

Key points
ACh is an important modulator of breathing, including at the level of the retrotrapezoid nucleus (RTN), where evidence suggests that ACh is essential for the maintenance of breathing. Despite this potentially important physiological role, little is known about the mechanisms responsible for the cholinergic control of RTN function.
In the present study, we show at the cellular level that ACh increases RTN chemoreceptor activity by a CO2/H+ independent mechanism involving M1/M3 receptor‐mediated inositol 1,4,5‐trisphosphate/Ca+2 signalling and downstream inhibition of KCNQ channels.
These results dispel the theory that ACh is required for RTN chemoreception by showing that ACh, similar to serotonin and other modulators, controls the activity of RTN chemoreceptors without interfering with the mechanisms by which these cells sense H+.
By identifying the mechanisms by which wake‐on neurotransmitters such as ACh modulate RTN chemoreception, the results of the present study provide a framework for understanding the molecular basis of the sleep–wake state‐dependent control of breathing.
Abstract
ACh has long been considered important for the CO2/H+‐dependent drive to breathe produced by chemosensitive neurons in the retrotrapezoid nucleus (RTN). However, despite this potentially important physiological role, almost nothing is known about the mechanisms responsible for the cholinergic control of RTN function. In the present study, we used slice‐patch electrophysiology and pharmacological tools to characterize the effects of ACh on baseline activity and CO2/H+‐sensitivity of RTN chemoreceptors, as well as to dissect the signalling pathway by which ACh activates these neurons. We found that ACh activates RTN chemoreceptors in a dose‐dependent manner (EC50 = 1.2 μm). The firing response of RTN chemoreceptors to ACh was mimicked by a muscarinic receptor agonist (oxotremorine; 1 μm), and blunted by M1‐ (pirezenpine; 2 μm) and M3‐ (diphenyl‐acetoxy‐N‐methyl‐piperidine; 100 nm) receptor blockers, but not by a nicotinic‐receptor blocker (mecamylamine; 10 μm). Furthermore, pirenzepine, diphenyl‐acetoxy‐N‐methyl‐piperidine and mecamylamine had no measurable effect on the CO2/H+‐sensitivity of RTN chemoreceptors. The effects of ACh on RTN chemoreceptor activity were also blunted by inhibition of inositol 1,4,5‐trisphosphate receptors with 2‐aminoethoxydiphenyl borate (100 μm), depletion of intracellular Ca2+ stores with thapsigargin (10 μm), inhibition of casein kinase 2 (4,5,6,7‐tetrabromobenzotriazole; 10 μm) and blockade of KCNQ channels (XE991; 10 μm). These results show that ACh activates RTN chemoreceptors by a CO2/H+ independent mechanism involving M1/M3 receptor‐mediated inositol 1,4,5‐trisphosphate/Ca+2 signalling and downstream inhibition of KCNQ channels. Identifying the components of the signalling pathway coupling muscarinic receptor activation to changes in chemoreceptor activity may provide new potential therapeutic targets for the treatment of respiratory control disorders.
Key points
ACh is an important modulator of breathing, including at the level of the retrotrapezoid nucleus (RTN), where evidence suggests that ACh is essential for the maintenance of breathing. Despite this potentially important physiological role, little is known about the mechanisms responsible for the cholinergic control of RTN function.
In the present study, we show at the cellular level that ACh increases RTN chemoreceptor activity by a CO2/H+ independent mechanism involving M1/M3 receptor‐mediated inositol 1,4,5‐trisphosphate/Ca+2 signalling and downstream inhibition of KCNQ channels.
These results dispel the theory that ACh is required for RTN chemoreception by showing that ACh, similar to serotonin and other modulators, controls the activity of RTN chemoreceptors without interfering with the mechanisms by which these cells sense H+.
By identifying the mechanisms by which wake‐on neurotransmitters such as ACh modulate RTN chemoreception, the results of the present study provide a framework for understanding the molecular basis of the sleep–wake state‐dependent control of breathing.
Abbreviations
- 2‐APB
2‐aminoethoxydiphenyl borate
- CaM
calmodulin
- CK2
casein kinase 2
- CNQX
6‐cyano‐7‐nitroquinoxaline‐2,3‐dione
- DAG
diacylglycerol
- 4‐DAMP
4‐diphenyl‐acetoxy‐N‐methyl‐piperidine
- IP3
inositol 1,4,5‐trisphosphate
- Mec
mecamylamine
- PIP2
phosphatidylinositol 4,5‐bisphosphate
- PKC
protein kinase C
- PPT
pedunculopontine tegmental nucleus
- RTN
retrotrapezoid nucleus
- SK
small conductance K+
- TBB
4,5,6,7‐tetrabromobenzotriazole
Introduction
Respiratory activity is maintained unconsciously by the activity of respiratory chemoreceptors, including those located in the retrotrapezoid nucleus (RTN), which control respiratory output in response to changes in tissue CO2/H+ (Mulkey et al. 2004; Nattie & Li, 2012; Guyenet, 2014; Huckstepp et al. 2015). Despite the importance of RTN chemoreceptors, little is known about the mechanisms that support their activity, particularly during sleep when disruption of chemoreceptor function most often results in respiratory failure. Cholinergic neurons are most active during wakefulness and rapid eye movement (REM) sleep (Kubin & Fenik, 2004) and, although ACh is excitatory at most levels of the respiratory circuit, activation of cholinergic centres such as the pedunculopontine tegmental nucleus (PPT) typically results in suppression of respiratory output (Lydic & Baghdoyan, 1993) and increased respiratory variability (Saponjic et al. 2003). These results suggest that our understanding of cholinergic control of breathing is far from complete. The RTN receives cholinergic input from regions including the PPT (Ruggiero et al. 1990; Yasui et al. 1990; Ruggiero et al. 1997) and a longstanding hypothesis contends that cholinergic neurotransmission is a requisite component of RTN chemoreception (Metz, 1966; Fukuda & Loeschcke, 1979; Dev & Loeschcke, 1979 a; Nattie et al. 1989; Monteau et al. 1990). For example, early studies centred on understanding how the brain controls breathing suggested that cholinergic transmission at the level of the ventral medullary surface, in a region that later became known as the RTN (Smith et al. 1989), is required for maintenance of respiratory activity and CO2/H+‐sensitivity in vitro (Fukuda & Loeschcke, 1979; Monteau et al. 1990; Burton et al. 1994) and in vivo (Dev & Loeschcke, 1979 a; Nattie et al. 1989). Despite this potentially important physiological role, almost nothing is known about the mechanisms underlying the cholinergic modulation of RTN chemoreceptors. Evidence does suggest that ACh modulates RTN chemoreceptors by activation of M1 and M3 Gq‐coupled receptors (Nattie et al. 1989; Nattie & Li, 1990; Burton et al. 1994), although the signalling molecules and downstream ion channel targets contributing to this response are entirely unknown.
It is well established in other brain regions that ACh increases neuronal excitability by activation of muscarinic Gq‐coupled receptors (e.g, M1 and M3) and inhibition of KCNQ channels (Brown & Adams, 1980; Delmas & Brown, 2005). Gq signalling may inhibit KCNQ channels by depletion of phospholipid phosphatidylinositol 4,5‐bisphosphate (PIP2), an essential co‐factor for channel activity (Zhang et al. 2003; Delmas & Brown, 2005; Suh et al. 2006), or indirectly by calmodulin (CaM) and Ca2+ (Gamper & Shapiro, 2003), casein kinase 2 (CK2) (Kang et al. 2014) or protein kinase C (PKC) (Kosenko et al. 2012) dependent effects on channel affinity for PIP2. Recent evidence indicates that KCNQ channels regulate the activity of RTN chemoreceptors (Hawryluk et al. 2012; Hawkins et al. 2015). Therefore, we aimed to investigate whether these channels also contribute to the cholinergic modulation of RTN chemoreceptors.
In the present study, at the cellular level, we show that chemosensitive RTN neurons are strongly activated by ACh, and this response could be reduced by blockers of M1 and M3 receptor but not nicotinic receptors. Importantly, the function of RTN neurons as chemoreceptors was not dependent on cholinergic signalling because CO2/H+‐sensitivity was wholly retained during nicotinic or muscarinic receptor blockade. Furthermore, we show that ACh‐sensitivity could be reduced by blocking inositol 1,4,5‐trisphosphate (IP3) receptors, depleting intracellular Ca2+ stores, inhibiting CK2 or blocking downstream KCNQ channels, although it was retained during PKC blockade. These results identify several components of the signalling pathway that couple muscarinic receptor activation to changes in chemoreceptor excitability.
Methods
Animals
Animal use was in accordance with guidelines approved by the University of Connecticut Institutional Animal Care and Use Committee. Brain slices were isolated from neonatal rat pups (7–11 days old; n = 91). All efforts were made to minimize animal discomfort, as well as the number of animals used.
Brain slice preparation and slice‐patch electrophysiology
Slices containing the RTN were prepared as described previously (Mulkey et al. 2004; Hawryluk et al. 2012). Briefly, neonatal rats were decapitated under ketamine/xylazine anaesthesia, and transverse brainstem slices (300 μm) were cut using a microslicer (DSK 1500E; Dosaka, Kyoto, Japan) in ice‐cold substituted Ringer solution containing (in mm): 260 sucrose, 3 KCl, 5 MgCl2, 1 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, 10 glucose and 1 kynurenic acid. Slices were incubated for ∼30 min at 37°C, and subsequently at room temperature in normal Ringer solution (in mm): 130 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 1.25 NaH2PO4, 26 NaHCO3 and 10 glucose. Both substituted and normal Ringer solutions were bubbled with 95% O2–5% CO2 (extracellular pH 7.35).
Slices containing the RTN were transferred to a recording chamber mounted on a fixed‐stage microscope (Axioskop FS; Carl Zeiss, Oberkochen, Germany) and perfused continuously (∼2 ml min−1) with normal Ringer solution bubbled with 95% O2–5% CO2. Slices were exposed to hypercapnia by equilibrating bath solution with 10% or 15% CO2. All recordings were made with an Axopatch 200B patch‐clamp amplifier, digitized with a Digidata 1322A A/D converter and recorded using pCLAMP, version 10.0 (Molecular Devices, Sunnyvale, CA, USA). Recordings were obtained at room temperature (∼22°C) with patch electrodes pulled from borosilicate glass capillaries (Harvard Apparatus, Molliston, MA, USA) on a two‐stage puller (P89; Sutter Instrument, Novato, CA, USA) to a DC resistance of 5–7 MΩ when filled with an internal solution containing (in mm): 120 KCH3SO3, 4 NaCl, 1 MgCl2, 0.5 CaCl2, 10 Hepes, 10 EGTA, 3 Mg‐ATP and 0.3 GTP‐Tris (pH 7.2); electrode tips were coated with Sylgard 184 (Dow Corning, Midland, MI, USA). All recordings of neuronal firing rate were performed using the cell‐attached configuration and firing rate histograms were generated by integrating action potential discharge in 10 s bins and plotted using Spike, version 5.0 (Cambridge Electronic Design, Cambridge, UK).
Drugs
All drugs were bath applied at the concentrations: ACh (0.05–20 μm; Sigma‐Aldrich Co., St Louis, MO, USA), oxotremorine (1 μm; Sigma‐Aldrich Co.), nicotine (5 μm; Tocris), pirenzepine (2 μm; Tocris), 4‐diphenyl‐acetoxy‐N‐methyl‐piperidine (4‐DAMP) (100 nm; Tocris), meacamylamine (10 μm; Tocris), 2‐aminoethoxydiphenyl borate (2‐APB) (100 μm; Sigma‐Aldrich Co.), thapsigargin (10 μm; Tocris), 4,5,6,7‐tetrabromobenzotriazole (TBB) (10 μm), apamin (100 μm; Sigma‐Aldrich Co.), calphostin C (1 μm; Tocris), XE991 (10 μm; Tocris), 6‐cyano‐7‐nitroquinoxaline‐2,3‐dione (CNQX) (10 μm; Sigma‐Aldrich Co.), strychnine (2 μm; Sigma‐Aldrich Co.) and gabazine (10 μm; Sigma‐Aldrich Co.). A low Ca2+, high Mg2+ synaptic block solution was used to block excitatory synaptic input to RTN chemoreceptors. The composition of the synaptic block medium used in the present study is similar to normal Ringer solution, except that MgCl2 was increased to 11.4 mm, CaCl2 was decreased to 0.2 mm and, to maintain osmolality, NaCl was decreased to 124 mm. The efficacy of this synaptic blocking medium is well established (Hatton, 1982), including at the level of the RTN, where is it has been shown to block excitatory synaptic currents measured in RTN chemoreceptors (Wenker et al. 2012).
Statistical analysis
Data are reported as the mean ± SEM. Statistical analysis was performed using SigmaStat, version 3.0 (Systat Software Inc., Chicago, IL, USA). All data had a normal distribution (Kolmogorov–Smirnov goodness‐of‐fit test) and statistical significance was determined using a paired t test or one‐way ANOVA with Tukey's multiple comparisons as appropriate (P < 0.05). The relevant values used for statistical analysis are provided in the Results as appropriate.
Results
Cell‐attached recordings were used to identify RTN chemoreceptors in acute brainstem slices by their characteristic response to CO2 (i.e. they show a low level of spontaneous activity under control conditions) (0.2 ± 0.1 Hz, 5% CO2) and an increase in firing rate in response to 10% (1.8 ± 0.1 Hz) and 15% CO2 (2.1 ± 0.1 Hz) (Fig. 1 C). This level of CO2/H+‐sensitivity is similar to that reported previously for RTN chemosensitive neurons (Wenker et al. 2012). Furthermore, considering that CO2/H+‐sensitive RTN neurons identified in this manner were shown to be located in the same region of the RTN and had a neurochemical phenotype (i.e. glutamatergic and express the transcription factor Phox2b) similar to that of neurons shown to function as RTN chemoreceptors in vivo (Mulkey et al. 2004; Mulkey et al. 2007), we consider this an appropriate way of functionally identifying RTN chemoreceptors. RTN neurons that showed a < 1 Hz firing response to 10% CO2 or a < 1.5 Hz firing response to 15% CO2 were considered non‐chemosensitive.
Figure 1. RTN chemoreceptors are strongly activated by ACh and a non‐specific muscarinic agonist .
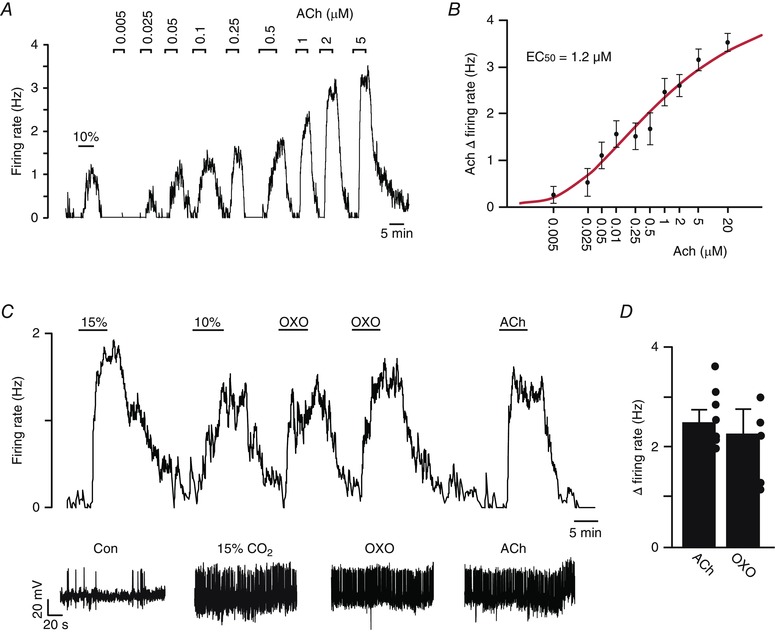
A, trace of firing rate showing the characteristic response of a chemosensitive RTN neuron to 10% CO2 and to graded increases in ACh. B, average firing rate at each concentration plotted (black dots) were fitted (continuous red line) to a logistic equation of the form: y = (a − c)/[1 + ([ACh]/EC50)b] + c, where a and c are the theoretical minimum and maximum, respectively, and b is a slope function (Li et al. 1998). Only neurons tested with at least two concentrations of ACh are included; each point represents data from three to 11 neurons. The calculated ACh EC50 was 1.2 μm. C, trace of firing rate from a chemoreceptor RTN neuron showing the firing response to 10 and 15% CO2. After return to control conditions (5% CO2), bath application of ACh (2 μm) or oxotremorine (OXO; 1 μm, n = 4), a non‐specific muscarinic receptor agonist, also increased activity of this chemosensitive neuron. D, summary graph (n = 4) to the right showing that ACh and OXO increased RTN chemoreceptor activity by 2.45 ± 0.3 and 1.95 ± 0.4, respectively.
Cholinergic modulation of RTN chemoreceptors involves M1 and M3 receptors
To identify cholinergic receptors that regulate RTN chemoreceptor function, we characterized the effects of cholinergic receptor agonists on the activity of RTN chemoreceptors under control conditions and during muscarinic or nicotinic receptor blockade. We found that bath application of ACh increased the activity of RTN chemoreceptors in a dose‐dependent manner with an EC50 of 1.2 μm (Fig. 1 A and B). For subsequent blocker experiments, we used 2 μm ACh to ensure that we obtained a near maximal response of RTN chemoreceptors to ACh (2.45 ± 0.2 Hz; T6 = –9.41, P = 0.003) (Fig. 1 D). We also found RTN neurons that did not respond to CO2/H+ also did not respond to ACh (0.4 ± 0.2 Hz; T2 = –1.57, P = 0.25), thus supporting the possibly that ACh discreetly activates H+‐sensitive RTN neurons (Dev & Loeschcke, 1979 a). In addition, the response of RTN chemoreceptors to ACh was mimicked by a muscarinic receptor agonist (Fig. 1 D); bath application of oxotremorine (1 μm) increased chemoreceptor activity by 1.9 ± 0.5 Hz (T4 = –3.93, P = 0.029).
To test whether cholinergic activation of RTN chemoreceptors is dependent on synaptic input, we exposed neurons to repeated bouts of ACh, first under control conditions, and then in the presence of a cocktail of neurotransmitter receptor blockers or in low Ca2+–high Mg2+ synaptic block medium. The blocker cocktail consisted of CNQX (10 μm) to block AMPA/kainite receptors, gabazine (10 μm) to block GABAA receptors and strychnine (2 μm) to block glycine receptors. Note that this concentration of strychnine can also block nicotinic receptors (Matsubayashi et al. 1998) but not muscarinic receptors (Bartolami et al. 1993); however, as noted below, nicotinic receptor blockade does not blunt the firing response to ACh (Fig. 4). As before, under control conditions, bath application of ACh (2 μm) increased the firing rate by 1.9 ± 0.14 Hz. After returning to control conditions, exposure to the blocker cocktail increased baseline activity from 1.0 ± 0.54 to 1.6 ± 0.47 Hz (T4 = –3.94, P = 0.017) (Fig. 2 A and B). In the continued presence of our blocker cocktail, exposure to ACh (2 μm) increased the firing rate by an amount similar to the control (1.8 ± 0.17; T4 = –2.33, P = 0.080) (Fig. 2 A and C). These results are consistent with the possibility that chemosensitive RTN neurons respond directly to ACh. However, RTN neurons also receive non‐glutamatergic excitatory input that would probably be preserved in our blocker cocktail. Therefore, we also tested ACh‐sensitivity when all excitatory input to RTN chemoreceptors was blocked with high Mg2+‐low Ca2+ synaptic block solution (Wenker et al. 2012). These experiments were performed in the same cells previously exposed to the blocker cocktail. Note that CNQX, gabazine and strychnine probably do not wash within the timeframe of these experiments. Exposure to low Ca2+‐high Mg2+ solution decreased baseline activity by 0.84 ± 0.10 (T3 = 8.99, P = 0.003), suggesting that RTN chemoreceptors may receive tonic non‐glutamatergic excitatory input. In low Ca2+‐high Mg2+ solution, exposure to ACh (2 μm) also increased the firing rate by an amount similar to the control (1.5 ± 0.21; T3 = 0.862, P = 0.452) (Fig. 2 A and C). Taken together, these results indicate that RTN chemoreceptors are directly activated by ACh.
Figure 4. RTN chemoreceptors are activated by nicotine but nicotinic receptor blockade did not blunt the firing response to ACh or CO2/H+ .
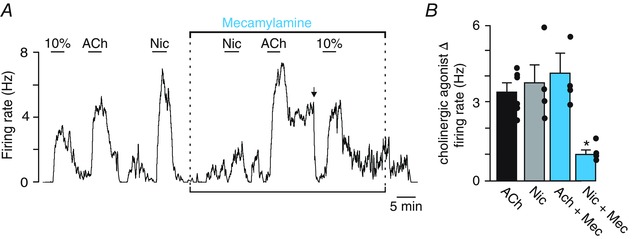
A, trace of firing rate from a chemosensitive RTN neuron showing typical responses to 10% CO2, ACh (2 μm) and nicotine (5 μm). Note that nicotine evoked a robust and reversible increase in excitability. Bath application of the nicotinic receptor antagonist mecamylamine (10 μm) had no effect on basal activity or the firing response to CO2/H+ or ACh; however, it did block nicotine responsiveness. B, graph to the right showing that mecamylamine did not affect the ACh responsiveness of these neurons (n = 6 cells) but it did attenuate the firing response to nicotine (n = 5 cells). *Significantly different from control (one‐way ANOVA followed by Tukey's multiple comparison test; P = 0.007).
Figure 2. Cholinergic activation of RTN chemoreceptors is not dependent on synaptic input .
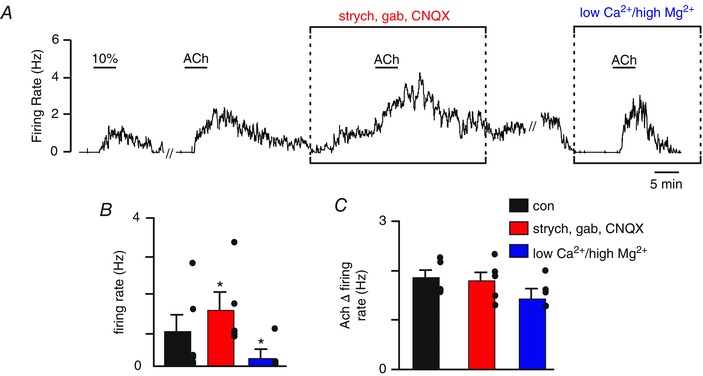
A, trace of firing rate showing the response of an RTN chemoreceptor to ACh (2 μm) under control conditions, in a neurotransmitter receptor blocker cocktail containing CNQX (10 μm), gabazine (gab; 10 μm) and strychnine (strych; 2 μm), and in high Mg2+‐low Ca2+ synaptic block solution. Under control conditions, bath application of ACh (2 μm) increased activity by ∼1.8 Hz. Subsequent exposure to the blocker cocktail increased baseline activity by ∼1 Hz. Once baseline activity in the blocker cocktail stabilized, a second exposure to ACh in the continued presence of blockers again increased activity by ∼1.7 Hz. After returning to control conditions, we then switched to a low Ca2+‐high Mg2+ solution to eliminate any residual excitatory synaptic input. Exposure to low Ca2+‐high Mg2+ solution suppressed baseline chemoreceptor activity but did not blunt the firing response to a third bout of ACh. B, summary data (n = 5) showing the effects of our blocker cocktail and low Ca2+‐high Mg2+ solution on baseline activity. C, summary data showing the ACh‐induced change in firing rate under control condition, in the blocker cocktail, and in low Ca2+‐high Mg2+ solution. // indicates a 20 min time break. *Significantly different from control (paired t test).
We consider M1 and M3 receptors as the leading candidates responsible for this response because previous evidence showed that M1 and M3 receptor blockers applied to the brainstem (Burton et al. 1994), or more specifically to the region of the RTN (Nattie et al. 1989; Nattie & Li, 1990; Nattie et al. 1994), decreased breathing and CO2 sensitivity. We tested this possibility using selective antagonists for M1 (pirenzepine, 2 μm) and M3 (4‐DAMP, 100 nm). We found that blockade of either M1 or M3 receptors had negligible effects on baseline activity or the firing response to CO2; however, each blocker decreased the ACh (2 μm) response of RTN chemoreceptors by 65% (2.4 ± 0.2 Hz vs. 0.9 ± 0.2 Hz in pirenzepine (F 2,6 = 17.24, P < 0.001); and 90% (3.1 ± 0.3 Hz vs. 0.3 ± 0.1 in 4‐DAMP; F 2,5 = 31.72, P < 0.001), respectively (Fig. 3). Furthermore, considering that nicotine applied to the ventral surface can mimic the stimulatory effects of ACh on breathing in anaesthetized cats (Dev & Loeschcke, 1979 a), and because ACh has been shown to activate inspiratory neurons in the nearby Pre‐Bötzinger complex by mechanisms involving both muscarinic and nicotinic receptors (Shao & Feldman, 2000, 2001, 2005, 2009), we also tested for the involvement of nicotinic receptors in the cholinergic modulation of RTN neurons. We found that application of nicotine (5 μm) increased RTN chemoreceptor activity by 3.6 ± 0.7 Hz (T3 = –4.57, P = 0.02) and bath application of the nicotinic ACh receptor antagonist mecamylamine (10 μm; Mec) decreased nicotine‐sensitivity (F 3,5 = 7.90, P = 0.007) (Fig. 4). However, Mec did not blunt the firing rate response elicited by ACh (3.9 ± 0.5 Hz vs. 4.0 ± 0.5 Hz in Mec) or 10% CO2 (Fig. 4). These results suggest that ACh modulates the activity of RTN chemoreceptors by a CO2/H+ independent mechanism involving M1‐ and M3‐receptors but not nicotinic receptors.
Figure 3. M1 and M3 receptor blockers suppressed the firing response of chemosensitive RTN neurons to ACh but not CO2/H+ .

A, trace of firing rate from a chemosensitive RTN neuron showing the firing response to 10% and 15% CO2. Under control conditions (5% CO2), bath application of ACh (2 μm) evoked a robust and reversible increase in firing rate. Bath application of the M1‐receptor antagonist pirenzepine (2 μm) had no effect on basal activity but significantly attenuated the firing rate response to ACh (2 μm). In the continued presence of pirezenpine, additional bouts of 10% and 15% CO2 increased the firing rate by an amount similar to control conditions. The firing response to ACh was potentiated after an ∼30 min wash in control media. Bar graph to right summarizes (n = 7 cells) the effects of pirenzepine on ACh responsiveness. B, firing rate trace from a chemosensitive RTN neuron showing typical responses to 15% CO2 and ACh (2 μm). Bath application of the M1/ M3‐receptor antagonist 4 ‐DAMP (100 nm) had no effect on basal activity but significantly attenuated the firing rate response to ACh. In the continued presence of 4‐DAMP, additional bouts of 15% CO2 increased the firing rate by an amount similar to control conditions. Bar graph to the right summarizes (n = 5 cells) the effect of 4‐DAMP on ACh responsiveness in these neurons. // indicates a 10 min time break. *Significantly different from control and wash (one‐way ANOVA followed by Tukey's multiple comparison test; P < 0.001).
Second messengers underlying the cholinergic modulation of RTN chemoreceptors
M1 and M3 are Gq‐coupled receptors that regulate neuronal excitability by activation of phospholipase C and subsequent hydrolysis of PIP2 to produce diacylglycerol (DAG) and IP3, which in turn triggers activation of PKC and Ca2+ release from intracellular stores, respectively (Delmas & Brown, 2005). As a first step towards identifying essential second messengers responsible for the cholinergic modulation of RTN chemoreceptors, we tested the effects of selective blockers of PKC and IP3 receptors on ACh responsiveness. A 10 min incubation in the PKC blocker calphostin C (1 μm) had no effect on basal activity or the firing rate response to ACh (1.8 ± 0.5 Hz vs. 2.9 ± 0.2 Hz in calphostin C; T2 = –2.33, P = 0.15). Conversely, IP3 receptor blockade with 2‐APB (100 μm) decreased ACh responsiveness by 55% (3.2 ± 0.6 Hz vs. 1.3 ± 0.2 Hz in 2‐APB; F 2,5 = 4.25, P = 0.038) (Fig. 5 A and C). These results identify IP3‐mediated Ca2+ release from intracellular stores as a key step in ACh modulation of RTN chemoreceptors. To test this possibility further, we re‐tested ACh‐sensitivity when intracellular Ca2+ stores were depleted with thapsigargin. Bath application of thapsigargin (10 μm) increased the baseline firing rate of RTN chemoreceptors by 1.7 ± 0.4 Hz (T5 = –4.24, P = 0.013) and essentially eliminated ACh‐sensitivity (Fig. 5 B and D). For example, in the continued presence of thapsigargin (with baseline adjusted by DC current injection), exposure to ACh increased the firing rate of RTN chemoreceptors by only 28% compared to control (3.5 ± 0.3 Hz vs. 1.7 ± 0.3 Hz first ACh in thapsigargin; F 4,6 = 9.90, P < 0.001) (Fig. 5 B and D). The firing rate response to ACh increased to 6.4 ± 0.5 Hz (T2 = –4.42, P = 0.05) after an ∼30 min wash in control media (Fig 5 D). Taken together, these results suggest that release of Ca2+ from intracellular stores is an important step in the cholinergic modulation of RTN neurons.
Figure 5. ACh activates RTN chemoreceptors by a mechanism involving IP3 receptor‐mediated Ca2+ release .
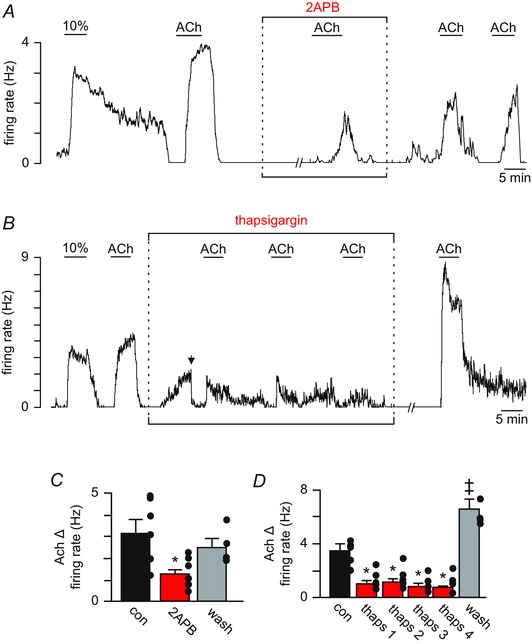
A, trace of firing rate from a chemosensitive RTN neuron showing that bath application of the IP3 receptor blocker 2‐APB (100 μm) had no effect on basal activity but significantly attenuated the firing rate response to ACh. B, trace of firing rate from a RTN chemosensitive neuron showing that bath application of thapsigargin (10 μm), a Ca2+‐ATPases blocker, increased the basal activity by ∼2.5 ± 0.3 Hz. In the continued presence of thapsigargin with baseline activity adjusted to near control levels by DC current injection (arrow), repeated exposures to ACh (2 μm) had minimal effect on cellular activity. The firing response to ACh increased ∼2‐fold after an ∼30 min wash in control media. C and D, summary data showing that 2‐APB (n = 6) (C) and thapsigargin (n = 7) (D) decreased the firing rate response of RTN chemoreceptors to ACh by 60% and 74%, respectively. // indicates a 10 min time break. *Significantly less than control or wash (one‐way ANOVA followed by Tukey's multiple comparison test; P < 0.05); ‡Significantly greater than control (paired t test; P < 0.05).
KCNQ channels contribute to the cholinergic modulation of RTN chemoreceptors
Voltage‐gated KCNQ channels have a high propensity for modulation by Gq‐signalling (Jentsch, 2000). In particular, activation of M1 or M3 receptors has been shown to inhibit KCNQ channel activity directly by PIP2 depletion and indirectly by Ca2+‐induced dissociation of calmodulin from KCNQ channels, which results in reduced channel affinity for PIP2 (Kosenko et al. 2012). Recent evidence also suggests that phosphorylation of CaM by CK2 strengthens its interaction with KCNQ, resulting in resistance to PIP2 depletion and increased channel activity (Kang et al. 2014). Based on this evidence and our finding that IP3‐mediated Ca2+ release is essential for the cholinergic modulation of RTN chemoreceptors, we tested for involvement of KCNQ channels and CK2 in the response of RTN neurons to ACh. We found that pharmacological blockade of KCNQ with XE991 (10 μm) increased baseline firing by ∼1 Hz and attenuated ACh responsiveness by ∼39% (3.1 ± 0.3 Hz vs. 1.9 ± 0.3 Hz in XE991; F 2,5 = 7.72, P = 0.014) (Fig. 6 A and B). Inhibition of CK2 with TBB (10 μm) had little effect on the resting activity of RTN chemoreceptors (T6 = –0.62, P = 0.57); however, TBB decreased the firing response to ACh (3.4 ± 0.7 Hz vs. 0.6 ± 0.1 Hz in TBB; F 2,6 = 9.86, P = 0.01) (Fig. 6 C and D). Considering that RTN chemoreceptors also express small conductance K+ (SK) channels (Hawryluk et al. 2012) and, in other cell types, Gq‐signalling has been shown to decrease SK channel activity by CK2‐mediated CaM phosphorylation, we also tested for the involvement of SK channels in the cholinergic modulation of RTN neurons. We found that cholinergic activation of RTN chemoreceptors was unaffected by SK channel blockade with 100 μm apamin (2.0 ± 0.2 Hz vs. 2.6 ± 0.2 Hz in apamin; T3 = –2.61, P = 0.08). These results highlight the importance of KCNQ channels in the regulation RTN chemoreceptor function, and provide a framework for understanding the molecular basis by which wake‐on neurotransmitters (e.g. serotonin and ACh) modulate breathing.
Figure 6. KCNQ channels are downstream targets of ACh in RTN chemoreceptors .
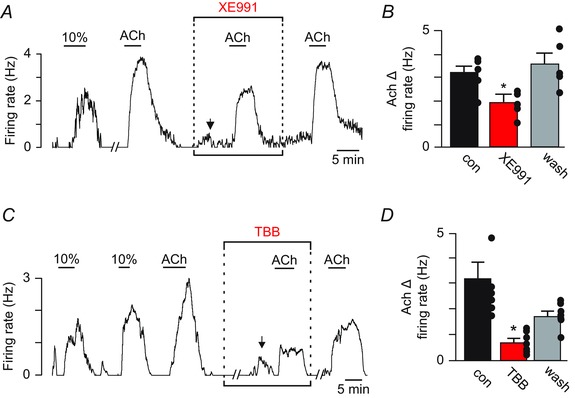
A, firing rate trace showing that bath application of the KCNQ channel blocker XE991 (10 μm) increased basal activity by ∼1.0 (n = 8). In the continued presence of XE991 with baseline activity was adjusted to near control levels by DC current injection (arrow), the firing response to ACh was reduced by 21% compared to control or after an ∼30 min wash in control solution. B, summary data (n = 5) showing that XE991 decreased ACh responsiveness of chemosensitive RTN neurons. Note that the molecular mechanisms contributing to the ACh response during KCNQ blockade remain undetermined. C, trace of firing rate from a RTN chemosensitive neuron showing that bath application of the CK2 inhibitor TBB (10 μm) had no effect on basal activity but significantly attenuated the firing rate response to ACh. D, summary data (n = 7) showing that TBB strongly suppressed the firing rate response of RTN chemoreceptors to ACh. // indicates a 10 min time break. *Significantly different from control (one‐way ANOVA followed by Tukey's multiple comparison test; P < 0.05).
Discussion
ACh is a powerful stimulus at multiple levels of the respiratory network including the RTN where cholinergic transmission is reportedly required for maintenance of respiratory activity and CO2/H+‐sensitivity (Fukuda & Loeschcke, 1979; Dev & Loeschcke, 1979 a; Nattie et al. 1989; Monteau et al. 1990; Burton et al. 1994). Despite this potentially important physiological role, almost nothing is known about the mechanisms underlying the cholinergic modulation of RTN chemoreceptors and breathing. In the present study, we show, at the cellular level, that ACh increases RTN chemoreceptor activity by a CO2/H+ independent mechanism involving M1/M3 receptor‐mediated IP3/Ca+2 signalling and downstream inhibition of KCNQ channels. These results identify components of the signalling pathway that couple activation of muscarinic receptors to changes in chemoreceptor excitability and, in doing so, provide a framework for understanding the molecular basis of state‐dependent control of breathing.
Experimental limitations
All experiments were conducted in the medullary brain slice preparation because it allows considerable control of the neuronal environment and easy access to neurons for patch‐clamp recording. However, it should be recognized that the tissue used in this preparation has been traumatized, subjected to oxidative stress in the form of hyperoxic incubation conditions (Mulkey et al. 2001) and neural network connections have been disrupted. Therefore, it will be important for future work to determine the role of cholinergic signalling in the RTN in the control of breathing in vivo, particularly across sleep–wake states. Our experiments were also limited to the use of animals less than ∼2 weeks of age. This is a potential issue because chemosensitive RTN neurons show a much larger firing response to CO2 in anaesthetized adult rats in vivo compared to in the neonatal slice preparation (Guyenet et al. 2005). Therefore, the results of the present study may not be fully representative of how chemosensitive RTN neurons respond to CO2 or neurotransmitters in adult animals in vivo. In addition, our experiments are limited by the use of exogenous drug application. This is a issue because bath application of cholinergic agonists may not mimic the discrete and rapid transient nature of endogenous neurotransmitter release, and differences in the spatiotemporal profile of cholinergic receptor activation can result in divergent neural responses (Unal et al. 2015). Therefore, it will be important for future studies to confirm that endogenous ACh modulates RTN chemoreceptor activity by a similar mechanism.
Cholinergic control of RTN chemoreceptor function
The RTN is an important locus of respiratory control. Neurons (Mulkey et al. 2004; Wang et al. 2013) and astrocytes (Gourine et al. 2010; Huckstepp et al. 2010; Wenker et al. 2010) in this region sense changes in CO2/H+ to produce an integrated CO2/H+‐dependent drive to other components of the respiratory circuit to regulate both inspiratory and expiratory activity (Guyenet, 2014). During the prenatal period, chemosensitive RTN neurons also contribute to inspiratory rhythmogenesis (Onimaru et al. 2008; Thoby‐Brisson et al. 2009) and, in adulthood, subsets of RTN neurons contribute to expiratory rhythm generation (Janczewski & Feldman, 2006; Huckstepp et al. 2015); thus, the RTN has a powerful influence on all aspects of breathing
A longstanding hypothesis contends that RTN chemoreceptor function is dependent on cholinergic transmission (Fukuda & Loeschcke, 1979; Dev & Loeschcke, 1979 a,b). This hypothesis is supported by evidence that hypercapnia increased ACh levels in putative brainstem chemosensitive areas (Metz, 1966), and application of ACh near the RTN stimulated respiratory activity, whereas atropine or more specific blockers of M1 and M3 receptors blunted the CO2 ventilatory response both in vitro (Fukuda & Loeschcke, 1979; Monteau et al. 1990; Eugenin & Nicholls, 1997; Coddou et al. 2009) and in anaesthetized animals (Dev & Loeschcke, 1979 a,b; Nattie et al. 1989; Nattie & Li, 1990). However, many of these early studies used high drug concentrations that may have had non‐specific effects contributing to the perceived cholinergic contribution to RTN chemosensitivity. Consistent with these previous studies, we found that ACh elicited a strong activation of chemosensitive RTN neurons even when glutamate, GABAA and glycine receptors were blocked and under experimental conditions that limited synaptic transmission (i.e. incubation in high Mg2+/low Ca2+ medium) (Figs 1 and 2). These results suggest that RTN chemoreceptors are directly activated by ACh. However, these experiments cannot exclude the involvement of RTN astrocytes, which are known to enhance the activity of chemosensitive neurons in a paracrine fashion by purinergic dependent mechanism (Gourine et al. 2010; Wenker et al. 2012). By contrast to previous work, we found that application of muscarinic receptor blockers at concentrations sufficient to attenuate the firing response to ACh had negligible effect on CO2/H+‐sensitivity of RTN chemoreceptors (Fig. 3). These results dispel the theory that ACh contributes to RTN chemoreception by showing that cholinergic signalling can modulate the activity of RTN neurons in a manner similar to serotonin, although it is not a requisite component of the mechanism by which these cells sense changes in CO2/H+.
Cholinergic neurons in the PPT are probably a source of cholinergic drive to the RTN (Ruggiero et al. 1990; Yasui et al. 1990; Ruggiero et al. 1997). These neurons exhibit wake‐ and REM‐dependent firing behaviour (Kubin & Fenik, 2004) and are known to participate in a wide range of state‐regulating functions including the control of breathing (Lydic & Baghdoyan, 1993; Saponjic et al. 2003). Therefore, connections between cholinergic PPT neurons and RTN chemoreceptors may serve as the anatomical basis for state‐dependent control of chemoreceptor activity. More generally, stimulation of the PPT elicits respiratory behaviour reminiscent of REM sleep, such as suppressed respiratory output (Lydic & Baghdoyan, 1993) and irregular breathing patterns (Saponjic et al. 2003). Although ACh is excitatory at most levels of the respiratory circuit including the RTN as shown in the present study, cholinergic neurons comprise only a minority of neurons in the PPT that are intermingled amongst a large number of GABAergic and glutamatergic neurons (Wang & Morales, 2009). Furthermore, PPT neurons innervate multiple levels of the respiratory circuit including pontine respiratory centres associated with expiration (e.g. parabrachial complex and Kölliker‐Fuse) (Saponjic et al. 2006). Therefore, PPT‐mediated respiratory depression (Lydic & Baghdoyan, 1993) and variability (Saponjic et al. 2003) probably results from either non‐cholinergic inhibition of respiratory centres or activation of regions associated with expiration. To fully understand the contribution of cholinergic drive to state‐dependent control of breathing, it will be important for future work to identify and selectively manipulate subsets of cholinergic PPT neurons with discrete projections to various levels of the respiratory system in vivo at the same time as measuring respiratory activity across natural sleep–wake states.
The molecular basis of cholinergic control of RTN function involves activation of M1 and M3 receptors and Gq‐mediated inhibition of KCNQ channels. Specifically, we show that chemosensitive RTN neurons are strongly activated by ACh (Fig. 1 A and B), and this response could be mimicked by application of oxotremorine (muscarinic receptor agonist) (Fig. 1 C and D) and blocked by pirenzepine (selective M1 receptor blocker) and 4‐DAMP (selective M3 receptor blocker) (Fig. 3). As noted above, these results are consistent with evidence that M1 and M3 receptors are expressed in the RTN (Nattie et al. 1994) and contribute to the cholinergic modulation of breathing in anaesthetized cats (Nattie et al. 1989; Nattie & Li, 1990). Also consistent with previous evidence that nicotine is a potent modulator of breathing (Shao & Feldman, 2001, 2005, 2009), including at the level of the RTN (Fukuda & Loeschcke, 1979; Dev & Loeschcke, 1979 a), we found that nicotine stimulated activity of RTN neurons (Fig. 4 A). However, because Mec, which blocks all nicotinic receptors except α7‐containing channels (Chamberlin et al. 2002), had no effect on ACh sensitivity (Fig. 4), we did not explore the role of these receptors further. M1 and M3 are Gq‐coupled receptors that, when stimulated, lead to the activation of phospholipase C β and subsequent hydrolysis of PIP2 to produce IP3 and DAG. Because depletion of PIP2 and production of IP3 and DAG all serve as potent intracellular signals (Kosenko et al. 2012), we considered these the most probable candidate messengers of cholinergic modulation of RTN chemoreceptors. Furthermore, because KCNQ channels are a classic downstream target of M1 and M3 signalling (Brown & Adams, 1980; Delmas & Brown, 2005) and these channels have been shown to control activity of RTN chemoreceptors (Hawryluk et al. 2012; Hawkins et al. 2015), we considered KCNQ channels to be the most probable effector coupling receptor activation to changes in neuronal excitability. Evidence suggests that KCNQ channels can be inhibited directly by PIP2 depletion (Jentsch, 2000; Zhang et al. 2003; Delmas & Brown, 2005; Suh et al. 2006), or indirectly by CaM and Ca2+ (Gamper & Shapiro, 2003) CK2 (Kang et al. 2014) or PKC (Kosenko et al. 2012) dependent effects of channel affinity for PIP2. Consistent with a role of IP3‐mediated Ca2+ release, we found that ACh‐sensitivity was strongly suppressed by blocking IP3‐receptors or by depleting intracellular Ca2+ stores (Fig. 5 A and D). These results are consistent with the involvement of KCNQ channels because these channels are known to be inhibited by increased intracellular Ca2+ (Gamper & Shapiro, 2003). The Ca2+ binding protein CaM is an essential co‐factor of KCNQ channel function, where an increase in Ca2+ causes CaM to dissociate from the channel and this in turn decreases the channel affinity for PIP2, resulting in decreased channel conductance (Kosenko & Hoshi, 2013). This mechanism has been established in homomeric KCNQ2 and heteromeric KCNQ2/3 channels, which are the KCNQ channels probably controlling RTN chemoreceptor activity (Hawryluk et al. 2012). In addition, CK2 has been shown to complex with KCNQ channels and phosphorylate CaM to facilitate its dissociation and channel closure (Kang et al. 2014). We showed that blocking either CK2 or KCNQ channels decreased cholinergic modulation of RTN chemoreceptor excitability (Fig. 6 A and D). These results clearly implicate KCNQ channels as downstream targets of cholinergic modulation of RTN chemoreceptors; however, because blocking CK2 can also inhibit KCNQ channel activity independently of cholinergic signalling (Kang et al. 2014), we do know whether CK2 is required for this transduction mechanism. Our evidence indicating that the disruption of IP3 receptors or Ca2+ release from intracellular stores almost eliminates the firing response to ACh suggests this as the main mechanism for the cholinergic modulation of RTN neurons. However, M1 and M3 receptor‐mediated PIP2 depletion can strongly inhibit KCNQ channels independent of Ca2+ or PKC signalling (Kosenko et al. 2012) and Ca2+ and CK2 regulate KCNQ activity by influencing channel affinity for PIP2. Therefore, PIP2 depletion likely contributes to the cholinergic modulation of RTN neurons.
In sum, we show that ACh increases RTN chemoreceptor activity in a dose‐dependent manner by mechanisms involving M1 and M3 receptors and Gq‐mediated inhibition of KCNQ channels. These results identify several components of the signalling pathway that couple muscarinic receptor activation to changes in chemoreceptor excitability and, in doing so, provide potential avenues for the therapeutic treatment of respiratory control problems associated with disordered breathing.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
CRS, TSM and DKM designed the experiments. CRS, FK and BFB collected and analysed data. TSM and DKM performed data analysis and drafted the article. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by funds from the National Institutes of Health Grants HL104101 (DKM) and public funding from the São Paulo Research Foundation (FAPESP) grants 2013/10573‐8 and 2009/54888‐7 (TSM); Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq); grant: 471283/2012‐6 (TSM). CNPq fellowship305533/2012‐6 (TSM).
References
- Bartolami S, Ripoll C & Eybalin M (1993). Anticholinergic effects of strychnine in the cochlea do not involve muscarinic receptors. Neuroreport 4, 1003–1006. [DOI] [PubMed] [Google Scholar]
- Brown DA & Adams PR (1980). Muscarinic suppression of a novel voltage‐sensitive K+ current in a vertebrate neurone. Nature 283, 673–676. [DOI] [PubMed] [Google Scholar]
- Burton MD, Nouri K, Baichoo S, Samuels‐Toyloy N & Kazemi H (1994). Ventilatory output and acetylcholine: perturbations in release and muscarinic receptor activation. J Appl Physiol (1985) 77, 2275–2284. [DOI] [PubMed] [Google Scholar]
- Chamberlin NL, Bocchiaro CM, Greene RW & Feldman JL (2002). Nicotinic excitation of rat hypoglossal motoneurons. Neuroscience 115, 861–870. [DOI] [PubMed] [Google Scholar]
- Coddou C, Bravo E & Eugenin J (2009). Alterations in cholinergic sensitivity of respiratory neurons induced by pre‐natal nicotine: a mechanism for respiratory dysfunction in neonatal mice. Philos Trans R Soc Lond B Biol Sci 364, 2527–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P & Brown DA (2005). Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci 6, 850–862. [DOI] [PubMed] [Google Scholar]
- Dev NB & Loeschcke HH (1979. a). A cholinergic mechanism involved in the respiratory chemosensitivity of the medulla oblongata in the cat. Pflügers Arch 379, 29–36. [DOI] [PubMed] [Google Scholar]
- Dev NB & Loeschcke HH (1979. b). Topography of the respiratory and circulatory responses to acetylcholine and nicotine on the ventral surface of the medulla oblongata. Pflügers Arch 379, 19–27. [DOI] [PubMed] [Google Scholar]
- Eugenin J & Nicholls JG (1997). Chemosensory and cholinergic stimulation of fictive respiration in isolated CNS of neonatal opossum. J Physiol 501, 425–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda Y & Loeschcke HH (1979). A cholinergic mechanism involved in the neuronal excitation by H+ in the respiratory chemosensitive structures of the ventral medulla oblongata of rats in vitro. Pflügers Arch 379, 125–135. [DOI] [PubMed] [Google Scholar]
- Gamper N & Shapiro MS (2003). Calmodulin mediates Ca2+‐dependent modulation of M‐type K+ channels. J Gen Physiol 122, 17–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine AV, Kasymov V, Marina N, Tang F, Figueiredo MF, Lane S, Teschemacher AG, Spyer KM, Deisseroth K & Kasparov S (2010). Astrocytes control breathing through pH‐dependent release of ATP. Science 329, 571–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG (2014). Regulation of breathing and autonomic outflows by chemoreceptors. Compr Physiol 4, 1511–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Mulkey DK, Stornetta RL & Bayliss DA (2005). Regulation of ventral surface chemoreceptors by the central respiratory pattern generator. J Neurosci 25, 8938–8947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton GI (1982). Phasic bursting activity of rat paraventricular neurones in the absence of synaptic transmission. J Physiol 327, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins VE, Hawryluk JM, Takakura AC, Tzingounis AV, Moreira TS & Mulkey DK (2015). HCN channels contribute to serotonergic modulation of ventral surface chemosensitive neurons and respiratory activity. J Neurophysiol 113, 1195–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawryluk JM, Moreira TS, Takakura AC, Wenker IC, Tzingounis AV & Mulkey DK (2012). KCNQ channels determine serotonergic modulation of ventral surface chemoreceptors and respiratory drive. J Neurosci 32, 16943–16952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckstepp RT, Cardoza KP, Henderson LE & Feldman JL (2015). Role of parafacial nuclei in control of breathing in adult rats. J Neurosci 35, 1052–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huckstepp RT, Id BR, Eason R, Spyer KM, Dicke N, Willecke K, Marina N, Gourine AV & Dale N (2010). Connexin hemichannel‐mediated CO2‐dependent release of ATP in the medulla oblongata contributes to central respiratory chemosensitivity. J Physiol 588, 3901–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janczewski WA & Feldman JL (2006). Distinct rhythm generators for inspiration and expiration in the juvenile rat. J Physiol 570, 407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ (2000). Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci 1, 21–30. [DOI] [PubMed] [Google Scholar]
- Kang S, Xu M, Cooper EC & Hoshi N (2014). Channel‐anchored protein kinase CK2 and protein phosphatase 1 reciprocally regulate KCNQ2‐containing M‐channels via phosphorylation of calmodulin. J Biol Chem 289, 11536–11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosenko A & Hoshi N (2013). A change in configuration of the calmodulin‐KCNQ channel complex underlies Ca2+‐dependent modulation of KCNQ channel activity. PLoS ONE 8, e82290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosenko A, Kang S, Smith IM, Greene DL, Langeberg LK, Scott JD & Hoshi N (2012). Coordinated signal integration at the M‐type potassium channel upon muscarinic stimulation. EMBO J 31, 3147–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubin L & Fenik V (2004). Pontine cholinergic mechanisms and their impact on respiratory regulation. Respir Physiol Neurobiol 143, 235–249. [DOI] [PubMed] [Google Scholar]
- Li YW, Guyenet PG & Bayliss DA (1998). Voltage‐dependent calcium currents in bulbospinal neurons of neonatal rat rostral ventrolateral medulla: modulation by alpha2‐adrenergic receptors. J Neurophysiol 79, 583–594. [DOI] [PubMed] [Google Scholar]
- Lydic R & Baghdoyan HA (1993). Pedunculopontine stimulation alters respiration and increases ACh release in the pontine reticular formation. Am J Physiol 264, R544–R554. [DOI] [PubMed] [Google Scholar]
- Matsubayashi H, Alkondon M, Pereira EF, Swanson KL & Albuquerque EX (1998). Strychnine: a potent competitive antagonist of alpha‐bungarotoxin‐sensitive nicotinic acetylcholine receptors in rat hippocampal neurons. J Pharmacol Exp Ther. 284, 904–913. [PubMed] [Google Scholar]
- Metz B (1966). Hypercapnia and acetylcholine release from the cerebral cortex and medulla. J Physiol 186, 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteau R, Morin D & Hilaire G (1990). Acetylcholine and central chemosensitivity: in vitro study in the newborn rat. Respir Physiol 81, 241–253. [DOI] [PubMed] [Google Scholar]
- Mulkey DK, Henderson RA 3rd, Olson JE, Putnam RW & Dean JB (2001). Oxygen measurements in brain stem slices exposed to normobaric hyperoxia and hyperbaric oxygen. J Appl Physiol (1985) 90, 1887–1899. [DOI] [PubMed] [Google Scholar]
- Mulkey DK, Stornetta RL, Weston MC, Simmons JR, Parker A, Bayliss DA & Guyenet PG (2004). Respiratory control by ventral surface chemoreceptor neurons in rats. Nat Neurosci 7, 1360–1369. [DOI] [PubMed] [Google Scholar]
- Mulkey DK, Talley EM, Stornetta RL, Siegel AR, West GH, Chen X, Sen N, Mistry AM, Guyenet PG & Bayliss DA (2007). TASK channels determine pH sensitivity in select respiratory neurons but do not contribute to central respiratory chemosensitivity. J Neurosci 27, 14049–14058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattie E & Li A (2012). Central chemoreceptors: locations and functions. Compr Physiol 2, 221–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattie EE, Li A, Mills J & Huang Q (1994). Retrotrapezoid nucleus muscarinic receptor subtypes localized by autoradiography. Respir Physiol 96, 189–197. [DOI] [PubMed] [Google Scholar]
- Nattie EE & Li AH (1990). Ventral medulla sites of muscarinic receptor subtypes involved in cardiorespiratory control. J Appl Physiol (1985) 69, 33–41. [DOI] [PubMed] [Google Scholar]
- Nattie EE, Wood J, Mega A & Goritski W (1989). Rostral ventrolateral medulla muscarinic receptor involvement in central ventilatory chemosensitivity. J Appl Physiol (1985) 66, 1462–1470. [DOI] [PubMed] [Google Scholar]
- Onimaru H, Ikeda K & Kawakami K (2008). CO2‐sensitive preinspiratory neurons of the parafacial respiratory group express Phox2b in the neonatal rat. J Neurosci 28, 12845–12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiero DA, Giuliano R, Anwar M, Stornetta R & Reis DJ (1990). Anatomical substrates of cholinergic‐autonomic regulation in the rat. J Comp Neurol 292, 1–53. [DOI] [PubMed] [Google Scholar]
- Ruggiero DA, Anwar M, Golanov EV & Reis DJ (1997). The pedunculopontine tegmental nucleus issues collaterals to the fastigial nucleus and rostral ventrolateral reticular nucleus in the rat. Brain Res 760, 272–276. [DOI] [PubMed] [Google Scholar]
- Saponjic J, Radulovacki M & Carley DW (2003). Respiratory pattern modulation by the pedunculopontine tegmental nucleus. Respir Physiol Neurobiol 138, 223–237. [DOI] [PubMed] [Google Scholar]
- Saponjic J, Radulovacki M & Carley DW (2006). Modulation of respiratory pattern and upper airway muscle activity by the pedunculopontine tegmentum: role of NMDA receptors. Sleep Breath 10, 195–202. [DOI] [PubMed] [Google Scholar]
- Shao XM & Feldman JL (2000). Acetylcholine modulates respiratory pattern: effects mediated by M3‐like receptors in preBotzinger complex inspiratory neurons. J Neurophysiol 83, 1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao XM & Feldman JL (2001). Mechanisms underlying regulation of respiratory pattern by nicotine in preBotzinger complex. J Neurophysiol 85, 2461–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao XM & Feldman JL (2005). Cholinergic neurotransmission in the preBotzinger complex modulates excitability of inspiratory neurons and regulates respiratory rhythm. Neuroscience 130, 1069–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao XM & Feldman JL (2009). Central cholinergic regulation of respiration: nicotinic receptors. Acta Pharmacol Sin 30, 761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Inoue T, Meyer T & Hille B (2006). Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science 314, 1454–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC, Morrison DE, Ellenberger HH, Otto MR & Feldman JL (1989). Brainstem projections to the major respiratory neuron populations in the medulla of the cat. J Comp Neurol 281, 69−96. [DOI] [PubMed] [Google Scholar]
- Thoby‐Brisson M, Karlén M, Wu N, Charnay P, Champagnat J & Fortin G (2009). Genetic identification of an embryonic parafacial oscillator coupling to the preBotzinger complex. Nat Neurosci 12, 1028–1035. [DOI] [PubMed] [Google Scholar]
- Unal CT, Pare D & Zaborszky L (2015). Impact of basal forebrain cholinergic inputs on basolateral amygdala neurons. J Neurosci 35, 853–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HL & Morales M (2009). Pedunculopontine and laterodorsal tegmental nuclei contain distinct populations of cholinergic, glutamatergic and GABAergic neurons in the rat. Eur J Neurosci 29, 340–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Shi Y, Shu S, Guyenet PG & Bayliss DA (2013). Phox2b‐expressing retrotrapezoid neurons are intrinsically responsive to H+ and CO2 . J Neurosci 33, 7756–7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenker IC, Kreneisz O, Nishiyama A & Mulkey DK (2010). Astrocytes in the retrotrapezoid nucleus sense H+ by inhibition of a Kir4.1‐Kir5.1‐like current and may contribute to chemoreception by a purinergic mechanism. J Neurophysiol 104, 3042–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenker IC, Sobrinho CR, Takakura AC, Moreira TS & Mulkey DK (2012). Regulation of ventral surface CO2/H+‐sensitive neurons by purinergic signalling. J Physiol 590, 2137–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui Y, Cechetto DF & Saper CB (1990). Evidence for a cholinergic projection from the pedunculopontine tegmental nucleus to the rostral ventrolateral medulla in the rat. Brain Res 517, 19–24. [DOI] [PubMed] [Google Scholar]
- Zhang H, Craciun LC, Mirshahi T, Rohacs T, Lopes CM, Jin T & Logothetis DE (2003). PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor‐mediated inhibition of M currents. Neuron 37, 963–975. [DOI] [PubMed] [Google Scholar]