

Abstract
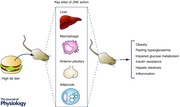
Obesity is currently at epidemic levels worldwide and is associated with a wide range of diseases such as type 2 diabetes, cardiovascular disease, fatty liver disease and certain forms of cancer. Obesity‐induced chronic inflammation is central to the disrupted metabolic homeostasis which underlies many of these conditions. While research over the past decade has identified many of the cells and signalling molecules that contribute to obesity‐induced inflammation, perhaps the best characterised are the stress‐activated c‐Jun NH2‐terminal kinases (JNKs). JNKs are activated in obesity in numerous metabolically important cells and tissues such as adipose tissue, macrophages, liver, skeletal muscle and regions of the brain and pituitary. Elegant in vivo mouse studies using Cre‐LoxP‐mediated recombination of the JNK1 and JNK2 genes have revealed the remarkably diverse roles that JNKs play in the development of obesity‐induced inflammation, impaired glucose homeostasis and hepatic steatosis. While JNK activation in classical metabolically active tissues such as skeletal muscle and adipose tissue only appears to play a minor role on the induction of the above‐mentioned pathologies, recent studies have clearly established the important roles JNK signalling fulfils in macrophages, the liver and cells of the anterior pituitary. Collectively, these studies place JNKs as important mediators of obesity and obesity‐associated disruptions to metabolic homeostasis.
Abbreviations
- BMI
body mass index
- BMT
bone marrow transplantation
- CA
constitutive activation
- Cre
recombinase from P1 bacteriophage
- DIO2
type 2 iodothyronine deiodinase
- HFD
high fat diet
- IRS
insulin receptor substrate
- JNK
c‐Jun NH2‐terminal kinase
- KO
knock out
- MAPK
mitogen‐activated protein kinase
- NCORC1
nuclear receptor corepressor
- PPAR
peroxisome proliferator‐activated receptor
- ROS
reactive oxygen species
- T2D
type‐2 diabetes
- TSH
thyroid‐stimulating hormone
- WT
wild type
Introduction
Over the last three decades changes to human lifestyle such as diet, physical activity and microbial exposure have led to epidemic levels of overweight and obesity worldwide. Current trends predict that the levels of overweight (BMI of 25–30 kg m−2) and obesity (BMI > 30 kg m−2) will increase from 33% of the global population in 2005 to 57.8% in 2030 (Chen et al. 2012). The health consequences of this are significant as obesity is associated with a wide range of health problems including type 2 diabetes (T2D), cardiovascular disease, neurodegenerative disorders, fatty liver disease and certain forms of cancer (Hotamisligil, 2006). In the case of T2D, it was estimated that greater than 250 million people had T2D in 2010, a figure that is predicted to rise to more than 400 million by 2030 (Chen et al. 2012). Obesity poses a major challenge to the maintenance of organismal metabolic homeostasis and many of the health problems and diseases listed above involve disruptions to metabolic homeostasis (Kotas & Medzhitov, 2015). For example, the development of impaired glucose tolerance and insulin resistance are key risk factors for the development of T2D.
A wealth of experimental evidence shows that chronic inflammation is inextricably linked to altered metabolic homeostasis in obesity (reviewed in Hotamisligil, 2006; Osborn & Olefsky, 2012; McNelis & Olefsky, 2014), While numerous cell types and signalling pathways have been identified that are activated in obesity and contribute to altered metabolic homeostasis, perhaps the most well‐characterised are the c‐Jun NH2‐terminal kinases (JNKs). JNKs are members of the mitogen‐activated protein kinase (MAPK) family, are activated by a wide variety of stress‐inducing stimuli, and, from Drosophila to mammals, play a number of important roles in metabolism and the development of obesity‐induced glucose intolerance and insulin resistance (Hirosumi et al. 2002; Hotamisligil, 2006; Woodcock et al. 2015). It has been shown that JNKs are activated in obese humans, highlighting the importance of unravelling the significance of JNK signalling on the metabolic consequences of obesity. In this review we will provide a brief overview of the molecular regulation of the JNK signalling cascade and a historical perspective on the role of JNK in obesity and metabolism. Then, in what will comprise the majority of this review, we will discuss the recent research that has revealed the many roles that JNKs play in mediating the metabolic disruptions caused by obesity.
JNK activation in obesity
The JNK family consists of three members, JNK1 (Mapk8), JNK2 (Mapk9) and JNK3 (Mapk10) and while JNK1 and JNK2 are ubiquitously expressed, JNK3 expression is principally restricted to regions of the brain, heart and testis (Davis, 2000; Seki et al. 2012). While a comprehensive discussion on the JNK signalling cascade is beyond the scope of the current review, a brief summary is included; for a more comprehensive discussion of the JNK signalling cascade see references (Davis, 2000; Seki et al. 2012; Sabio & Davis, 2014). JNKs are key effector serine/threonine protein kinases that belong to the stress‐activated MAPK family. JNKs are activated by two upstream MAP2Ks, MKK4 and MKK7, which are in turn activated by a range of upstream MAP3Ks, which are themselves regulated by a number of different upstream factors (Sabio & Davis, 2014). What this apparent complexity achieves is an elegant system in which a wide variety of stimuli, e.g. growth factors, cytokines, heat shock, osmotic stress, UV radiation, reactive oxygen species and fatty acids, can be sensed by unique cellular mechanisms but all resulting in the activation of JNKs. In the context of obesity, it is well known that high circulating levels of pro‐inflammatory cytokines such as tumour necrosis factor α (TNFα) and interleukin‐1β (IL‐1β), and increases in free saturated fatty acids such as palmitate, activate JNK signalling in insulin target cells. Additionally, the induction of endoplasmic reticulum stress leads to activation of the JNK pathway (Urano et al. 2000) and this is proposed to be a key driver of JNK activation in obesity (Ozcan et al. 2004). Another potentially important factor for the activation of JNK during obesity are reactive oxygen species (ROS) (Houstis et al. 2006). Increased accumulation of ROS results in oxidative stress, which activates stress‐kinase signalling such as JNK (Nakano et al. 2006). Once activated, JNKs can regulate several nuclear and extra‐nuclear substrates. Perhaps best characterised is the transcription factor activator protein 1 (AP1) which controls the expression of numerous genes including the pro‐inflammatory cytokines. Furthermore, early studies provided evidence that that JNKs might directly interfere with the insulin signalling pathway.
The central role of JNK in metabolism and insulin resistance – a brief history of JNKs
In the mid‐1990s, Hotamisligil and colleagues made the seminal observations that the expression of the pro‐inflammatory cytokine TNFα was elevated in the adipose tissue of obese mice and humans (Hotamisligil et al. 1993, 1995) and that ablation of TNFα’s actions improved insulin sensitivity in rodent models of obesity (Hotamisligil et al. 1993; Uysal et al. 1997). At around the same time, JNK1 was identified as the major protein kinase that bound to and phosphorylated the AP1 family member c‐Jun (Hibi et al. 1993; Derijard et al. 1994), and, as discussed above, it was subsequently shown that JNK1 was activated by a wide range of stress‐inducing stimuli. Since obesity is associated with a number of stressful stimuli on cells and tissues, such as increased TNFα and free fatty acid concentrations, many of which had been shown to activate JNK, it was hypothesised that JNKs may be a central mediator of many of the deleterious consequences of obesity, such as impaired glucose metabolism and insulin resistance (Hirosumi et al. 2002). Indeed, it was shown that global JNK1 knockout (KO) mice fed a high fat diet (HFD) were protected from the development of impaired glucose tolerance and insulin resistance (Hirosumi et al. 2002). However, perhaps most strikingly, when fed a HFD or crossed to the genetically obese ob/ob background, the deletion of JNK1 profoundly protected against the development of obesity (Hirosumi et al. 2002). These findings raised the question of what signalling events might be responsible for the protective phenotype in JNK1 KO mice. In earlier in vitro studies it was shown that JNK is able to phosphorylate IRS1 at its S307 site (Aguirre et al. 2000). Phosphorylation at S307 promotes impaired insulin action and thereby presents a mechanism by which the activation of JNK can directly antagonise insulin action. Indeed, when investigating IRS1 S307 phosphorylation, it was shown that upon high fat feeding JNK1 KO mice display reduced levels of IRS1 S307 phosphorylation when compared to wild‐type (WT) control animals. The notion that phosphorylation of IRS1 by activated JNK in obesity is a major mechanism of insulin resistance and impaired glucose metabolism is now widespread. Hence, this is one of many possible mechanisms by which JNK activation links insulin resistance during the course of obesity. It is worth noting that the same studies have been performed in JNK2 deficient mice, but probably due to compensatory action by JNK1, JNK2 KO mice show a similar increase in obesity and insulin resistance as WT control mice. However, later studies have also implicated a more prominent role for the JNK2 isoform to partially be responsible for the development of obesity‐associated insulin resistance. The latter finding requires the absence of JNK1 signalling, which normally would compensate for the loss of JNK2 (Tuncman et al. 2006). While it was proposed that the deletion of JNK1 improves glucose tolerance and insulin sensitivity via reduced serine phosphorylation of the key insulin signalling pathway intermediate IRS1 (Hirosumi et al. 2002), subsequent research using mouse models that harbour tissue specific deletion of JNK1 and/or JNK2 have identified numerous mechanisms by which JNKs are likely to impact on metabolism in obesity. A crucial tool in the elucidation of the cell and tissue specific effects of JNKs has been the generation of conditional JNK1 and JNK2 alleles harbouring LoxP sites. When these so‐called ‘floxed’ mice are crossed with mice expressing the enzyme Cre recombinase under the control of cell/tissue specific promoters it allows for targeted excision of the gene of interest, i.e. Jnk1 and Jnk2. These mice have been invaluable in elucidating the different roles that JNKs play and much of the succeeding discussion pertains to studies that have used these mice to assess the roles of JNKs in specific cells and tissues.
The role of JNKs in adipose tissue
Chronic inflammation within adipose tissue is a hallmark of obesity and plays an important role in the impaired glucose metabolism and insulin resistance that occurs in obesity (Osborn & Olefsky, 2012; McNelis & Olefsky, 2014). Adipose tissue inflammation is highly multifactorial, consisting of the recruitment of many immune cell types, the production of a wide array of secreted products including pro‐inflammatory cytokines, e.g. TNFα, IL‐1β, interferon γ (IFNγ) and IL‐6, proteases, nitric oxide and an increase in the local concentration of free fatty acids (Osborn & Olefsky, 2012; McNelis & Olefsky, 2014). This pro‐inflammatory environment impairs adipocyte insulin action, which has effects at peripheral tissues such as the liver (Perry et al. 2015). Given that JNK activation is downstream of many of these pro‐inflammatory factors, the activation of JNK within the adipocyte may be an important mechanism of inflammation‐induced adipocyte insulin resistance. Indeed, the deletion of JNK1 specifically within adipocytes using a mouse model in which Cre‐recombinase is expressed under the control of the AP2 promoter improved adipocyte insulin action in HF fed mice (Sabio et al. 2008). Furthermore, adipocyte‐specific JNK1 deletion improved hepatic insulin action in a mechanism that was dependent on the production of IL‐6 (Sabio et al. 2008). Specifically, adipocyte deletion of JNK1 in HF fed mice reduced the expression of IL‐6 within the adipose tissue and decreased plasma concentrations of IL‐6. This was associated with a reduction in the expression of the classical IL‐6 target gene suppressor of cytokine signalling 3 (SOCS3) in the liver (Sabio et al. 2008), an effect that was reversed upon the administration of recombinant IL‐6 to adipocyte JNK1 KO mice (Sabio et al. 2008). Importantly, SOCS3 has previously been linked to the development of hepatic insulin resistance (Galic et al. 2014). Of note, deletion of JNK1 from adipocytes did not affect the development of obesity, demonstrating that adipocyte JNK1 is not responsible for the protection against obesity observed in global JNK1 KO mice (Sabio et al. 2008). (A summary of these findings is shown in Fig. 1.) However, the detrimental effects of the above described hepatic IL‐6 signalling on insulin resistance have been challenged using mice that lack the IL‐6 receptor α specifically in the liver (Wunderlich et al. 2010). It should be noted that the use of the AP2 promoter to drive Cre‐mediated gene recombination is not specific to white adipose tissue and results in efficient deletion of JNK1 from brown adipose tissue (Sabio et al. 2008). Furthermore, AP2‐Cre has shown to be active in macrophages and regions of the peripheral and central nervous systems (CNS) (Furuhashi & Hotamisligil, 2008; Martens et al. 2010). Given the important roles of both brown adipose tissue and regions of the CNS in whole body metabolism, a degree of caution should be exercised in attributing the effects observed by Sabio et al. (2008) to the deletion of JNK1 solely within adipocytes of the white adipose tissue. Findings for the role of JNK1 in adipose tissue will need to be confirmed by the use of the AdipoQ‐Cre mouse, which results in ablation of JNK1 signalling specifically in adipose tissue.
Figure 1. The roles of macrophage and adipocyte JNK in the development of obesity‐associated metabolic disruption .
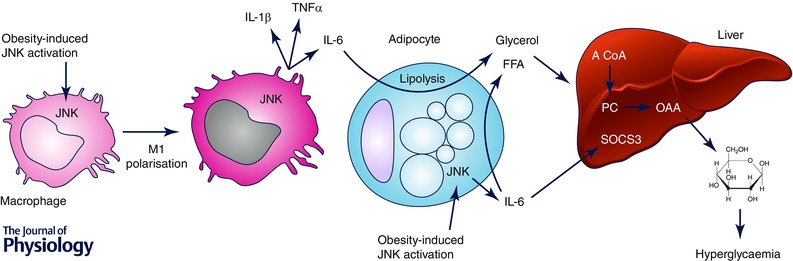
Macrophage JNK plays a critical role in the polarisation of macrophages to the M1, pro‐inflammatory state, leading to enhanced production of pro‐inflammatory factors such as IL‐1β, TNFα and IL‐6. These factors are likely to induce impaired glucose metabolism and insulin resistance in a number of ways. For example, macrophage JNK‐dependent IL‐6 production induces adipocyte lipolysis, resulting in enhanced liver glucose production (see text for details). JNK activation within adipocytes similarly increases IL‐6 production, potentially impacting on systemic metabolism in a similar manner to that of macrophage JNK activation. The up‐regulation of SOCS3 by JNK‐dependent, adipocyte‐derived IL‐6 may also lead to hepatic insulin resistance.
The role of JNKs in macrophages
Macrophages were the first immune cell type to be implicated in the development of obesity‐associated adipose tissue inflammation (Weisberg et al. 2003), and, given the importance of JNKs as regulators of inflammatory cytokine production, it was hypothesised that JNKs in macrophages may play an important role in the development of obesity‐induced inflammation, impaired glucose metabolism and insulin resistance. Initial studies on the role of JNKs in macrophages used the bone marrow transplantation (BMT) model in which bone marrow from donor mice, i.e. JNK1 KO, is transplanted into lethally irradiated recipient WT mice (Solinas et al. 2007). An important consideration with the BMT approach is that it results in the replacement of many types of immune cells, and, given that nearly all cells of the immune system are now implicated in the development of obesity‐associated inflammation and impaired glucose metabolism (Osborn & Olefsky, 2012), BMT is not a specific means of targeting macrophages. Nonetheless, BMT studies revealed that the loss of JNK1 within the immune system improves glucose and insulin tolerance and reduces adipose tissue inflammation following a HFD (Solinas et al. 2007). Of note, these improvements were independent of obesity, as the loss of JNK1 within the immune system did not affect the HFD‐induced accrual of fat mass (Solinas et al. 2007). However, the results of subsequent studies, using both the BMT approach (Sabio et al. 2008; Vallerie et al. 2008) and myeloid cell‐specific deletion of JNK1 using Cre‐LoxP technology (Sabio et al. 2008) have failed to fully support these initial results (Solinas et al. 2007), questioning the importance of JNK1 in macrophages to the development of obesity‐associated inflammation and insulin resistance. As discussed above, in most tissues, two JNK isoforms are present, JNK1 and JNK2, and, importantly, a large degree of functional redundancy exists between JNK1 and JNK2. Thus, it has been shown that both JNK1 and JNK2 play a role in adipose tissue inflammation. Accordingly, to definitively address the contribution of macrophage JNKs to the development of obesity, inflammation and insulin resistance, mice with a myeloid cell‐specific combined deletion of both JNK1 and JNK2 (macrophage JNK KO) have been generated (Han et al. 2013). Importantly, the combined loss of both JNK1 and JNK2 from macrophages results in a profound protection from obesity‐induced impairments to glucose metabolism and the development of insulin resistance, but has no effect on HFD‐induced obesity (Han et al. 2013). Insulin action in adipose tissue, liver and skeletal muscle is improved in macrophage JNK KO mice, highlighting the important role of macrophage JNK activation in mediating the deleterious effects of a HFD in multiple insulin target tissues (Han et al. 2013). Mechanistically, both in vitro and in vivo, macrophage JNK KO skews the polarisation of macrophages from the classically activated, M1 state, towards the anti‐inflammatory, M2 state. As a consequence, the levels of pro‐inflammatory cytokines such as TNFα, IL‐1β and IL‐6 within the adipose tissue, liver and circulation are decreased in macrophage JNK KO mice. Collectively, these results, which have recently been confirmed (Perry et al. 2015), demonstrate a critical role for macrophage JNK in the development of many of the deleterious consequences of a HFD, such as impaired glucose metabolism, insulin resistance and inflammation.
With regard to how pro‐inflammatory macrophages mediate the development of insulin resistance, it has conventionally been considered that the secreted products of M1 macrophages, such as TNFα, IL‐1β and IL‐6, induce insulin resistance in target tissues by antagonising key components of the insulin signalling cascade such as IRS1 (Glass & Olefsky, 2012; Osborn & Olefsky, 2012; McNelis & Olefsky, 2014). However, the relevance of IRS1 serine phosphorylation to the development of impaired glucose metabolism and insulin resistance in vivo has been questioned (Hoehn et al. 2008; Copps et al. 2010). Excitingly, a very recent study sheds new light on the role of inflammatory macrophages and macrophage JNK activation in the development of hepatic insulin resistance. Fasting hyperglycaemia and an inability of insulin to suppress hepatic glucose production are a hallmark of impaired glucose metabolism and T2D. In very elegant work, Perry and colleagues (2015) show that the inability of insulin to suppress hepatic glucose production, resulting in hyperglycaemia, is not due to impaired insulin action in the liver, but is the result of an inability of insulin to suppress lipolysis in the adipose tissue. Thus, in the context of a HFD, pro‐inflammatory cytokines within the adipose tissue are likely to contribute to elevated lipolysis in two principle ways: firstly, by directly initiating lipolysis, and, secondly, by impairing insulin's ability to suppress lipolysis. The consequence of this inflammation‐dependent increase in adipose tissue lipolysis is increased glycerol release which serves as a substrate in gluconeogenesis, and an increase in hepatic acetyl CoA content which allosterically activates pyruvate carboxylase leading to the generation of oxaloacetate and subsequent conversion to glucose. Remarkably, in mice fed a HFD, the deletion of JNK from macrophages reduces hepatic acetyl CoA content, pyruvate carboxylase activity and dramatically improves the ability of insulin to suppress hepatic glucose production. Collectively, the data support the hypothesis that the activation of JNK in macrophages has a crucial role in mediating obesity‐associated inflammation, leading to impaired glucose metabolism, insulin resistance and hyperglycaemia. (A summary of the effects of JNK in macrophages is provided in Fig. 1.)
The role of JNKs in the liver
The liver is a crucial glucoregulatory organ, producing glucose during times of fasting to maintain blood glucose concentrations. Obesity results in elevated lipid storage and inflammation within the liver as well as impairing the ability of insulin to suppress hepatic glucose production leading to fasting hyperglycaemia. Because global JNK1 deficiency in HF fed and hyperphagic mice lowers fasting blood glucose levels and improves glucose tolerance (Hirosumi et al. 2002), the liver may be an important site of JNK action. Indeed, HFD‐induced obesity causes marked JNK activation in the liver (Hirosumi et al. 2002; Sabio et al. 2009; Vernia et al. 2014). Early studies using adenoviral delivery of shRNA against JNK1 (Yang et al. 2007) or dominant‐negative JNK (Nakatani et al. 2004) showed that disrupting JNK signalling in the liver improved hepatic insulin action and lowered fasting blood glucose levels. However, it is important to note that these approaches (e.g. injected adenovirus are rapidly taken up by Kupffer cells) potentially affect JNK expression/activation in non‐parenchymal cells, and, therefore, it is difficult to dissect the action of JNK1 signalling in hepatocytes from potential effects in other cell types. Accordingly, to assess JNK function in hepatocytes per se, a mouse model in which Cre‐recombinase is expressed under the control of the albumin promoter was used to selectively delete JNK1 from hepatocytes (Sabio et al. 2009). Surprisingly when mice were placed on a HFD, hepatocyte‐specific deletion of JNK1 did not prevent glucose intolerance, fasting hyperglycaemia or hepatic insulin resistance, compared with WT mice. However, in mice fed a standard chow diet, hepatocyte‐specific deletion of JNK1 impaired glucose tolerance, an effect that was due to enhanced insulin clearance as a result of increased hepatic insulin receptor expression. Furthermore, standard chow fed mice lacking hepatic JNK1 displayed liver insulin resistance and steatosis. Collectively, these data suggest that obesity‐induced JNK1 activation in hepatocytes is not required for the development of hepatic insulin resistance. Since the deletion of JNK1 and JNK2 in adipocytes (Sabio et al. 2008) and macrophages (Han et al. 2013) prevents HFD‐induced hepatic insulin resistance, it is possible that obesity‐induced activation of JNK in non‐hepatic cells mediates HFD‐induced hepatic insulin resistance.
Nonetheless, as discussed above, considerable functional redundancy exists between JNK isoforms and, therefore, it is possible that in the absence of hepatic JNK1 (Sabio et al. 2009), JNK2 may compensate. Alternately, JNK2, and not JNK1, may play a predominant role in modulating liver metabolism. To definitively address the role of hepatic JNK in metabolism, mice with compound deletion of both JNK1 and JNK2 in the liver were generated (Vernia et al. 2014). Firstly, hepatocyte‐specific deletion of JNK1 and JNK2 led to a small but significant decrease in fat mass following 16 weeks of HF feeding (Vernia et al. 2014). Importantly, and in contrast to hepatocyte‐specific JNK1 deletion (Sabio et al. 2009), hepatocyte‐specific deletion of both JNK1 and JNK2 significantly improved glucose and insulin tolerance, increased hepatic insulin action and lowered fasting blood glucose levels in HF fed mice (Vernia et al. 2014). Moreover, while the sole inactivation of JNK1 in hepatocytes led to hepatic steatosis this was not observed in the double KO model. While hepatocyte‐specific deletion of JNK1 alone was without effect on HFD‐induced impaired glucose metabolism (Sabio et al. 2009), deletion of JNK2 alone led to a modest improvement in glucose and pyruvate tolerance, although these effects were markedly less than that observed in hepatocyte‐specific double JNK1 and JNK2 KO mice (Vernia et al. 2014). These data demonstrate that while JNK2, to some extent, has a non‐redundant role in mediating the deleterious metabolic consequences of obesity, both JNK1 and JNK2 have crucial roles in this response. To gain insight into how hepatic deletion of JNK improves liver insulin action Vernia and colleagues (2014) performed RNA‐sequencing and gene ontology analysis in livers from WT and hepatocyte‐specific JNK1 and JNK2 KO mice and identified an association between JNK1 and JNK2 deficiency and an up‐regulation in the expression of genes involved in oxidative metabolism and the peroxisome proliferator‐activated receptor (PPAR) pathway. The consequence of these changes in gene expression is enhanced mitochondrial and peroxisomal β‐oxidation, an increased capacity to oxidise fatty acids and, in vivo, reduced HFD‐induced hepatic steatosis (Vernia et al. 2014). Given that excess lipid accumulation within the liver can impair liver insulin action, this increased capacity of the liver to oxidise fatty acids and the attendant decrease in lipid storage is likely to contribute to the improved hepatic insulin action observed in mice lacking JNK1 and JNK2 in the liver. While transcription factors such as PPARs control the expression of many genes involved in fatty acid metabolism and are critical to enhancing oxidative metabolism, transcriptional co‐repressors such as nuclear receptor corepressor 1 (NCoR1) have an equally important role in controlling transcriptional responses (Yamamoto et al. 2011). Accordingly, deletion of NCoR1 in skeletal muscle (Yamamoto et al. 2011) derepresses PPARγ target genes leading to the reprogramming of muscles fibres from a glycolytic to an oxidative profile and increasing oxidative mitochondrial metabolism (Yamamoto et al. 2011). In mice with hepatic deletion of JNK1 and JNK2, NCoR1 expression is decreased and this decrease is largely responsible for the increased hepatic PPARα target gene expression and improved glucose tolerance of mice lacking JNK1 and JNK2 in the liver (Vernia et al. 2014). Collectively, these data suggest that the aberrant activation of liver JNK in obesity may, directly or indirectly, modulate NCoR1 expression/function and thereby restrain the expression of PPARα‐target genes involved in oxidative metabolism, limiting the ability of the liver to oxidise dietary fatty acids and resulting in hepatic steatosis and impaired hepatic insulin action. Conversely, deletion of liver JNK in the context of obesity allows for an up‐regulation of PPARα target genes, such as those involved in oxidative metabolism, fibroblast growth factor (FGF)21 and ketogenesis via a decrease in NCoR1 expression, resulting in enhanced liver oxidative capacity, decreased liver lipid storage and improved hepatic insulin action. (A summary of the effects of JNK deletion in the liver is provided in Fig. 2.)
Figure 2. The effect of liver‐specific JNK deletion on obesity‐associated metabolic disruption .
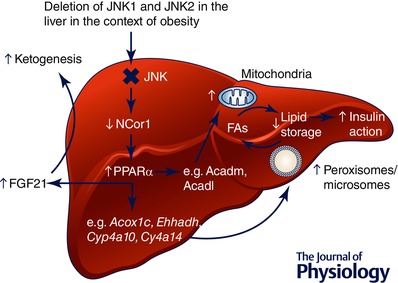
Combined JNK1 and JNK2 deletion in hepatocytes leads to a derepression of PPARα target gene expression as a result of decreased NCor1 expression. The consequence of this increase in PPARα target genes is enhanced fatty acid oxidation, ketogenesis and improved insulin sensitivity.
The role of JNKs in the skeletal muscle
Skeletal muscle is one of the largest tissues of the human body, is a major site of post‐absorptive glucose disposal, and, accordingly, plays a key role in the control of whole body glucose metabolism. It is worth noting that skeletal muscle, unlike adipose tissue, liver and macrophages, appears somewhat resistant to the pro‐inflammatory effects of obesity. For example, activation of nuclear factor κB (NFκB), a transcription factor that controls pro‐inflammatory gene expression, in skeletal muscle has no effect on glucose tolerance or insulin sensitivity, but rather induces a skeletal muscle wasting phenotype (Cai et al. 2004). Similarly, while endoplasmic reticulum (ER) stress, an important cellular process that promotes obesity‐induced inflammation, is prevalent in adipose tissue and liver of obese mice, skeletal muscle appears to be resistant to the induction of ER stress in obesity (Ozcan et al. 2004). Nonetheless, JNKs are activated by obesity in skeletal muscle and, therefore, may potentially impact on skeletal muscle glucose metabolism (Hirosumi et al. 2002; Sabio et al. 2010 b; Pal et al. 2013). In an initial study, the over‐expression of a constitutively active (CA) JNK, but not WT JNK, construct specifically in the tibialis anterior muscle of mice led to a reduction in insulin‐stimulated glucose clearance (Henstridge et al. 2012). However, using a transgenic mouse model in which CA JNK is specifically expressed only in skeletal muscle tissues, resulting in comparable levels of JNK activity to that observed in the skeletal muscle of HF fed mice, no effect of skeletal muscle‐specific CA JNK on the development of obesity, glucose tolerance or insulin sensitivity could be observed (Pal et al. 2013). However, both of these studies present a somewhat non‐physiological experimental model, the results from which may not reflect the function of endogenous JNKs in obesity. Accordingly, to define the role of skeletal muscle JNKs in obesity, two studies from independent laboratories have used mouse models in which Cre recombinase is expressed under the control of the muscle creatine kinase (Mck) promoter to delete JNK1 specifically from skeletal muscles (Sabio et al. 2010 b; Pal et al. 2013). Firstly, in both studies, skeletal muscle‐specific deletion of JNK1 did not affect the development of HFD‐induced obesity (Sabio et al. 2010 b; Pal et al. 2013). However, while Pal et al. observed no effect of skeletal muscle‐specific JNK1 deletion on any indices of glucose metabolism or insulin sensitivity, Sabio et al. observed a more complex phenotype. Consistent with the idea that the activation of JNK in skeletal muscle may negatively impact on skeletal muscle insulin action, mice lacking JNK1 in their skeletal muscles were more insulin tolerant and had improved muscle insulin sensitivity (Sabio et al. 2010 b). However, skeletal muscle‐specific JNK1 KO mice had increased hepatic steatosis following either a standard chow diet or HFD as well as an increase in macrophage recruitment into adipose tissue (Sabio et al. 2010 b), both of which would be expected to impair insulin action at these tissue sites. Overall, these findings from separate laboratories and using models of JNK1 deletion and over‐expression present an ambiguous picture of the role of skeletal muscle JNKs in obesity and metabolic regulation. These discrepancies may reflect differences in the gene targeting strategies used to generate the ‘floxed’ JNK1 allele or differences in laboratory/experimental conditions. Of note, skeletal muscle specific disruption of IKKβ signalling had no effect on the development of obesity and glucose intolerance, indicating a minor role for stress‐signalling pathways in skeletal muscle tissue under obese conditions (Rohl et al. 2004). As has been observed in many other tissues in which JNKs have been deleted, compensation by remaining JNK isoforms may mask the effects of deletion of a single JNK isoform. Accordingly, deletion of both JNK1 and JNK2 in skeletal muscle may be required to elucidate whether JNKs in skeletal muscle play an important role in obesity‐induced metabolic disruption.
Finally, consistent with the idea that JNK1 in skeletal muscle may not play an important role in obesity‐induced muscle insulin resistance, intramyocellular JNK signalling appears to play an important role during exercise (Whitham et al. 2012). During contraction, IL‐6 is produced in skeletal muscle and released into the circulation as a myokine to alter the metabolism of many organs including the adipose tissue and liver and is responsible for the observed insulin‐sensitizing effects following exercise (for review, see Pedersen & Febbraio, 2008). Importantly, the increase in skeletal muscle IL‐6 gene expression following exercise is dependent on intact signalling via JNK1 (Whitham et al. 2012).
The role of JNKs in the hypothalamus and pituitary
While it is well established that obesity leads to JNK activation in a number of peripheral tissues, such as the adipose tissue (Hirosumi et al. 2002; Han et al. 2013), skeletal muscle (Hirosumi et al. 2002; Sabio et al. 2010 b; Pal et al. 2013) and liver (Hirosumi et al. 2002; Sabio et al. 2009; Vernia et al. 2014), more recent studies clearly show that JNKs are also activated in the hypothalamus (Belgardt et al. 2010; Tsaousidou et al. 2014) and pituitary (Belgardt et al. 2010) in obese mice. From a metabolic perspective, perhaps the most pronounced phenotypic aspects of global JNK1 KO mice are the profound protection from HFD‐induced obesity and impaired glucose metabolism. While the development of obesity and impaired glucose metabolism are often inextricably linked, studies of the selective deletion of JNK1 and JNK2 in adipose tissue, macrophages, liver and skeletal muscle have revealed that while the tissue‐specific deletion of JNKs can completely prevent the deleterious effects of obesity on impaired glucose metabolism, they do not affect the development of obesity per se, and raise the question in which tissue/cell type does the deletion of JNKs prevent obesity? Given the important roles of both the hypothalamus and pituitary in the control of whole body metabolism, two early studies addressed the contribution of JNK1 in the central nervous system and pituitary to the development of obesity and impaired glucose metabolism (Belgardt et al. 2010; Sabio et al. 2010 a). Importantly, deletion of JNK1 within central nervous system and pituitary did not prevent HFD‐induced obesity, rather, the most prominent feature of CNS/pituitary‐specific JNK1 KO mice was decreased somatic growth, as indicated by both decreased fat and lean mass, irrespective of diet, as well as decreased body length (Belgardt et al. 2010; Sabio et al. 2010 a). It is likely that CNS/pituitary‐specific JNK1 deletion mediates these effects via decreased growth hormone (GH) and, consequently, reduced insulin‐like growth factor (IGF)1 production from the liver (Belgardt et al. 2010; Sabio et al. 2010 a), critical factors in somatic growth. Consistent with altered pituitary function, CNS/pituitary‐specific JNK1 KO mice displayed activation of the hypothalamus–pituitary–thyroid axis, as indicated by increased circulating thyroid stimulating hormone (TSH), T3 and T4 levels, as well as increased expression of TSHβ and the thyroid releasing hormone receptor (TRHR) within the pituitary. Collectively these data point to a major role of JNK1 in the control of the pituitary–thyroid axis. With regard to the effects of CNS/pituitary‐specific JNK1 deletion on whole body metabolism, CNS/pituitary‐specific JNK1 KO mice have improved glucose and insulin tolerance, increased energy expenditure, enhanced liver insulin action, reduced fasting blood glucose concentrations and reduced lipid accumulation in both brown adipose tissue and liver (Belgardt et al. 2010; Sabio et al. 2010 a). However, given the wide ranging effects of CNS/pituitary‐specific JNK1 deletion, including effects on somatic growth, thyroid function and hypothalamic insulin sensitivity, it is difficult to establish which of these effects has a primary role in mediating the observed protection against the deleterious metabolic consequences of a HFD.
Given the increased activation of the pituitary–thyroid axis that was observed in CNS/pituitary‐specific JNK1 KO mice (Belgardt et al. 2010; Sabio et al. 2010 a), and the known wide‐ranging effects of thyroid hormone (TH) on metabolism (Mullur et al. 2014), Vernia and colleagues examined the function of JNK specifically within the anterior pituitary gland. Importantly, compound deletion of both JNK1 and JNK2 in the pituitary using the glycoprotein hormone α‐subunit promoter to drive Cre‐recombinase expression did not affect JNK1 or JNK2 expression in the hypothalamus or other regions of the brain (Vernia et al. 2013). Remarkably, anterior pituitary‐specific deletion of JNK1 and JNK2 largely prevented HFD‐induced obesity (Vernia et al. 2013), phenocopying the protection from obesity observed in global JNK1 KO mice (Hirosumi et al. 2002). Importantly, lean mass and animal length was not altered in pituitary‐specific JNK KO mice, demonstrating that somatic growth was unaffected. In addition to effects on obesity, pituitary‐specific deletion of JNK improved glucose tolerance and reduced fasting glucose and insulin levels. It may be questioned why deletion of JNK1 and JNK2 within the anterior pituitary prevented HFD‐induced obesity but JNK1 within both the pituitary and CNS did not. Again, as described above, this is likely to be a case of redundancy, with loss of one JNK isoform being compensated for by the other. Indeed, loss of either JNK1 or JNK2 specifically within the anterior pituitary did not prevent HFD‐induced obesity (Vernia et al. 2013). Consistent with previous studies (Belgardt et al. 2010; Sabio et al. 2010 a), the deletion of JNK specifically within the anterior pituitary increased circulating TSH, T3 and T4 levels (Vernia et al. 2013). Importantly, in support of a causal role of increased TH levels in preventing HFD‐induced obesity and impaired glucose metabolism, treatment of anterior pituitary‐specific JNK KO mice with propylthiouracil, an inhibitor of thyroperoxidase and thereby reducing TH production, completely prevented protection from HFD‐induced obesity and impaired glucose metabolism (Vernia et al. 2013). TH has many effects on metabolism, including effects on basal metabolic rate and adaptive thermogenesis, with TH stimulating both (Mullur et al. 2014). Given the important contributions of basal metabolic rate and adaptive thermogenesis to energy expenditure it is significant that energy expenditure is increased in anterior pituitary‐specific JNK KO mice, independently of physical activity and food intake, and that this increase is prevented by propylthiouracil (Vernia et al. 2013). Collectively, these data argue that the deletion of JNK within the anterior pituitary prevents HFD‐induced obesity and its associated metabolic disruptions as a result of TH‐dependent increases in energy expenditure. (A summary of the effects of JNK in the anterior pituitary is provided in Fig. 3.)
Figure 3. The role of JNK activation in the anterior pituitary .
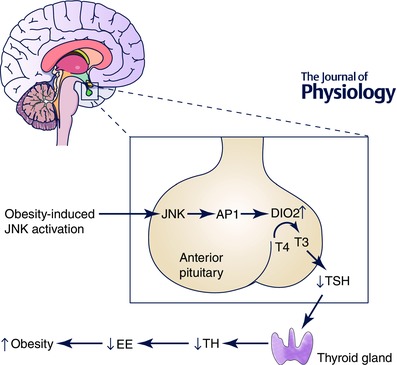
Obesity activates JNKs in the anterior pituitary, leading to activation of the AP1 family of transcription factors and increased expression of the AP1 target gene, type 2 iodothyronine deiodinase (DIO2). Increased DIO2 leads to increased negative feedback regulation of thyroid‐stimulating hormone (TSH) production, resulting in decreased thyroid hormone (TH) production, increased energy expenditure and the development of obesity.
While the Nestin‐Cre transgenic mice used to mediate deletion of JNK1 within the CNS and pituitary results in JNK1 deletion in multiple regions of the brain such as the cortex, cerebellum, hippocampus, medulla oblongata and hypothalamus, insulin action on hypothalamic neurons in particular is known to be a critical regulator of food intake and plays a key role in body weight regulation and glucose homeostasis. Of note, CNS/pituitary‐specific JNK1 KO mice have increased hypothalamic insulin sensitivity as shown by an increased sensitivity to the anorectic effects of intracerebroventricular insulin administration as well as enhanced hypothalamic phosphorylation of protein kinase B (AKT) (Belgardt et al. 2010), indicating that JNK1 within hypothalamic neurons may play a role in the regulation of feeding behaviour and potentially whole body metabolism.
By responding to hormonal cues, agouti‐related peptide (AgRP)‐expressing neurons within the hypothalamus play a key role in the regulation of body weight and glucose homeostasis and, interestingly, Tsaousidou and colleagues recently demonstrated that a HFD triggers the activation of JNK within AgRP neurons (Tsaousidou et al. 2014). To examine the consequences of JNK activation within AgRP neurons, mice expressing constitutively active (CA) JNK specifically within AgRP neutrons were generated (Tsaousidou et al. 2014). Importantly, mice expressing AgRP neuron‐specific CA JNK showed reduced activation of signal transducer and activator of transcription 3 (STAT3) and a failure to suppress food intake and reduce body weight following leptin administration, indicative of leptin resistance (Tsaousidou et al. 2014). Furthermore, AgRP neuron‐specific CA JNK expressing mice had an increase in body weight and fat mass when fed a standard chow diet, although this did not result in impaired insulin or glucose tolerance (Tsaousidou et al. 2014). Collectively, these data demonstrate that HFD activates JNK signalling in the hypothalamus within neurons that play a critical role in feeding behaviour and the control of body weight.
JNK activation in obesity in humans
While activated JNKs undoubtedly play a number of important roles in mediating the deleterious metabolic consequences associated with obesity in mice, whether JNKs play similar roles in humans is unknown. However, evidence from human studies demonstrates that JNKs are activated in several metabolically important sites in human obesity. Specifically, increased JNK activation has been reported in skeletal muscle biopsies obtained from obese, insulin‐resistant individuals compared with lean, insulin‐sensitive controls (Chung et al. 2008; Carvalho et al. 2013). Additionally, biopsies of subcutaneous adipose tissue have shown increased levels of the active, phosphorylated form of JNK in obese individuals compared to lean controls (Boden et al. 2008; Carvalho et al. 2013). Furthermore, weight loss and concomitantly improved insulin sensitivity, induced via gastric bypass surgery, resulted in a decrease in active, phosphorylated JNK levels in subcutaneous abdominal adipose tissue biopsies (Gregor et al. 2009; Carvalho et al. 2013). Finally, active, phosphorylated JNK levels in subcutaneous adipose tissue display a significant inverse relationship with insulin sensitivity as assessed by hyperinsulinaemic–euglycaemic clamp (Sourris et al. 2009). These data are all supportive of a role for JNKs in human obesity and its associated metabolic consequences.
Summary and future directions
The initial description of protection from obesity, impaired glucose metabolism and insulin resistance in mice lacking JNK1 in all cells and tissues was a harbinger for a great many studies that have subsequently identified mechanisms by which JNKs are activated in obesity as well as those that have attempted to identify precise roles for JNKs in numerous different tissues. A summary of the described JNK KO animals is provided in Table 1. Human studies have furthermore revealed that the JNK pathway is also activated in obese and insulin‐resistant humans. However, animal work has clearly established the mechanisms by which increased JNK activity contributes to metabolic disorders. JNK activation primarily in the anterior pituitary but also the liver contributes to the development of obesity. JNK activation within the liver also represses activation of the nuclear hormone receptor PPARα, thereby decreasing the expression of genes that mediate fatty acid oxidation and ketogenesis. JNK activation in macrophages leads to polarisation towards the pro‐inflammatory M1 state resulting in increased pro‐inflammatory cytokine production and metabolic disruption to several tissues. While the studies to date have provided a remarkably detailed analysis of the role of JNKs in obesity and disrupted metabolic homeostasis, one key site that has not been examined are the pancreatic β‐cells. For T2D to develop the pancreatic β‐cells must fail to fully compensate for the decline in insulin sensitivity and the continuing progression of T2D is largely the result of a continual fall in pancreatic β‐cell function (Kahn et al. 2006). JNKs are activated in pancreatic islets by numerous stimuli and several studies using cultured islets have demonstrated that inhibition of JNK prevents pancreatic β‐cell apoptosis (Ammendrup et al. 2000; Major & Wolf, 2001; Maedler et al. 2008; Subramanian et al. 2012); however, formal in vivo genetic proof of the pathogenic role of JNKs in the loss of pancreatic β‐cells in models of T2D is lacking. Traditional wild type strains of mice, e.g. HF fed C57Bl/6 mice or genetically obese ob/ob mice, even though they become very obese and severely insulin‐resistant, do not develop pancreatic β‐cell failure. A hallmark of T2D in humans is the accumulation of islet amyloid which is associated with loss of pancreatic β‐cell mass and function. Mice expressing the amyloidogenic human form of islet amyloid polypeptide (IAPP) have a loss of pancreatic β‐cell mass and deletion of JNK1 and/or JNK2 in this rodent model of T2D would be of considerable interest.
Table 1.
Metabolic phenotype of tissue‐specific JNK KO mice when fed a HFD
| Global JNK | Adipose‐JNK | Macrophage‐JNK | Liver‐JNK | Muscle‐JNK | Pituitary‐JNK | |
|---|---|---|---|---|---|---|
| Phenotype | KO1 | KO2 | KO3 | KO4 | KO5 , 6 | KO7 |
| Adiposity | ↓↓↓ | ↔ | ↔ | ↓ | ↔ | ↓↓↓ |
| Glucose tolerance | ↑↑↑ | ↔ | ↑↑↑ | ↑↑ | ↔ | ↑↑↑ |
| Fasting hyperglycaemia | ↓↓↓ | ↔ | ↓↓↓ | ↓↓ | ↔ | ↓↓↓ |
| Hepatic steatosis | — | ↓↓ | ↓↓ | ↓↓ | ↑ | ↓↓↓ |
1Hirosumi et al. (2002); 2Sabio et al. (2008); 3Han et al. (2013); 4Vernia et al. (2014); 5Sabio et al. (2010); 6Pal et al. (2013); 7Vernia et al. (2013). The number of arrows (↑↓) indicates the magnitude of the effect on the indicated parameters. — indicates parameter was not investigated. ↔ indicates no difference.
Finally, as discussed above, JNKs have a major role in macrophage polarisation and skewing towards the pro‐inflammatory M1 phenotype plays an important role in obesity‐induced inflammation. Given that a wide range of immune cells are now known to be involved in obesity‐associated inflammation and the development of impaired glucose metabolism, such as CD8+ T lymphocytes (Nishimura et al. 2009), B lymphocytes (Winer et al. 2011), neutrophils (Talukdar et al. 2012) and NK cells (Wensveen et al. 2015), and that analogous states of polarisation exist in many of these cell types, e.g. Th1 and Th2 lymphocytes, it will be of interest to determine whether the activation of JNK in these cells is critical to their deleterious functions in the context of obesity and disrupted metabolic homeostasis.
Additional information
Competing interests
The authors declare no conflicts of interest.
Author contributions
All authors wrote and revised the manuscript. All authors approved the final version of the manuscript. All persons designated as authors qualify for authorship.
Funding
This work was supported by Project Grants, Development Grants and Fellowships from the National Health and Medical Research Council of Australia (NHMRC), by the Australian Research Council and Diabetes Australia Research Trust. M.A.F. is a Senior Principal Research Fellow of the NHMRC. M.P. is supported by a research fellowship from the German Research Foundation (PA 2459/1‐1).
Acknowledgements
We would like to thank all past and present members of the Cellular and Molecular Metabolism Laboratory and all of our collaborators over the years.
Biography
Mark A. Febbraio is a Senior Principal Research Fellow of the NHMRC and Head of the Cellular and Molecular Metabolism Laboratory and the Division of Diabetes and Metabolism at The Garvan Institute of Medical Research in Sydney, Australia. His research is focused on understanding cellular and molecular mechanisms associated obesity and type 2 diabetes and his aim is to develop novel drugs to treat metabolic disease. Martin Pal received his PhD in genetics from the University of Cologne, Germany, for generating transgenic novel mouse strains to investigate stress‐signalling pathways in the context of obesity‐associated insulin resistance. He then moved to Australia to perform his post‐doctoral training in the laboratory of Professor Mark Febbraio at the Garvan Institute of Medical Research in Sydney, Australia, mainly focusing on deciphering the beneficial effects of exercise on health and disease, such as metabolic disease. Graeme Lancaster received his PhD degree from the University of Birmingham, UK, in 2004 before moving to Australia to undertake post‐doctoral studies with Professor Mark Febbraio at RMIT University (Melbourne) and the Baker IDI Heart and Diabetes Institute (Melbourne). Dr Lancaster's work now focuses on understanding the molecular basis by which obesity initiates inflammation.

References
- Aguirre V, Uchida T, Yenush L, Davis R & White MF (2000). The c‐Jun NH2‐terminal kinase promotes insulin resistance during association with insulin receptor substrate‐1 and phosphorylation of Ser307 . J Biol Chem 275, 9047–9054. [DOI] [PubMed] [Google Scholar]
- Ammendrup A, Maillard A, Nielsen K, Aabenhus Andersen N, Serup P, Dragsbaek Madsen O, Mandrup‐Poulsen T & Bonny C (2000). The c‐Jun amino‐terminal kinase pathway is preferentially activated by interleukin‐1 and controls apoptosis in differentiating pancreatic β‐cells. Diabetes 49, 1468–1476. [DOI] [PubMed] [Google Scholar]
- Belgardt BF, Mauer J, Wunderlich FT, Ernst MB, Pal M, Spohn G, Bronneke HS, Brodesser S, Hampel B, Schauss AC & Bruning JC (2010). Hypothalamic and pituitary c‐Jun N‐terminal kinase 1 signaling coordinately regulates glucose metabolism. Proc Natl Acad Sci USA 107, 6028–6033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden G, Duan X, Homko C, Molina EJ, Song W, Perez O, Cheung P & Merali S (2008). Increase in endoplasmic reticulum stress‐related proteins and genes in adipose tissue of obese, insulin‐resistant individuals. Diabetes 57, 2438–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Frantz JD, Tawa NE, Jr. , Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ & Shoelson SE (2004). IKKβ/NF‐κB activation causes severe muscle wasting in mice. Cell 119, 285–298. [DOI] [PubMed] [Google Scholar]
- Carvalho BM, Oliveira AG, Ueno M, Araujo TG, Guadagnini D, Carvalho‐Filho MA, Geloneze B, Lima MM, Pareja JC, Carvalheira JB & Saad MJ (2013). Modulation of double‐stranded RNA‐activated protein kinase in insulin sensitive tissues of obese humans. Obesity 21, 2452–2457. [DOI] [PubMed] [Google Scholar]
- Chen L, Magliano DJ & Zimmet PZ (2012). The worldwide epidemiology of type 2 diabetes mellitus – present and future perspectives. Nat Rev Endocrinol 8, 228–236. [DOI] [PubMed] [Google Scholar]
- Chung J, Nguyen AK, Henstridge DC, Holmes AG, Chan MH, Mesa JL, Lancaster GI, Southgate RJ, Bruce CR, Duffy SJ, Horvath I, Mestril R, Watt MJ, Hooper PL, Kingwell BA, Vigh L, Hevener A & Febbraio MA (2008). HSP72 protects against obesity‐induced insulin resistance. Proc Natl Acad Sci USA 105, 1739–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copps KD, Hancer NJ, Opare‐Ado L, Qiu W, Walsh C & White MF (2010). Irs1 serine 307 promotes insulin sensitivity in mice. Cell Metab 11, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ (2000). Signal transduction by the JNK group of MAP kinases. Cell 103, 239–252. [DOI] [PubMed] [Google Scholar]
- Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M & Davis RJ (1994). JNK1: a protein kinase stimulated by UV light and Ha‐Ras that binds and phosphorylates the c‐Jun activation domain. Cell 76, 1025–1037. [DOI] [PubMed] [Google Scholar]
- Furuhashi M & Hotamisligil GS (2008). Fatty acid‐binding proteins: role in metabolic diseases and potential as drug targets. Nature Rev Drug Discov 7, 489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galic S, Sachithanandan N, Kay TW & Steinberg GR (2014). Suppressor of cytokine signalling (SOCS) proteins as guardians of inflammatory responses critical for regulating insulin sensitivity. Biochem J 461, 177–188. [DOI] [PubMed] [Google Scholar]
- Glass CK & Olefsky JM (2012). Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab 15, 635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregor MF, Yang L, Fabbrini E, Mohammed BS, Eagon JC, Hotamisligil GS & Klein S (2009). Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes 58, 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MS, Jung DY, Morel C, Lakhani SA, Kim JK, Flavell RA & Davis RJ (2013). JNK expression by macrophages promotes obesity‐induced insulin resistance and inflammation. Science 339, 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henstridge DC, Bruce CR, Pang CP, Lancaster GI, Allen TL, Estevez E, Gardner T, Weir JM, Meikle PJ, Lam KS, Xu A, Fujii N, Goodyear LJ & Febbraio MA (2012). Skeletal muscle‐specific overproduction of constitutively activated c‐Jun N‐terminal kinase (JNK) induces insulin resistance in mice. Diabetologia 55, 2769–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibi M, Lin A, Smeal T, Minden A & Karin M (1993). Identification of an oncoprotein‐ and UV‐responsive protein kinase that binds and potentiates the c‐Jun activation domain. Genes Dev 7, 2135–2148. [DOI] [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M & Hotamisligil GS (2002). A central role for JNK in obesity and insulin resistance. Nature 420, 333–336. [DOI] [PubMed] [Google Scholar]
- Hoehn KL, Hohnen‐Behrens C, Cederberg A, Wu LE, Turner N, Yuasa T, Ebina Y & James DE (2008). IRS1‐independent defects define major nodes of insulin resistance. Cell Metab 7, 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS (2006). Inflammation and metabolic disorders. Nature 444, 860–867. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Arner P, Caro JF, Atkinson RL & Spiegelman BM (1995). Increased adipose tissue expression of tumor necrosis factor‐α in human obesity and insulin resistance. J Clin Invest 95, 2409–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS & Spiegelman BM (1993). Adipose expression of tumor necrosis factor‐α: direct role in obesity‐linked insulin resistance. Science 259, 87–91. [DOI] [PubMed] [Google Scholar]
- Houstis N, Rosen ED & Lander ES (2006). Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440, 944–948. [DOI] [PubMed] [Google Scholar]
- Kahn SE, Hull RL & Utzschneider KM (2006). Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444, 840–846. [DOI] [PubMed] [Google Scholar]
- Kotas ME & Medzhitov R (2015). Homeostasis, inflammation, and disease susceptibility. Cell 160, 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNelis JC & Olefsky JM (2014). Macrophages, immunity, and metabolic disease. Immunity 41, 36–48. [DOI] [PubMed] [Google Scholar]
- Maedler K, Schulthess FT, Bielman C, Berney T, Bonny C, Prentki M, Donath MY & Roduit R (2008). Glucose and leptin induce apoptosis in human β‐cells and impair glucose‐stimulated insulin secretion through activation of c‐Jun N‐terminal kinases. FASEB J 22, 1905–1913. [DOI] [PubMed] [Google Scholar]
- Major CD & Wolf BA (2001). Interleukin‐1β stimulation of c‐Jun NH2‐terminal kinase activity in insulin‐secreting cells: evidence for cytoplasmic restriction. Diabetes 50, 2721–2728. [DOI] [PubMed] [Google Scholar]
- Martens K, Bottelbergs A & Baes M (2010). Ectopic recombination in the central and peripheral nervous system by aP2/FABP4‐Cre mice: implications for metabolism research. FEBS Lett 584, 1054–1058. [DOI] [PubMed] [Google Scholar]
- Mullur R, Liu YY & Brent GA (2014). Thyroid hormone regulation of metabolism. Physiol Rev 94, 355–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano H, Nakajima A, Sakon‐Komazawa S, Piao JH, Xue X & Okumura K (2006). Reactive oxygen species mediate crosstalk between NF‐κB and JNK. Cell Death Differ 13, 730–737. [DOI] [PubMed] [Google Scholar]
- Nakatani Y, Kaneto H, Kawamori D, Hatazaki M, Miyatsuka T, Matsuoka TA, Kajimoto Y, Matsuhisa M, Yamasaki Y & Hori M (2004). Modulation of the JNK pathway in liver affects insulin resistance status. J Biol Chem 279, 45803–45809. [DOI] [PubMed] [Google Scholar]
- Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, Yoshimura K, Kadowaki T & Nagai R (2009). CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 15, 914–920. [DOI] [PubMed] [Google Scholar]
- Osborn O & Olefsky JM (2012). The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med 18, 363–374. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH & Hotamisligil GS (2004). Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461. [DOI] [PubMed] [Google Scholar]
- Pal M, Wunderlich CM, Spohn G, Bronneke HS, Schmidt‐Supprian M & Wunderlich FT (2013). Alteration of JNK‐1 signaling in skeletal muscle fails to affect glucose homeostasis and obesity‐associated insulin resistance in mice. PLoS One 8, e54247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BK & Febbraio MA (2008). Muscle as an endocrine organ: focus on muscle‐derived interleukin‐6. Physiol Rev 88, 1379–1406. [DOI] [PubMed] [Google Scholar]
- Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang XM, Ruan HB, Yang X, Caprio S, Kaech SM, Sul HS, Birnbaum MJ, Davis RJ, Cline GW, Petersen KF & Shulman GI (2015). Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 160, 745–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohl M, Pasparakis M, Baudler S, Baumgartl J, Gautam D, Huth M, De Lorenzi R, Krone W, Rajewsky K & Bruning JC (2004). Conditional disruption of IκB kinase 2 fails to prevent obesity‐induced insulin resistance. J Clin Invest 113, 474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G, Cavanagh‐Kyros J, Barrett T, Jung DY, Ko HJ, Ong H, Morel C, Mora A, Reilly J, Kim JK & Davis RJ (2010. a). Role of the hypothalamic–pituitary–thyroid axis in metabolic regulation by JNK1. Genes Dev 24, 256–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G, Cavanagh‐Kyros J, Ko HJ, Jung DY, Gray S, Jun JY, Barrett T, Mora A, Kim JK & Davis RJ (2009). Prevention of steatosis by hepatic JNK1. Cell Metab 10, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, Barrett T, Kim JK & Davis RJ (2008). A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 322, 1539–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G & Davis RJ (2014). TNF and MAP kinase signalling pathways. Semin Immunol 26, 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio G, Kennedy NJ, Cavanagh‐Kyros J, Jung DY, Ko HJ, Ong H, Barrett T, Kim JK & Davis RJ (2010. b). Role of muscle c‐Jun NH2‐terminal kinase 1 in obesity‐induced insulin resistance. Mol Cell Biol 30, 106–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki E, Brenner DA & Karin M (2012). A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology 143, 307–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinas G, Vilcu C, Neels JG, Bandyopadhyay GK, Luo JL, Naugler W, Grivennikov S, Wynshaw‐Boris A, Scadeng M, Olefsky JM & Karin M (2007). JNK1 in hematopoietically derived cells contributes to diet‐induced inflammation and insulin resistance without affecting obesity. Cell Metab 6, 386–397. [DOI] [PubMed] [Google Scholar]
- Sourris KC, Lyons JG, de Courten MP, Dougherty SL, Henstridge DC, Cooper ME, Hage M, Dart A, Kingwell BA, Forbes JM & de Courten B (2009). c‐Jun NH2‐terminal kinase activity in subcutaneous adipose tissue but not nuclear factor‐κB activity in peripheral blood mononuclear cells is an independent determinant of insulin resistance in healthy individuals. Diabetes 58, 1259–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian SL, Hull RL, Zraika S, Aston‐Mourney K, Udayasankar J & Kahn SE (2012). cJUN N‐terminal kinase (JNK) activation mediates islet amyloid‐induced β cell apoptosis in cultured human islet amyloid polypeptide transgenic mouse islets. Diabetologia 55, 166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukdar S, Oh da Y, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, Ofrecio J, Lin M, Brenner MB & Olefsky JM (2012). Neutrophils mediate insulin resistance in mice fed a high‐fat diet through secreted elastase. Nat Med 18, 1407–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsaousidou E, Paeger L, Belgardt BF, Pal M, Wunderlich CM, Bronneke H, Collienne U, Hampel B, Wunderlich FT, Schmidt‐Supprian M, Kloppenburg P & Bruning JC (2014). Distinct roles for JNK and IKK activation in agouti‐related peptide neurons in the development of obesity and insulin resistance. Cell Rep 9, 1495–1506. [DOI] [PubMed] [Google Scholar]
- Tuncman G, Hirosumi J, Solinas G, Chang L, Karin M & Hotamisligil GS (2006). Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc Natl Acad Sci USA 103, 10741–10746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP & Ron D (2000). Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666. [DOI] [PubMed] [Google Scholar]
- Uysal KT, Wiesbrock SM, Marino MW & Hotamisligil GS (1997). Protection from obesity‐induced insulin resistance in mice lacking TNF‐α function. Nature 389, 610–614. [DOI] [PubMed] [Google Scholar]
- Vallerie SN, Furuhashi M, Fucho R & Hotamisligil GS (2008). A predominant role for parenchymal c‐Jun amino terminal kinase (JNK) in the regulation of systemic insulin sensitivity. PLoS One 3, e3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernia S, Cavanagh‐Kyros J, Barrett T, Jung DY, Kim JK & Davis RJ (2013). Diet‐induced obesity mediated by the JNK/DIO2 signal transduction pathway. Genes Dev 27, 2345–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernia S, Cavanagh‐Kyros J, Garcia‐Haro L, Sabio G, Barrett T, Jung DY, Kim JK, Xu J, Shulha HP, Garber M, Gao G & Davis RJ (2014). The PPARα–FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metab 20, 512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL & Ferrante AW, Jr (2003). Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112, 1796–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensveen FM, Jelencic V, Valentic S, Sestan M, Wensveen TT, Theurich S, Glasner A, Mendrila D, Stimac D, Wunderlich FT, Bruning JC, Mandelboim O & Polic B (2015). NK cells link obesity‐induced adipose stress to inflammation and insulin resistance. Nat Immunol 16, 376–385. [DOI] [PubMed] [Google Scholar]
- Whitham M, Chan MH, Pal M, Matthews VB, Prelovsek O, Lunke S, El‐Osta A, Broenneke H, Alber J, Bruning JC, Wunderlich FT, Lancaster GI & Febbraio MA (2012). Contraction‐induced interleukin‐6 gene transcription in skeletal muscle is regulated by c‐Jun terminal kinase/activator protein‐1. J Biol Chem 287, 10771–10779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, Tsui H, Wu P, Davidson MG, Alonso MN, Leong HX, Glassford A, Caimol M, Kenkel JA, Tedder TF, McLaughlin T, Miklos DB, Dosch HM & Engleman EG (2011). B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 17, 610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock KJ, Kierdorf K, Pouchelon CA, Vivancos V, Dionne MS & Geissmann F (2015). Macrophage‐derived upd3 cytokine causes impaired glucose homeostasis and reduced lifespan in Drosophila fed a lipid‐rich diet. Immunity 42, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich FT, Strohle P, Konner AC, Gruber S, Tovar S, Bronneke HS, Juntti‐Berggren L, Li LS, van Rooijen N, Libert C, Berggren PO & Bruning JC (2010). Interleukin‐6 signaling in liver‐parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab 12, 237–249. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Williams EG, Mouchiroud L, Canto C, Fan W, Downes M, Heligon C, Barish GD, Desvergne B, Evans RM, Schoonjans K & Auwerx J (2011). NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell 147, 827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Wilcox DM, Haasch DL, Jung PM, Nguyen PT, Voorbach MJ, Doktor S, Brodjian S, Bush EN, Lin E, Jacobson PB, Collins CA, Landschulz KT, Trevillyan JM, Rondinone CM & Surowy TK (2007). Liver‐specific knockdown of JNK1 up‐regulates proliferator‐activated receptor γ coactivator 1β and increases plasma triglyceride despite reduced glucose and insulin levels in diet‐induced obese mice. J Biol Chem 282, 22765–22774. [DOI] [PubMed] [Google Scholar]