

Abstract
Systemic amyloidosis is characterized by amyloid deposition throughout the body and subsequent dysfunction of various organs. Although pulmonary amyloidosis does occur, pulmonary hypertension (PH) caused by amyloidosis is extremely rare. In most of these cases, amyloid deposition occurred diffusely in alveolar septa, indicating that PH was due to lung disease and/or hypoxia. On the other hand, the mechanism of PH due to amyloid deposition in the pulmonary arteries has never been demonstrated. Here, we report the first case of PH due to amyloid deposition in pulmonary elastic arteries and muscular artery, which was complicated by multiple myeloma (MM). In the autopsy specimen of the patient, amyloid deposition was found mainly in the pulmonary arterial media, along with intimal thickening with luminal narrowing. PH thus appeared to be caused by marked decrease of pulmonary elasticity due to the amyloid deposition in the arterial media that resulted in stasis of the blood flow and subsequent luminal narrowing. Our present data demonstrates a new concept of PH caused by amyloidosis, namely, pulmonary arterial hypertension due to amyloidosis.
Keywords: AL amyloidosis, pulmonary arterial hypertension, multiple myeloma, pulmonary artery
Introduction
Systemic amyloidosis is caused by the deposition of amyloid in various organs, subsequently causing their dysfunction. However, pulmonary dysfunction due to amyloid deposition in the lungs is rare [1,2], and in particular, pulmonary hypertension (PH) caused by amyloidosis is an extremely rare condition; only 13 cases have been reported in the English literature to date (Table 1) [3-11]. In most of these cases, amyloid deposition was found diffusely in alveolar septa; amyloid deposition on the media of the pulmonary elastic and muscular arteries causing pulmonary arterial hypertension (PAH) has never been reported. Here, we present a first case of PAH due to pulmonary vascular amyloid deposition in a patient with multiple myeloma (MM), and demonstrate a new mechanism of PAH due to pulmonary amyloid deposition.
Table 1.
Previous reports of cases of pulmonary hypertension with amyloidosis
| Case | Author (year) | Age (sex) | Pattern of pulmonary amyloid deposition (method of detection) | PAP (mmHg) | Amyloidosis type | MM |
|---|---|---|---|---|---|---|
| 1 | Shiue ST, et al (3) (1988) | 65 (F) | Diffuse vascular deposition with little in alveolar capillaries (open lung biopsy) | 39 | AL | (+) |
| 2 | Johnson WJ, et al (4) (1991) | 48 (F) | Small blood vessels including alveolar capillaries (autopsy) | 97 | AA-FMF | (-) |
| 3 | Lutz AE, et al (5) (1995) | 61 (F) | Small peripheral vessels (autopsy) | 75* | β2M | (-) |
| 4 | Chapman AD, et al (6) (1999) | 91 (F) | Alveolar septum (autopsy) | ND | AL | (-) |
| 5 | Dingli D, et al (7) (2001) | 61 (F) | ND | 58 | AL-IgGκ | (+) |
| 6 | Dingli D, et al (7) (2001) | 64 (F) | ND | 36* | AL-IgGλ | (+) |
| 7 | Dingli D, et al (7) (2001) | 82 (M) | ND | 76* | AL-IgGλ | (-) |
| 8 | Dingli D, et al (7) (2001) | 54 (F) | Wide spread vascular deposition (autopsy) | 48 | AL-IgGκ | (-) |
| 9 | Dingli D, et al (7) (2001) | 48 (F) | Wide spread vascular deposition (autopsy) | 62 | AA-FMF | (-) |
| 10 | Erdem H, et al (8) (2006) | 60 (F) | Alveolar septum (lung biopsy) | 40* | AA-FMF | (-) |
| 11 | Eder L, et al (9) (2007) | 73 (F) | ND | 90* | AL | (+) |
| 12 | Lehtonen J, et al (10) (2007) | 48 (M) | ND | 67 | AL-IgGκ | (+) |
| 13 | Arslan S, et al (11) (2015) | 26 (M) | Vasculature of bronchus (transbronchial biopsy) | 65* | AA | (-) |
| 14 | Present case (2015) | 85 (F) | Pulmonary vascular deposition with little in alveolar capillaries (autopsy) | 60* | AL-IgGκ | (+) |
PAP: pulmonary arterial pressure, ND: not described, FMF: familial Mediterranean fever, β2M: beta-2 microglobulin, MM: multiple myeloma.
Estimated by echocardiography.
Clinical history
An 83-year-old woman was admitted to hospital owing to pneumonia. She had previously had pneumonia twice, at 74-years and 76-years of age. Her previous doctor suspected MM from her data of hypergammaglobulinemia, hypercalcemia, elevated serum creatinine level, and anemia. After treatment of the pneumonia, she was referred to our hospital. At admission, her serum hemoglobin level was 8.7 g/dL, calcium level corrected by her albumin level was 9.8 mg/dL, creatinine level was 1.50 mg/dL, and beta-2 microglobulin was 8.9 mg/dL (normal: 0.9-1.9). Her total protein level was high at 9.5 g/dL, and the M protein was observed by analysis of the protein fraction, and immunoelectrophoresis revealed immunoglobulin (Ig) G-κ type. X-ray photography displayed multiple punched-out lesions in her bones. Thirty-four percent of the nucleated cells in her bone marrow aspiration material were monoclonal plasmacytoid cells. These data led to a diagnosis of stage III MM according to the International Staging System.
After the diagnosis, the patient was treated with chemotherapy. At the beginning of chemotherapy, her right ventricular pressure estimated by cardiac echography was 39 mmHg, showing slight high pressure; however no other cardiac dysfunction was observed. Initially, the patient was treated with 5 courses of melphalan-prednisolone therapy. Although stable disease was obtained, severe myelosuppression terminated the therapy. Thereafter, she was treated with lenalidomide-dexamethasone (Rd) therapy, high-dose dexamethasone therapy, combination therapy of bortezomib and dexamethasone, and combination therapy of cyclophosphamide, vincristine, and dacarbazine for 2 years and 5 months. Severe side effects or restricted therapeutic effects resulted in the changes in treatment regimens. The patient was then re-admitted because of severe myelosupression and a worsened state of MM. She was treated by Rd therapy, which was highly effective despite her past history of severe side effects.
At this time, her cardiac echography demonstrated an ejection fraction of 79%, indicating normal left ventricular contractile activity, along with normal left ventricular expansion ability. However, her estimated right ventricular pressure was 60 mmHg, suggesting PH (Figure 1). Further investigation for PH was difficult because of her poor general state. The patient’s advanced status of MM, decreased blood pressure, and renal dysfunction worsened her general status, and she died 1 month after the last admission. Subsequently, an autopsy was performed.
Figure 1.
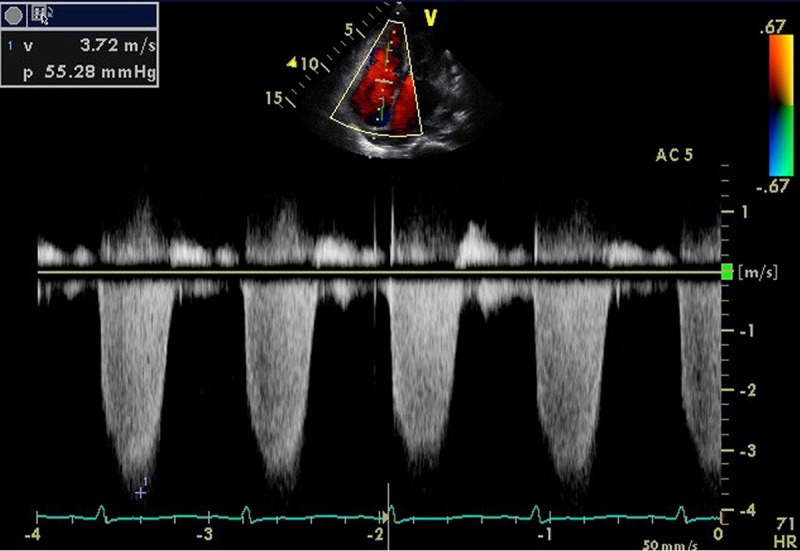
Antemortem cardiac echography. Echocardiography demonstrated that the pressure gap between the right atrium and right ventricle estimated by the speed of tricuspid regurgitation was 55 mmHg, suggesting pulmonary hypertension.
Autopsy findings
At autopsy, multiple osteolytic lesions containing massive plasmacytoid cells were found in the patient’s ileum, ribs, and vertebrae. Immunohistochemical analysis showed that the plasmacytoid cells were positive for CD138. Furthermore, polymerase chain reaction analysis of the genome of these plasmacytoid cells, to amplify the complementarity determining 3 region of the Ig heavy chain showed a monoclonal band upon electrophoresis, revealing that they were residual myeloma cells. In addition, the deposition of amorphous eosinophilic material was found in systemic blood vessels, mainly in the vascular media of elastic arteries and relatively thick muscular arteries (outer media > 100 μm). The deposition of eosinophilic material was also seen in muscles, mainly smooth muscles of multiple organs. These materials were stained with the direct fast scarlet stain, and double refraction was confirmed by polarization, which indicated amyloid deposition. Immunohistochemical analysis showed that these amyloid deposits were positive for amyloid P component, IgG, and Igκ, and negative for amyloid A component, Igλ, beta-2 microglobulin, and transthyretin. Thus, the patient was also diagnosed as having systemic amyloid light chain (AL) amyloidosis.
The pulmonary arteries in the pulmonary hilar region were dilated, and her right ventricle was also slightly dilated, which was compatible with PH. Furthermore, amyloid deposition was also found in the pulmonary arteries and veins; in the pulmonary elastic arteries, amyloid deposition was seen on the vascular walls, mainly on the media (Figure 2A), along with torn medial elastic fibers (Figure 2B). Amyloid deposition was also observed in the relatively thick muscular arteries and veins (thicker than interlobular veins). In muscular arteries, amyloid diffusely deposited on medial muscles (Figure 3A), accompanied by intimal thickening with luminal narrowing (Figure 3B). Furthermore, recanalization of organized thrombus was partially observed (Figure 3C). But amyloid deposition was rarely detected in the thin vessels, capillary or alveolar septa. Plexiform lesions, fibrinoid necrosis, and vasculitis were not identified.
Figure 2.

Pulmonary hilar elastic arteries. A. Direct fast scarlet staining of pulmonary hila demonstrated amyloid deposition in the pulmonary elastic arterial media and muscular arterial wall without alveolar involvement. B. Elastica-Masson staining of the pulmonary arteries demonstrated that the patient’s medial elastic fibers were torn by amyloid deposition.
Figure 3.
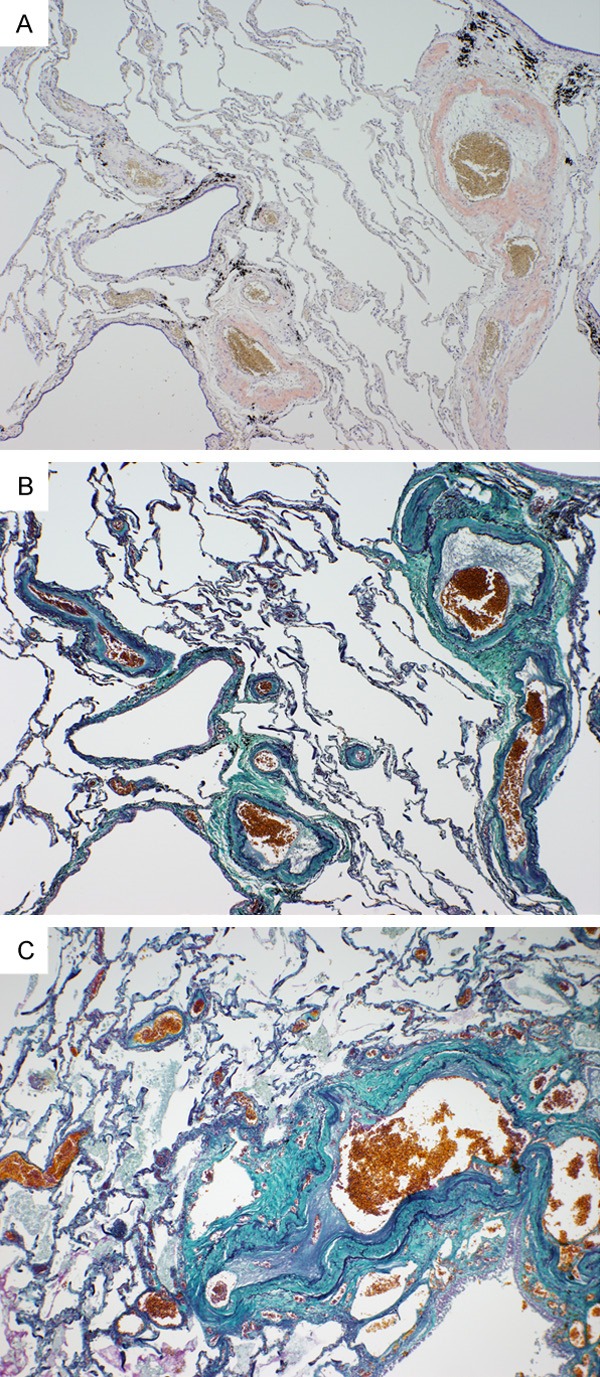
Pulmonary muscular arteries with luminal narrowing. A. Direct fast scarlet staining showed diffuse amyloid deposition on medial muscles of the relatively thick muscular arteries. B. Intimal thickening with luminal narrowing along with amyloid deposition on media of muscular artery was revealed by Elastica-Masson staining. C. Recanalization of organized thrombus was partially observed.
The cause of the renal dysfunction was concluded to be myeloma kidney and decreased blood flow caused by amyloidosis in the kidneys without deposition in the glomeruli.
The patient’s cause of death was considered to be a worsened general status caused by the MM, as well as amyloidosis that caused the PH and renal dysfunction.
Discussion
We presented a case of a patient with MM accompanying systemic AL amyloidosis and PH. The unique finding of this case was the pattern of pulmonary amyloid deposition. Pulmonary amyloid deposition has been classified into 3 main types: nodular lesion, tracheobronchial deposition, and diffuse alveolar septal deposition [12]. The present case lacked all these 3 patterns. Instead, amyloid deposition in the media of the elastic and muscular pulmonary arteries was found.
PH is generally classified into different groups by their mechanism, and most of the reported 13 cases of PH associated with systemic amyloidosis, with amyloid deposition in alveolar septa, would be classified into the group of PH due to lung disease and/or hypoxia [13]. On the other hand, the present case should be classified into the group of PAH, which has never been reported in association with pulmonary amyloidosis. The mechanism of PAH was thought to be attributed to marked decrease in pulmonary elasticity due to the destruction of pulmonary medial elastic fibers and muscles by amyloid deposition, leading to stasis of the blood flow, subsequently causing thrombus with recanalization or luminal narrowing. Luminal narrowing is one of the most important factors presented by Poiseulle equation of flow rate [14]. Other diseases that cause PH, such as idiopathic PAH, left ventricular systolic or diastolic dysfunction, and lung disease were excluded by antemortem echography or autopsy findings.
The first case of PH caused by amyloidosis was reported in 1988, in which the patient was originally diagnosed with MM [3]. In this patient, amyloid deposition in the muscular arterial wall and in a few alveolar septa was demonstrated by open lung biopsy. The authors hypothesized that MM-associated amyloidosis causing PH might have different characteristics to the other types of amyloidosis. However, amyloid distribution could not be investigated as postmortem examinations were not performed. Since then, 4 additional cases of PH with amyloidosis in patients with MM have been reported [7,9,10], but autopsy findings have not been described so far (Table 1).
Here, we present the first case of PAH due to amyloidosis, and demonstrate a new mechanism of PH caused by amyloidosis. The findings of this case shed light on a new concept of pulmonary amyloidosis.
Acknowledgements
We would like to thank Dr. Kei Hara for his academic advice, and Asako Shindo, Yoshiko Miyazaki, Goichiro Yanagi, and the clinical technologists of NTT Medical Center Tokyo for their technical assistance.
Disclosure of conflict of interest
None.
References
- 1.Utz JP, Swensen SJ, Gertz MA. Pulmonary amyloidosis: the Mayo Clinic experience from 1980 to 1993. Ann Intern Med. 1996;124:407–413. doi: 10.7326/0003-4819-124-4-199602150-00004. [DOI] [PubMed] [Google Scholar]
- 2.Travis WD, Colby TV, Koss MN, Rosado-de-Christenson ML, Müller NL, King TE. Miscellaneous disease of uncertain etiology. In: King DW, editor. Atlas of nontumor pathology: non-neoplastic disorders of the lower respiratory tract. 1st edition. Washington DC: American Registry of Pathology and Armed Forced Institute of Pathology; 2002. pp. 857–900. [Google Scholar]
- 3.Shiue ST, McNally DP. Pulmonary hypertension from prominent vascular involvement in diffuse amyloidosis. Arch Intern Med. 1988;148:687–689. [PubMed] [Google Scholar]
- 4.Johnson WJ, Lie JT. Pulmonary hypertension and familial Mediterranean fever: a previously unrecognized association. Mayo Clin Proc. 1991;66:919–925. doi: 10.1016/s0025-6196(12)61579-1. [DOI] [PubMed] [Google Scholar]
- 5.Lutz AE, Scneider U, Ehlerding G, Frenzel H, Koch KM, Kühn K. Right ventricular cardiac failure and pulmonary hypertension in a longterm dialysis patient-unusual presentation of visceral beta 2-microglobulin amyloidosis. Nephrol Dial Transplant. 1995;10:555–558. doi: 10.1093/ndt/10.4.555. [DOI] [PubMed] [Google Scholar]
- 6.Chapman AD, Brown PA, Kerr KM. Right heart failure as the dominant clinical picture in a case of primary amyloidosis affecting the pulmonary vasculature. Scot Med J. 1999;44:116–117. doi: 10.1177/003693309904400407. [DOI] [PubMed] [Google Scholar]
- 7.Dingli D, Utz JP, Gertz MA. Pulmonary hypertension in patients with amyloidosis. Chest. 2001;120:1735–1738. doi: 10.1378/chest.120.5.1735. [DOI] [PubMed] [Google Scholar]
- 8.Erdem H, Simsek I, Pay S. Diffuse pulmonary amyloidosis that mimics interstitial lung disease in a patient with Familial Mediterranean fever. J Clin Rheumatol. 2006;12:34–36. doi: 10.1097/01.rhu.0000200424.58122.38. [DOI] [PubMed] [Google Scholar]
- 9.Eder L, Zisman D, Wolf R, Bitterman H. Pulmonary hypertension and amyloidosis--an uncommon association: a case report and review of the literature. J Gen Intern Med. 2007;22:416–419. doi: 10.1007/s11606-006-0052-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lehtonen J, Kettunen P. Pulmonary hypertension as a dominant clinical picture in a case of amyloidosis and smoldering multiple myeloma. Int J Cardiol. 2007;115:e29–e30. doi: 10.1016/j.ijcard.2006.07.059. [DOI] [PubMed] [Google Scholar]
- 11.Arslan S, Ucar R, Yavsan DM, Esen H, Maden E, Reisli I, Calıskaner AZ. Common variable immunodeficiency and pulmonary amyloidosis: a case report. J Clin Immunol. 2015;35:344–347. doi: 10.1007/s10875-015-0151-7. [DOI] [PubMed] [Google Scholar]
- 12.Gillmore JD, Hawkins PN. Amyloidosis and the respiratory tract. Thorax. 1999;54:444–451. doi: 10.1136/thx.54.5.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, Olschewski H, Robbins IM, Souza R. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–D41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 14.Badeer HS. Hemodynamics for medical students. Adv Physiol Educ. 2001;25:44–52. doi: 10.1152/advances.2001.25.1.44. [DOI] [PubMed] [Google Scholar]