

Key points
Chronic hypoxia has a direct effect in down‐regulating the BKCa channel β1 subunit and inhibiting the BKCa channel activity in uterine arteries of pregnant sheep.
Oxidative stress plays a causal role in hypoxia‐mediated suppression of BKCa channel function.
The steroid hormone‐induced effect on BKCa channels is a target of hypoxia‐mediated oxidative stress.
Inhibition of oxidative stress ameliorates the adverse effect of hypoxia both ex vivo and in vivo in pregnant sheep exposed to long‐term high‐altitude hypoxia.
Our findings provide novel evidence of a causative role of oxidative stress in hypoxia‐mediated inhibition of the BKCa channel activity in uterine arteries and new insights in understanding and alleviating pregnancy complications associated with gestational hypoxia such as pre‐eclampsia and fetal growth restriction.
Abstract
Uterine arteries of pregnant sheep acclimatized to long‐term high‐altitude hypoxia were associated with a decrease in large‐conductance Ca2+‐activated K+ (BKCa) channel activity. The present study tested the hypothesis that prolonged hypoxia has a direct effect in suppressing BKCa channel activity by increasing oxidative stress. Uterine arteries were isolated from non‐pregnant and near‐term (∼142 days) pregnant sheep, and were treated ex vivo with 21.0 or 10.5% O2 for 48 h. The hypoxia treatment significantly increased the production of reactive oxygen species in uterine arteries, which was blocked by N‐acetylcysteine. In uterine arteries of pregnant sheep, hypoxia significantly inhibited BKCa channel current density, decreased NS1619‐induced relaxations and increased pressure‐dependent tone, which were annulled by N‐acetylcysteine. In accordance, hypoxia resulted in down‐regulation of BKCa channel β1 subunit, which was restored in the presence of N‐acetylcysteine. In addition, the N‐acetylcysteine treatment significantly increased BKCa channel β1 subunit abundance and BKCa channel current density in uterine arteries from pregnant sheep exposed to high‐altitude hypoxia (3801 m, : 60 mmHg) for 110 days. In uterine arteries of non‐pregnant animals, hypoxia inhibited steroid hormone‐induced up‐regulation of BKCa channel current density and NS1619‐mediated relaxations, which were reversed by N‐acetylcysteine. Furthermore, the synthetic superoxide dismutase and catalase mimetic EUK‐134 also ablated the effects of hypoxia on BKCa channel currents in uterine arteries. The results demonstrate a direct effect of hypoxia in inhibiting the BKCa channel activity in uterine arteries via increased oxidative stress.
Key points
Chronic hypoxia has a direct effect in down‐regulating the BKCa channel β1 subunit and inhibiting the BKCa channel activity in uterine arteries of pregnant sheep.
Oxidative stress plays a causal role in hypoxia‐mediated suppression of BKCa channel function.
The steroid hormone‐induced effect on BKCa channels is a target of hypoxia‐mediated oxidative stress.
Inhibition of oxidative stress ameliorates the adverse effect of hypoxia both ex vivo and in vivo in pregnant sheep exposed to long‐term high‐altitude hypoxia.
Our findings provide novel evidence of a causative role of oxidative stress in hypoxia‐mediated inhibition of the BKCa channel activity in uterine arteries and new insights in understanding and alleviating pregnancy complications associated with gestational hypoxia such as pre‐eclampsia and fetal growth restriction.
Abbreviations
- BKCa
large conductance Ca2+‐activated K+
- DCF
2′,7′‐dichlorodihydrofluorescein
- DCFH‐DiOxyQ
dichlorodihydrofluorescin DiOxyQ
- DHE
dihydroethidium
- DMEM
Dulbecco's Modified Eagle's medium
- ER‐α
oestrogen receptor alpha
- ER‐β
oestrogen receptor beta
- Nox
NADPH oxidase
- IKCa
intermediate conductance Ca2+‐activated K+
- KCNMB 1
Ca2+‐activated K+ subunit beta‐1
- ONOO−
peroxynitrite anion
partial pressure of arterial oxygen
- PKC
protein kinase C
- PSS
physiological saline solution
- RNS
reactive nitrogen species
- ROO
peroxyl radical
- ROS
reactive oxygen species
- SKCa
small conductance Ca2+‐activated K+
- SOD
superoxide dismutase
- VSMCs
vascular smooth muscle cells
Introduction
Marked changes in the cardiovascular system occur during normal pregnancy to accommodate fetal growth; one of major haemodynamic adaptions is the dramatic increase in uterine blood flow (Rosenfeld, 1977; Palmer et al. 1992). Adequate uterine blood supply during pregnancy is essential for the development/growth of the placenta and fetus, as well as the well‐being of the mother. Aberrant uterine perfusion is associated with pregnancy complications such as pre‐eclampsia and eclampsia (Lang et al. 2003; Browne et al. 2015). These complications are often associated with significant maternal morbidity and mortality and fetal growth restriction (Lambert et al. 2014). Gestational hypoxia is a notorious insult to maternal cardiovascular homeostasis and increases the incidence of pre‐eclampsia and fetal growth restriction by impairing the uteroplacental circulation (Zamudio et al. 1995 a,b; Palmer et al. 1999). The large conductance Ca2+‐activated K+ (BKCa) channel plays a pivotal role in regulating the membrane potential of vascular smooth muscle cells (VSMCs) and thus vascular tone (Hill et al. 2010; Hu & Zhang, 2012). The channel opening, predominantly stimulated by an increase in intracellular Ca2+ concentrations, results in membrane hyperpolarization and reduces the activity of voltage‐dependent Ca2+ channels. Increased expression of the BKCa channel β1 subunit and enhanced channel activity in uterine arteries during pregnancy attribute to decreased uterine vascular tone and increased uterine blood flow (Rosenfeld et al. 2001, 2009; Hu et al. 2011). Oestrogen has been found to play a critical role in the upregulation of the BKCa channel β1 subunit and reduced pressure‐dependent tone in uterine arteries during pregnancy (Nagar et al. 2005; Hu et al. 2011; Chen et al. 2014). Our recent studies demonstrated that long‐term high‐altitude hypoxia during gestation in sheep was associated with a decrease in the BKCa channel activity, leading to an increase in pressure‐dependent myogenic tone in uterine arteries (Hu et al. 2012; Zhu et al. 2014). Furthermore, chronic hypoxia nullified steroid hormone‐mediated upregulation of BKCa channel activity and increased pressure‐dependent tone of uterine arteries (Hu et al. 2012; Zhu et al. 2014).
Whereas these initial observations of decreased BKCa channel activity in uterine arteries of pregnant sheep acclimatized to long‐term high‐altitude hypoxia were of high interest, the question arose is to what extent this effect observed in the in vivo studies was a direct effect of hypoxia on uterine arteries or was a response to indirect mediators resulting from the hypoxic stress in animals. In addition, the mechanisms underlying the hypoxia‐mediated inhibition of BKCa channel activity in uterine arteries remain largely elusive. Hypoxia has been shown to promote production of reactive oxygen species (ROS) and to increase oxidative stress in the cardiovascular system (Giordano, 2005; Fresquet et al. 2006; Liu et al. 2006). The excessive production of ROS and increased oxidative stress in the placenta and vasculature have been observed in pregnancy complications including pre‐eclampsia (Raijmakers et al. 2004; Siddiqui et al. 2010). The NADPH oxidase (Nox) family is of critical importance in the regulation of ROS production (Drummond et al. 2011). Our recent studies demonstrated that long‐term high‐altitude hypoxia during gestation increased ROS production in ovine uterine arteries, in part due to increased expression and activity of Nox2 (Xiao et al. 2013). It has been shown that ROS are implicated in down‐regulating BKCa channels, as well as small (SKCa) and intermediate (IKCa) conductance Ca2+‐activated K+ channels in the cardiovascular system (Lu et al. 2012; Zhao et al. 2014; Yi et al. 2015). Herein, we present novel evidence that prolonged hypoxia has a direct effect in inhibiting the BKCa channel activity in uterine arteries by increasing oxidative stress.
Methods
Ethical approval
All procedures and protocols were approved by the Institutional Animal Care and Use Committee of Loma Linda University and followed the guidelines by the National Institutes of Health Guide for the Care and Use of Laboratory Animals. After tissue collection, animals were killed via intravenous injection of 15 ml T‐61 solution (Hoechst‐Rousel, Somervile, NJ, USA), according to American Veterinary Medical Association guidelines.
Tissue preparation and treatment
Uterine arteries were harvested from non‐pregnant and near‐term (∼142–145 days of gestation) pregnant sheep (Ovis aries) maintained at ∼300 m above sea level (low altitude) or exposed to high‐altitude (3801 m) hypoxia (: 60 mmHg) for 110 days (starting from 30 days of gestation for pregnant animals) (Chang et al. 2010). Animals were anaesthetized via iv injection of propofol (2 mg kg−1) followed by intubation, and anaesthesia was maintained on 1.5–3.0% isoflurane balanced in O2 throughout the surgery. An incision was made in the abdomen and the uterus was exposed. The fourth generation branches of main uterine arteries and resistance‐sized uterine arteries were isolated and removed without stretching and placed into a Krebs solution containing (in mm) 130.0 NaCl, 10.0 Hepes, 6.0 glucose, 4.0 KCl, 4.0 NaHCO3, 1.8 CaCl2, 1.2 MgSO4, 1.18 KH2PO4 and 0.025 EDTA (pH 7.4). To determine the direct effect of hypoxia, uterine arteries were treated ex vivo under normoxic or hypoxic conditions for 48 h. Given an ∼50% decrease in arterial observed in high‐altitude hypoxic sheep, uterine arteries were placed in a culture dish containing 5 ml of phenol red‐free Dulbecco's Modified Eagle's medium (DMEM) supplemented with 1% charcoal‐stripped fetal bovine serum, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin and incubated at 37°C in humidified incubators for 48 h with oxygen levels at either 21.0% O2 for normoxic or 10.5% O2 for hypoxic conditions, as described previously (Chang et al. 2010; Dasgupta et al. 2012; Hu et al. 2012). For the hormonal treatment, uterine arteries from non‐pregnant sheep were incubated in phenol red‐free DMEM with 1% charcoal‐stripped fetal bovine serum for 48 h at 37°C in a humidified CO2 incubator under 21.0% O2 and 10.5% O2 in the absence or presence of 17β‐estradiol (0.3 nm; Sigma, St Louis, MO, USA) and progesterone (100.0 nm; Sigma), as reported previously (Xiao et al. 2009; Chang et al. 2010; Hu et al. 2011). Phases of the ovarian cycle and systemic blood steroid levels were not determined in non‐pregnant animals in the present study. The potential pre‐exposure to 17β‐estradiol and/or progesterone of uterine arteries from non‐pregnant animals and their priming effects on the vessels could not be excluded. Each experimental group had four to five animals.
Relaxation studies
Uterine arteries were separated from surrounding tissues and cut into 2 mm ring segments. Isometric tension was measured in the Krebs solution in a tissue bath system (Radnoti, Monrovia, CA, USA) at 37°C as described previously (Chang et al. 2010; Xiao et al. 2013). Briefly, each ring segment was equilibrated for 60 min and then gradually stretched to the optimal resting tension determined by responses to three 120 mm KCl challenges. Tissues were then pre‐contracted with submaximal concentrations of noradrenaline that produced ∼70–80% of the maximal contraction, followed by additions of the BKCa channel opener NS1619 in a cumulative manner.
Measurement of pressure‐dependent tone
Pressure‐dependent tone of resistance‐sized uterine arteries was measured as described previously (Chang et al. 2010; Hu et al. 2011). Briefly, the arterial segments (diameter ∼150 μm) were mounted and pressurized in an organ chamber (Living Systems Instruments, Burlington, VT, USA). The intraluminal pressure was controlled by a servo‐system to set transmural pressures, and arterial diameter was recorded using the SoftEdge Acquisition Subsystem (IonOptix LLC, Milton, MA, USA). After the equilibration period, the intraluminal pressure was increased in a stepwise manner from 10 to 100 mmHg in 10 mmHg increments, and each pressure was maintained for 5 min to allow vessel diameter to stabilize before the measurement. The passive pressure–diameter relationship was determined in Ca2+‐free physiological saline solution (PSS) containing 3.0 mm EGTA to determine the maximum passive diameter. The following formula was used to calculate the percentage of pressure‐dependent tone at each pressure step: % tone = (D1 − D2)/D1 × 100, where D1 is the passive diameter in Ca2+‐free PSS (0 Ca2+ with 3.0 mm EGTA), and D2 is the active diameter with normal PSS in the presence of extracellular Ca2+.
Measurement of ROS/RNS productions
Total ROS/reactive nitrogen species (RNS) production in uterine arteries was measured with the Oxiselect in vitro ROS/RNS assay kit (Cell Biolabs, Inc., San Diego, CA, USA) following the manufacturer's instruction, as described previously (Patterson et al. 2012; Xiong et al. 2012; Xiao et al. 2013). The principle of this assay is that dichlorodihydrofluorescin DiOxyQ (DCFH‐DiOxyQ) reacts with ROS/RNS such as hydrogen peroxide (H2O2), peroxyl radical (ROO·), nitric oxide (NO) and peroxynitrite anion (ONOO−), and the complex is rapidly oxidized to the highly fluorescent 2′,7′‐dichlorodihydrofluorescein (DCF) in the cytosol. The intensity of DCF fluorescence is proportional to the amount of ROS/RNS in the biological specimen. Briefly, tissues were homogenized in PBS followed by centrifugation at 4°C for 10 mins at 10,000 g, and the supernatants were collected for ROS/RNS assay. A given amount of sample was incubated with the probe for ∼30 min, and the fluorescence was determined with a fluorescence plate reader at 480 nm excitation/530 nm emission. It appears that all techniques currently available to measure ROS/RNS have their own drawbacks (Dikalov et al. 2007; Chen et al. 2010). DCF may trigger an increase in ROS itself (Dikalov et al. 2007). Thus, ROS contents measured in the present study might be somewhat overestimated in tissues exposed to both normoxic and hypoxic environments. However, the DCF assay is still commonly used to measure ROS (Chen et al. 2010). Our previous studies revealed that assays using DCF and dihydroethidium (DHE) yielded comparable measurements in cells and tissues of various animal sources (Patterson et al. 2012; Xiong et al. 2012; Xiao et al. 2013).
Western immunoblotting
Protein abundance of the BKCa channel β1 subunit in uterine arteries was measured as described previously (Hu et al. 2011, 2012). Briefly, tissues were homogenized in lysis buffer followed by centrifugation at 4°C for 10 min at 10,000 g, and the supernatants were collected. Samples with equal proteins were loaded onto 7.5% polyacrylamide gel with 0.1% SDS and were separated by electrophoresis at 100 V for 2 h. Proteins were then transferred onto nitrocellulose membranes. After blocking non‐specific binding sites by dry milk, membranes were incubated with primary antibodies against the BKCa channel β1 subunit (Santa Cruz Biotechnology, Santa Cruz, CA, USA). After washing, membranes were incubated with secondary HRP‐conjugated antibodies. Proteins were visualized with enhanced chemiluminescence reagents, and blots were exposed to Hyperfilm. Results were quantified with the Kodak electrophoresis documentation and analysis system and Kodak ID image analysis software (Kodak, Rochester, NY, USA). The target protein abundance was normalized to the abundance of β‐actin as a protein loading control.
Measurement of BKCa channel current
Arterial smooth muscle cells were enzymatically dissociated from resistance‐sized uterine arteries, and whole‐cell K+ currents were recorded using an EPC 10 patch‐clamp amplifier with Patchmaster software (HEKA, Lambrecht/Pfalz, Germany) at room temperature as previously described (Hu et al. 2011, 2012). Briefly, cell suspension drops were placed in a recording chamber, and adherent cells were continuously superfused with Hepes‐buffered PSS containing (in mm) 140.0 NaCl, 5.0 KCl, 1.8 CaCl2, 1.2 MgCl2, 10.0 Hepes and 10.0 glucose (pH 7.4). Only relaxed and spindle‐shaped myocytes were used for recording. Micropipettes were pulled from borosilicate glass and had resistances of 2–5 mΩ when filled with the pipette solution containing (in mm) 140.0 KCl, 1.0 MgCl2, 5.0 Na2ATP, 5.0 EGTA and 10.0 Hepes (pH 7.2). CaCl2 was added to bring free Ca2+ concentrations to 100 nm as determined using WinMAXC software (Chris Patton, Stanford University). Cells were held at –50 mV, and whole‐cell K+ currents were evoked by voltage steps from −60 to +80 mV by stepwise 10 mV depolarizing pulses (350–ms duration, 10 s intervals) in the absence and presence of 1 mm BKCa channel blocker tetraethylammonium (Hu et al. 2011, 2012). BKCa currents, determined as the difference between whole‐cell K+ currents in the absence of tetraethylammonium and that in the presence of tetraethylammonium, were normalized to cell capacitance and expressed as picoamps per picofarad (pA pF–1).
Pharmacological tools
To ascertain roles of ROS in the aberrant regulation of BKCa channel functions in uterine arteries, pharmacological tools N‐acetylcysteine and EUK 134 were used in the present study. N‐Acetylcysteine is a precursor of glutathione and functions as a free radical scavenger (Zafarullah et al. 2003; Sun, 2010), whereas EUK 134 is a superoxide dismutase (SOD) and catalase mimetic (Rong et al. 1999). Both compounds have been extensively used as tools to probe the role of ROS in physiological and pathophysiological processes (Zafarullah et al. 2003; Ajith & Jayakumar, 2014). Our previous studies demonstrated that N‐acetylcysteine at 1 mm effectively ablated hypoxia‐ and noradrenaline‐stimulated ROS generation in H9c2 cells and intact fetal hearts (Patterson et al. 2012; Xiong et al. 2012). Moreover, we demonstrated that N‐acetylcysteine at 1 mm inhibited ROS‐mediated suppression of BKCa channel activity in uterine arteries of pregnant sheep acclimatized to long‐term high‐altitude hypoxia (Zhu et al. 2014). Therefore, the same concentration of N‐acetylcysteine was used in the present study. In addition, the functional roles of BKCa channels was probed using the BKCa channel opener NS1619 (Olesen et al. 1994; Holland et al. 1996). The specificity of NS1619 on BKCa channels in uterine arteries has been demonstrated previously using iberitoxin (Zhu et al. 2013).
Statistics
Data are expressed as means ± SEM obtained from the number of experimental animals given. Concentration–response curves were analysed by computer‐assisted non‐linear regression to fit the data using GraphPad Prism (GraphPad Software, San Diego, CA, USA). Differences were evaluated for statistical significance (P < 0.05) by ANOVA or t test where appropriate; and the concentration–response relationship was analysed with repeated measures ANOVA.
Results
Hypoxia increased ROS/RNS production in uterine arteries
Uterine arteries isolated from pregnant sheep were treated under normoxia (21.0% O2) or hypoxia (10.5% O2) for 48 h in the absence or presence of N‐acetylcysteine. As shown in Fig. 1, the prolonged hypoxic treatment significantly increased ROS/RNS production in uterine arteries in the absence of N‐acetylcysteine. N‐Acetylcysteine had no significant effect on ROS/RNS production under normoxic conditions, but it ablated the hypoxia‐induced increase in ROS/RNS production in uterine arteries (Fig. 1).
Figure 1. Hypoxia increased ROS in uterine arteries .
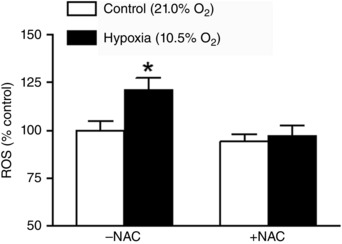
Uterine arteries of pregnant sheep were treated under control (21.0% O2) and hypoxia (10.5% O2) for 48 h in the absence or presence of 1 mm N‐acetylcysteine (NAC). ROS were measured with a DCF‐based quantitative assay kit. Data are means ± SEM of four tissues from four animals of each group. Data were analysed with two‐way ANOVA. *P < 0.05, hypoxia vs. control.
Antioxidants blocked hypoxia‐mediated inhibition of BKCa channel current density in uterine arteries
BKCa channel currents were recorded in myocytes isolated from uterine arteries of pregnant sheep, treated with normoxia (21.0% O2) or hypoxia (10.5% O2) for 48 h in the absence or presence of antioxidants. As illustrated in Fig. 2 A, hypoxia markedly inhibited BKCa channel current density (10.3 ± 0.8 vs. 28.0 ± 3.5 pA pF–1 at +80 mV; P < 0.05) in the absence of antioxidants. BKCa channel currents were not significantly altered by antioxidants in uterine arterial myocytes under normoxic conditions. However, N‐acetylcysteine reduced the effect of hypoxia and partially restored the BKCa channel current density from 8.2 ± 0.9 to 19.3 ± 0.9 pA pF–1 at +80 mV (P < 0.05, Fig. 2 B). Moreover, EUK 134 also ablated hypoxia‐induced suppression of BKCa channel and the BKCa channel current density at +80 mV was elevated from 12.3 ± 1.6 to 29.4 ± 3.3 pA pF–1 (P < 0.05, Fig. 2 C).
Figure 2. Antioxidants alleviated hypoxia‐mediated inhibition of BKCa channel current density in uterine arteries .
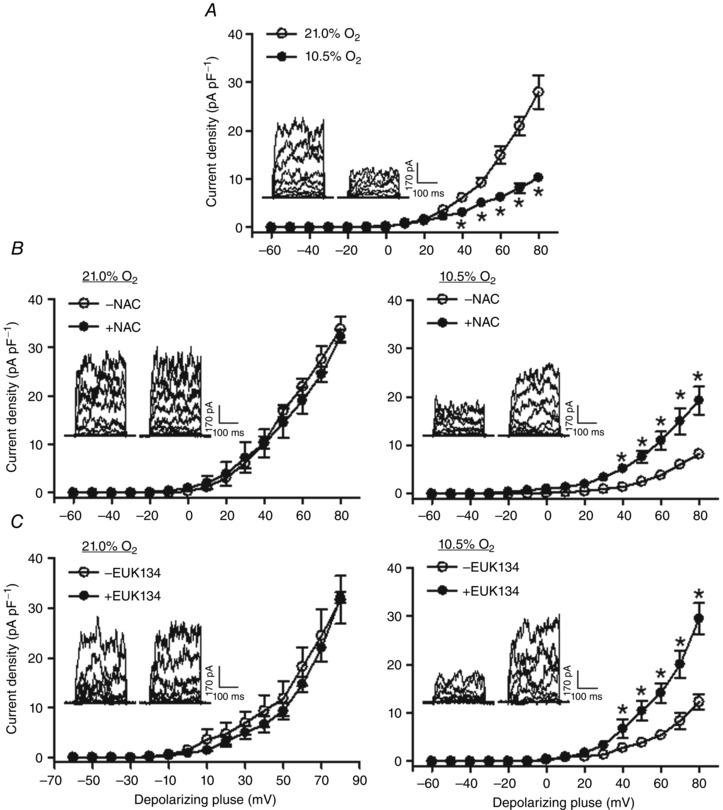
Uterine arteries of pregnant sheep were treated under control (21.0% O2) and hypoxia (10.5% O2) for 48 h in the absence or presence of 1 mm N‐acetylcysteine (NAC) or 20 μM EUK 134. BKCa currents were normalized to cell capacitance and expressed as picoamps per picofarad (pA pF–1; y axis), as a function of stepwise 10 mV depolarizing pulses (x axis). Data are means ± SEM of 5–7 cells from five animals of each group. Data were analysed with repeated measures ANOVA. *P < 0.05, hypoxia vs. control in A; +NAC vs. –NAC in B; +EUK 134 vs. –EUK 134 in C.
N‐Acetylcysteine ablated hypoxia‐mediated decrease in NS1619‐induced relaxations in uterine arteries
Uterine arteries isolated from pregnant sheep were treated under normoxia (21.0% O2) or hypoxia (10.5% O2) for 48 h in the absence or presence of N‐acetylcysteine. The BKCa channel opener NS1619 produced concentration‐dependent relaxations of uterine arteries (Fig. 3). In the absence of N‐acetylcysteine, the hypoxic treatment significantly decreased NS1619‐induced relaxations (Fig. 3 A). In the presence of N‐acetylcysteine, the hypoxic effect was abrogated and there were no significant differences in NS1619‐induced relaxations between normoxic and hypoxic conditions (Fig. 3 B).
Figure 3. N‐Acetylcysteine ablated hypoxia‐mediated decrease in NS1619‐induced relaxations in uterine arteries .
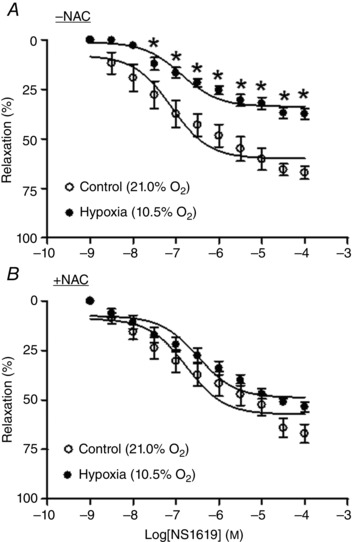
Uterine arteries of pregnant sheep were treated under control (21.0% O2) and hypoxia (10.5% O2) for 48 h in the absence or presence of 1 mm N‐acetylcysteine (NAC). NS1619‐induced relaxations were determined after the treatments. Data are means ± SEM of five tissues from five animals of each group. Data were analysed with repeated measures ANOVA. *P < 0.05, hypoxia vs. control.
N‐Acetylcysteine inhibited hypoxia‐mediated increase in pressure‐dependent tone in uterine arteries
BKCa channel activity plays a critical role in determining pressure‐dependent tone in uterine arteries (Hu et al. 2011, 2012). We next determined whether the inhibition of ROS by N‐acetylcysteine affected uterine arterial pressure‐dependent response. As shown in Fig. 4 A, in the absence of N‐acetylcysteine, the hypoxic treatment resulted in a significant increase in pressure‐dependent tone in uterine arteries of pregnant animals. This hypoxia‐mediated effect was inhibited by N‐acetylcysteine (Fig. 4 B).
Figure 4. N‐Acetylcysteine inhibited hypoxia‐mediated increase in pressure‐dependent tone in uterine arteries .
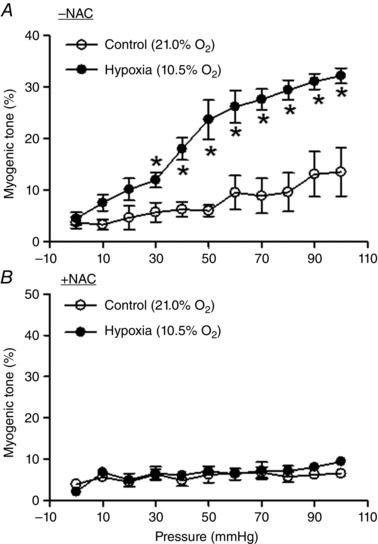
Uterine arteries of pregnant sheep were treated under control (21.0% O2) and hypoxia (10.5% O2) for 48 h in the absence or presence of 1 mm N‐acetylcysteine (NAC). Pressure‐dependent tone was determined after the treatments. Data are mean ± SEM of four tissues from four animals of each group. Data were analysed with repeated measures ANOVA. *P < 0.05, hypoxia vs. control.
N‐Acetylcysteine restored hypoxia‐induced down‐regulation of the BKCa channel β1 subunit in uterine arteries
The β1 subunit plays a vital role in regulating BKCa channel activity in vascular smooth muscle by increasing the Ca2+ sensitivity of the channel (Hu & Zhang, 2012). Previous study revealed that long‐term high‐ altitude hypoxia was associated with a selective down‐regulation of the BKCa channel β1 subunit in uterine arteries of pregnant sheep (Hu et al. 2012). Consistent with this finding, the ex vivo treatment of uterine arteries of pregnant sheep with prolonged hypoxia showed a direct effect of hypoxia in suppressing expression of the BKCa channel β1 subunit protein in uterine arteries (Fig. 5). Of importance, the inhibition of ROS by N‐acetylcysteine blocked the hypoxic effect and restored protein expression of the β1 subunit (Fig. 5). To confirm this finding of an ex vivo hypoxia‐mediated effect in animals exposed to chronic hypoxia, uterine arteries from pregnant sheep acclimatized to long‐term high‐altitude hypoxia were treated with N‐acetylcysteine. As shown in Fig. 6 A, N‐acetylcysteine significantly increased protein abundance of the BKCa channel β1 subunit in uterine arteries of pregnant sheep that had been exposed to long‐term high‐altitude hypoxia. Accordingly, the BKCa channel activity in uterine arterial myocytes of the hypoxic animals was significantly increased by N‐acetylcysteine (Fig. 6 B).
Figure 5. N‐acetylcysteine restored hypoxia‐induced down‐regulation of the BKCa channel β1 subunit in uterine arteries .
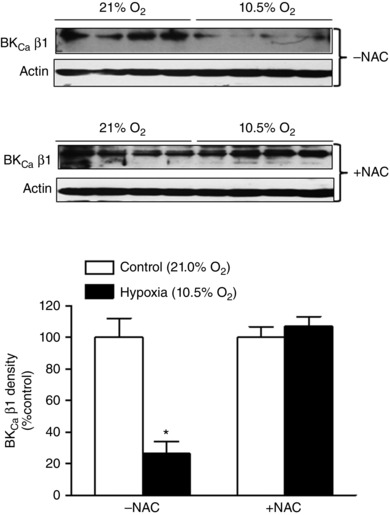
Uterine arteries of pregnant sheep were treated under control (21.0% O2) and hypoxia (10.5% O2) for 48 h in the absence or presence of 1 mm N‐acetylcysteine (NAC). Protein abundance of the β1 subunit was determined by Western blot. Data are means ± SEM of four tissues from four animals of each group. Data were analysed by two‐way ANOVA. *P < 0.05, hypoxia vs. control.
Figure 6. N‐acetylcysteine increased the BKCa channel β1 subunit and BKCa channel in uterine arteries of high‐altitude pregnant sheep .
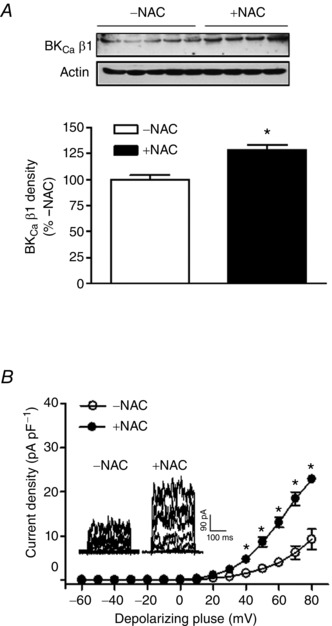
Uterine arteries isolated from pregnant sheep acclimatized to long‐term high‐altitude hypoxia were treated under 10.5% O2 for 48 h in the absence or presence of 1 mm N‐acetylcysteine (NAC). A, protein abundance of the β1 subunit was determined by Western blot. Data are means ± SEM of 4–5 tissues from 4–5 animals of each group. Data were analysed with t test. *P < 0.05, +NAC vs. –NAC. B, BKCa currents in smooth muscle cells were recorded. Data are means ± SEM of six cells from five animals of each group. Data were analysed with repeated measures ANOVA. *P < 0.05, +NAC vs. –NAC.
Antioxidants reinstated steroid hormone‐mediated up‐regulation of BKCa channel activity under hypoxia
Steroid hormones, 17β‐estradiol and progesterone, play a key role in up‐regulating BKCa channel activity in uterine arteries during pregnancy (Hu et al. 2011). Under normoxic conditions, the steroid hormone treatment of uterine arteries from non‐pregnant sheep for 48 h significantly increased BKCa channel current density (Fig. 7 A). In contrast, under hypoxic conditions (10.5% O2) in the absence of antioxidants, the hormonal treatment had no significant effect on BKCa channel activity (Fig. 7 B). However, N‐acetylcysteine and EUK 134 restored steroid hormone‐mediated up‐regulation of BKCa channel current density in uterine arteries under hypoxia (Fig. 7 C and D). Accordingly, the previous study demonstrated that the hormonal treatment of uterine arteries from normoxic, non‐pregnant sheep significantly increased NS1619‐induced relaxations (Zhu et al. 2014). In contrast, the hormone treatment of uterine arteries from non‐pregnant animals under hypoxic conditions had no significant effect on NS1619‐induced relaxations (Fig. 8 A). Of importance, N‐acetylcysteine rescued the hormonal effect on the up‐regulation of NS1619‐induced relaxations under hypoxia (Fig. 8 B).
Figure 7. Antioxidants restored steroid hormone‐mediated up‐regulation of BKCa channel activity under hypoxia .
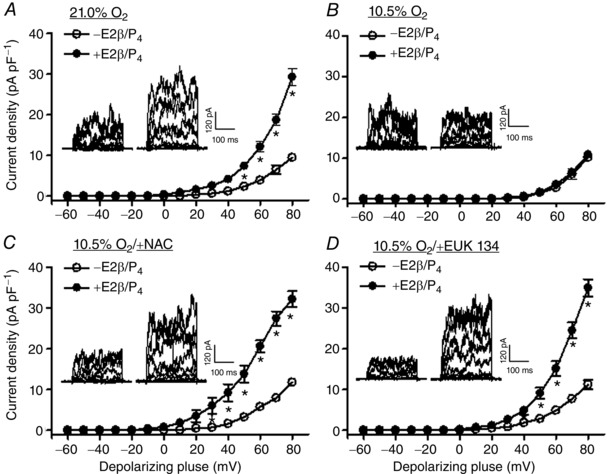
Uterine arteries of non‐pregnant sheep were treated with or without 17β‐estradiol (E2β; 0.3 nm)/progesterone (P4; 100 nm) under control (21.0% O2) or hypoxia (10.5% O2) for 48 h, in the absence or presence of 1 mm N‐acetylcysteine (NAC) or 20 μm EUK 134. BKCa currents were normalized to cell capacitance and expressed as picoamps per picofarad (pA pF–1; y axis), as a function of stepwise 10 mV depolarizing pulses (x axis). Data are means ± SEM of six cells from five animals of each group. Data were analysed with repeated measures ANOVA. *P < 0.05; +E2β/P4 vs. –E2β/P4.
Figure 8. N‐acetylcysteine recovered steroid hormone‐mediated up‐regulation of NS1619‐induced relaxations in uterine arteries .
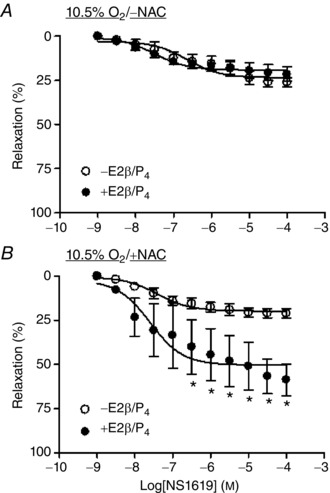
Uterine arteries of non‐pregnant sheep were treated with or without 17β‐estradiol (E2β; 0.3 nm)/progesterone (P4; 100 nm) under hypoxia (10.5% O2) for 48 h, in the absence or presence of 1 mm N‐acetylcysteine (NAC). NS1619‐induced relaxations were determined after the treatments. Data are means ± SEM of five tissues from five animals of each group. Data were analysed with repeated measures ANOVA. *P < 0.05; +E2β/P4 vs. –E2β/P4.
Discussion
The present study presents several novel findings. (1) The ex vivo treatment of uterine arteries with prolonged hypoxia increased oxidative stress in the vessel, mimicking the effect observed in vivo in pregnant sheep acclimatized to long‐term high‐altitude hypoxia. (2) Hypoxia had a direct effect on the down‐regulation of BKCa channel β1 subunit expression and BKCa channel activity in uterine arteries, emulating the adverse effect of long‐term high‐altitude hypoxia on pregnant sheep. (3) Antioxidants ablated oxidative stress and blocked the hypoxia‐induced inhibition of BKCa channels, providing a mechanistic understanding of oxidative stress as a causal role in the hypoxia‐mediated effect. (4) Steroid hormone‐induced up‐regulation of BKCa channel function in uterine arteries was inhibited by hypoxia and this direct hypoxia‐mediated effect was recovered by antioxidants, suggesting a target of hormonal effect by oxidative stress. (5) The impaired BKCa channel β1 subunit expression and channel activity in uterine arteries of pregnant sheep exposed to long‐term high‐altitude hypoxia were ameliorated by inhibiting oxidative stress, linking the findings of ex vivo and in vivo hypoxia‐mediated effects and physiological significance in animals.
Oxygen is a major determinant of gene expression in the cardiovascular system (Ratcliffe et al. 1998; Peers, 2002). Chronic hypoxia has been shown to cause down‐regulation of BKCa channels in VSMCs (Bonnet et al. 2003; Navarro‐Antolin et al. 2005). Consistent with these observations, we demonstrated that prolonged exposure of uterine arteries from pregnant sheep to hypoxia resulted in down‐regulation of the BKCa channel β1 subunit. BKCa channels in VSMCs are primarily activated by an increase in intracellular Ca2+ concentrations; and the association of the β1 subunit to the pore forming α subunit dramatically increases the channel's sensitivity to Ca2+ (Brenner et al. 2000; Hu & Zhang, 2012). The primary role of BKCa channels in the vasculature is to hyperpolarize the VSMC membrane and to promote vasorelaxation (Hu & Zhang, 2012). The impaired expression of the BKCa channel β1 subunit would in turn decrease the activation of BKCa channels, which could explain the reduced vasorelaxation mediated by BKCa channels and increased vascular pressure‐dependent tone in uterine arteries of pregnant animals exposed to ex vivo hypoxia. However, the mechanism by which the BKCa channel β1 subunit is down‐regulated following exposure to prolonged hypoxia is not completely understood. The similarity of adverse effects of long‐term high‐altitude hypoxia (Hu et al. 2012) and ex vivo hypoxia on BKCa channels suggests a direct effect of hypoxia in regulating BKCa channels in uterine arteries and that the ex vivo studies may provide an effective model to explore cellular and molecular mechanisms underlying aberrant uterine vascular function in response to high‐altitude chronic hypoxia.
It is well established that ROS and oxidative stress play critical roles in the physiological and pathophysiological processes in the cardiovascular system (Taniyama & Griendling, 2003; Papaharalambus & Griendling, 2007). ROS include oxygen radicals such as superoxide (O2 •−) and hydroxyl (•OH) and non‐radical hydrogen peroxide (H2O2); and superoxide and H2O2 are the major ROS in the cardiovascular system (Bedard & Krause, 2007; Papaharalambus & Griendling, 2007). ROS are generated through a cascade of chemical reactions that starts with the production of superoxide by the Nox family. Superoxide is short‐lived and rapidly dismutated by SOD to H2O2, which is then converted by catalase to H2O. Oxidative stress occurs as the result of an imbalance of production and removal of ROS. Chronic hypoxia promotes oxidative stress in vitro and in vivo and increases ROS in cultured VSMCs (Taniyama & Griendling, 2003; Papaharalambus & Griendling, 2007; Parraguez et al. 2011). In the present study, we demonstrated that ex vivo prolonged hypoxia treatment also promoted oxidative stress in uterine arteries. The level of oxidative stress in uterine arteries exposed to ex vivo hypoxia reported in the present study is comparable to the previous measurements in the vessels isolated from animals acclimatized to long‐term high‐altitude hypoxia (Xiao et al. 2013). Thus, our ex vivo culture condition largely mimicked high‐altitude impacts on this vessel. High‐altitude hypoxia elevated maternal oxidative stress in ewes (Parraguez et al. 2011), and gestational hypoxia promoted both maternal and placental oxidative stress in rats (Richter et al. 2012). It may be difficult to compare directly the data obtained from ex vivo studies and this in vivo study as different oxidative biomarkers were determined. Moreover, it is probable that locally produced ROS may play a predominant role in altering organ/tissue function. N‐Acetylcysteine functions as an ROS scavenger through increasing intracellular glutathione and its thiol‐disulfide exchange activity (Zafarullah et al. 2003). Although N‐acetylcysteine may exert its action through non‐antioxidant activities (Sun, 2010), the observations that the free radical scavenger N‐acetylcysteine and SOD/catalase mimetic EUK 134 partially or completely reversed the effects of chronic hypoxia on the BKCa channel suggest that hypoxia‐induced enhancement of ROS generation was primarily responsible for the aberrant BKCa channel function in uterine arteries. EUK 134 is a SOD/catalase mimetic and may not be able to discern oxidative species such as superoxide radicals and H2O2 that may be involved (Rong et al. 1999). Our recent findings that uterine arteries of high‐altitude pregnant sheep exhibited increased oxidative stress in part by enhanced expression and activity of Nox2, and that an SOD mimetic tempol inhibited hypoxia‐mediated increase in myogenic reactivity of uterine arteries (Xiao et al. 2013), would suggest a possible role of superoxide radicals in suppressing BKCa channel function in response to hypoxia. However, the possible involvement of H2O2 in impairing BKCa channel activity could not be excluded.
Increased ROS productions in cells could modify ion channel function through multiple mechanisms including post‐translational modifications of key amino acid residues in channel proteins by oxidation and regulation of gene expression (Matalon et al. 2003). Our previous findings that acute application of N‐acetylcysteine or the Nox inhibitor apocynin enhanced BKCa channel activity and the BKCa channel opener NS1619 induced relaxations in uterine arteries of high‐altitude pregnant sheep (Xiao et al. 2013; Zhu et al. 2014) suggest that ROS could alter BKCa channel function by post‐translational modification of the channel. The impairment of BKCa channel‐mediated relaxation is unlikely to be due to endothelial damage as endothelium‐dependent relaxation of this vessel was increased by chronic hypoxia, whereas endothelium‐independent relaxation was not altered by chronic hypoxia (Xiao et al. 2001). BKCa channels in uterine arteries are inhibited by protein kinase C (PKC), and chronic hypoxia‐induced upregulation of PKC has been shown to attenuate BKCa channel‐mediated relaxation of uterine arteries in pregnancy (Hu et al. 2011; Xiao et al. 2014). Of interest, a chronic hypoxia‐induced increase in PKC‐mediated myogenic reactivity in uterine arteries of pregnant sheep was blocked by apocynin and tempol (Xiao et al. 2013).
In the present study, we extended our previous investigations and assessed the impact of oxidative stress induced by ex vivo hypoxia on BKCa channel expression and function in uterine arteries. We demonstrated that the direct effect of prolonged hypoxia induced an increase in oxidative stress and inhibition of BKCa channel β1 subunit expression in uterine arteries, and these effects were blocked by antioxidants. These findings suggest that oxidative stress caused by hypoxia plays a causal role in the down‐regulation of the BKCa channel β1 subunit in uterine arteries. Furthermore, the findings that hypoxia‐mediated suppression of BKCa channel activity decreases in BKCa channel‐mediated vasorelaxation and increases in pressure‐dependent tone all recovered by antioxidants suggest that the dysfunction of BKCa channels in uterine arteries is probably secondary to BKCa channel repression conferred by increased oxidative stress. These observations support the notion that ROS play an important causative role in chronic hypoxia‐induced BKCa channel repression and dysfunction in uterine arteries. Consistent with the present findings, oxidative stress was attributed to down‐regulation of the BKCa channel β1 subunit in arteries, as well as SKCa and IKCa channels in atria of diabetic rodents (Lu et al. 2012; Zhao et al. 2014; Yi et al. 2015). Taken together, these studies provide evidence that the direct effect of chronic hypoxia regulates BKCa channel function in uterine arteries of pregnant animals via increased oxidative stress, involving both post‐translational modification and gene expression. Downregulation of the BKCa channel β1 subunit by heightened oxidative stress may also result from alterations in protein turnover (i.e. translation and degradation). However, our recent finding that long‐term high altitude drastically reduced Ca2+‐activated K+ subunit beta‐1 (KCNMB 1) mRNA level in uterine arteries from pregnant sheep (Chen et al. 2014) suggests that ROS generated during hypoxic exposure probably caused repression of the KCNMB 1 gene.
Both in vivo and ex vivo studies demonstrated that oestrogen alone or in conjunction with progesterone was able to imitate pregnancy‐induced up‐regulation of BKCa channel β1 subunit expression at both protein and mRNA levels and BKCa channel function in uterine arteries of non‐pregnant sheep (Nagar et al. 2005; Hu et al. 2011; Zhu et al. 2014). However, the up‐regulation of BKCa channel β1 subunit expression/function by steroid hormones was inhibited by chronic hypoxia (Hu et al. 2012; Chen et al. 2014; Zhu et al. 2014). In the present study, we showed that the oxidative stress promoted by chronic hypoxia in uterine arteries was responsible for dampening steroid hormone‐stimulated up‐regulation of BKCa channel function, as antioxidants were able to reinstate the hormonal upregulation of the BKCa channel. Similarly, we previously demonstrated that co‐treatment of uterine arteries of high‐altitude non‐pregnant animals with steroid hormones and N‐acetylcysteine restored BKCa channel activity and BKCa channel‐mediated vasorelaxation (Zhu et al. 2014). N‐Acetylcysteine probably restored the genomic actions of steroid hormones in upregulating BKCa channel expression and function in this study, although acute applications of oestrogen (≥ 100 nm) could upregulate BKCa channel activity via non‐genomic effects in VSMCs (White et al. 1995; Rosenfeld et al. 2000). The regulation of gene expression by oestrogen is primarily mediated by oestrogen receptors ER‐α and ER‐β in the cardiovascular system (Murphy, 2011). ER‐α plays a critical role in oestrogen‐mediated modulation of uterine vascular function, and both in vivo and ex vivo chronic hypoxia caused down‐regulation of ER‐α (Chang et al. 2010; Dasgupta et al. 2012). Oxidative stress has been demonstrated to down‐regulate ER‐α in endothelial cells (Chakrabarti & Davidge, 2009) and in a cancer cell line (Weitsman et al. 2009). This down‐regulation could be triggered by post‐translational modifications of ER‐α serine residues (Weitsman et al. 2009) and/or epigenetic modification of the ER‐α gene (Dasgupta et al. 2012). The diminished effect of steroid hormones in regulating BKCa channel function following exposure to chronic hypoxia is probably the consequence of the down‐regulation of ER‐α. In addition, oxidative stress has been shown to inhibit the binding of ERs to DNA (Liang et al. 1998), thus disrupting oestrogen‐stimulated transcription of the KCNMB 1 gene. Nevertheless, reducing oxidative stress may prevent the adverse effects of chronic hypoxia on ER‐α and reinstate the ability of steroid hormones to upregulate BKCa channel expression and function in uterine arteries.
The finding that the antioxidants were able to suppress chronic hypoxia‐induced down‐regulation of BKCa channel expression and function in uterine arteries is appealing. High‐altitude hypoxia has been shown to reduce uterine blood flow and is associated with increased incidence of pre‐eclampsia and fetal growth restriction (Zamudio et al. 1995 a,b; Palmer et al. 1999; Browne et al. 2011). Compelling evidence indicates that oxidative stress plays an important role in the pathogenesis of pregnancy complications including pre‐eclampsia and fetal growth restriction (Roggensack et al. 1999; Raijmakers et al. 2004; Siddiqui et al. 2010; Stanley et al. 2012). Recent studies revealed that Nox 2 expression was upregulated in umbilical vessels and primary cultured human umbilical vein endothelial cells from pre‐eclamptic pregnancy (Choi et al. 2013; Lim et al. 2015). It appeared that Nox 2‐derived ROS caused down‐regulation of KCa3.1 in these vessels (Choi et al. 2013). Interestingly, we also demonstrated upregulation of Nox 2 in uterine arteries from sheep acclimatized to long‐term high altitude (Xiao et al. 2013) and development of pre‐eclampsia‐like symptoms in pregnant rats following gestational hypoxia (Zhou et al. 2013). Our findings raise the possibility that ROS‐induced impairment of BKCa channel function in uterine arteries could be attributed to the pathogenesis of these complications. Whereas it may be relatively difficult to directly test this link in vivo by inhibiting Nox 2‐derived ROS in sheep exposed to long‐term high‐altitude hypoxia, rodents may provide an alternative model to further investigate this mechanism in vivo in future studies. Our recent study demonstrated that gestational hypoxia induced pre‐eclampsia‐like symptoms in pregnant rats (Zhou et al. 2013). Interestingly, chronic treatment with the ROS scavenger tempol in mouse models of fetal growth restriction and pre‐eclampsia improved fetal growth, which was associated with an increase in uterine artery blood flow (Hoffmann et al. 2008; Stanley et al. 2012). Moreover, supplementation with antioxidant oxidants such as vitamins C and E during pregnancy effectively prevents maternal oxidative stresses triggered by high‐altitude and gestational hypoxia, improving pregnancy outcomes (Parraguez et al. 2011; Richter et al. 2012). Similarly, the antioxidants N‐acetylcysteine and tempol have been shown to alleviate hypertension produced by reduced uterine perfusion in pregnant rats, an animal model mimicking pre‐eclampsia (Chang et al. 2005; Sedeek et al. 2008). Additionally, ROS‐scavenging therapies improved voltage‐gated K+ (KV) channel function in pulmonary arteries of newborn pigs with progressive hypoxia‐induced pulmonary hypertension (Fike et al. 2013) and reduced hypoxia‐induced pulmonary hypertension and right ventricular hypertrophy in rats (Hoshikawa et al. 2001). Thus, antioxidant treatment may provide a useful intervention to improve impaired uteroplacental function associated with pregnancy complications. However, note that minimal levels of ROS are necessary for proper cell function in early fetal development, and wide use of antioxidants should be avoided at this stage.
Additional information
Conflict of interest
None.
Author contributions
X.Q.H.: conceived, designed and performed experiments, analysed data, and wrote the manuscript. X.H.: performed experiments and analysed data. D.X.: performed experiments, analysed data and participated in writing the manuscript. L.Z.: provided animals and all other support for the studies, conceived and designed the experiments, interpreted data and wrote the manuscript. All contributors have read and approved the final manuscript for submission.
Funding
This work was supported by National Institutes of Health grants HD031226 (L.Z.), HL089012 (L.Z.) and HL110125 (L.Z.).
Acknowledgements
We thank Shriley Hu for ROS measurement.
References
- Ajith TA & Jayakumar TG (2014). Mitochondria‐targeted agents: future perspectives of mitochondrial pharmaceutics in cardiovascular diseases. World J Cardiol 6, 1091–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K & Krause KH (2007). The NOX family of ROS‐generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87, 245–313. [DOI] [PubMed] [Google Scholar]
- Bonnet S, Savineau JP, Barillot W, Dubuis E, Vandier C & Bonnet P (2003). Role of Ca2+‐sensitive K+ channels in the remission phase of pulmonary hypertension in chronic obstructive pulmonary diseases. Cardiovasc Res 60, 326–336. [DOI] [PubMed] [Google Scholar]
- Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT & Aldrich RW (2000). Vasoregulation by the β1 subunit of the calcium‐activated potassium channel. Nature 407, 870–876. [DOI] [PubMed] [Google Scholar]
- Browne VA, Julian CG, Toledo‐Jaldin L, Cioffi‐Ragan D, Vargas E & Moore LG (2015). Uterine artery blood flow, fetal hypoxia and fetal growth. Philos Trans R Soc Lond B Biol Sci 370, 20140068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne VA, Toledo‐Jaldin L, Davila RD, Lopez LP, Yamashiro H, Cioffi‐Ragan D, Julian CG, Wilson MJ, Bigham AW, Shriver MD, Honigman B, Vargas E, Roach R & Moore LG (2011). High‐end arteriolar resistance limits uterine artery blood flow and restricts fetal growth in preeclampsia and gestational hypertension at high altitude. Am J Physiol Regul Integr Comp Physiol 300, R1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S & Davidge ST (2009). High glucose‐induced oxidative stress alters estrogen effects on ERα and ERβ in human endothelial cells: reversal by AMPK activator. J Steroid Biochem Mol Biol 117, 99–106. [DOI] [PubMed] [Google Scholar]
- Chang EY, Barbosa E, Paintlia MK, Singh A & Singh I (2005). The use of N‐acetylcysteine for the prevention of hypertension in the reduced uterine perfusion pressure model for preeclampsia in Sprague‐Dawley rats. Am J Obstet Gynecol 193, 952–956. [DOI] [PubMed] [Google Scholar]
- Chang K, Xiao D, Huang X, Xue Z, Yang S, Longo LD & Zhang L (2010). Chronic hypoxia inhibits sex steroid hormone‐mediated attenuation of ovine uterine arterial myogenic tone in pregnancy. Hypertension 56, 750–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Dasgupta C, Xiong F & Zhang L (2014). Epigenetic upregulation of large‐conductance Ca2+‐activated K+ channel expression in uterine vascular adaptation to pregnancy. Hypertension 64, 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zhong Z, Xu Z, Chen L & Wang Y (2010). 2′,7′‐Dichlorodihydrofluorescein as a fluorescent probe for reactive oxygen species measurement: forty years of application and controversy. Free Radic Res 44, 587–604. [DOI] [PubMed] [Google Scholar]
- Choi S, Kim JA, Na HY, Kim JE, Park S, Han KH, Kim YJ & Suh SH (2013). NADPH oxidase 2‐derived superoxide downregulates endothelial KCa3.1 in preeclampsia. Free Radic Biol Med 57, 10–21. [DOI] [PubMed] [Google Scholar]
- Dasgupta C, Chen M, Zhang H, Yang S & Zhang L (2012). Chronic hypoxia during gestation causes epigenetic repression of the estrogen receptor‐α gene in ovine uterine arteries via heightened promoter methylation. Hypertension 60, 697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikalov S, Griendling KK & Harrison DG (2007). Measurement of reactive oxygen species in cardiovascular studies. Hypertension 49, 717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GR, Selemidis S, Griendling KK & Sobey CG (2011). Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov 10, 453–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fike CD, Aschner JL, Kaplowitz MR, Zhang Y & Madden JA (2013). Reactive oxygen species scavengers improve voltage‐gated K+ channel function in pulmonary arteries of newborn pigs with progressive hypoxia‐induced pulmonary hypertension. Pulm Circ 3, 551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fresquet F, Pourageaud F, Leblais V, Brandes RP, Savineau JP, Marthan R & Muller B (2006). Role of reactive oxygen species and gp91phox in endothelial dysfunction of pulmonary arteries induced by chronic hypoxia. Br J Pharmacol 148, 714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano FJ (2005). Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest 115, 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MA, Yang Y, Ella SR, Davis MJ & Braun AP (2010). Large conductance, Ca2+‐activated K+ channels (BKCa) and arteriolar myogenic signaling. FEBS Lett 584, 2033–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann DS, Weydert CJ, Lazartigues E, Kutschke WJ, Kienzle MF, Leach JE, Sharma JA, Sharma RV & Davisson RL (2008). Chronic tempol prevents hypertension, proteinuria, and poor feto‐placental outcomes in BPH/5 mouse model of preeclampsia. Hypertension 51, 1058–1065. [DOI] [PubMed] [Google Scholar]
- Holland M, Langton PD, Standen NB & Boyle JP (1996). Effects of the BKCa channel activator, NS1619, on rat cerebral artery smooth muscle. Br J Pharmacol 117, 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshikawa Y, Ono S, Suzuki S, Tanita T, Chida M, Song C, Noda M, Tabata T, Voelkel NF & Fujimura S (2001). Generation of oxidative stress contributes to the development of pulmonary hypertension induced by hypoxia. J Appl Physiol 90, 1299–1306. [DOI] [PubMed] [Google Scholar]
- Hu XQ, Xiao D, Zhu R, Huang X, Yang S, Wilson S & Zhang L (2011). Pregnancy upregulates large‐conductance Ca2+‐activated K+ channel activity and attenuates myogenic tone in uterine arteries. Hypertension 58, 1132–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XQ, Xiao D, Zhu R, Huang X, Yang S, Wilson SM & Zhang L (2012). Chronic hypoxia suppresses pregnancy‐induced upregulation of large‐conductance Ca2+‐activated K+ channel activity in uterine arteries. Hypertension 60, 214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XQ & Zhang L (2012). Function and regulation of large conductance Ca2+‐activated K+ channel in vascular smooth muscle cells. Drug Discov Today 17, 974–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert G, Brichant JF, Hartstein G, Bonhomme V & Dewandre PY (2014). Preeclampsia: an update. Acta Anaesthesiol Belg 65, 137–149. [PubMed] [Google Scholar]
- Lang U, Baker RS, Braems G, Zygmunt M, Kunzel W & Clark KE (2003). Uterine blood flow – a determinant of fetal growth. Eur J Obstet Gynecol Reprod Biol 110 Suppl 1, S55–61. [DOI] [PubMed] [Google Scholar]
- Liang X, Lu B, Scott GK, Chang CH, Baldwin MA & Benz CC (1998). Oxidant stress impaired DNA‐binding of estrogen receptor from human breast cancer. Mol Cell Endocrinol 146, 151–161. [DOI] [PubMed] [Google Scholar]
- Lim R, Acharya R, Delpachitra P, Hobson S, Sobey CG, Drummond GR & Wallace EM (2015). Activin and NADPH‐oxidase in preeclampsia: insights from in vitro and murine studies. Am J Obstet Gynecol 212, 86.e81–12. [DOI] [PubMed] [Google Scholar]
- Liu JQ, Zelko IN, Erbynn EM, Sham JS & Folz RJ (2006). Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox). Am J Physiol Lung Cell Mol Physiol 290, L2–10. [DOI] [PubMed] [Google Scholar]
- Lu T, Chai Q, Yu L, d'Uscio LV, Katusic ZS, He T & Lee HC (2012). Reactive oxygen species signaling facilitates FOXO‐3a/FBXO‐dependent vascular BK channel β1 subunit degradation in diabetic mice. Diabetes 61, 1860–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matalon S, Hardiman KM, Jain L, Eaton DC, Kotlikoff M, Eu JP, Sun J, Meissner G & Stamler JS (2003). Regulation of ion channel structure and function by reactive oxygen–nitrogen species. Am J Physiol Lung Cell Mol Physiol 285, L1184–1189. [DOI] [PubMed] [Google Scholar]
- Murphy E (2011). Estrogen signaling and cardiovascular disease. Circ Res 109, 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagar D, Liu XT & Rosenfeld CR (2005). Estrogen regulates β1‐subunit expression in Ca2+‐activated K+ channels in arteries from reproductive tissues. Am J Physiol Heart Circ Physiol 289, H1417–1427. [DOI] [PubMed] [Google Scholar]
- Navarro‐Antolin J, Levitsky KL, Calderon E, Ordonez A & Lopez‐Barneo J (2005). Decreased expression of maxi‐K+ channel β1‐subunit and altered vasoregulation in hypoxia. Circulation 112, 1309–1315. [DOI] [PubMed] [Google Scholar]
- Olesen SP, Munch E, Moldt P & Drejer J (1994). Selective activation of Ca2+‐dependent K+ channels by novel benzimidazolone. Eur J Pharmacol 251, 53–59. [DOI] [PubMed] [Google Scholar]
- Palmer SK, Moore LG, Young D, Cregger B, Berman JC & Zamudio S (1999). Altered blood pressure course during normal pregnancy and increased preeclampsia at high altitude (3100 meters) in Colorado. Am J Obstet Gynecol 180, 1161–1168. [DOI] [PubMed] [Google Scholar]
- Palmer SK, Zamudio S, Coffin C, Parker S, Stamm E & Moore LG (1992). Quantitative estimation of human uterine artery blood flow and pelvic blood flow redistribution in pregnancy. Obstet Gynecol 80, 1000–1006. [PubMed] [Google Scholar]
- Papaharalambus CA & Griendling KK (2007). Basic mechanisms of oxidative stress and reactive oxygen species in cardiovascular injury. Trends Cardiovasc Med 17, 48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parraguez VH, Atlagich M, Araneda O, Garcia C, Munoz A, De Los Reyes M & Urquieta B (2011). Effects of antioxidant vitamins on newborn and placental traits in gestations at high altitude: comparative study in high and low altitude native sheep. Reprod Fertil Dev 23, 285–296. [DOI] [PubMed] [Google Scholar]
- Patterson AJ, Xiao D, Xiong F, Dixon B & Zhang L (2012). Hypoxia‐derived oxidative stress mediates epigenetic repression of PKCε gene in foetal rat hearts. Cardiovasc Res 93, 302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peers C (2002). The G. L. Brown Prize Lecture. Hypoxic regulation of ion channel function and expression. Exp Physiol 87, 413–422. [DOI] [PubMed] [Google Scholar]
- Raijmakers MT, Dechend R & Poston L (2004). Oxidative stress and preeclampsia: rationale for antioxidant clinical trials. Hypertension 44, 374–380. [DOI] [PubMed] [Google Scholar]
- Ratcliffe PJ, O'Rourke JF, Maxwell PH & Pugh CW (1998). Oxygen sensing, hypoxia‐inducible factor‐1 and the regulation of mammalian gene expression. J Exp Biol 201, 1153–1162. [DOI] [PubMed] [Google Scholar]
- Richter HG, Camm EJ, Modi BN, Naeem F, Cross CM, Cindrova‐Davies T, Spasic‐Boskovic O, Dunster C, Mudway IS, Kelly FJ, Burton GJ, Poston L & Giussani DA (2012). Ascorbate prevents placental oxidative stress and enhances birth weight in hypoxic pregnancy in rats. J Physiol 590, 1377–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roggensack AM, Zhang Y & Davidge ST (1999). Evidence for peroxynitrite formation in the vasculature of women with preeclampsia. Hypertension 33, 83–89. [DOI] [PubMed] [Google Scholar]
- Rong Y, Doctrow SR, Tocco G & Baudry M (1999). EUK‐134, a synthetic superoxide dismutase and catalase mimetic, prevents oxidative stress and attenuates kainate‐induced neuropathology. Proc Natl Acad Sci USA 96, 9897–9902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld CR (1977). Distribution of cardiac output in ovine pregnancy. Am J Physiol 232, H231–235. [DOI] [PubMed] [Google Scholar]
- Rosenfeld CR, Cornfield DN & Roy T (2001). Ca2+‐activated K+ channels modulate basal and E2β‐induced rises in uterine blood flow in ovine pregnancy. Am J Physiol Heart Circ Physiol 281, H422–431. [DOI] [PubMed] [Google Scholar]
- Rosenfeld CR, Liu XT & DeSpain K (2009). Pregnancy modifies the large conductance Ca2+‐activated K+ channel and cGMP‐dependent signaling pathway in uterine vascular smooth muscle. Am J Physiol Heart Circ Physiol 296, H1878–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld CR, White RE, Roy T & Cox BE (2000). Calcium‐activated potassium channels and nitric oxide coregulate estrogen‐induced vasodilation. Am J Physiol Heart Circ Physiol 279, H319–328. [DOI] [PubMed] [Google Scholar]
- Sedeek M, Gilbert JS, LaMarca BB, Sholook M, Chandler DL, Wang Y & Granger JP (2008). Role of reactive oxygen species in hypertension produced by reduced uterine perfusion in pregnant rats. Am J Hypertens 21, 1152–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui IA, Jaleel A, Tamimi W & Al Kadri HM (2010). Role of oxidative stress in the pathogenesis of preeclampsia. Arch Gynecol Obstet 282, 469–474. [DOI] [PubMed] [Google Scholar]
- Stanley JL, Andersson IJ, Hirt CJ, Moore L, Dilworth MR, Chade AR, Sibley CP, Davidge ST & Baker PN (2012). Effect of the antioxidant tempol on fetal growth in a mouse model of fetal growth restriction. Biol Reprod 87, 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SY (2010). N‐acetylcysteine, reactive oxygen species and beyond. Cancer Biol Ther 9, 109–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniyama Y & Griendling KK (2003). Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension 42, 1075–1081. [DOI] [PubMed] [Google Scholar]
- Weitsman GE, Weebadda W, Ung K & Murphy LC (2009). Reactive oxygen species induce phosphorylation of serine 118 and 167 on estrogen receptor alpha. Breast Cancer Res Treat 118, 269–279. [DOI] [PubMed] [Google Scholar]
- White RE, Darkow DJ & Lang JL (1995). Estrogen relaxes coronary arteries by opening BKCa channels through a cGMP‐dependent mechanism. Circ Res 77, 936–942. [DOI] [PubMed] [Google Scholar]
- Xiao D, Bird IM, Magness RR, Longo LD & Zhang L (2001). Upregulation of eNOS in pregnant ovine uterine arteries by chronic hypoxia. Am J Physiol Heart Circ Physiol 280, H812–820. [DOI] [PubMed] [Google Scholar]
- Xiao D, Hu XQ, Huang X, Zhou J, Wilson SM, Yang S & Zhang L (2013). Chronic hypoxia during gestation enhances uterine arterial myogenic tone via heightened oxidative stress. PloS ONE 8, e73731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao D, Huang X, Yang S & Zhang L (2009). Direct chronic effect of steroid hormones in attenuating uterine arterial myogenic tone: role of protein kinase C/extracellular signal‐regulated kinase 1/2. Hypertension 54, 352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao D, Zhu R & Zhang L (2014). Gestational hypoxia up‐regulates protein kinase C and inhibits calcium‐activated potassium channels in ovine uterine arteries. Int J Med Sci 11, 886–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong F, Xiao D & Zhang L (2012). Norepinephrine causes epigenetic repression of PKCε gene in rodent hearts by activating Nox1‐dependent reactive oxygen species production. FASEB J 26, 2753–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F, Ling TY, Lu T, Wang XL, Li J, Claycomb WC, Shen WK & Lee HC (2015). Down‐regulation of the small‐conductance calcium‐activated potassium channels in diabetic mouse atria. J Biol Chem 290, 7016–7026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafarullah M, Li WQ, Sylvester J & Ahmad M (2003). Molecular mechanisms of N‐acetylcysteine actions. Cell Mol Life Sci 60, 6–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamudio S, Palmer SK, Dahms TE, Berman JC, Young DA & Moore LG (1995. a). Alterations in uteroplacental blood flow precede hypertension in preeclampsia at high altitude. J Appl Physiol 79, 15–22. [DOI] [PubMed] [Google Scholar]
- Zamudio S, Palmer SK, Droma T, Stamm E, Coffin C & Moore LG (1995. b). Effect of altitude on uterine artery blood flow during normal pregnancy. J Appl Physiol 79, 7–14. [DOI] [PubMed] [Google Scholar]
- Zhao LM, Wang Y, Ma XZ, Wang NP & Deng XL (2014). Advanced glycation end products impair KCa3.1‐ and KCa2.3‐mediated vasodilatation via oxidative stress in rat mesenteric arteries. Pflugers Arch 466, 307–317. [DOI] [PubMed] [Google Scholar]
- Zhou J, Xiao D, Hu Y, Wang Z, Paradis A, Mata‐Greenwood E & Zhang L (2013). Gestational hypoxia induces preeclampsia‐like symptoms via heightened endothelin‐1 signaling in pregnant rats. Hypertension 62, 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu R, Hu XQ, Xiao D, Yang S, Wilson SM, Longo LD & Zhang L (2013). Chronic hypoxia inhibits pregnancy‐induced upregulation of SKCa channel expression and function in uterine arteries. Hypertension 62, 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu R, Huang X, Hu XQ, Xiao D & Zhang L (2014). Gestational hypoxia increases reactive oxygen species and inhibits steroid hormone‐mediated upregulation of Ca2+‐activated K+ channel function in uterine arteries. Hypertension 64, 415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]