

Abstract
Key points
Bradykinin may play a role in the autodigestive disease acute pancreatitis, but little is known about its pancreatic actions.
In this study, we have investigated bradykinin‐elicited Ca2+ signal generation in normal mouse pancreatic lobules.
We found complete separation of Ca2+ signalling between pancreatic acinar (PACs) and stellate cells (PSCs). Pathophysiologically relevant bradykinin concentrations consistently evoked Ca2+ signals, via B2 receptors, in PSCs but never in neighbouring PACs, whereas cholecystokinin, consistently evoking Ca2+ signals in PACs, never elicited Ca2+ signals in PSCs.
The bradykinin‐elicited Ca2+ signals were due to initial Ca2+ release from inositol trisphosphate‐sensitive stores followed by Ca2+ entry through Ca2+ release‐activated channels (CRACs). The Ca2+ entry phase was effectively inhibited by a CRAC blocker.
B2 receptor blockade reduced the extent of PAC necrosis evoked by pancreatitis‐promoting agents and we therefore conclude that bradykinin plays a role in acute pancreatitis via specific actions on PSCs.
Abstract
Normal pancreatic stellate cells (PSCs) are regarded as quiescent, only to become activated in chronic pancreatitis and pancreatic cancer. However, we now report that these cells in their normal microenvironment are far from quiescent, but are capable of generating substantial Ca2+ signals. We have compared Ca2+ signalling in PSCs and their better studied neighbouring acinar cells (PACs) and found complete separation of Ca2+ signalling in even closely neighbouring PACs and PSCs. Bradykinin (BK), at concentrations corresponding to the slightly elevated plasma BK levels that have been shown to occur in the auto‐digestive disease acute pancreatitis in vivo, consistently elicited substantial Ca2+ signals in PSCs, but never in neighbouring PACs, whereas the physiological PAC stimulant cholecystokinin failed to evoke Ca2+ signals in PSCs. The BK‐induced Ca2+ signals were mediated by B2 receptors and B2 receptor blockade protected against PAC necrosis evoked by agents causing acute pancreatitis. The initial Ca2+ rise in PSCs was due to inositol trisphosphate receptor‐mediated release from internal stores, whereas the sustained phase depended on external Ca2+ entry through Ca2+ release‐activated Ca2+ (CRAC) channels. CRAC channel inhibitors, which have been shown to protect PACs against damage caused by agents inducing pancreatitis, therefore also inhibit Ca2+ signal generation in PSCs and this may be helpful in treating acute pancreatitis.
Key points
Bradykinin may play a role in the autodigestive disease acute pancreatitis, but little is known about its pancreatic actions.
In this study, we have investigated bradykinin‐elicited Ca2+ signal generation in normal mouse pancreatic lobules.
We found complete separation of Ca2+ signalling between pancreatic acinar (PACs) and stellate cells (PSCs). Pathophysiologically relevant bradykinin concentrations consistently evoked Ca2+ signals, via B2 receptors, in PSCs but never in neighbouring PACs, whereas cholecystokinin, consistently evoking Ca2+ signals in PACs, never elicited Ca2+ signals in PSCs.
The bradykinin‐elicited Ca2+ signals were due to initial Ca2+ release from inositol trisphosphate‐sensitive stores followed by Ca2+ entry through Ca2+ release‐activated channels (CRACs). The Ca2+ entry phase was effectively inhibited by a CRAC blocker.
B2 receptor blockade reduced the extent of PAC necrosis evoked by pancreatitis‐promoting agents and we therefore conclude that bradykinin plays a role in acute pancreatitis via specific actions on PSCs.
Abbreviations
- 2‐APB
2‐aminoethoxydiphenylborate
- ACE
angiotensin‐converting enzyme
- AM
acetoxymethyl
- BK
bradykinin
- B2
bradykinin type 2
- CCK
cholecystokinin
- CCKR
cholecystokinin receptor
- CFTR
cystic fibrosis transmembrane conductance regulator
- CRAC
Ca2+ release‐activated Ca2+ channel
- DAPI
4′,6‐diamidino‐2 phenylindole
- ER
endoplasmic reticulum
- IP3
inositol trisphosphate
- IP3R
inositol trisphosphate receptor
- PAC
pancreatic acinar cell
- PI
propidium iodide
- PSC
pancreatic stellate cell
Introduction
It has been known for more than 40 years that pancreatic enzyme secretion is regulated by Ca2+ signals in the pancreatic acinar cells (PACs) and that the primary event is acetylcholine‐ (ACh) or cholecystokinin‐ (CCK) elicited intracellular Ca2+ release followed by Ca2+ entry (Case & Clausen, 1973; Matthews et al. 1973; Kondo & Schulz, 1976; Petersen & Ueda, 1976). The ACh‐evoked intracellular Ca2+ release is mediated by inositol trisphosphate (IP3) (Streb et al. 1983; Wakui et al. 1990), whereas the CCK‐elicited Ca2+ release is mediated by nicotinic acid adenine dinucleotide phosphate (Cancela et al. 2000; Gerasimenko et al. 2015). As high K+ depolarization of the PAC membrane does not evoke enzyme secretion or Ca2+ movement (Argent et al. 1971; Matthews et al. 1973), the Ca2+ entry does not occur through voltage‐gated channels (Petersen, 1992), but is due to opening of Ca2+ release‐activated Ca2+ (CRAC) channels (Parekh & Putney, 2005; Gerasimenko et al. 2013). The acinar Ca2+ signals also regulate acinar fluid secretion via Ca2+‐activated Cl− and K+ channels (Petersen, 1992; Park et al. 2001). However, the major component of pancreatic fluid secretion is contributed by the ducts and this is regulated by secretin‐elicited intra‐ductal cyclic AMP formation controlling cystic fibrosis transmembrane conductance regulator (CFTR) channels (Argent, 2006), whose openings are also regulated by the Cl− concentration in the luminal fluid (Broadbent et al. 2015).
The often fatal human disease acute pancreatitis, in which the pancreas digests itself and its surroundings, is initiated by excessive intracellular Ca2+ release in the PACs followed by excessive Ca2+ entry, mostly elicited by combinations of alcohol and fatty acids or by bile acids (Petersen & Sutton, 2006; Gerasimenko et al. 2014). Pancreatitis‐inducing agents also inhibit ductal fluid and bicarbonate secretion, exacerbating the disease (Pallagi et al. 2011; Maleth et al. 2015).
We have extensive knowledge of the mechanisms generating Ca2+ signals in the PACs, which has been built up over many decades (Petersen & Tepikin, 2008; Gerasimenko et al. 2014). In contrast, the more recently discovered pancreatic stellate cells (PSCs) (Watari et al. 1982), located in the peri‐acinar space – with elongated processes around the base of the acinus – have received much less attention and it has essentially been the functional properties of cultured cells that have been investigated (Apte et al. 1998, 2012; Bachem et al. 1998; Wells & Crawford, 1998). The prevailing, and so far unchallenged, view has been that normal PSCs are quiescent and the focus in all published studies has been on the activation of PSCs and their role – in the activated state – in chronic pancreatitis and pancreatic cancer (Bachem et al. 1998; Wells & Crawford, 1998; Sherman et al. 2014). Activation of PSCs – during pancreatic injury or culturing of quiescent PSCs – induces proliferation as well as secretion of extracellular matrix components, thereby playing an important role in the fibrosis that occurs in chronic pancreatitis and pancreatic cancer (Sherman et al. 2014). Work on the so‐called quiescent PSCs in culture has shown that they can generate substantial cytosolic Ca2+ signals in response to stimulation with high concentrations of the blood pressure‐lowering nona‐peptide bradykinin (BK) and some other substances (Won et al. 2011).
Because BK has long been known as an important player in inflammatory disease, including acute pancreatitis (Ryan et al. 1964; Hirata et al. 2002), we have now investigated BK‐induced Ca2+ signal generation, and its underlying mechanism, in normal PSCs in their normal microenvironment. We find that normal PSCs are much more sensitive to BK than PSCs in culture, generating substantial Ca2+ signals in response to a BK concentration as low as 100 pm, with maximal effect at 1 nm, orders of magnitude lower than what has been observed in cultured cells (Won et al. 2011). This has important implications, as the threshold for activating normal PSCs (100 pm) is close to the normal plasma level of BK (40–70 pm) (Blais et al. 1999; Hirata et al. 2002) and any increase in the plasma or tissue BK levels, which occurs under several conditions – including acute pancreatitis and use of angiotensin‐converting‐enzyme (ACE) inhibitors (Liu et al. 1997; Blais et al. 1999; Tsutsumi et al. 1999; Hirata et al. 2002; Su, 2014) – would therefore elicit Ca2+ signals in PSCs. We have explored the Ca2+ signalling events and their underlying mechanisms in normal PSCs and show that BK activates bradykinin type 2 (B2) receptors, which causes primary Ca2+ release from internal stores. This effect can be abolished by phospholipase C inhibition or blockade of IP3 receptors (IP3Rs). Following the initial intracellular Ca2+ release, there is opening of conventional CRAC channels (Parekh & Putney, 2005; Parekh, 2010) and we show that a recently employed CRAC channel inhibitor, which was protective against the destruction of PACs evoked by agents inducing pancreatitis (Gerasimenko et al. 2013, 2014), also reduces BK‐induced Ca2+ signal generation in PSCs. We show that B2 receptor blockade protects against the necrosis evoked by pancreatitis‐inducing agents and suggest that the protective effect of CRAC channel blockade against pancreatitis may in part be due to inhibition of Ca2+ signal generation in PSCs. Overall, our new data indicate that the so far generally accepted notion of normal PSCs being quiescent is potentially misleading as they are in fact exquisitely sensitive to relatively small changes in BK concentrations found in vivo.
Methods
Ethical approval
All procedures were carried out in accordance with UK Home Office regulations.
Preparation of pancreatic lobules
Pancreatic lobules and big clusters were isolated from the pancreas of adult C57Bl/6 male mice. Animals were killed according to UK Schedule 1 regulations. The pancreas was rapidly dissected, transferred to collagenase Na‐Hepes‐based solution (Sigma, Poole, UK) and incubated at 37°C for 5–6 min. After digestion, the tissue was kept in Na‐Hepes‐based extracellular media, containing (in mm): NaCl, 140; KCl, 4.7; Hepes (KOH), 10; MgCl2, 1; glucose 10; CaCl2 1; pH 7.2. Pancreatic lobules were then incubated with fluorescent dye following the manufacturer's description. All experiments were carried out with freshly prepared pancreatic lobules, attached to the coverslip of the perfusion chamber at room temperature (23°C). Penetration of various substances deep into the pancreatic lobule was highly dependent on the distance from the surface (Fig. 1 A, B). It was therefore necessary, in several cases, to use relatively high concentrations (up to 5‐fold higher than would have been necessary in experiments on isolated cells or very small cell clusters) and relatively long pre‐incubation times.
Figure 1. Pancreatic lobule preparation: penetration of applied substances into deep layers .
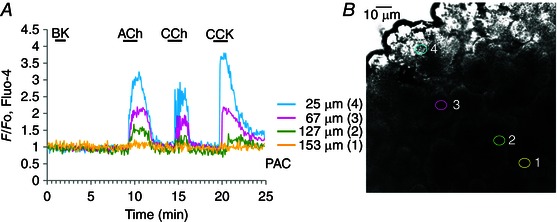
Experiment illustrating the difficulties of attaining the full concentration level deep inside the pancreatic tissue of substances applied to the surface of a lobule. A, [Ca2+]i traces obtained from PACs at different depths from the surface of the lobule (signposted by the colour coding shown). Responses to ACh (10 μm), carbachol (CCh) (10 μm) and CCK (10 nm) are shown. B, transmitted light image of the lobule from which the recordings shown in A were obtained. The different recording positions are indicated using the colour coding shown in A.
[Ca2+] measurements in intact cells
Intact cells were loaded with 5 μm Fluo‐4 acetoxymethyl ester (AM), for 10 min at room temperature. Cells were transferred to a flow chamber and perfused with the Na‐Hepes‐based extracellular solution as described above. Cells were visualized using a Leica SP5 MPII two‐photon confocal microscope, with a 63× 1.2 NA objective lens. Fluo‐4 was excited with a 488 nm Ar laser, at 1–4% power, and emitted light was collected at 510–580 nm. Generally, a series of images was recorded at 512×512 pixel resolution (at a speed of 0.3 frames s−1), and analysed using Leica Confocal Software (Leica, Mannheim, Germany). Fluorescence signals were plotted as F/F 0 (F 0 is the initial level of fluorescence). Statistical analysis was performed using ANOVA or Student's t‐test.
Measurements of necrosis level in pancreatic lobules
Pancreatic lobules were exposed to 350 mm ethanol, or 500 μm palmitoleic acid ethyl ester (POAEE) or 0.5% sodium choleate for 2 h in the presence or absence of 0.5 μm or 10 μm WIN 64338. Ethanol‐ or bile‐induced pancreatic necrosis was visualized by staining the lobules with propidium iodide (PI) and compared with the control necrosis level (without any treatment). Simultaneously lobules were stained with the nuclear dye Hoechst 33342 to enable cell counting. Both stainings were performed according to the manufacturer's protocols. Fluorescence of Hoechst 33342 and PI was recorded using confocal microscopy, with a 63× 1.2 NA objective lens (excitations 355 and 543 nm; and emissions 390–480 and 570–650 nm, respectively).
Immunocytochemistry
Immunocytochemistry in pancreatic lobules and big clusters was performed as described by Lur et al. (2009) with some modifications. Following blocking with 1% BSA and 10% goat serum, the isolated pancreatic lobules were incubated for 1 h at room temperature with primary antibody (CCK‐AR) in 5% goat serum in PBS. The pancreatic lobules were subsequently incubated with CruzFluor (CFL) 594‐conjugated secondary antibody for 30 min at room temperature. Cells were attached to the glass coverslips covered with poly‐l‐lysin. For immunochemical staining with desmin antibody, pancreatic lobules were fixed with 4% paraformaldehyde followed by permeabilization with Triton X100, blocking and incubation with primary (desmin) and then with secondary (CFL) antibodies as described above.
Reagents
Chemicals, unless otherwise indicated, were obtained from Sigma or Calbiochem (Merck, UK). BK, R‐715 and WIN64338 were purchased from Tocris Biosciences (Bristol, UK). Fluo‐4 AM, PI and Hoechst 33342 were purchased from Invitrogen (Life Technologies, Carlsbad, CA, USA). Antibodies against CCK‐A (CCK1) receptor (sc‐16172), desmin antibody (sc‐7559) and donkey anti‐goat IgG‐CFL 594 (sc‐362275) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Results
Separate Ca2+ signalling events in neighbouring PSCs and PACs
It has been shown that desmin is a good marker for PSCs (Apte et al. 1998) and we found that immunochemical staining with desmin antibody identified small elongated cells situated at the periphery of the dominant acinar cells (Fig. 2 A–C). The presence of vitamin A in lipid droplets is another characteristic of PSCs (Bachem et al. 1998) and we could visualize this by intrinsic multiphoton fluorescence (Fig. 2 D, E).
Figure 2. Identification of PSCs in lobule preparation .
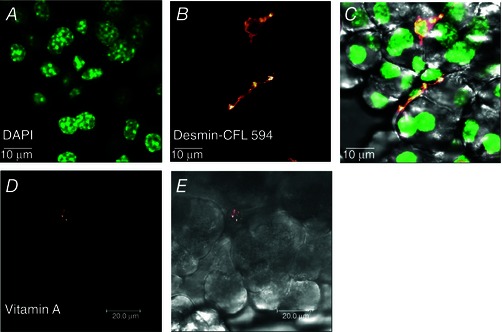
A, localization of nuclei is shown by staining with DAPI. B, staining with desmin antibody, visualized with conjugated secondary antibody IgG‐CruzFluor 594 (CFL 594). C, overlay of transmitted light image with DAPI and desmin antibody (n = 6). D, visualization of lipid droplets containing vitamin A by multiphoton intrinsic fluorescence (n = 6). E, transmitted light image of the pancreatic lobule shown in D.
Cells situated in the position characteristic of desmin‐containing cells (Fig. 2 C) took up the Ca2+‐sensitive fluorescent probe Fluo‐4 much more avidly than PACs (Fig. 3 A–C). In large lobules stained with Fluo‐4 (Fig. 3 A, B), it was often possible to observe PSCs as bright ‘strings’, whereas in smaller cell clusters, they typically appeared as individual bright cells at the periphery of acinar units (Fig. 3 Ca).
Figure 3. Complete separation of Ca2+ signalling in neighbouring PSCs and PACs .
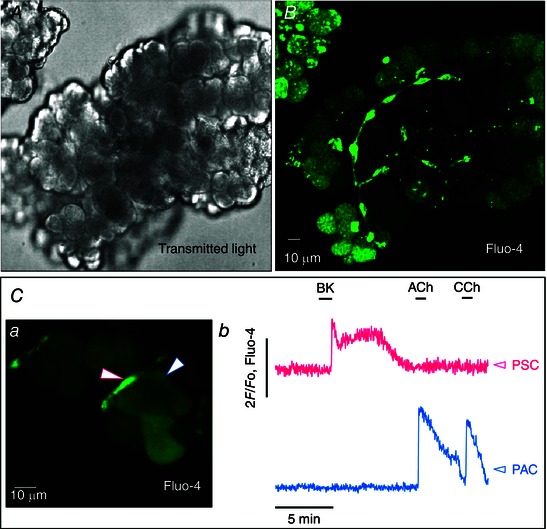
A, transmitted light image of live mouse pancreatic lobule. B, fluorescence image of the same lobule (Fluo‐4). The small elongated cells with long processes (PSCs) are much better loaded with Fluo‐4 than the larger PACs. C, [Ca2+]i measurements in neighbouring PSCs and PACs in a small cell cluster. a, fluorescence image of the live small cell cluster from which recordings were made. A highly fluorescent PSC is signposted by the white arrow outlined in red, whereas the much less fluorescent neighbouring PAC is signposted by the white arrow with blue outline. b, [Ca2+]i traces from the two cells signposted in a. Red trace from the PSC and blue trace from the PAC. BK (1 nm) evokes a typical biphasic Ca2+ signal in the PSC but not in the neighbouring PAC, whereas ACh (10 μm) and CCh (10 μm) evoke Ca2+ signals in the PAC but not in the PSC.
Cultured PSCs produce cytosolic Ca2+ signals in response to a micromolar concentration of BK (Won et al. 2011). In our experiments, normal PSCs in their normal micro‐environment typically responded to a short‐lasting exposure to a much lower BK concentration (1 nm) with a sharp transient rise in [Ca2+]i, quickly followed by a longer lasting (plateau) phase of elevated [Ca2+]i (Figs 3 Cb and 4 A). Neighbouring PACs never displayed any change in [Ca2+]i during the BK‐induced PSC Ca2+ signals (>100 experiments) (Figs 3 Cb and 4 A), indicating lack of functional BK receptors on PACs and lack of direct communication between neighbouring PACs and PSCs. The typical, and physiologically important, PAC stimulants ACh and CCK evoked Ca2+ signals in PACs (Petersen & Tepikin, 2008), which were not transmitted to the neighbouring PSCs (n = 14 and 17, respectively) (Figs 3 Cb and 4 A). ATP (100 μm) elicited Ca2+ signals in a proportion of PSCs (41 of 107 cells), but failed to do so in many others (Fig. 4 A). When ATP evoked a [Ca2+]i rise in a PSC, it was never transmitted to the neighbouring PAC (Fig. 4 A).
Figure 4. BK, but not CCK, evokes Ca2+ signal in PSCs .
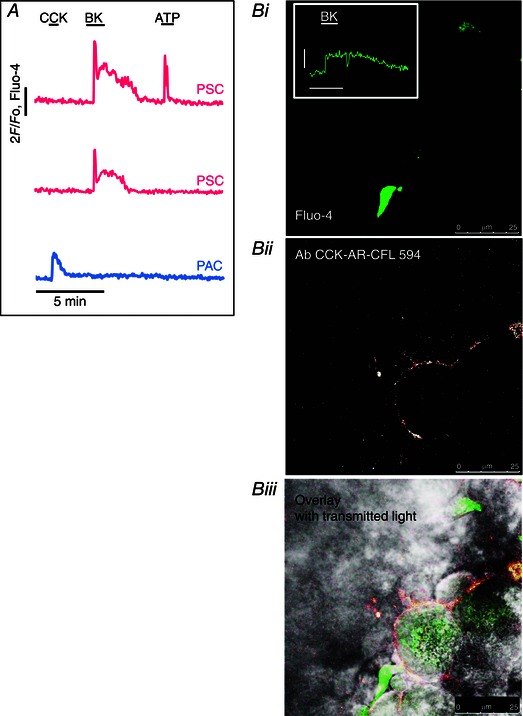
A, simultaneous [Ca2+]i traces from two PSCs and one PAC in the same cell cluster. CCK (10 nm) only evoked a Ca2+ signal in the PAC but not in the two PSCs, whereas BK (1 nm) elicited Ca2+ signals in both PSCs but not the PAC. ATP (100 μm) evoked a response in one PSC but not in the other and not in the PAC. B, immunostaining of CCK receptors in a pancreatic lobule. i, a PSC identified by bright staining with Fluo‐4. The response to BK (5 nm) is shown in the inset (amplitude scale bar 1 F/F 0, time scale bar 2 min). ii, visualization of CCK1 receptors by antibody against the CCK1 receptor with subsequent application of secondary antibody (CFL 594). iii, overlay of i and ii showing the localization of CCK1 receptors on the surface of PACs, but not the PSC.
Phillips et al. (2010) have suggested that CCK‐evoked release of ACh from PSCs could in turn activate PACs and they proposed that this could be the normal mechanism for CCK‐elicited secretion from human PACs. However, we did not find any evidence for this hypothesis as CCK never evoked any Ca2+ signals in PSCs in our preparations (n = 17; at [CCK] = 10 nm) and Ca2+ signals in PSCs consistently failed to be transmitted to neighbouring PACs (Fig. 4 A). Furthermore, although a fluorescent antibody to CCK1 receptors clearly identified the presence of CCK1 receptors on PACs, it failed to do so with regard to PSCs (four experiments) (Fig. 4 B).
BK evokes Ca2+ signals in PSCs at pico‐ and low nanomolar concentrations
It seemed important to explore the levels of BK needed to evoke Ca2+ signals (Fig. 5) in relation to what is known about BK concentrations in plasma. From measurements in humans and rats, it is known that normal plasma BK concentrations are in the range 40–70 pm (Blais et al. 1999; Hirata et al. 2002). However, in a bile duct obstruction model of acute pancreatitis induced in rats, it has been shown that the BK plasma concentration rose to ∼140 pm (Hirata et al. 2002). As the BK release in acute pancreatitis primarily comes from the pancreas, the local (intra‐pancreatic) BK levels could be higher (Ryan et al. 1964; Schachter, 1969). We found that a BK concentration as low as 100 pm (n = 14) could elicit PSC Ca2+ signals and that maximal responses were obtained at 1 nm (n = 9, in the series of experiments represented by Fig. 5 C). Overall, in the course of this investigation, BK responses to 1 nm have been observed in >100 experiments. At the low BK concentrations we used, there was no sign of desensitization (Fig. 5 A). Within the time frame of our protocols, the [Ca2+]i elevation was maintained as long as the stimulus was maintained (n = 19) (Fig. 5 A). It would therefore appear that normal PSCs are sufficiently sensitive to BK to be able to sense relatively small increases in the surrounding BK level.
Figure 5. BK concentration–response relationship .
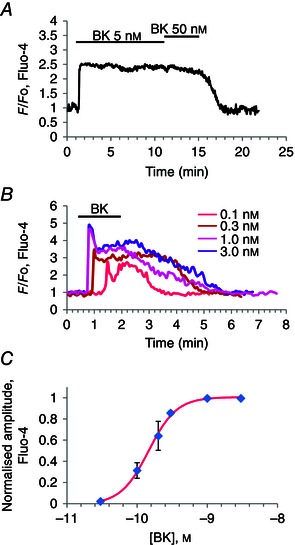
A, [Ca2+]i trace from PSC showing that 5 nm BK evokes maximal sustained response, which is not enhanced by increasing the BK concentration to 50 nm. B, family of [Ca2+]i traces all obtained from the same PSC, showing responses to BK concentrations from 0.1 to 3 nm. C, BK concentration–response (amplitude of plateau phase) relationship. Half‐maximal response is at a BK concentration of 200 pm. A very similar relationship was obtained for the initial peak response.
Although the PSCs in our lobule preparation were much more sensitive to BK than those previously studied in culture (Won et al. 2011), they do have some characteristics in common with quiescent PSCs in culture which, unlike activated PSCs, do not respond to trypsin and thrombin. As seen in Fig. 6 there was no response to thrombin (n = 6) (Fig. 6 A) or trypsin (n = 18) (Fig. 6 B) in cells that responded to BK with a clear Ca2+ signal.
Figure 6. Thrombin and trypsin do not evoke Ca2+ signals in PSCs .
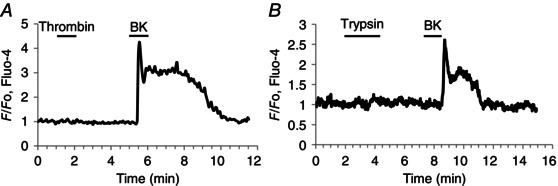
A, thrombin (10 mU ml−1) does not evoke a Ca2+ signal in a PSC, but a subsequent BK application (1 nm) evokes the usual response. B, trypsin (20 μm) fails to evoke a Ca2+ signal in a PSC, but a subsequent BK application (1 nm) does so. The apparently delayed and somewhat smaller response to BK (although still within the normal range) in B (as compared to A) is probably due to the fact that the cell cluster in this case was relatively large and the PSC deep in the tissue (see Fig. 1).
The BK‐elicited Ca2+ signals in PSCs are due to activation of B2 receptors
The BK receptor sub‐type responsible for generating Ca2+ signals in PSCs has not previously been investigated. We employed the B2 receptor blocker WIN64338 (Hu et al. 2004) and showed that this agent reversibly blocked BK‐elicited Ca2+ signal generation (n = 27) (Fig. 7 A). In contrast, the B1 receptor blocker R‐715 (Abdouh et al. 2008) failed to inhibit BK‐induced Ca2+ signalling (n = 8) (Fig. 7 B). Furthermore, the specific B1 agonist Sar‐des‐Arg‐BK did not evoke any changes in [Ca2+]i of PSCs (n = 8) (Fig. 7 C). It would appear that the plateau phase of the BK‐induced response to some extent depends on continued B2 receptor activation, because application of WIN64338 immediately after the initial BK‐elicited Ca2+ spike shortened the duration of the plateau phase (Fig. 7 D).
Figure 7. BK receptor pharmacology .
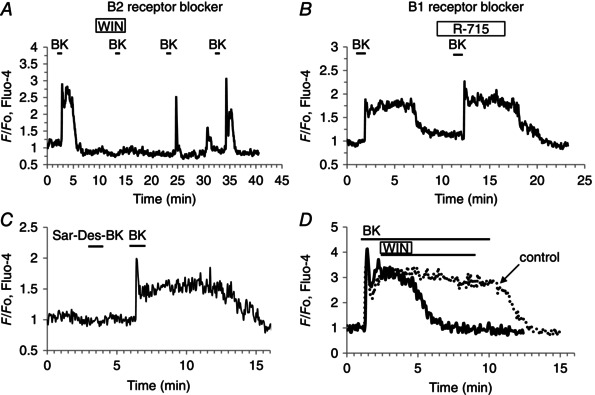
A, the B2 receptor antagonist WIN64338 (10 μm) reversibly and completely blocked the BK‐ (1 nm) elicited Ca2+ signal, both peak (P < 0.000003) and plateau (P < 0.00004). B, the B1 receptor antagonist R‐715 (10 μm) failed to block the BK‐ (1 nm) elicited Ca2+ signal. C, the B1 receptor agonist Sar‐des‐Arg‐BK (Sar‐Des‐BK) (1 μm) does not evoke a Ca2+ signal in a PSC, whereas a subsequent application of BK (1 nm) elicits such a signal. D, the B2 receptor antagonist WIN64338 applied after the initial BK‐evoked Ca2+ spike reduces markedly the length of the plateau phase.
As a previous study showed that BK levels increase in acute pancreatitis and that blockade of B2 receptors attenuated the cellular changes underlying the development of acute pancreatitis evoked by obstruction of the pancreatico‐biliary duct in rats (Hirata et al. 2002), our results raise the possibility that B2 receptor‐mediated Ca2+ signals in PSCs contribute to the negative outcome of this disease. We therefore tested whether B2 receptor blockade could protect against cellular changes relevant to the development of acute pancreatitis, which is most frequently related to alcohol abuse or biliary disease (Petersen & Sutton, 2006). In alcohol‐related acute pancreatitis, the pancreas is exposed not only to alcohol, but also to fatty acid ethyl esters (non‐oxidative combinations of alcohol and fatty acids), which have been shown to be particularly effective releasers of intracellular Ca2+ (Criddle et al. 2006; Gerasimenko et al. 2009). In biliary disease, the pancreas will be exposed to high concentrations of bile acids, which have also been shown to be effective liberators of stored Ca2+ (Voronina et al. 2002; Gerasimenko et al. 2006). We therefore tested the effects of B2 receptor blockade on the level of necrosis elicited by alcohol, POAEE and a bile acid mixture. As seen in Fig. 8, B2 receptor blockade markedly reduced the extent of acinar cell necrosis induced by either a high alcohol concentration (350 mm), POAEE (500 μm) or bile acids (0.5% sodium choleate: a crude ox bile extract which contains the sodium salts of taurocholic, glycocholic, deoxycholic and cholic acids).
Figure 8. The extent of PAC death elicited by pancreatitis‐inducing agents is markedly reduced by B2 receptor blockade .
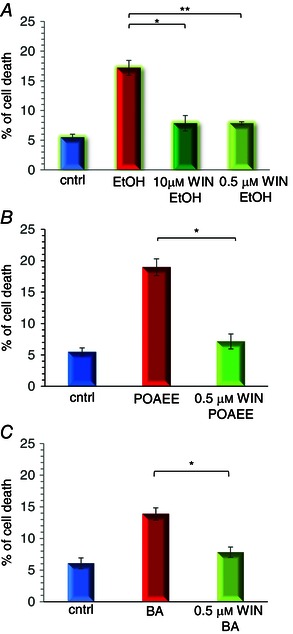
A, treatment of pancreatic lobules with ethanol (350 mm) for 2 h at room temperature increased significantly the percentage of PAC necrosis from the control level of 5.47 ± 0.54% (n = 7) to 17 ± 1% (n = 7) and this was significantly reduced to 7.85 ± 1.3% (n = 3) by 10 μm WIN64338 and to 7.84 ± 0.25% (n = 4) by 0.5 μm WIN64338 (*P = 0.0025 and **P = 0.00043, respectively, >1500 cells in each experimental group). B, treatment of pancreatic lobules with POAEE (500 μm) for 2 h at room temperature increased significantly the percentage of PAC necrosis from the control level of 5.46 ± 1.25% (n = 3) to 18.96 ± 1.76% (n = 3) and this was reduced to 7.15 ± 0.67% (n = 3) by 0.5 μm WIN64338 (*P = 0.003, >990 cells in each experimental group). C, treatment of pancreatic lobules with 0.5% sodium choleate for 2 h at room temperature also increased significantly the percentage of PAC necrosis from the control level of 6.1 ± 0.85% (n = 3) to 14 ± 1% (n = 3) and this was significantly reduced to 7.8 ± 0.8% (n = 3) by 0.5 μm WIN64338 (*P = 0.0087, >1600 cells in each experimental group). Necrosis was measured by staining of lobules with PI. Cell count was performed using nuclear staining with Hoechst 33342. Bars represent ±SEM.
BK‐elicited Ca2+ signals in PSCs are primarily due to release of Ca2+ from internal stores, but is followed by store‐operated Ca2+ entry
As shown in Figs 3, 4, 5, the BK‐elicited Ca2+ signals consist of a brief transient rise in [Ca2+]i, followed by a prolonged plateau phase of elevated [Ca2+]i. Acute removal of external Ca2+ did not reduce the initial phase of BK‐evoked Ca2+ signals, but eliminated the following plateau phase (Fig. 9 A, B). Re‐admission of Ca2+ resulted in a transient increase in [Ca2+]i and enabled a subsequent BK application to evoke a normal response (Fig. 9 A, B). We tested the ability of PSCs to generate store‐operated Ca2+ entry, by using the now ‘classical’ protocol for assessing this phenomenon (Fig. 9 C), employing cyclopiazonic acid (CPA) to block the Ca2+ pumps in the endoplasmic reticulum (ER). As shown in Fig. 9 B and C, re‐admission of external Ca2+ after a period of external Ca2+ deprivation, and BK stimulation, resulted in a transient rise in [Ca2+]i, but, if the Ca2+ re‐admission occurred after and during a period of continued ER Ca2+ pump inhibition, the [Ca2+]i elevation was sustained (Fig. 9 C). In this situation, Ca2+ entering store‐operated Ca2+ channels cannot be taken up into the ER, but will only slowly be extruded by the plasma membrane Ca2+ pumps.
Figure 9. Mechanisms of BK‐elicited Ca2+ signal generation in PSCs .
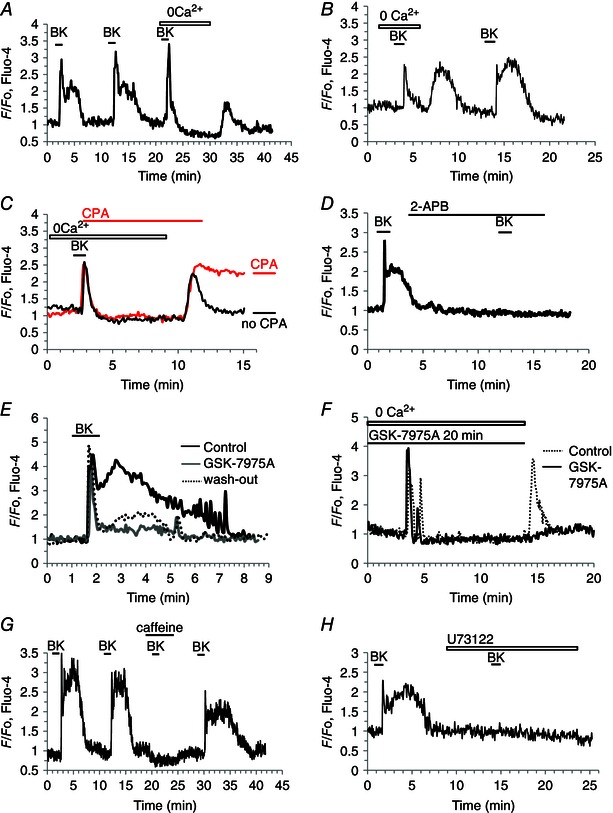
A and B, removal of external Ca2+ has no effect on the initial [Ca2+]i rise evoked by BK (1 nm, P > 0.7), but abolishes the following plateau phase (P < 0.0003). Readmitting external Ca2+, in the absence of BK stimulation, causes a transient rise in [Ca2+]i. C, when the SERCA pump inhibitor CPA (20 μm) is present, the [Ca2+]i rise upon readmission of external Ca2+ is prolonged. D, 2‐APB (100 μm) (IP3R antagonist and inhibitor of CRAC channels) blocks Ca2+ signalling elicited by BK (1 nm). E, the CRAC channel blocker GSK‐7975A (10 μm) reduces markedly the plateau phase of the BK‐ (1 nm) elicited response (P < 0.0015). Washout of GSK‐7975A partially restored the response (P < 0.009). F, GSK (10 μm) does not inhibit the initial BK‐elicited Ca2+ signal occurring in the absence of external Ca2+ but prevents the [Ca2+]i rise normally occurring upon external Ca2+ readmission. G, caffeine (30 mm) reversibly blocks BK‐ (1 nm) elicited Ca2+ signal. H, the phospholipase C inhibitor U73122 (30 μm) abolished Ca2+ signal generation elicited by BK (1 nm), both peak (P < 0.0002) and plateau (P < 0.00001).
We tried to block the store‐operated Ca2+ entry pharmacologically. 2‐APB, a well‐known, but relatively unspecific, blocker of CRAC channels (Parekh & Putney, 2005), abolished BK‐induced Ca2+ signal generation (Fig. 9 D), which may be due to blockade of IP3Rs (Ma et al. 2000). We also used a more specific CRAC channel blocker, GSK‐7975A, which has recently been shown to block CRAC channel currents in PACs (Gerasimenko et al. 2013). In these experiments, we were able to show that GSK‐7975A reversibly blocked the plateau phase of the BK‐induced [Ca2+]i elevation, without affecting the initial spike (n = 9)(Fig. 9 E). GSK‐7975A also blocked the Ca2+ entry normally occurring when external Ca2+ was re‐admitted after a period of external Ca2+ deprivation (n = 14) (Fig. 9 F).
Finally, we examined the mechanism underlying the initial Ca2+ spike in response to BK stimulation. Having established that it is due to release of Ca2+ from internal stores (Fig. 9 A), we tested the hypothesis that this Ca2+ liberation occurs through IP3Rs. It is now well stablished that caffeine inhibits opening of IP3Rs (Wakui et al. 1990; Ehrlich et al. 1994; Foskett et al. 2007). As seen in Fig. 9 G, caffeine did reversibly block BK‐elicited Ca2+ signalling (n = 29). BK probably activates phospholipase C, as U73122 (Bleasdale et al. 1990; Smith et al. 1990) blocked the ability of BK to evoke Ca2+ signals (n = 14) (Fig. 9 H).
Discussion
Our results demonstrate for the first time that BK, in concentrations close to those measured in normal plasma (Blais et al. 1999; Hirata et al. 2002), elicits substantial cytosolic Ca2+ signals in normal PSCs in their normal micro‐environment and therefore cast doubt on the hitherto prevailing concept of the quiescent PSC (Apte et al. 1998, 2012; Sherman et al. 2014).
Given that an important role for BK in the development of acute pancreatitis was proposed by Ryan et al. (1964), that there is an increase in the plasma level of BK in acute pancreatitis (Hirata et al. 2002) and that a number of studies have shown that pharmacological blockade of B2 receptors is helpful in suppressing the cellular changes in several pancreatitis disease models (Griesbacher & Lembeck 1992; Griesbacher et al. 1993; Hoffmann et al. 1996; Bloechle et al. 1998; Hirata et al. 2002), it would seem important to identify the target for the action of BK in the pancreas and its mechanism of action.
Won et al. (2011) demonstrated that BK – in high (micromolar) concentrations – evoked transient Ca2+ signals in quiescent and activated PSCs in culture. We focused our attention on the effect of low quasi‐physiological concentrations of BK on normal PSCs in their normal micro‐environment, the mechanism of action and its consequence. Our results show that whereas BK consistently evokes bi‐phasic Ca2+ signals in PSCs, it consistently fails to do so even in closely neighbouring PACs (Figs 3 and 4). We have therefore now identified a specific cellular site for the action of BK, which may partly or fully explain its importance in pancreatitis. Furthermore, we have established that the BK‐elicited Ca2+ signals in PSCs are due to activation of B2 receptors, providing a plausible explanation for the suppressive effect of B2 blockade on the development of acute pancreatitis (Griesbacher & Lembeck 1992; Griesbacher et al. 1993; Hoffmann et al. 1996; Bloechle et al. 1998; Hirata et al. 2002). We have also shown that PAC necrosis elicited by alcohol, POAEE or bile can be significantly reduced by B2 receptor blockade (Fig. 8).
We failed to observe any effects of CCK on PSCs, although this hormone evoked its usual effect (Petersen & Tepikin, 2008) on neighbouring PACs (Fig. 4 A). Therefore, our results do not provide support for the hypothesis that CCK acting on PSCs will increase ACh secretion from these cells, which in turn would activate PACs (Phillips et al. 2010). We worked on normal mouse pancreatic tissue, whereas Phillips et al. (2010) studied ACh secretion from cultured human PSCs. Cultured PSCs clearly have different properties from normal PSCs, because in the normal mouse pancreas we have observed Ca2+ signal generation in PSCs in response to 0.1 nm BK (Fig. 5), whereas micromolar BK concentrations were required to obtain such responses in cultured mouse PSCs (Won et al. 2011). With regard to the mechanism of CCK action in the human pancreas, the simplest hypothesis remains direct action on PACs, as shown in isolated acinar cell clusters from human pancreas (Murphy et al. 2008).
Although the initiating event elicited by pancreatitis‐inducing agents in PACs is Ca2+ release from intracellular stores (Gerasimenko et al. 2014), we have previously shown that the cellular damage only happens if there is secondary Ca2+ entry from the extracellular fluid (Raraty et al. 2000). We have recently shown that the CRAC channel blocker GSK‐7975A markedly inhibits the store‐operated Ca2+ entry that sustains the [Ca2+]i elevation in PACs evoked by a pancreatitis‐inducing agent (Gerasimenko et al. 2013, 2014), a finding recently confirmed by Voronina et al. (2015). We have previously shown that the CRAC channel blockade by GSK‐7975A provides effective protection of the PACs from alcohol‐related intracellular protease activation and necrosis (Gerasimenko et al. 2013) and these results have also very recently been confirmed in a study employing three different in vivo mouse models of acute pancreatitis (Wen et al. 2015). The BK‐elicited Ca2+ signal generation in PSCs is due to initial release of Ca2+ from internal stores followed by activation of Ca2+ entry via store‐operated channels. Our new results show that GSK‐7975A is also effective in reducing the plateau phase of the BK‐elicited [Ca2+]i elevation in PSCs (Fig. 9 E). Given that B2 receptor blockade protects against pancreatitis‐like cellular changes (Fig. 8), the inhibitory effect of GSK‐7975A on BK responses in PSCs may represent an additional potential benefit of treating cases of severe acute pancreatitis with a CRAC channel blocker. The inhibitory action of GSK‐7975A on the BK‐induced plateau elevation of [Ca2+]i in PSCs may well have contributed to the impressive protective effects of this agent against acute pancreatitis in vivo (Wen et al. 2015).
Our new data are also relevant when considering the now widespread use of ACE inhibitors in the treatment of hypertension because ACE inhibitors inhibit the breakdown of BK, causing an increase in the tissue and plasma levels of BK (Israili & Hall, 1992; Liu et al. 1997; Tsutsumi et al. 1999; Su, 2014). Previous studies have shown that use of ACE inhibitors is associated with a significantly increased risk of developing acute pancreatitis (Tilkemeier & Thompson, 1988; Eland et al. 2006), but the mechanism was unknown. As we have now shown that any increase in the BK level will elicit Ca2+ signals in the PSCs via B2 receptors and that blockade of these receptors protects against acute pancreatitis, it is likely that Ca2+ signal generation in PSCs mediated by BK is at least in part responsible for the increased risk of developing acute pancreatitis during ACE inhibitor treatment.
Additional information
Competing interests
None declared.
Author contributions
Conception and design of the experiments: O.H.P., O.V.G., O.G. and J.V.G. Collection, analysis and interpretation of data: O.G., J.V.G. and O.V.G. Drafting the article or revising it critically for important intellectual content: O.H.P., J.V.G., O.V.G. and O.G.
Funding
This work was supported by Medical Research Council Programme Grant MR/J002771/1. O.H.P. is a Medical Research Council Professor (G19/22/2).
Translational Perspective
Our work indicates that bradykinin‐elicited Ca2+ signals in pancreatic stellate cells may influence negatively the outcome of acute pancreatitis. We tested the hypothesis that blockage of bradykinin type 2 (B2) receptors would offer protection against the pancreatic acinar cell necrosis caused by pancreatitis‐inducing agents, such as alcohol, fatty acid ethyl esters or bile acids. The results showed that specific B2 receptor blockade markedly reduced the extent of necrosis observed after treatment with ethanol, POAEE or a mixture of bile acids. This suggests that B2 receptor blockade in the early stage of acute pancreatitis may be helpful in reducing the severity of the disease. We also show that the bradykinin‐elicited sustained elevation of the cytosolic Ca2+ concentration in pancreatic stellate cells can be inhibited by a specific inhibitor of Ca2+ release – activated Ca2+ (CRAC) channels. We have recently shown that CRAC inhibition in PACs offers remarkable protection against necrosis induced by fatty acid ethyl esters. In the intact pancreas, CRAC inhibition would reduce excessive Ca2+ signal generation both in the acinar cells (brought about by, for example, fatty acid ethyl esters) and in the stellate cells (brought about by bradykinin). Both effects would be beneficial. Our work therefore indicates that combined treatment with a CRAC inhibitor and a B2 receptor – blocking agent should be tested in vivo as a potentially useful therapy against acute pancreatitis.
O. Gryshchenko and J. V. Gerasimenko contributed equally to this work.
References
- Abdouh M, Talbot S, Couture R & Hassessian HM (2008) Retinal plasma extravasation in streptozotocin‐diabetic rats mediated by kinin B1 and B2 receptors. Br J Pharmacol 154, 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, Pirola RC & Wilson JS (1998). Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut 43, 128–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte MV, Pirola RC & Wilson JS (2012). Pancreatic stellate cells: a starring role in normal and diseased pancreas. Front Physiol 3, Article 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argent BE (2006). Cell physiology of pancreatic ducts In: Physiology of the gastrointestinal tract, vol 2, ed. Johnson LR, pp. 1376–1396. Elsevier, San Diego. [Google Scholar]
- Argent BE, Case RM & Scratcherd T (1971). Stimulation of amylase secretion from the perfused cat pancreas by potassium and other alkali metal ions. J Physiol 216, 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachem MG, Schneider E, Gross H, Weidenbach H, Schmid RM, Menke A, Siech M, Beger H, Grunert A & Adler G (1998). Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 115, 412–432. [DOI] [PubMed] [Google Scholar]
- Blais C, Adam A, Massicotte D & Peronnet F (1999). Increase in blood bradykinin concentration after eccentric weight‐training exercise in men. J Appl Physiol 87, 1197–1201. [DOI] [PubMed] [Google Scholar]
- Bleasdale JE, Thakur NR, Gremban RS, Bundy GL, Fitzpatrick FA, Smith RJ & Bunting S (1990). Selective inhibition of receptor‐coupled phospholipase C‐dependent processes in human platelets and polymorphonuclear neutrophils. J Pharmacol Exp Ther 255, 756–768. [PubMed] [Google Scholar]
- Bloechle C, Kusterer K, Kuehn RM, Schneider C, Knoefel WT & Izbicki JR (1998). Inhibition of bradykinin B2 receptor preserves microcirculation in experimental pancreatitis in rats. Am J Physiol 274, G42–G51. [DOI] [PubMed] [Google Scholar]
- Broadbent SD, Ramjeesingh M, Bear CE, Argent BE, Linsdell P & Gray MA (2015). The cystic fibrosis transmembrane conductance regulator is an extracellular chloride sensor. Pflügers Arch 467, 1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancela JM, Gerasimenko OV, Gerasimenko JV, Tepikin AV & Petersen OH (2000). Two different but converging messenger pathways to intracellular Ca2+ release: the roles of NAADP, cADPR and IP3 . EMBO J 19, 2549–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case RM & Clausen T (1973). The relationship between calcium exchange and enzyme secretion in the isolated rat pancreas. J Physiol 235, 75–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criddle D, Murphy J, Fistetto G, Barrow S, Tepikin AV, Neoptolemos JP, Sutton R & Petersen OH (2006). Fatty acid ethyl esters cause pancreatic calcium toxicity via inositol trisphosphate receptors and loss of ATP synthesis. Gastroenterology 130, 781–793. [DOI] [PubMed] [Google Scholar]
- Eland IA, Sundstrom A, Velo GP, Andersen M, Sturkenboom MCJM, Langman MJS, Stricker BHC & Wiholm B (2006). Antihypertensive medication and the risk of acute pancreatitis: the European case‐control study on drug‐induced acute pancreatitis. Scand J Gastroenterol 41, 1484–1490. [DOI] [PubMed] [Google Scholar]
- Ehrlich BE, Kaftan E, Bezprozvannaya S & Bezprozvanny I (1994). The pharmacology of intracellular Ca2+ release channels. Trends Pharmacol Sci 15, 145–149. [DOI] [PubMed] [Google Scholar]
- Foskett JK, White C, Cheung KH & Mak DOD (2007). Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87, 593–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimenko JV, Charlesworth RM, Sherwood MW, Ferdek PE, Mikoshiba K, Parrington J, Petersen OH & Gerasimenko OV (2015). Both RyRs and TPCs are required for NAADP‐induced intracellular Ca2+ release. Cell Calcium 58, 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimenko JV, Flowerdew SE, Voronina SG, Sukhomlin TK, Tepikin AV, Petersen OH & Gerasimenko OV (2006). Bile acids induce Ca2+ release from both the endoplasmic reticulum and acidic intracellular calcium stores through activation of inositol trisphosphate receptors and ryanodine receptors. J Biol Chem 281, 40154–40163. [DOI] [PubMed] [Google Scholar]
- Gerasimenko JV, Gerasimenko OV & Petersen OH (2014). The role of Ca+ in the pathophysiology of pancreatitis. J Physiol 592, 269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimenko JV, Gryshchenko O, Ferdek PE, Stapleton E, Hebert TOG, Bychkova S, Peng S, Begg M, Gerasimenko OV & Petersen OH (2013). Ca2+ release‐activated Ca2+ channel blockade as a potential tool in antipancreatitis therapy. Proc Natl Acad Sci USA 110, 13186–13191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimenko JV, Lur G, Sherwood MW, Ebisui E, Tepikin AV, Mikoshiba K, Gerasimenko OV & Petersen OH (2009). Pancreatic protease activation by alcohol metabolite depends on Ca2+ release via acid store IP3 receptors. Proc Natl Acad Sci USA 106, 10758–10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbacher T & Lembeck F (1992). Effects of the bradykinin antagonist, HOE 140, in experimental acute pancreatitis. Br J Pharmacol 107, 356–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbacher T, Tiran B & Lembeck F (1993). Pathological events in experimental acute pancreatitis prevented by the bradykinin antagonist Hoe140. Br J Pharmacol 108, 405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata M, Hayashi I, Yoshimura K, Ishi K, Soma K, Ohwada T, Kakita A & Majima M (2002). Blockade of bradykinin B2 receptor suppresses acute pancreatitis induced by obstruction of the pancreaticobiliary duct in rats. Br J Pharmacol 135, 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann TF, Waldner H & Messmer K (1996). The bradykinin antagonist CP0597 can limit the progression of post‐ischemic pancreatitis. Immunopharmacology 33, 243–246. [DOI] [PubMed] [Google Scholar]
- Hu H‐Z, Gao N, Liu S, Ren J, Wang X, Xia Y & Wood JD (2004). Action of bradykinin in the submucosal plexus of guinea pig small intestine. J Pharmacol Exp Ther 309, 320–327. [DOI] [PubMed] [Google Scholar]
- Israili ZH & Hall WD (1992). Cough and angioneurotic edema associated with angiotensin‐converting enzyme inhibitor therapy. A review of the literature and pathophysiology. Ann Intern Med 117, 234–242. [DOI] [PubMed] [Google Scholar]
- Kondo S & Schulz I (1976). Calcium ion uptake in isolated pancreas cells induced by secretagogues. Biochim Biophys Acta 419, 76–92. [DOI] [PubMed] [Google Scholar]
- Liu YH, Yang XP, Sharov VG, Nass O, Sabbah HN, Peterson E & Carretero OA (1997). Effects of angiotensin‐converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure – role of kinins and angiotensin II type 2 receptors. J Clin Invest 99, 1926–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lur G, Haynes LP, Prior IA, Gerasimenko OV, Feske S, Petersen OH, Burgoyne RD & Tepikin AV (2009). Ribosome‐free terminals of rough ER allow formation of STIM1 puncta and segregation of STIM1 from IP3 receptors. Curr Biol 19, 1648–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H‐T, Patterson RL, van Rossum DB, Birnbaumer L, Mikoshiba K & Gill DL (2000). Requirement of the inositol trisphosphate receptor for activation of store‐operated Ca2+ channels. Science 287, 1647–1651. [DOI] [PubMed] [Google Scholar]
- Maleth J, Balazs A, Pallagi P, Balla Z, Kui B, Katona M, Judak L, Nemeth I, Kemeny LV, Lajos V, Rakonczay Z, Venglovecz V, Foldesi I, Peto Z, Somoracz A, Borka K, Perdomo D, Lukacs GL, Gray MA, Monterisi S, Zaccolo M, Sendler M, Mayerle J, Kuhn JP, Lerch MM, Sahin‐Toth M & Hegyi P (2015). Alcohol disrupts levels and function of the cystic fibrosis conductance regulator to promote development of pancreatitis. Gastroenterology 148, 427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews EK, Petersen OH & Williams JA (1973). Pancreatic acinar cells: acetylcholine‐induced membrane depolarization, calcium efflux and amylase release. J Physiol 234, 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JA, Criddle DN, Sherwood M, Chvanov M, Mukherjee R, McLaughlin E, Booth D, Gerasimenko JV, Raraty MGT, Ghaneh P, Neoptolemos JP, Gerasimenko OV, Tepikin AV, Green GM, Reeve JR Jr, Petersen OH & Sutton R (2008). Direct activation of cytosolic Ca2+ signaling and enzyme secretion by cholecystokinin in human pancreatic acinar cells. Gastroenterology 135, 632–641. [DOI] [PubMed] [Google Scholar]
- Pallagi P, Venglovecz V, Rakonczay Z, Borka K, Korompay A, Ozsvari B, Judak L, Sahin‐Toth M, Geisz A, Schnur A, Maleth J, Takacs T, Gray MA, Argent BE, Mayerle J, Lerch MM, Wittmann T & Hegyi P (2011). Trypsin reduces pancreatic ductal bicarbonate secretion by inhibiting CFTR Cl– channels and luminal anion exchangers. Gastroenterology 141, 2228–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB (2010). Store‐operated CRAC channels: function in health and disease. Nat Rev Drug Disc 9, 399–410. [DOI] [PubMed] [Google Scholar]
- Parekh AB & Putney JW Jr (2005). Store‐operated calcium channels. Physiol Rev 85, 757–810. [DOI] [PubMed] [Google Scholar]
- Park MK, Lomax RB, Tepikin AV & Petersen OH (2001). Local uncaging of caged Ca2+ reveals distribution of Ca2+‐activated Cl− channels in pancreatic acinar cells. Proc Natl Acad Sci USA 98, 10948–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen OH (1992). Stimulus‐secretion coupling: cytoplasmic calcium signals and the control of ion channels in exocrine acinar cells. J Physiol 448, 1–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen OH & Sutton R (2006). Ca2+ signalling and pancreatitis: effects of alcohol, bile and coffee. Trends Pharmacol Sci 27, 113–120. [DOI] [PubMed] [Google Scholar]
- Petersen OH & Tepikin AV (2008). Polarized calcium signaling in exocrine gland cells. Annu Rev Physiol 70, 273–299. [DOI] [PubMed] [Google Scholar]
- Petersen OH & Ueda N (1976). Pancreatic acinar cells: the role of calcium in stimulus‐secretion coupling. J Physiol 254, 583–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips PA, Yang L, Shulkes A, Vonlaufen A, Poljak A, Bustamante S, Warren A, Xu ZH, Guilhaus M, Pirola R, Apte & Wilson JS (2010). Pancreatic stellate cells produce acetylcholine and may play a role in pancreatic exocrine secretion. Proc Natl Acad Sci USA 107, 17397–17402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raraty M, Ward J, Erdemli G, Vaillant C, Neoptolemos JP, Sutton R & Petersen OH (2000). Calcium‐dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc Natl Acad Sci USA 97, 13126–13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan JW, Moffat JG & Thompson AG (1964). Role of bradykinin in the development of acute pancreatitis. Nature 204, 1212–1213. [DOI] [PubMed] [Google Scholar]
- Schachter M (1969). Kallikreins and kinins. Physiol Rev 49, 509–547. [DOI] [PubMed] [Google Scholar]
- Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S, Martin P, Tseng TW, Dawson DW, Donahue TR, Masamune A, Shimosegawa T, Apte MV, Wilson JS, Ng B, Lau SL, Gunton JE, Wahl GM, Hunter T, Drebin JA, O'Dwyer PJ, Liddle C, Tuveson DA, Downes M & Evans RM (2014). Vitamin D receptor‐mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 159, 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RJ, Sam LM, Justen JM, Bundy JL, Bala GA & Bleasdale JE (1990). Receptor‐coupled signal transduction in human polymorphonuclear neutrophils – effects of a novel inhibitor of phospholipase C‐dependent processes on cell responsiveness. J Pharmacol Exp Ther 253, 688–697. [PubMed] [Google Scholar]
- Streb H, Irvine RF, Berridge MJ & Schulz I (1983). Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol‐1,4,5‐trisphosphate. Nature 306, 67–69. [DOI] [PubMed] [Google Scholar]
- Su JB (2014). Different cross‐talk sites between the renin‐angiotensin and the kallikrein‐kinin systems. J Renin Angiotensin Aldosterone Syst 15, 319–328. [DOI] [PubMed] [Google Scholar]
- Tilkemeier P & Thompson PD (1988). Acute pancreatitis possibly related to enalapril. N Engl J Med 318, 1275–1276. [DOI] [PubMed] [Google Scholar]
- Tsutsumi Y, Matsubara H, Masaki H, Kurihara H, Murasawa S, Takai S, Miyazaki M, Nozawa Y, Ozono R, Nakagawa K, Miwa T, Kawada N, Mori Y, Shibasaki Y, Tanaka Y, Fujiyama S, Koyama Y, Fujiyama A, Takahashi H & Iwasaka T (1999). Angiotensin II type 2 receptor overexpression activates the vascular kinin system and causes vasodilation. J Clin Invest 104, 925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronina SG, Collier D, Chvanov M, Middlehurst B, Beckett AJ, Prior IA, Criddle DN, Begg M, Mikoshiba K, Sutton R & Tepikin AV (2015). The role of Ca2+ influx in endocytic vacuole formation in pancreatic acinar cells. Biochem J 465, 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronina S, Longbottom R, Sutton R, Petersen OH & Tepikin AV (2002). Bile acids induce calcium signals in mouse pancreatic acinar cells. Implications for bile‐induced pancreatic pathology. J Physiol 540, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakui M, Osipchuk YV & Petersen OH (1990). Receptor‐activated cytoplasmic Ca2+ spiking mediated by inositol trisphosphate is due to Ca2+‐induced Ca2+ release. Cell 63, 1025–1032. [DOI] [PubMed] [Google Scholar]
- Watari H, Hotta Y & Mabuchi Y (1982). Morphological studies on a vitamin A storing cell and its complex with macrophage observed in mouse pancreatic tissues following excess vitamin A administration. Okajimas Folia Anat Jpn 58, 837–858. [DOI] [PubMed] [Google Scholar]
- Wells RG & Crawford JM (1998). Pancreatic stellate cells: the new stars of chronic pancreatitis? Gastroenterology 115, 491–493. [DOI] [PubMed] [Google Scholar]
- Wen L, Voronina S, Javed MA, Awais M, Szatmary P, Latawiec D, Chvanov M, Collier D, Huang W, Barrett J, Begg M, Stauderman K, Roos J, Grigoryev S, Ramos S, Rogers E, Whitten J, Velicelebi G, Dunn M, Tepikin AV, Criddle DN & Sutton R (2015). Inhibitors of ORAI1 prevent cytosolic calcium‐associated injury of human pancreatic acinar cells and acute pancreatitis in 3 mouse models. Gastroenterology 149, 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won JH, Zhang Y, Baoan JI, Logsdon CD & Yule DI (2011). Phenotypic changes in mouse pancreatic stellate cell Ca2+ signaling events following activation in culture and in a disease model of pancreatitis. Mol Biol Cell 22, 412–436. [DOI] [PMC free article] [PubMed] [Google Scholar]