmed and their expressions were consistent with the iTRAQ result. Moreover, the protein myosin 11, collagen, HSPA12A, RAC1, Myl9 and Mylk protein were also altered in the mitral valve and may play an important role in the pathogenesis of ARHD.
In our early research, we compared three immune methods induced in animal model of RHD and selected an optimal one. So in this current study, we applied the method of continuous GAS stimulation on Lewis rats to duplicate the RHD model. In ARHD group, at 12 weeks diffuse inflammatory cell infiltrate and the Aschoff like cells were detected, and at 24 weeks, Masson staining also showed more collagen in valve than in control group. Our animal model proved to be more suitable in terms of the growth state of rats and pathological changes of myocarditis and valvulitis in this study. The pathological changes of valve at 12 weeks were similar to the ARHD. So we choose the valve of rat sacrificed at 12 weeks for the proteomic analysis of ARHD.
Because of the highest fold change of GAPDH (6.793 times higher than the control group), we verified the changes with western blotting. Except their role in glycolysis and as a reference gene, GAPDH plays significant role in processes of apoptosis and cellular signaling [19]. Apoptotic stimuli, for instance oxidative or genotoxic stress, results in GAPDH translocation and accumulation into nuclei [20,21] to induce apoptosis. And cell apoptosis have essential role in cardiac valve calcification [22-26]. The apoptosis resulted in the initiation of calcification of mitral valve and probably even extended to the chronic phase [27,28]. Other studies also demonstrated that GAPDH might be a candidate Ag for an autoimmune response to neurons and axons in the autoimmune disease such as MS [29,30]. Based on these reports, we concluded that GAPDH may play an essential role in the development of ARHD via apoptosis.
CD9 antigen was 2.63 times higher than the control group. The tetraspanin CD9, also called as motility related protein-1 (MRP-1), has been shown to be involved with cell motility, growth, immune response and adhesion [31-33]. It may be ignored that CD9 regulated the formation of human multinucleated giant cell although the precise regulatory mechanism was not clear [34]. However, the appearance of multinucleated giant cells, known as Aschoff body strongly indicated granulomatous inflammation at proliferative stage of ARHD. Moreover, CD9 contacting with the surface-exposed RA-A47 might induce inflammatory reactions and autoantibodies in autoimmune disease such as rheumatoid arthritis [35]. In our immunohistochemical analysis and study, we noticed overexpression of CD9 in valves, especially in the pericardial inflammatory cells. Therefore, CD9 is possibly associated with the inflammatory reactions in the valvular tissue of ARHD.
In addition, we likewise found elevated Myosin11, collagen I and V with the fold changes of 1.419, 1.665 and 1.392 respectively. It was widely believed for so long that, Myosin and Collagen were autoantigens initiated in the pathogenic progression in RF and RHD [7,8]. A further study showed that, in acute rheumatic fever with carditis in a patient, collagen I was the major autoimmune response antigen between the putative heart valve components [9]. So our research verified previous work and underlined the role of myosin and collagen in the RF and ARHD as the autoantigen target of antibodies.
Heat shock 70 kDa protein 12A (HSPA12A) is an atypical member of HSP70 family, altered with the fold change of 1.282. HSP70 belonging to the stress protein have also shown to regulate some functions of immune response and also acting as an antioxidant [36]. It was reported that, the antibodies reacting with stress protein hsp70, hsp60 were detected in dilated cardiomyopathy [37]. In other investigation about RHD, HSPA5, hsp78 and hsp60 were considered to induce the autoimmune reaction and eventually lead to valve permanent damage [10,11,38]. Combining with the early study, we can speculate that, stress proteins including HSPA12A may be implicated in the inflammatory pathogenesis of ARHD.
In addition, molecular functions of identified proteins were involved in binding (48.78%). The KEGG pathway analysis showed that, the up-regulated RAC1, RAP1, Chondroadherin, Myl9 protein, Protein Mylk and Collagen I were closely related to the focal adhesion. With the development of ARHD, cross-reactive antibody binding with valvular endothelium could provide signal to accelerate the overexpression of adhesion molecules, which in turn could enhance the lymphocytes infiltration to valve [39].
In conclusion, with iTRAQ-coupled 2D LC-ESI-MS/MS we firstly identified 295 different proteins and formed the altered protein spectrum data of ARHD. The significantly altered protein such as GAPDH, CD9, myosin, collagen and RAC1 may be potential biomarkers for ARHD. Moreover, the mitral valve protein profile we delineated in this study will provide useful information for further understanding and investigating ARHD.
Acknowledgements
This work is supported by National Natural Science Foundation of China (NSFC; No. 81260067).
Disclosure of conflict of interest
None.
References
- 1.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 2.Iung B, Vahanian A. Epidemiology of acquired valvular heart disease. Can J Cardiol. 2014;30:962–970. doi: 10.1016/j.cjca.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 3.Manjunath CN, Srinivas P, Ravindranath KS, Dhanalakshmi C. Incidence and patterns of valvular heart disease in a tertiary care highvolume cardiac center: a single center experience. Indian Heart J. 2014;66:320–326. doi: 10.1016/j.ihj.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marijon E, Mirabel M, Celermajer DS, Jouven X. Rheumatic heart disease. Lancet. 2012;379:953–964. doi: 10.1016/S0140-6736(11)61171-9. [DOI] [PubMed] [Google Scholar]
- 5.Bland EF, Duckett Jones T. Rheumatic fever and rheumatic heart disease; a twenty year report on 1000 patients followed since childhood. Circulation. 1951;4:836–843. doi: 10.1161/01.cir.4.6.836. [DOI] [PubMed] [Google Scholar]
- 6.Sanyal SK, Thapar MK, Ahmed SH, Hooja V, Tewari P. The initial attack of acute rheumatic fever during childhood in North India; a prospective study of the clinical profile. Circulation. 1974;49:7–12. doi: 10.1161/01.cir.49.1.7. [DOI] [PubMed] [Google Scholar]
- 7.Tandon R, Sharma M, Chandrashekhar Y, Kotb M, Yacoub MH, Narula J. Revisiting the pathogenesis of rheumatic fever and carditis. Nat Rev Cardiol. 2013;10:171–177. doi: 10.1038/nrcardio.2012.197. [DOI] [PubMed] [Google Scholar]
- 8.Cunningham MW. Rheumatic fever revisited. Nat Rev Cardiol. 2014;11:123. doi: 10.1038/nrcardio.2012.197-c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martins TB, Hoffman JL, Augustine NH, Phansalkar AR, Fischetti VA, Zabriskie JB, Cleary PP, Musser JM, Veasy LG, Hill HR. Comprehensive analysis of antibody responses to streptococcal and tissue antigens in patients with acute rheumatic fever. Int Immunol. 2008;20:445–452. doi: 10.1093/intimm/dxn004. [DOI] [PubMed] [Google Scholar]
- 10.Fae KC, da Silva DD, Bilate AMB, Tanaka AC, Pomerantzeff PM, Kiss MH, Silva CA, Cunha-Neto E, Kalil J, Guilherme L. PDIA3, HSPA5 and vimentin, proteins identified by 2-DE in the valvular tissue, are the target antigens of peripheral and heart infiltrating T cells from chronic rheumatic heart disease patients. J Autoimmun. 2008;31:136–141. doi: 10.1016/j.jaut.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 11.Zheng D, Xu L, Sun L, Feng Q, Wang Z, Shao G, Ni Y. Comparison of the ventricle muscle proteome between patients with rheumatic heart disease and controls with mitral valve prolapse: HSP 60 may be a specific protein in RHD. Biomed Res Int. 2014;2014:151726. doi: 10.1155/2014/151726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao G, Xuan C, Yang Q, Liu XC, Liu ZG, He GW. Identification of altered plasma proteins by proteomic study in valvular heart diseases and the potential clinical significance. PLoS One. 2013;8:e72111. doi: 10.1371/journal.pone.0072111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cunningham MW. Streptococcus-induced myocarditis in mice. Autoimmunity. 2001;34:193–197. doi: 10.3109/08916930109007384. [DOI] [PubMed] [Google Scholar]
- 14.Quinn A, Kosanke S, Fischetti VA, Factor SM, Cunningham MW. Induction of autoimmune valvular heart disease by recombinant streptococcal m protein. Infect Immun. 2001;69:4072–4078. doi: 10.1128/IAI.69.6.4072-4078.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie X, Zhou H, Huang J, Huang H, Feng Z, Mei K, Yu B, Su Z, Gu J. An animal model of chronic rheumatic valvulitis induced by formalin-killed streptococci. Rheumatol Int. 2010;30:1621–1625. doi: 10.1007/s00296-009-1246-3. [DOI] [PubMed] [Google Scholar]
- 16.Chang YH, Ye L, Cai W, Lee Y, Guner H, Lee Y, Kamp TJ, Zhang J, Ge Y. Quantitative Proteomics Reveals Differential Regulation of Protein Expression in Recipient Myocardium after Trilineage Cardiovascular Cell Transplantation. Proteomics. 2015;15:2560–7. doi: 10.1002/pmic.201500131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oda T, Matsumoto KI. Proteomic analysis in cardiovascular research. Surg Today. 2015 doi: 10.1007/s00595-015-1169-4. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mukherjee S, Jagadeeshaprasad MG, Banerjee T, Ghosh SK, Biswas M, Dutta S, Kulkarni MJ, Pattari S, Bandyopadhyay A. Proteomic analysis of human plasma in chronic rheumatic mitral stenosis reveals proteins involved in the complement and coagulation cascade. Clin Proteomics. 2014;11:35. doi: 10.1186/1559-0275-11-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nicholls C, Li H, Liu JP. GAPDH: a common enzyme with uncommon functions. Clin Exp Pharmacol Physiol. 2012;39:674–679. doi: 10.1111/j.1440-1681.2011.05599.x. [DOI] [PubMed] [Google Scholar]
- 20.Brown VM, Krynetski EY, Krynetskaia NF, Grieger D, Mukatira ST, Murti KG, Slaughter CA, Park HW, Evans WE. A novel CRM1-mediated nuclear export signal governs nuclear accumulation of glyceraldehyde-3-phosphate dehydrogenase following genotoxic stress. J Biol Chem. 2004;279:5984–5992. doi: 10.1074/jbc.M307071200. [DOI] [PubMed] [Google Scholar]
- 21.Dastoor Z, Dreyer JL. Potential role of nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase in apoptosis and oxidative stress. J Cell Sci. 2001;114:1643–1653. doi: 10.1242/jcs.114.9.1643. [DOI] [PubMed] [Google Scholar]
- 22.Jian B, Narula N, Li QY, Mohler ER 3rd, Levy RJ. Progression of aortic valve stenosis: TGFbeta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75:457–465. doi: 10.1016/s0003-4975(02)04312-6. discussion 465-456. [DOI] [PubMed] [Google Scholar]
- 23.Zayzafoon M. Calcium/calmodulin signaling controls osteoblast growth and differentiation. J Cell Biochem. 2006;97:56–70. doi: 10.1002/jcb.20675. [DOI] [PubMed] [Google Scholar]
- 24.Farzaneh-Far A, Proudfoot D, Shanahan C, Weissberg PL. Vascular and valvar calcification: recent advances. Heart. 2001;85:13–17. doi: 10.1136/heart.85.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Proudfoot D, Skepper JN, Hegyi L, Farzaneh-Far A, Shanahan CM, Weissberg PL. The role of apoptosis in the initiation of vascular calcification. Z Kardiol. 2001;90(Suppl 3):43–46. doi: 10.1007/s003920170041. [DOI] [PubMed] [Google Scholar]
- 26.Knight RL, Wilcox HE, Korossis SA, Fisher J, Ingham E. The use of acellular matrices for the tissue engineering of cardiac valves. Proc Inst Mech Eng H. 2008;222:129–143. doi: 10.1243/09544119JEIM230. [DOI] [PubMed] [Google Scholar]
- 27.Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res. 2000;87:1055–1062. doi: 10.1161/01.res.87.11.1055. [DOI] [PubMed] [Google Scholar]
- 28.Khoynezhad A, Jalali Z, Tortolani AJ. A synopsis of research in cardiac apoptosis and its application to congestive heart failure. Tex Heart Inst J. 2007;34:352–359. [PMC free article] [PubMed] [Google Scholar]
- 29.Kolln J, Ren HM, Da RR, Zhang Y, Spillner E, Olek M, Hermanowicz N, Hilgenberg LG, Smith MA, van den Noort S, Qin Y. Triosephosphate isomerase- and glyceraldehyde-3-phosphate dehydrogenase-reactive autoantibodies in the cerebrospinal fluid of patients with multiple sclerosis. J Immunol. 2006;177:5652–5658. doi: 10.4049/jimmunol.177.8.5652. [DOI] [PubMed] [Google Scholar]
- 30.Kolln J, Zhang Y, Thai G, Demetriou M, Hermanowicz N, Duquette P, van den Noort S, Qin Y. Inhibition of glyceraldehyde-3-phosphate dehydrogenase activity by antibodies present in the cerebrospinal fluid of patients with multiple sclerosis. J Immunol. 2010;185:1968–1975. doi: 10.4049/jimmunol.0904083. [DOI] [PubMed] [Google Scholar]
- 31.Wang HX, Li Q, Sharma C, Knoblich K, Hemler ME. Tetraspanin protein contributions to cancer. Biochem Soc Trans. 2011;39:547–552. doi: 10.1042/BST0390547. [DOI] [PubMed] [Google Scholar]
- 32.Powner D, Kopp PM, Monkley SJ, Critchley DR, Berditchevski F. Tetraspanin CD9 in cell migration. Biochem Soc Trans. 2011;39:563–567. doi: 10.1042/BST0390563. [DOI] [PubMed] [Google Scholar]
- 33.Rana S, Zoller M. Exosome target cell selection and the importance of exosomal tetraspanins: a hypothesis. Biochem Soc Trans. 2011;39:559–562. doi: 10.1042/BST0390559. [DOI] [PubMed] [Google Scholar]
- 34.Hulme RS, Higginbottom A, Palmer J, Partridge LJ, Monk PN. Distinct regions of the large extracellular domain of tetraspanin CD9 are involved in the control of human multinucleated giant cell formation. PLoS One. 2014;9:e116289. doi: 10.1371/journal.pone.0116289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hattori T, von der Mark K, Kawaki H, Yutani Y, Kubota S, Nakanishi T, Eberspaecher H, de Crombrugghe B, Takigawa M. Downregulation of rheumatoid arthritis-related antigen RAA47 (HSP47/colligin-2) in chondrocytic cell lines induces apoptosis and cell-surface expression of RA-A47 in association with CD9. J Cell Physiol. 2005;202:191–204. doi: 10.1002/jcp.20112. [DOI] [PubMed] [Google Scholar]
- 36.Wieten L, van der Zee R, Spiering R, Wagenaar-Hilbers J, van Kooten P, Broere F, van Eden W. A novel heat-shock protein coinducer boosts stress protein Hsp70 to activate T cell regulation of inflammation in autoimmune arthritis. Arthritis Rheum. 2010;62:1026–1035. doi: 10.1002/art.27344. [DOI] [PubMed] [Google Scholar]
- 37.Portig I, Pankuweit S, Maisch B. Antibodies against stress proteins in sera of patients with dilated cardiomyopathy. J Mol Cell Cardiol. 1997;29:2245–2251. doi: 10.1006/jmcc.1997.0463. [DOI] [PubMed] [Google Scholar]
- 38.Tontsch D, Pankuweit S, Maisch B. Autoantibodies in the sera of patients with rheumatic heart disease: characterization of myocardial antigens by two-dimensional immunoblotting and N-terminal sequence analysis. Clin Exp Immunol. 2000;121:270–274. doi: 10.1046/j.1365-2249.2000.01283.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberts S, Kosanke S, Terrence Dunn S, Jankelow D, Duran CM, Cunningham MW. Pathogenic mechanisms in rheumatic carditis: focus on valvular endothelium. J Infect Dis. 2001;183:507–511. doi: 10.1086/318076. [DOI] [PubMed] [Google Scholar]