

Abstract
Objectives: Idiopathic pulmonary fibrosis (IPF) is a group of lung diseases that cause irreversible architectural distortion and impair gas, and finally progressive pulmonary functional decline and death, in which the common variant in the promoter region of the mucin 5B (MUC5B) gene may be involved. The present study aims to investigate whether variants within the MUC5B gene rs35705950 contributed to IPF susceptibility and severity in Chinese Han Population. Methods: A total of 187 patients diagnosed with IPF and 250 healthy controls were enrolled in this study. All subjects were genotyped for MUV5B SNP rs35705950. The demographic, comorbidity, clinical and functional data were recorded. Results: The rs35705950 of MUC5B were found significantly associated with increased risk of IPF susceptibility. One way ANOVA analysis found that there was a significant decreased FVC (P < 0.0001) and DLco (P < 0.0001) in correction with the minor allele of the SNP rs35705950. In the 5 years’ follow-up, the carriers of the minor allele T increased mortality (P = 0.0294). Conclusion: This study demonstrated that the MUC5B polymorphism rs35705950 is associated with increased risk of idiopathic pulmonary fibrosis susceptibility, severity, and the decreased overall survival.
Keywords: MUC5B, SNP, idiopathic pulmonary fibrosis
Introduction
The Interstitial Lung Diseases (ILDs) or diffuse interstitial lung diseases (DILD) are a heterogeneous group of lung diseases that cause irreversible architectural distortion and impair gas, and finally result in progressive pulmonary functional decline and death. Idiopathic pulmonary fibrosis (IPF) is the most common idiopathic ILD, with an estimated 100,000 Americans affected and about 0.7 per 100,000 affected in East Asia [1]. The incidence of IPF increases with advancing age, and the peaks in those over 75 years of age. Aberrant remodeling and repair is an archetypal feature of this chronic, unrelenting inflammatory disorder [2,3], which is resulting from various patterns of inflammation and fibrosis of unknown cause, which results in a loss of mechanical and functional properties of the lungs with consequent respiratory failure and death.
This patient group has a shorter clinical history before diagnosis, even more, it is difficult to identify patients who are at significant risk of death at initial diagnosis. This prevents early referral for lung transplantation and optimum clinical management. As a result, several risk prediction models have been developed, like CC chemokine ligand-18 (CCL-18), KL-6, intercellular adhesion molecule-1, matrix metalloproteinase-7, IL-8, and vascular cell adhesion molecule-1 (VCAM-1).
Most cases of IPF are sporadic, but also familial form is common in clinic, which is defined as IPF occurring in two or more first-degree relatives within the same family. Consequently, current theory suggests that fibrotic lung disease occurs when genetically susceptible individuals are exposed to environmental triggers. In the largest genome-wide association study to date of fibrotic idiopathic interstitial pneumonias, a total of 10 genetic loci were identified as significantly associated with IPF [4]. In another study, Noth et al. identified two additional SNPs that were associated with idiopathic pulmonary fibrosis [5]. Among these recently reported associations, a common variant in the promoter region of the mucin 5B (MUC5B) gene rs35705950 was found to be associated with the development of idiopathic pulmonary fibrosis as well an increased production of MUC5B, an airway mucin. The gene MUC5B encodes a member of the mucin family of proteins, which are highly glycosylated macromolecular components of mucus secretions. This family member is the major gel-forming mucin in mucus. It is a major contributor to the lubricating and viscoelastic properties of whole saliva, normal lung mucus and cervical mucus. This gene has been found to be up-regulated in some human diseases, including sinus mucosa of chronic rhinosinusitis (CRS), CRS with nasal polyposis, chronic obstructive pulmonary disease (COPD) and H. pylori-associated gastric disease, and it may be involved in the pathogenesis of these diseases. The association between MUC5B polymorphism and IPF susceptibility has been widely researched around the word, however, few is based on the Han population. And no study is performed with regard to the severity and over-all survival with the MUC5B polymorphism rs35705950. Hence, the present study aims to determine whether the MUC5B SNP rs35705950 was associated with idiopathic pulmonary fibrosis susceptibility and severity in Chinese Han Populations.
Methods and materials
Study subjects
Subjects were recruited from Qilu Hospital of Shandong University between 2004 and 2009. This study was approved by the Qilu Hospital, Shandong University. The diagnosis of IPF was established according to the criteria of the American Thoracic Society (ATS)/European Respiratory Society published in 2002 [6]. All subjects provided a written informed consent for participation. A total of 187 patients diagnosed with IPF and 250 age and sex matched healthy controls were enrolled in this study. All subjects included in this study were Chinese Han Population. All demographic, comorbidity, clinical and functional data were recorded. The HRCT investigation was performed using a SOMATOM Sensation 40 machine (Siemens AG, Berlin and Munich, Germany). The results were evaluated by an experienced consultant using interstitial and alveolar score scales, which were based on the IPF HRCT description system of Gay et al.
Genotyping
DNA samples were obtained from all the participants from peripheral blood with the Chelex-100 method [19]. The SNP was then genotyped using a competitive allele specific PCR system (Kaspar genotyping, Kbioscience, Hoddeston, UK) and Taqman SNP genotyping assay-allelic discrimination method (Applied Biosystem, Foster City, CA). The primer and probe were designed and synthesized by Sigma (Sigma-Proligo, The Woodlands, TX). Genotyping was performed by independent laboratory personnel who were blinded to the study, and three authors independently reviewed the genotyping results, data entry, and statistical analyses. In addition, we randomly selected 5% samples of case and control subjects for reproducibility tests at least twice in different days and yielded a 100% concordant.
Statistical analysis
The Statistical Package for Social Sciences software for Windows (SPSS, version 16.0, Inc., Chicago, IL, USA). To analyze baseline characteristics, the continuous variables was presented as Mean ± SD and compared between groups by the Student’s t-tests, and the categorical data compared by Chi-square tests. The genotype and allelic frequencies were evaluated by Hardy-Weinberg equilibrium and compared by the Chi-square test. The association between the MUC5B gene SNP rs35705950 and IPF susceptibility was assessed under the following genetic models, which were treated as a dichotomous variable: (i) T-allele versus C-allele for allele level comparison; (ii) CT + TT versus CC for a dominant model of the T allele; (iii) TT versus CT + CC for a recessive model of the T-allele; and (iv) TT versus CC for the extreme genotype. The P < 0.05 was consiered to indicate a statistically significant difference. Additionally, associations between MUC5B SNP and over survival of the patients with IPF were estimated using adjusted relative risks and 95% confidence intervals (95% CIs) from multivariate logistic regression. Survival time was calculated from the date of IPF diagnosis to the date of death or last follow-up. Survival analysis was estimated using the Kaplan-Meier method, log-rank test, and Cox-proportional hazards regression model.
Results
Patient characteristics
A total of 187 patients diagnosed with IPF and 250 age and sex matched healthy controls were enrolled in this study. Demographic data of the population studied and the number of individuals in each group were shown in Table 1. There were no significant differences between groups in terms of age and gender. The smoking rate of the cases is marked higher than the controls (OR = 1.819, 95% CI = 1.211 to 2.733, P = 0.0046). The percentage of predicted forced vital capacity (FVC) of the cases is 78.8 ± 7.1, and the percentage of predicted diffusion capacity of lung for carbon monoxide (DLCO) is 46.2 ± 6.2.
Table 1.
The summary of the basic characteristics of the groups
| IPF | Control | ||
|---|---|---|---|
| No. of patients | 187 | 250 | |
| Age (year) | 69.7 ± 4.3 | 67.7 ± 7.3 | |
| Gender | Male | 138 | 172 |
| Female | 49 | 78 | |
| Smoking (n) | Yes | 135 | 147 |
| No | 52 | 103 | |
| Pulmonary Fonction (Percentage of predicted, %) | FVC | 78.8 ± 7.1 | / |
| DLCO | 46.2 ± 6.2 | / | |
FVC = forced vital capacity. DLCO = diffusion capacity of lung for carbon monoxide.
Association of MUC5B polymorphism with IPF susceptibility
As expected, the distribution of the genotypes of SNP rs35705950 of MUC5B conformed to the Hardy-Weinberg equilibrium and the genotyping success rate was 100%. Table 2 listed the genotyped and allele distributions of the SNP for the cases and controls. The increased T allele was found significantly associated with increased susceptibility of IPF with the allele level comparison (P = 0.0007), with dominant model comparison (P = 0.0293), with recessive model comparison (P = 0.0018), and with the extreme genotype comparison (P = 0.0015).
Table 2.
The genotype and allele distributions of the SNP rs35705950 of MUC5B for the cases and controls
| Group | rs35705950 | ||||
|---|---|---|---|---|---|
|
| |||||
| TT vs. GG | GT + TT vs. GG | TT vs. GG + GT | G (%) | T (%) | |
| Control | 7 vs. 202 | 48 vs. 202 | 7 vs. 243 | 89.0 | 11.0 |
| Case | 19 vs. 134 | 53 vs. 134 | 19 vs. 168 | 80.7 | 19.3 |
| OR (95% CI) | 4.092 (1.674 to 10.00) | 1.664 (1.064 to 2.604) | 3.926 (1.614 to 9.549) | / | 1.929 (1.319 to 2.822) |
| P | 0.0015 | 0.0293 | 0.0018 | / | 0.0007 |
Association of MUC5B polymorphism with IPF severity
The assocition between the SNP and the IPF severity was analyzed based on the value of the FVC and DLco. One way ANOVA analysis found that there was a significant decreased FVC (Figure 1A, P < 0.0001) and DLco (Figure 1B, P < 0.0001) in correction with the minor allele of the SNP rs35705950.
Figure 1.
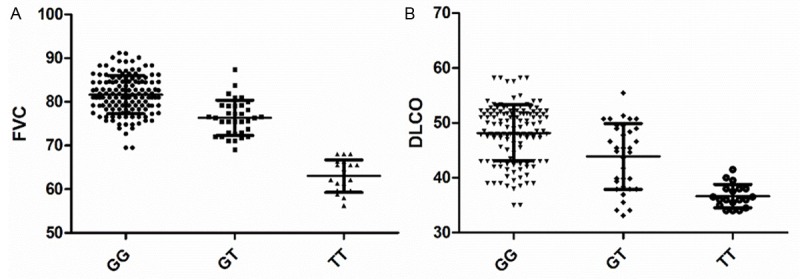
The association between the SNP and the value of the FVC and DLco.
Association of MUC5B polymorphism with survival in patients with IPF
In the 5 years’ follow-up, the association between rs35705950 and prognosis was detected using Kaplan-Meier method and log-rank test. Carriers of the minor allele rs35705950T that increased mortality (P = 0.0294, Figure 2).
Figure 2.
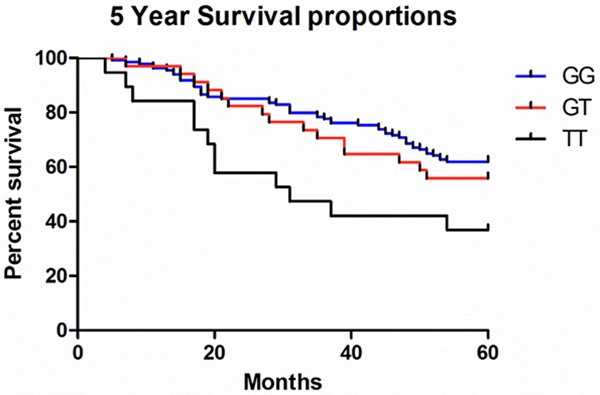
Analysis of the overall survival rate of the patients with SNP rs35705950 using Kaplan-Meier method and log-rank test.
Discussion
This study investigates the relationship between the MUC5B polymorphism rs35705950 and risk of idiopathic pulmonary fibrosis susceptibility, severity, and the overall survival. We observed that the minor allele T was associated with increased IPF susceptibility. In addtion, the increased minor allele T was also significantly correlated with aggressive clinicopathological features, namely, the decreased FVC and DLco. The results of Kaplan-Meier analyses showed that IPF patients with the increased T allele tend to have shorter overall survival.
IPF is associated with abnormal invasive fibroproliferation and enhanced myofibroblast transformation [7]. This results in the excessive deposition of collagen and other extracellular matrix components within the lung interstitium, which promotes the progressive loss of alveolar and interstitial [7,8] architecture, impaired gas exchange, and respiratory failure. Despite recent therapeutic advances, IPF remains refractory to pharmacologic therapy and is invariably fatal with amedian survival of 2-3 years.
The single-nucleotide polymorphism (SNP) rs35705950 is located 3 kb upstream of the MUC5B transcription start site on the gene encoding the Mucin 5 subtype B, which is a gel-forming mucin and a major component of mucus in the respiratory tract [9,10]. Seibold et al. first used linkage and fine mapping to identify a region of interest on the p-terminus of chromosome 11 that included gel-forming mucin genes [11]. The polymorphism of the MUC5B gene has a profound effect on the risk of familial interstitial pneumonia and sporadic IPF in the American population [11,12] and was recently confirmed in 2 genome wide association studies [4,13]. The strong assotion of the MUC5B variant with IPF was also recently confirmed in other European Caucasian populations, including Italian, French, and British cohorts. The subjects who were heterozygous or homozygous for the minor allele of this MUC5B polymorphism rs35705950 have a significantly increased risk for IPF (OR = 6.8 and 20.8) and for sporadic IPF (OR = 9.0 and 21.8), respectively [11]. Interestingly a trend was observed by Stock and colleagues between the MUC5B variant and slower decline in forced vital capacity, whereas no difference was evidenced in this cohort regarding age or severity at diagnosis between carriers or non carriers of the T allele risk [14]. Borie et al. reported that the distribution of the rs35705950 T-allele risk did not differ by gender in Caucasian populations. However, recent gene association studies in the Caucasian population also confirmed an association with IPF but a lack of association with systemic sclerosis-ILD or sarcoidosis [14-16]. The MUC5B rs35705950 T allele has been recently shown to be associated with better survival among patients with IPF [17].
The exact role of the polymorphism rs35705950 in IPF pathophysiology remains to be determined, Seibold et al. suggested it was functional [11]. MUC5B is the major gel-forming mucin in the normal distal airway epithelium. Indeed, the carriage of the T allele was found associated with a 37 fold increased expression of MUC5B gene in the lung in unaffected subjects [11], and with a 14-fold increased expression in IPF patients when compared to controls [14]. Also in contrast to chronic obstructive pulmonary disease, MUC5B was found the predominant mucin in the abnormal mucus cells in patients with IPF [18,19], which may be related to the abnormal differentiation of the respiratory epithelium. MUC5B may interfere with the normal repair process of the alveolar epithelium. For example, MUC5B overexpression introduces an aggressive behavior of breast cancer MCF7 cells, like increased proliferation and invasion in vitro [20].
There are some limitations in this study. First, the sample size is relatively small. Given the infrequency of the T allele of rs35705950, further investigations with larger number of subjects are needed for further validation. Second, there are distinctions in smoking histories between patients and health controls. Third, it is only an observation study with limited evidence level, and no functional study is performed. Despite these limitations, the results from our study are important in that we can successfully validate the association between the MUC5B polymorphism and IPF susceptibility and severity in the Chinese Han population.
In conclusion, data from this study demonstrated that the MUC5B polymorphism rs35705950 is associated with increased risk of idiopathic pulmonary fibrosis susceptibility, severity, and the decreased overall survival.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81170136, 81100147, 81300103, 81300219), Taishan Scholar Program of Shandong Province (ts20130911), Specialized Research Fund for the Doctoral Program of Higher Education (20130131110048), Key Technology Research and Development Program of Science and Technology of Shandong Province (2014kjhm0102), Roadmap for Science and Technology Development of Binzhou City (2014ZC0102).
Disclosure of conflict of interest
None.
References
- 1.Lai CC, Wang CY, Lu HM, Chen L, Teng NC, Yan YH, Wang JY, Chang YT, Chao TT, Lin HI, Chen CR, Yu CJ, Wang JD. Idiopathic pulmonary fibrosis in Taiwan-a population-based study. Respir Med. 2012;106:1566–1574. doi: 10.1016/j.rmed.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 2.Donnelly SC. Review Series--Inflammation & Fibrosis. Introduction. QJM. 2012;105:503. doi: 10.1093/qjmed/hcs067. [DOI] [PubMed] [Google Scholar]
- 3.Mahendran S, Sethi T. Treatments in idiopathic pulmonary fibrosis: time for a more targeted approach? QJM. 2012;105:929–934. doi: 10.1093/qjmed/hcs076. [DOI] [PubMed] [Google Scholar]
- 4.Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, Loyd JE, Cosgrove GP, Lynch D, Groshong S, Collard HR, Wolters PJ, Bradford WZ, Kossen K, Seiwert SD, du Bois RM, Garcia CK, Devine MS, Gudmundsson G, Isaksson HJ, Kaminski N, Zhang Y, Gibson KF, Lancaster LH, Cogan JD, Mason WR, Maher TM, Molyneaux PL, Wells AU, Moffatt MF, Selman M, Pardo A, Kim DS, Crapo JD, Make BJ, Regan EA, Walek DS, Daniel JJ, Kamatani Y, Zelenika D, Smith K, McKean D, Pedersen BS, Talbert J, Kidd RN, Markin CR, Beckman KB, Lathrop M, Schwarz MI, Schwartz DA. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45:613–620. doi: 10.1038/ng.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, Broderick SM, Wade MS, Hysi P, Scuirba J, Richards TJ, Juan-Guardela BM, Vij R, Han MK, Martinez FJ, Kossen K, Seiwert SD, Christie JD, Nicolae D. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. 2013;1:309–317. doi: 10.1016/S2213-2600(13)70045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr, Kondoh Y, Myers J, Müller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schünemann HJ ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y, Jiang D, Liang J, Meltzer EB, Gray A, Miura R, Wogensen L, Yamaguchi Y, Noble PW. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J Exp Med. 2011;208:1459–1471. doi: 10.1084/jem.20102510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 9.Thornton DJ, Devine PL, Hanski C, Howard M, Sheehan JK. Identification of two major populations of mucins in respiratory secretions. Am J Respir Crit Care Med. 1994;150:823–832. doi: 10.1164/ajrccm.150.3.8087358. [DOI] [PubMed] [Google Scholar]
- 10.Kirkham S, Kolsum U, Rousseau K, Singh D, Vestbo J, Thornton DJ. MUC5B is the major mucin in the gel phase of sputum in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;178:1033–1039. doi: 10.1164/rccm.200803-391OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, Fingerlin TE, Zhang W, Gudmundsson G, Groshong SD, Evans CM, Garantziotis S, Adler KB, Dickey BF, du Bois RM, Yang IV, Herron A, Kervitsky D, Talbert JL, Markin C, Park J, Crews AL, Slifer SH, Auerbach S, Roy MG, Lin J, Hennessy CE, Schwarz MI, Schwartz DA. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364:1503–1512. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Noth I, Garcia JG, Kaminski N. A variant in the promoter of MUC5B and idiopathic pulmonary fibrosis. N Engl J Med. 2011;364:1576–1577. doi: 10.1056/NEJMc1013504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, Broderick SM, Wade MS, Hysi P, Scuirba J, Richards TJ, Juan-Guardela BM, Vij R, Han MK, Martinez FJ, Kossen K, Seiwert SD, Christie JD, Nicolae D, Kaminski N, Garcia JG. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. 2013;1:309–317. doi: 10.1016/S2213-2600(13)70045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stock CJ, Sato H, Fonseca C, Banya WA, Molyneaux PL, Adamali H, Russell AM, Denton CP, Abraham DJ, Hansell DM, Nicholson AG, Maher TM, Wells AU, Lindahl GE, Renzoni EA. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax. 2013;68:436–441. doi: 10.1136/thoraxjnl-2012-201786. [DOI] [PubMed] [Google Scholar]
- 15.Peljto AL, Steele MP, Fingerlin TE, Hinchcliff ME, Murphy E, Podlusky S, Carns M, Schwarz M, Varga J, Schwartz DA. The pulmonary fibrosis-associated MUC5B promoter polymorphism does not influence the development of interstitial pneumonia in systemic sclerosis. Chest. 2012;142:1584–1588. doi: 10.1378/chest.12-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borie R, Crestani B, Dieude P, Nunes H, Allanore Y, Kannengiesser C, Airo P, Matucci-Cerinic M, Wallaert B, Israel-Biet D, Cadranel J, Cottin V, Gazal S, Peljto AL, Varga J, Schwartz DA, Valeyre D, Grandchamp B. The MUC5B variant is associated with idiopathic pulmonary fibrosis but not with systemic sclerosis interstitial lung disease in the European Caucasian population. PLoS One. 2013;8:e70621. doi: 10.1371/journal.pone.0070621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peljto AL, Zhang Y, Fingerlin TE, Ma SF, Garcia JG, Richards TJ, Silveira LJ, Lindell KO, Steele MP, Loyd JE, Gibson KF, Seibold MA, Brown KK, Talbert JL, Markin C, Kossen K, Seiwert SD, Murphy E, Noth I, Schwarz MI, Kaminski N, Schwartz DA. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA. 2013;309:2232–2239. doi: 10.1001/jama.2013.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plantier L, Crestani B, Wert SE, Dehoux M, Zweytick B, Guenther A, Whitsett JA. Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax. 2011;66:651–657. doi: 10.1136/thx.2010.151555. [DOI] [PubMed] [Google Scholar]
- 19.Seibold MA, Smith RW, Urbanek C, Groshong SD, Cosgrove GP, Brown KK, Schwarz MI, Schwartz DA, Reynolds SD. The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS One. 2013;8:e58658. doi: 10.1371/journal.pone.0058658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valque H, Gouyer V, Gottrand F, Desseyn JL. MUC5B leads to aggressive behavior of breast cancer MCF7 cells. PLoS One. 2012;7:e46699. doi: 10.1371/journal.pone.0046699. [DOI] [PMC free article] [PubMed] [Google Scholar]