

Abstract
We investigated c-Met overexpression and MET gene amplification in gliomas to determine their incidence and prognostic significance. c-Met immunohistochemistry and MET gene fluorescence in situ hybridization were carried out on tissue microarrays from 250 patients with gliomas (137 grade IV GBMs and 113 grade II and III diffuse gliomas). Clinicopathological features of these cases were reviewed. c-Met overexpression and MET gene amplification were detected in 13.1% and 5.1% of the GBMs, respectively. All the MET-amplified cases showed c-Met overexpression, but MET amplification was not always concordant with c-Met overexpression. None of grade II and III gliomas demonstrated c-Met overexpression or MET gene amplification. Mean survival of the GBM patients with MET amplification was not significantly different from patients without MET amplification (P=0.155). However, GBM patients with c-Met overexpression survived longer than patients without c-Met overexpression (P=0.035). Although MET amplification was not related to poor GBM prognosis, it is partially associated with the aggressiveness of gliomas, as MET amplification was found only in grade IV, not in grade II and III gliomas. We suggest that MET inhibitor therapy may be beneficial in about 5% GBMs, which was the incidence of MET gene amplification found in the patients included in this study.
Keywords: Brain tumor, c-Met, glioblastoma, FISH, immunohistochemistry, target therapy
Introduction
Glioblastomas (GBMs) are the most common type of malignant primary brain tumor, comprising about 15% of all gliomas. Approximately 450 and 9000 new cases are diagnosed each year in Korea and the United States, respectively, according to the Brain Tumor Registry of Korea and Central Brain Tumor Registry of the United States (www.cbtrus.org). In South Korea, GBMs accounted for 5.9% of all primary CNS tumors and 33.5% of gliomas [1]. Despite the use of intensive treatment modalities GBM still has a poor prognosis. Receptor tyrosine kinase (RTK) c-Met (proto-oncogene proteins) overexpression and MET (hepatocyte growth factor receptor) gene amplification was found in GBMs as well as in other cancers, such as lung, pancreas, ovary, salivary gland, and breast cancers. c-Met overexpression and gene amplification promote malignancy and are associated with poor clinical outcome in gliomas [2,3]. Aberrant MET activation can occur through various mechanisms, such as autocrine or paracrine stimulation, transcriptional regulation, ligand-independent or mutational activation [4,5]. MET amplification can be a major driver after acquired resistance to epidermal growth factor receptor (EGFR) inhibitors, because of the cross-talk with other RTK family members [6]. Hepatocyte growth factor (HGF) may induce resistance to EGFR tyrosine kinase inhibitors in EGFR mutant lung cancer cells by Met/PI3K/Akt signaling [7]. However, one recent study reported a patient with MET-amplified GBM who showed clinical improvement when treated with the MET inhibitor crizotinib. Moreover, targeting the MET pathway potentiates the responsiveness of GBM to γ-radiation [8]. The purpose of our study is to investigate the positive rate, expression pattern, and significance of c-Met overexpression and MET gene amplification in gliomas.
Materials and methods
c-Met immunohistochemistry (IHC) and MET gene fluorescence in situ hybridization (FISH) were carried out on tissue microarrays made from formalin-fixed paraffin-embedded (FFPE) glioma tissues obtained from 250 patients (137 GBMs and 113 grade II and III diffuse gliomas). We reviewed clinicopathological features of the 137 GBMs and compared these findings with other protein expression and molecular genetic factors to ascertain the significance of c-Met overexpression and MET gene amplification patterns. Clinical feature of the 137 GBM patients are summarized in Table 1. The mean age was 48 years (range: 19-73 years old) and the male to female ratio was 1.6:1. Patients underwent gross total resection (37.1% of patients), partial resection (53.6%), or biopsy only (9.3%). Grade II and III diffuse gliomas were composed of 13 diffuse astrocytomas, 36 anaplastic astrocytomas, 30 low-grade oligodendrogliomas, and 34 anaplastic oligodendrogliomas. This research was approved by the Institutional Review Board of Seoul National University Hospital (H-1010-059-434).
Table 1.
Clinicopathologic characteristics of glioblastoma patients
| Characteristics | No. |
|---|---|
| Patients | 137 (100%) |
| Sex | |
| Male | 85 (62.0%) |
| Female | 52 (38.0%) |
| Mean age (range) | 47.9 (19-73) years |
| Mean overall survival | 24.6 months |
| Surgical resection | |
| Gross total resection | 69 (50.4%) |
| Partial resection | 59 (43.1%) |
| Biopsy only | 9 (6.5%) |
Immunohistochemistry (IHC)
Immunohistochemical staining was performed on FFPE tissues of 3 μm thickness. All IHC procedures were carried out using a Leica BOND-MAX (Leica, Heidelberg, Germany) according to routine laboratory procedures and the manufacturer’s protocols. The primary antibodies, antigen retrieval methods, and dilution ratios used for IHC analysis are listed in Table 2. Positivity was measured by Aperio membrane algorithm after scanning with Aperio Scanscope, which appeared as positive %.
Table 2.
Primary antibodies used in this study
| Name | Manufacturer | Antigen retrieval | Dilution |
|---|---|---|---|
| c-Met | Ventana (SP44) | RTU MILD/16’ | Prediluted |
| PTEN | DAKO, Glostrup, Denmark | EDTA, microwave | 1:100 |
| IDH1 (H09) | Dianova, Hamburg, Germany | Microwave | 1:50 |
Fluorescence in situ hybridization (FISH)
Analysis of MET and EGFR gene status was carried out by FISH using Vysis probes
Tissue sections of 4 μm thickness were deparaffinized using xylene, incubated with 0.3% pepsin in 10 mM HCl at 37°C for 10 min, boiled with citrate buffer (pH 6.0) in a microwave, incubated in 1 M NaSCN for 35 min at 80°C, immersed in the pepsin solution, and fixed in 10% neutral-buffered formalin. Labeled locus-specific (LSI) EGFR/CEP7 dual-color probes (Abbott Molecular) and LSI MET/CEP9 dual color probes (Abbott Molecular) were used for recognizing the EGFR-gene and MET gene status according to the manufacturer’s protocol. We applied the probe mixture to slides and incubated them in a humidified atmosphere with HYBrite™ (Abbott Molecular) at 73°C for 5 min for simultaneous denaturation of the probe and the target DNA. Next, we changed the temperature to 37°C for 19 hours to hybridize the probes and target DNAs. Slides were then soaked in 0.4× SSC buffer/0.3% NP-40 for 2 min at room temperature, followed by 2× SSC/0.1% NP-40 for 5 min at 73°C. The processing and analysis of the FISH studies were conducted as described previously [9,10]. The signals on 100 non-overlapping intact nuclei were counted.
Statistical analysis
Kaplan-Meier analysis and other statistical analyses were carried out using SPSS version 21. Student t-test was used to calculate P-values.
Results
IHC and FISH results
Results of the IHC and FISH studies are shown in Table 3 and Figure 1. c-Met overexpression was detected in 13.1% (18 of 137) of GBMs, but not in any of the 113 cases of grade II and III astrocytic and oligodendroglial tumors. All MET-amplified GBMs also had c-Met overexpression, but among the c-Met-overexpressing GBMs, only 38.9% (7/18) showed MET gene amplification. IHC showed that only one out of seven cases with MET gene amplification also had phosphatase and tensin homolog deleted on chromosome 10q (PTEN) (Table 3). c-Met overexpression or MET amplification was not associated with IDH mutation. Only 16.7% (3/18) of c-Met expressed GBM and 28.5% (2/7) of MET amplified GBM were positive for IDH1, respectively. Loss of PTEN expression was detected in 13.9% (20 of 137) of GBMs, but only four out of 18 c-MET expressed cases (22.2%) and one out of seven cases (14.3%) with MET gene amplification had loss of PTEN expression.
Table 3.
c-Met overexpression (measured by IHC) and MET gene amplification (measured by FISH) in a cohort of glioblastoma patients
| c-Met IHC | MET FISH | Total | PTEN IHC | Total | ||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Amplification | No amplification | Loss | No loss | |||
| Positive | 7 (5.1%) | 11 (8.0%) | 18 (13.1%) | 1 (0.7%) | 7 (5.1%) | 8 (5.8%) |
| Negative | 0 | 119 (86.9%) | 119 (86.9%) | 19 (13.9%) | 110 (80.3%) | 129 (94.2%) |
| Total | 7 (5.1%) | 130 (94.9%) | 137 (100%) | 20 (14.6%) | 117 (85.4%) | 137 (100%) |
IHC: immunohistochemistry, FISH: fluorescence in situ hybridization.
Figure 1.
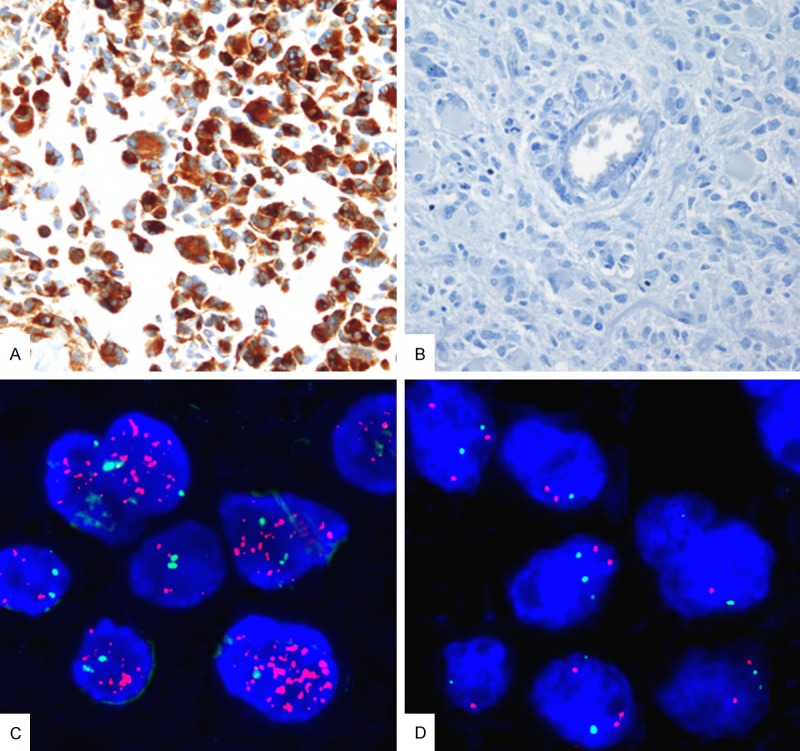
Representative images of glioblastomas with (A) diffuse c-Met overexpression, (B) lack of c-Met immunopositivity, (C) MET gene amplification, and (D) no MET gene amplification. (A, B: c-Met immunohistochemistry, ×400; C, D: Labeled locus-specific dual-color Vysis probe of MET gene (7q31), spectrum orange/CEP7 Spectrum green, ×1000).
Clinical outcomes
It had been reported that the mean survival of GBM patients with MET amplification was not shorter than patients without MET amplification; however, in our cohort, the patients with c-MET positive GBM survived for longer (35.7 months versus 24.0 months for c-Met positive and c-MET negative GBMs, respectively, P=0.035, Figure 2B). Among MET gene-amplified GBMs, two were IDH1 positive, which were secondary GBMs, initially an anaplastic astrocytoma and a diffuse astrocytoma, respectively. At that time, neither c-Met overexpression nor MET gene amplification was detected (Table 4). Kaplan Meier survival analyses revealed that survival did not differ between patients with MET amplification and those without MET amplification (P=0.155, Figure 2A).
Figure 2.
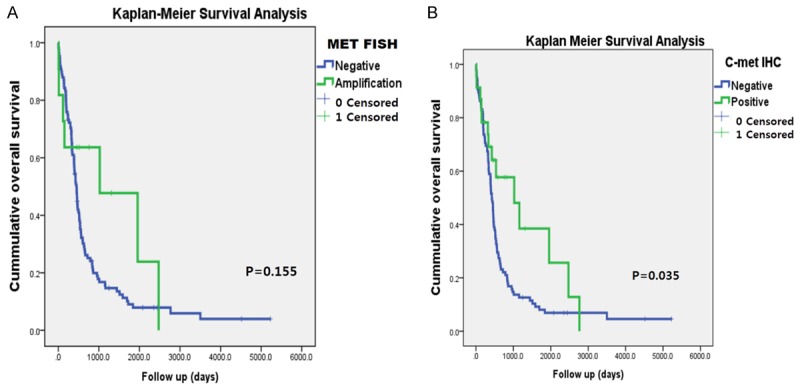
(A) Kaplan-Meier survival analysis revealed that patient survival was not related with MET gene amplification, but that (B) patients with c-Met overexpression had better survival than those without c-Met overexpression.
Table 4.
Clinical outcome of seven glioblastomas patients with MET amplification
| Sex/Age | Operation | Initial Dx | Final Dx | Recur | PFS (months) | OS (months) | Outcome |
|---|---|---|---|---|---|---|---|
| F/40 | GTR | GBM, conventional | GBM-IDHw | No | 5.1 | 5.1 | DOD |
| M/65 | Biopsy | GBM, conventional | GBM-IDHw | No | 3.9 | 3.9 | DOD |
| M/49 | GTR | GBM, conventional | GBM-IDHw | Yes | 81.2 | 82.4 | DOD |
| M/77 | Biopsy | GBM, conventional | GBM-IDHw | No | 15.4 | 15.4 | DOD |
| F/21 | PR | GBM, epithelioid | GBM-IDHw | Yes | 7.7 | 43.8 | Alive |
| F/41 | PR | LO | AA→GBM-IDHm | Yes | 89.6 (from oligo to GBM) | 124 (from LO to DOD) | DOD |
| 29 (from GBM to DOD) | DOD | ||||||
| F/47 | PR | DA | DA→GBM-IDHm | Yes | 19.8 (from DA to AA) | 65.2 (from DA to DOD) | DOD |
| 40 (from AA to GBM) | 7 (from GBM to DOD) |
GBM: glioblastoma, IDHw: isocitrate dehydrogenase wild type, IDHm: IDH mutant, GTR: gross total removal, PR: partial removal, PFS: progression free survival, OS: overall survival, DOD: dead of disease, LO: low grade (grade II) oligodendroglioma, DA: diffuse astrocytoma, AA: anaplastic astrocytoma.
Discussion
GBM is the most common malignant glioma, comprising 15% of primary brain tumors. Outcomes for patients with GBM are dismal, due to its highly malignant nature and infiltrative growth. There is no suitable chemotherapy regimen, even though CCRT with temozolomide can extend the longevity of patients. Recently, personalized targeted therapies have been trialed in various tumors, such as non-small cell lung carcinoma, gastrointestinal stromal tumors, and melanoma. However, the genetic abnormalities found in gliomas, including GBMs, are remarkably different from extracranial malignant tumors. Therefore, more research is needed to identify suitable GBM tumor markers and therapeutic targets, such as the MET signaling pathway.
MET is a proto-oncogene that encodes a protein known as HGF receptor. c-Met is an integral plasma membrane RTK, which is normally expressed by epithelial cells [11]. Its ligand, HGF, is the only ligand to bind c-Met, and is usually secreted by mesenchymal cells. HGF binds to c-Met and activates the c-Met signaling pathway, resulted in fetal morphogenesis and increased cell motility, infiltrative growth, angiogenesis and resistance to apoptotic signals. Tumor cells can secrete HGF, and HGF has been shown to stimulate the motility and invasiveness of several types of cancer cells and to induce angiogenesis [12]. In tumors, HGF binding to c-Met triggers leads to tumor cell proliferation, invasion, survival, and angiogenesis by activating the RAS, PI3K, STAT, beta-catenin and NOTCH signal transduction pathways. Aberrant c-Met activation can occur through numerous mechanisms, such as autocrine or paracrine stimulation, or transcriptional activation [4]. The identification of tumor subgroups that are suitable targets for MET inhibitors is of great clinical importance, this question was addressed by Xie et al. [13]. They found that an HGF paracrine environment did not indicate sensitivity to MET inhibitors, although it may enhance GBM growth in vivo [4]. Among 18 cell lines investigated by in vivo and in vitro studies, only two cell lines (U87 and U87M2) expressed autocrine HGF at a significant level; however, c-Met expression was not affected by these autocrine HGF levels. HGF-autocrine GBMs bear an activated c-Met signaling pathway that may predict sensitivity to MET inhibitors [4]. The same study also suggests that serum HGF levels may serve as a biomarker for the presence of autocrine tumors and their responsiveness to MET therapeutics [4].
Somatic mutations/deletions of PTEN are commonly found in a wide variety of solid tumors, and are detected in 20-50% of GBMs [14,15]. PTEN is known to co-regulate RTK-mediated gene expression in glioma models and influences their response to targeted kinase inhibitors [16]. Previous studies have suggested that the therapeutic efficacy of RTK inhibitors is predicted by PTEN activity, with PTEN loss rendering tumors [17,18]. Therefore, here we investigated c-Met expression and MET gene amplification as well as PTEN loss in gliomas.
Among our cases, MET amplification and c-Met overexpression was detected in 5.1% (7/137) and 13.1% (18 of 137) of GBMs, respectively, but in none of 113 grade II and III gliomas. c-Met overexpression and MET gene amplification were not perfectly concordant. Only 38.9% (7/18) of c-Met immunopositive cases showed MET gene amplification. The incidence of c-Met overexpression in our study is far lower than reported by Kong et al., who reported c-Met overexpression rate in 29.0% of GBMs [3]. Pierscianek et al. studied MET copy number gain using quantitative polymerase chain reaction and found that MET gain was present in 47% of primary GBMs, 44% of secondary GBMs, 38% of diffuse astrocytomas, and 16% of oligodendrogliomas; however, a prognostic association was found only in diffuse astrocytomas, in which MET gain was associated with shorter survival [2]. However, MET gain was not associated with survival in GBM patients [2]. Therefore, the prognostic value of MET gain or amplification remains controversial.
In our cohort, c-Met overexpression or MET amplification was not associated with IDH1 mutation. Loss of PTEN expression was detected in 13.9% (20 of 137) of GBMs, but only four out of 18 c-MET expressed cases and one out of seven cases with MET gene amplification had loss of PTEN expression; such PTEN lost cases may have increased susceptibility to c-Met inhibitors. Paradoxically, GBM patients with c-Met overexpression had better survival than those without c-Met expression (P=0.035). However MET gene amplification was not associated with patient survival (P=0.155) in our patients. Therefore, early recognition of GBMs with c-Met overexpression and MET amplification and a trial of treatment with a MET inhibitor may be necessary next steps for research and provision of personalized targeted therapy. However, MET amplification was not detected in our series of grade II and III gliomas, suggesting that MET inhibition may not be a viable therapeutic option in these cases.
Conclusion
This study clearly showed that MET amplification is not associated with poor prognosis in GBMs, but is partially associated with the aggressiveness of gliomas, as MET amplification was found only in grade IV GBMs, not in grade II or III gliomas. We predict that MET might play a key role in tumorigenesis and cancer progression in about 5% of GBMs. Accordingly, our study implies that the administration of a MET inhibitor may be a promising targeted therapy for GBMs with MET amplification. Further studies are required to verify the exact incidence of MET gene amplification and to determine if a MET inhibitor is a precision medicine for gliomas.
Acknowledgements
This work was supported by a grant of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (Grant No. HI13C1468).
Disclosure of conflict of interest
None.
References
- 1.Lee CH, Jung KW, Yoo H, Park S, Lee SH. Epidemiology of primary brain and central nervous system tumors in Korea. J Korean Neurosurg Soc. 2010;48:145–152. doi: 10.3340/jkns.2010.48.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pierscianek D, Kim YH, Motomura K, Mittelbronn M, Paulus W, Brokinkel B, Keyvani K, Wrede K, Nakazato Y, Tanaka Y, Mariani L, Vital A, Sure U, Ohgaki H. MET gain in diffuse astrocytomas is associated with poorer outcome. Brain Pathol. 2013;23:13–18. doi: 10.1111/j.1750-3639.2012.00609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kong DS, Song SY, Kim DH, Joo KM, Yoo JS, Koh JS, Dong SM, Suh YL, Lee JI, Park K, Kim JH, Nam DH. Prognostic significance of c-Met expression in glioblastomas. Cancer. 2009;115:140–148. doi: 10.1002/cncr.23972. [DOI] [PubMed] [Google Scholar]
- 4.Xie Q, Bradley R, Kang L, Koeman J, Ascierto ML, Worschech A, De Giorgi V, Wang E, Kefene L, Su Y, Essenburg C, Kaufman DW, DeKoning T, Enter MA, O’Rourke TJ, Marincola FM, Vande Woude GF. Hepatocyte growth factor (HGF) autocrine activation predicts sensitivity to MET inhibition in glioblastoma. Proc Natl Acad Sci U S A. 2012;109:570–575. doi: 10.1073/pnas.1119059109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uchinokura S, Miyata S, Fukushima T, Itoh H, Nakano S, Wakisaka S, Kataoka H. Role of hepatocyte growth factor activator (HGF activator) in invasive growth of human glioblastoma cells in vivo. Int J Cancer. 2006;118:583–592. doi: 10.1002/ijc.21362. [DOI] [PubMed] [Google Scholar]
- 6.Xu L, Kikuchi E, Xu C, Ebi H, Ercan D, Cheng KA, Padera R, Engelman JA, Janne PA, Shapiro GI, Shimamura T, Wong KK. Combined EGFR/MET or EGFR/HSP90 inhibition is effective in the treatment of lung cancers codriven by mutant EGFR containing T790M and MET. Cancer Res. 2012;72:3302–3311. doi: 10.1158/0008-5472.CAN-11-3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kang XH, Xu ZY, Gong YB, Wang LF, Wang ZQ, Xu L, Cao F, Liao MJ. Bufalin Reverses HGFInduced Resistance to EGFR-TKIs in EGFR Mutant Lung Cancer Cells via Blockage of Met/PI3k/Akt Pathway and Induction of Apoptosis. Evid Based Complement Alternat Med. 2013;2013:243859. doi: 10.1155/2013/243859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lal B, Xia S, Abounader R, Laterra J. Targeting the c-Met pathway potentiates glioblastoma responses to gamma-radiation. Clin Cancer Res. 2005;11:4479–4486. doi: 10.1158/1078-0432.CCR-05-0166. [DOI] [PubMed] [Google Scholar]
- 9.Jeon YK, Park K, Park CK, Paek SH, Jung HW, Park SH. Chromosome 1p and 19q status and p53 and p16 expression patterns as prognostic indicators of oligodendroglial tumors: a clinicopathological study using fluorescence in situ hybridization. Neuropathology. 2007;27:10–20. doi: 10.1111/j.1440-1789.2006.00735.x. [DOI] [PubMed] [Google Scholar]
- 10.Kim MA, Lee HS, Lee HE, Jeon YK, Yang HK, Kim WH. EGFR in gastric carcinomas: prognostic significance of protein overexpression and high gene copy number. Histopathology. 2008;52:738–746. doi: 10.1111/j.1365-2559.2008.03021.x. [DOI] [PubMed] [Google Scholar]
- 11.Mughal A, Aslam HM, Sheikh A, Khan AM, Saleem S. c-Met inhibitors. Infect Agent Cancer. 2013;8:13. doi: 10.1186/1750-9378-8-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lamszus K, Laterra J, Westphal M, Rosen EM. Scatter factor/hepatocyte growth factor (SF/HGF) content and function in human gliomas. Int J Dev Neurosci. 1999;17:517–530. doi: 10.1016/s0736-5748(99)00008-8. [DOI] [PubMed] [Google Scholar]
- 13.Xie Q, Gao CF, Shinomiya N, Sausville E, Hay R, Gustafson M, Shen Y, Wenkert D, Vande Woude GF. Geldanamycins exquisitely inhibit HGF/SF-mediated tumor cell invasion. Oncogene. 2005;24:3697–3707. doi: 10.1038/sj.onc.1208499. [DOI] [PubMed] [Google Scholar]
- 14.Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol. 2005;64:479–489. doi: 10.1093/jnen/64.6.479. [DOI] [PubMed] [Google Scholar]
- 15.Kim B, Myung JK, Seo JH, Park CK, Paek SH, Kim DG, Jung HW, Park SH. The clinicopathologic values of the molecules associated with the main pathogenesis of the glioblastoma. J Neurol Sci. 2010;294:112–118. doi: 10.1016/j.jns.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 16.Abounader R, Reznik T, Colantuoni C, Martinez-Murillo F, Rosen EM, Laterra J. Regulation of c-Met-dependent gene expression by PTEN. Oncogene. 2004;23:9173–9182. doi: 10.1038/sj.onc.1208146. [DOI] [PubMed] [Google Scholar]
- 17.Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH, Chute DJ, Riggs BL, Horvath S, Liau LM, Cavenee WK, Rao PN, Beroukhim R, Peck TC, Lee JC, Sellers WR, Stokoe D, Prados M, Cloughesy TF, Sawyers CL, Mischel PS. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 18.Bianco R, Shin I, Ritter CA, Yakes FM, Basso A, Rosen N, Tsurutani J, Dennis PA, Mills GB, Arteaga CL. Loss of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitors. Oncogene. 2003;22:2812–2822. doi: 10.1038/sj.onc.1206388. [DOI] [PubMed] [Google Scholar]