

Summary
During retroviral infection, viral capsids are subject to restriction by the cellular factor TRIM5α. Here, we show that dendritic cells (DCs) derived from human and non-human primate species lack efficient TRIM5α-mediated retroviral restriction. In DCs, endogenous TRIM5α accumulates in nuclear bodies (NB) that partly co-localize with Cajal bodies in a SUMOylation-dependent manner. Nuclear sequestration of TRIM5α allowed potent induction of type I interferon (IFN) responses during infection, mediated by sensing of reverse transcribed DNA by cGAS. Overexpression of TRIM5α or treatment with the SUMOylation inhibitor ginkgolic acid (GA) resulted in enforced cytoplasmic TRIM5α expression and restored efficient viral restriction but abrogated type I IFN production following infection. Our results suggest that there is an evolutionary trade-off specific to DCs in which restriction is minimized to maximize sensing. TRIM5α regulation via SUMOylation-dependent nuclear sequestration adds to our understanding of how restriction factors are regulated.
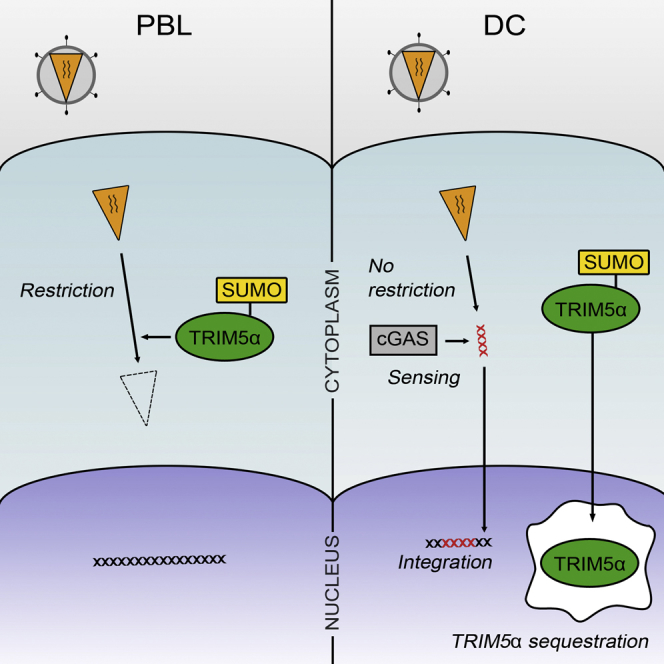
Graphical Abstract
Highlights
-
•
Primate dendritic cells (DCs) lack efficient TRIM5α-mediated retroviral restriction
-
•
In DCs TRIM5α is sequestered in the nucleus in a SUMOylation-dependent manner
-
•
TRIM5α nuclear sequestration allows DC sensing of retroviral DNA by cGAS
Retroviruses can be efficiently blocked by a cellular restriction factor called TRIM5α. Intriguingly, TRIM5α is inactive in dendritic cells (DCs). Portilho et al. show that in DCs TRIM5α is sequestered in the nucleus in a SUMOylation-dependent manner, favoring innate sensing of retroviruses in the cytoplasm by cGAS and thus an antiviral response.
Introduction
After entry in target cells, retroviral capsids are subject to host restriction by the “alpha” spliced variant of tripartite motif protein 5 (TRIM5α) (Stremlau et al., 2004). Restriction operates by direct recognition of the viral capsid in the cytoplasm, leading to a premature and accelerated uncoating and to abortive infection (Stremlau et al., 2006). TRIM5α proteins restrict retroviruses in a species-specific manner and thus constitute an effective barrier to cross-species transmissions of retroviruses. For instance, rhesus macaque (RM) TRIM5α restricts HIV-1, SIVagm, and N-MLV, but not SIVmac, whereas human TRIM5α can block N-MLV and equine infectious anemia virus (EIAV), but is less effective against HIV-1 (Bieniasz, 2004, Song et al., 2005).
TRIM5α-mediated restriction was identified in a number of macaque primary cells and cell lines (LLC-MK2, FRhK-4, FrHL-2, Rh.F and RM primary lung fibroblasts) (Besnier et al., 2002, Cowan et al., 2002, Münk et al., 2002, Stremlau et al., 2004, Yap et al., 2004). However, while RM T lymphocytes efficiently block HIV-1 infection, we have previously shown that RM dendritic cells (DCs) lack TRIM5α-mediated restriction and are permissive to HIV-1 infection (Arhel et al., 2008). We further showed that TRIM5α RNA levels are normal in DCs and there is no detectable increase in the competitive inhibitor TRIM5γ. These findings suggest that the endogenous TRIM5α protein is likely to be dysfunctional, a possibility supported by the finding that the overexpression of RM TRIM5α in DCs restores restriction (Arhel et al., 2008).
Recent work proposed that TRIM5α restriction may be modulated by the small ubiquitin-related modifier (SUMO) machinery (Arriagada et al., 2011, Dutrieux et al., 2015, Lukic et al., 2013, Nepveu-Traversy and Berthoux, 2014). The human SUMO family consists of four members, SUMO1 to SUMO4. SUMO can either be directly conjugated to protein substrates by covalent addition of a SUMO moiety to a lysine residue within the ΨKxE consensus sequence (where Ψ is a large hydrophobic residue and x any amino acid) (Sampson et al., 2001), or can interact non-covalently with target proteins through SUMO-interaction motifs (SIMs) (Hecker et al., 2006, Minty et al., 2000). Many viral proteins are SUMOylated or influence the SUMOylation of cellular proteins, and the infection of some viruses is dependent on pathways that are regulated by SUMOylation (Everett et al., 2013). SUMO1 overexpression enhances restriction of N-MLV and HIV-1 by human and RM TRIM5α, respectively, while knockdown of SUMO1 or the Ubc9 E2 SUMO-conjugating enzyme reduces HIV-1 restriction by RM TRIM5α (Arriagada et al., 2011, Dutrieux et al., 2015, Lukic et al., 2013). We recently showed that human and RM TRIM5α are substrates for SUMO modification in vitro and in cellulo, and the putative SUMO conjugation consensus site at residue K10 is the main site for SUMO modification (Dutrieux et al., 2015). However, although K10R point mutants abrogated SUMOylation at this position, TRIM5α antiviral activity was not impaired, arguing that non-covalent interaction with SUMO or SUMO-modified proteins rather than direct SUMOylation of TRIM5α might be responsible for TRIM5α restriction. Three SIMs that are able to bind SUMO1 were identified within the PRYSPRY domain necessary for capsid recognition and are shown to contribute to the ability of human TRIM5α to restrict N-MLV infection (Arriagada et al., 2011, Lukic et al., 2013). Given that the MLV capsid (CA) was shown to undergo SUMO1 conjugation (Yueh et al., 2006), this suggests that human TRIM5α might interact with SUMOylated MLV CA via SIMs within the PRYSPRY domain. However, two of the three SIMs in RM TRIM5α are reportedly buried internally within the PRYSPRY domain and are therefore unlikely to mediate SUMO-SIM interactions (Brandariz-Nuñez et al., 2013).
The aim of this study was to define the mechanism responsible for the lack of TRIM5α function in DCs and to clarify the effects on viral immune sensing by these potent accessory cells. We first show that absence of TRIM5α restriction is observed in DCs from all tested human and non-human primates, pointing to a common trait of humans and old world monkeys. Endogenous TRIM5α in DCs was found to be sequestered in the nucleus within NB that co-localize with Cajal bodies, a phenotype that was reversed upon treatment with ginkgolic acid (GA), an inhibitor of the E1 SUMO activating enzyme. Finally, induced cytoplasmic localization of TRIM5α in DCs by its overexpression or by GA treatment restored restriction and abrogated type I IFN production following retroviral infection. Together, our data show that TRIM5α is unable to restrict incoming retroviruses in DCs because it is absent from the cytoplasm. The resulting unhampered retroviral infection triggers innate sensing by host cell sensors and robust type I IFN production, suggesting that the lack of TRIM5α restriction in DCs is an anti-viral mechanism that has evolved to promote immune responses necessary to control viral spread in the infected host.
Results
Human and Non-human Primate DCs Lack TRIM5α-Mediated Restriction
We previously showed that RM (Macaca mulatta) DCs, contrary to all other tested RM cells (Arhel et al., 2008, Besnier et al., 2002, Cowan et al., 2002, Münk et al., 2002, Stremlau et al., 2004, Yap et al., 2004), are permissive to HIV-1 infection, indicating an absence of functional TRIM5α-mediated restriction in this cell type (Arhel et al., 2008). Given that immature DCs are highly phagocytic, we carried out retroviral transductions in the presence of a reverse transcription inhibitor to ascertain whether uptake might be the result of non-specific pseudo-transduction. RM peripheral blood lymphocytes (PBLs) and monocyte-derived DCs were characterized with phenotypic markers (Figure S1A) and transduced with either SIVmac (not restricted) or HIV-1 (restricted). RM PBLs were not permissive to HIV-1, and treatment with a specific TRIM5α small interfering RNA (siRNA) restored transduction, confirming that TRIM5α is responsible for restriction in these cells (Figure 1A). In contrast, RM DCs were equally permissive to SIVmac and HIV-1, indicating an absence of efficient TRIM5α restriction in DCs. HIV-1 infection of RM DCs was also observed using R5-tropic HIV-1 virus (Figure S1B). Treatment with nevirapine (Nev) reduced HIV-1 infection to levels similar to those seen in uninfected samples, validating that HIV-1 infection of RM DCs resulted in bona fide productive transduction events (Figure 1A).
Figure 1.

Lack of TRIM5α-Mediated Restriction in Human and Non-human Primate Myeloid DCs
(A–C) PBLs and DCs from (A) rhesus macaque, (B) human, and (C) African green monkey were transduced at MOI 5, ± nevirapine (5 μM), tenofovir disoproxil fumarate (TDF, 100 μM), heat-treated, or treated with a specific siRNA against human and RM TRIM5α (siHM3). Representative flow cytometry charts at 3 dpt are shown left, while graphs show the mean of three independent experiments with log10 scale ± SD using different primate donors (RM, n = 2; human, n = 4; AGM, n = 2) and different vector batches.
(D) PBLs and DCs from cynomolgus macaque (n = 2), human (n = 3) and pigtailed macaque (n = 3) were transduced with increasing MOI ranging from 0.5 to 50. Graphs show GFP expression 3 dpt (3 independent experiments ± SD).
See also Figure S1.
To determine whether the absence of TRIM5α restriction in DCs is a common phenomenon, human PBLs and DCs were transduced with either HIV-1 (not restricted) or EIAV (restricted) vectors (Figure 1B). DCs were ∼2-fold less permissive than PBLs to HIV-1 infection (12.0% ± 6.9% versus 24.1% ± 8.7% infected cells, n = 4), as reported in the literature (Berger et al., 2011, Laguette et al., 2011). As expected, human PBLs were refractory to EIAV infection (0.9% ± 0.3% infected cells) and knockdown of TRIM5α in these cells restored transduction, confirming that restriction is mediated by TRIM5α. Human DCs, on the other hand, were efficiently transduced with EIAV (28.7% ± 3.8%), pointing to absence of restriction. Infection in the presence of tenofovir (TDF), which blocks both HIV-1 and EIAV reverse transcription (Figure S1C), reduced infection to levels comparable to uninfected samples (0.5% and 1.6% for PBLs and DCs, respectively, Figure 1B).
Similarly, African green monkeys (AGM, sabaeus species) PBLs were found to be non-permissive to HIV-1 transduction with <1% GFP-positive PBLs and weakly permissive to SIVmac, with ∼8% transduction. In contrast, >30% Chlorocebus sabaeus DCs were efficiently transduced by HIV-1 and ∼20% by SIVmac, indicating an absence of efficient TRIM5α restriction in AGM DCs (Figure 1C).
Since restriction by TRIM5α is saturable, the addition of sufficient quantities of virus-like particles overcomes its activity. To ensure that the transduction of DCs did not saturate TRIM5α activity, we performed dose-response studies. PBLs and DCs from cynomolgus macaques (CM) (Macaca fascicularis), whose TRIM5α restricts HIV-1 but not SIVmac, were infected with increasing MOI from 0.5 to 25. Cells from pigtailed macaques (PTM) (Macaca nemestrina), which, in the place of TRIM5α, express a TRIMCyp fusion protein that cannot bind to or restrict HIV-1 (Brennan et al., 2008, Liao et al., 2007, Newman et al., 2008, Virgen et al., 2008), and cells from human donors were included as controls. PBLs from all three species were permissive to SIVmac transduction (Figure 1D). However, as expected, CM PBLs were poorly permissive to HIV-1 transduction compared with PTM and human PBLs, even at high MOI, indicating efficient restriction of HIV-1. In contrast, CM DCs were as permissive to HIV-1 infection as PTM DCs and more permissive than human DCs, even at a MOI as low as 0.5 (Figure 1D).
Thus, our results demonstrate a lack of efficient TRIM5α-mediated restriction in DCs from rhesus, cynomolgus, and pigtailed macaques, in humans, and in African green monkeys, suggesting that this phenomenon is a widespread trait of primate DCs. Infection of DCs was dependent on efficient reverse transcription and was also observed at low MOI, indicating that it was not a result of pseudotransduction (non-specific uptake of GFP protein) or saturation of the TRIM5α restriction factor.
Endogenous TRIM5α Localizes to Nuclear Bodies in DCs
TRIM5α proteins from humans and RM have been reported to shuttle between the cytoplasm and the nucleus (Diaz-Griffero et al., 2011). We therefore hypothesized that TRIM5α might be mislocalized in DCs and thus unable to interact with incoming cytoplasmic retroviral cores. To test this hypothesis, we used a rabbit polyclonal antibody raised against a C-terminal fragment of TRIM5α (Zhang et al., 2008) to detect endogenous TRIM5α. Reactivity against rhesus TRIM5α (TRIM5αrh) was previously established by western blotting (Zhang et al., 2008), and we confirmed that it also recognizes endogenous human TRIM5α (TRIM5αhu) (Figure S2A). Similarly to commercial antibodies, the rabbit TRIM5α antibody used for this study recognizes higher molecular weight bands that likely correspond to TRIM5α multimers (Li et al., 2011, Nepveu-Traversy et al., 2009). Specific TRIM5α knockdown was associated with a marked decrease in the TRIM5α signal detected by immunofluorescence (Figure S2B). Conversely, transduction of MDTF cells that do not encode a TRIM5 ortholog (reviewed in Lee and KewalRamani, 2004) with TRIM5αrh-HA or TRIM5αhu-HA led to positive TRIM5α labeling in HA-positive cells, while negative cells showed minimal background staining (Figure S2C).
PBLs and DCs from human and RM donors were labeled with the TRIM5α-specific antibody to assess endogenous localization in these cells (Figure 2A). Whereas TRIM5α localized mainly in the cytoplasm of PBLs, it aggregated within the nucleus of DC in structures reminiscent of NB, a phenotype that was not observed in PBLs. Although some cytoplasmic signal was also detected in DCs, the majority co-localized with the Golgi marker GM-130 (Figure 2B). Golgi-localized TRIM5α, which may correspond to a step in TRIM5α proteasome-independent degradation (Sastri and Campbell, 2011), was also observed in PBLs. In contrast, dense nuclear aggregates were never observed in PBLs. Whole-cell extracts revealed no difference in total levels of TRIM5α between PBLs and DCs, but subcellular fractionation confirmed the differences in subcellular localization observed by microscopy (Figure 2C). Given that TRIM5α restriction is operational in PBLs but not DCs, we hypothesized that the accumulation of TRIM5α in structures resembling NB in DCs might account for its lack of antiretroviral activity.
Figure 2.

Endogenous TRIM5α Is Cytoplasmic in PBLs and Forms Nuclear Bodies in Human and RM DCs
(A) Endogenous TRIM5α localization in human and RM PBLs and DCs. Insets show higher magnification images for each sample. Scale bars represent 10 μm.
(B) TRIM5α (green) and Golgi marker GM-130 (red) co-localization in human PBLs and DCs. Scale bar represents 5 μm.
(C) TRIM5α protein levels from whole cell extracts (RM and human) and following fractionation (human). Left-hand graph shows quantification from six different samples (ImageJ).
See also Figure S2.
DC Nuclear TRIM5α Aggregates in Cajal Bodies but Not in PML-NB
Previous work showed that exogenously expressed TRIM5α in HeLa cells can be induced to co-localize with PML-NB by blocking CRM1-dependent protein nuclear export with leptomycin B treatment (LMB) (Diaz-Griffero et al., 2011). We therefore tested whether nuclear TRIM5α aggregates co-localize with PML-NB. In PBLs, TRIM5α is mainly cytoplasmic and no co-localization with PML-NB was detected (Figure 3A). In DCs, TRIM5α NB and PML-NB were both readily observed, but the signals did not overlap (Manders’ coefficient of co-localization M < 0.1). Treatment with IFNα, previously shown to enhance TRIM5α and PML expression (Carthagena et al., 2008, Sakuma et al., 2007), increased the number and size of PML-NB, as previously described (Chelbi-Alix et al., 1995), but did not induce co-localization between TRIM5α and PML-NB. LMB treatment, shown to trap TRIM5α in the nucleus (Diaz-Griffero et al., 2011), induced diffuse nuclear accumulation of TRIM5α in PBLs, but not co-localization with PML-NB.
Figure 3.

Endogenous TRIM5α Co-localizes with Cajal Bodies in Human and RM DCs
(A and B) Co-localization of TRIM5α with (A) PML-NB and (B) the Cajal body marker Coilin in human PBLs and DCs ± 100 U IFNα for 24 hr, 5 or 50 μg/ml leptomycin B (LMB) for 4 hr. Scale bars represent 5 μm. Co-localization was assessed using the Image J plugin Just Another Colocalization Plugin (JACoP) with threshold set at 20 for all images in both fluorescence channels. Graph shows Manders’ coefficients for the fraction of PML-NB overlapping with TRIM5α, mean of three independent experiments ± SEM. In total, a minimum of ∼50 cells were analyzed for each condition.
(C) Proximity between TRIM5α and coilin was assessed by Duolink proximity ligation assay (red dots). Graph shows the average number of spots over the number of Hoechst-stained nuclei in eight random fields per cell type ± SEM (Photoshop CS5).
Scale bars represent 5 μm. Statistical significance was assessed by non-parametric unpaired t test (∗∗∗p ≤ 0.001; ∗p ≤ 0.05; ns, non-significant).
Next, we tested for TRIM5α co-localization with another nuclear body, Cajal bodies, using the marker p80 coilin to identify these subnuclear structures (Figure 3B). In DCs, TRIM5α NB were found to partially co-localize with coilin (M ∼0.3), indicating that in DCs ∼30% of TRIM5α might be trapped in Cajal bodies. This was observed both in human and RM DCs and was not further induced by LMB treatment (Figure 3B). To confirm the interaction of endogenous TRIM5α with Cajal bodies, we performed a Duolink in situ proximity ligation assay (Figure 3C). In PBLs, signal detection was low (0.27 ± 0.06 spots/cell) indicating a lack of proximity between TRIM5α and coilin. In DCs, however, signals of proximity were about seven times more frequent (1.86 ± 0.35 spots/cell). Given that the number of Cajal bodies detected in DCs is one to five per nucleus, this result is concordant with the co-localization of TRIM5α with Cajal bodies.
Mislocalization of TRIM5α in DCs Accounts for Lack of Restriction
We previously showed that overexpression of TRIM5αrh in RM DCs restores efficient restriction of HIV-1 infection (Arhel et al., 2008). We therefore tested the effect of TRIM5α overexpression on its localization. Human DCs were either labeled with TRIM5α antibody to reveal endogenous protein, or transduced with a lentiviral vector coding for mRFP-TRIM5αhu fusion protein. While endogenous TRIM5α formed nuclear aggregates in DCs, its overexpression resulted in its accumulation as dense cytoplasmic aggregates, characteristic of cytoplasmic bodies as previously reported (Stremlau et al., 2004) (Figure 4A). Control or TRIM5α-overexpressing human DCs were challenged with EIAV and infection was assessed by flow cytometry at 48 hr post-infection (hpi) (Figure 4B). Forced cytoplasmic expression of TRIM5α by overexpression restored restriction against EIAV in human DCs, suggesting that TRIM5α nuclear localization in DCs accounts for the lack of efficient restriction. To confirm this, TRIM5αrh was fused at its N terminus to the Cajal body targeting domain (CBD) of human coilin (Figure 4C). P4-CCR5 cells were transduced with TRIP-TRIM5αrh or TRIP-Coilin-TRIM5αrh and challenged with HIV-1. Infection was efficiently restricted by TRIM5α but forced nuclear targeting abrogated restriction, confirming that mislocalization of TRIM5α accounts for the lack of restriction.
Figure 4.

SUMOylation-Dependent Mislocalization of TRIM5α in DCs Accounts for Lack of Restriction
(A) Human DCs were labeled with anti-TRIM5α antibody (above), or transduced with mRFP-TRIM5αhu for 48 hr (below). Two representative confocal images are shown for each condition. Scale bar represents 10 μm.
(B) Human DCs were transduced or not with mRFP-TRIM5αhu for 2 days and infected with EIAV-eGFP vector at MOI 1. Representative flow cytometry charts at 3 dpt are shown left, while the graph shows the mean of three independent experiments ± SEM.
(C) RM TRIM5α was fused in frame at its amino terminus with the first 112 aa of human coilin, comprising the Cajal body targeting domain (CBD) and a NLS. P4-CCR5 cells transduced with TRIP-TRIM5αrh, TRIP-Coilin-TRIM5αrh, or untransduced (NT) were labeled for TRIM5α for immunofluorescence detection and challenged with HIV-1 LAI with either wild-type envelope or VSV-G. β-galactosidase activity at 48 hpi is shown normalized by Bradford as the mean of two independent experiments carried out in triplicate ± SD.
(D) Co-localization of TRIM5α and Coilin was measured as in Figure 3B in human PBLs and DCs ± 10 or 100 μM GA for 24 hr. Graph shows Manders’ coefficients for the fraction of coilin-labeled Cajal bodies overlapping with TRIM5α, mean of three independent experiments ± SEM with a minimum of ∼50 cells analyzed for each condition. Statistical significance was assessed by unpaired t test (∗∗∗p ≤ 0.001 ∗∗p ≤ 0.01 ∗p ≤ 0.05; ns, non-significant). White arrows point to areas of co-localization.
(E) Human DCs were treated or not with 100 μM GA for 24 hr and infected with EIAV-eGFP vector at MOI 1. Representative flow cytometry charts at 3 dpt are shown left, while the graph shows the mean of three independent experiments ± SEM.
(F) Human DCs were lipofected or not with two different siRNAs targeting human TRIM5α (HM1 and HM3) or control siLuc (day 0) ± 100 μM GA (day 1) and EIAV-eGFP at MOI 1 (day 2). TRIM5α mRNA expression on day 3 is normalized for housekeeping transcript RPL13A (60S ribosomal protein L13a) with siLuc arbitrarily set to 1. GFP expression on day 4 is the mean of two experiments ± SD.
TRIM5α Localizes to Cajal Bodies in a SUMOylation-Dependent Manner in DCs
Recent work showed that human and RM TRIM5α are substrates for SUMO modification (Dutrieux et al., 2015), and that putative SIM domains in TRIM5α may mediate interaction with SUMO-modified proteins (Arriagada et al., 2011). SUMO modification has been shown to modulate sub-nuclear localization and function of TRIM19/PML (Müller et al., 1998). We therefore examined whether SUMO modification might account for TRIM5α accumulation in NB of DCs. Human PBLs and DCs were treated with 10 and 100 μM GA for 24 hr and labeled for endogenous TRIM5α and either PML or coilin. GA treatment did not induce TRIM5α co-localization with PML in either cell type (Figure 4D). Similarly, no co-localization between TRIM5α and coilin was detected in PBLs and this was not altered following GA treatment. In DCs, however, GA treatment led to a dose-dependent reduction of TRIM5α co-localization with coilin, without reducing the number of Cajal bodies per cell. Pre-treatment with 10 μM GA led to a 5-fold decrease in TRIM5α/coilin co-localization, and co-localization was abolished following treatment with 100 μM GA. Given the obtained Manders’ coefficients, we estimate that approximately one-third of nuclear TRIM5α is found associated with Cajal bodies in DCs and that this is dependent on SUMO modification. Localization of TRIM5α in Golgi was not sensitive to GA treatment in either DCs or PBLs, indicating that it is unlikely to contribute to the SUMOylation-dependent functional inactivation of TRIM5α in DCs.
Given that inhibition of SUMOylation counteracts TRIM5α nuclear localization in DCs, we next tested whether inhibition of SUMOylation restored TRIM5α restriction in DCs. Human DCs were treated with 100 μM GA for 24 hr and infected with EIAV. Untreated human DCs were permissive to EIAV, whereas GA treatment restored efficient restriction, leading to a 3- to 8-fold reduction in infectivity in three independent experiments (Figure 4E), suggesting that SUMOylation-dependent nuclear sequestration of TRIM5α accounts for the observed lack of restriction in DCs. To determine whether GA-induced restriction is TRIM5α-dependent, we performed GA treatment in the presence of siRNAs targeting TRIM5α. Twenty-four hours prior to GA treatment, human DCs were DOTAP transfected with two different siRNAs targeting either TRIM5α or Luc and then transduced with EIAV (Figure 4F). GA treatment induced strong anti-EIAV restriction in DCs, and this phenotype was reversed by TRIM5α knockdown, indicating that GA-induced restriction in DCs is TRIM5α-dependent. Further exploration is required to confirm this phenotype using replicative virus, however, no replicative EIAV was available at the time of the study. Together, our results show that the lack of restriction in DCs is explained by the SUMOylation-dependent re-localization of TRIM5α to the nucleus and to Cajal bodies, thereby preventing contact with incoming retroviral capsids.
Nuclear-Sequestered TRIM5α Is DeSUMOylated
SUMO modification of human and RM TRIM5α was demonstrated using immobilized metal affinity chromatography on extracts prepared from HeLa cells overexpressing HA-tagged TRIM5α, Ubc9, and 6 × His-SUMO1 (Dutrieux et al., 2015). To assess SUMO modification of TRIM5α in primary DCs, we were limited by the fact that TRIM5α overexpression cannot be used, since it reverses observed phenotypes, and endogenous TRIM5α is present in quantities that are too small for pull-down assays to be effective. We therefore analyzed post-translational modifications of endogenous TRIM5α by western blotting. Preparation of PBLs and DCs lysates in the presence of iodoacetamide (IAA), a cysteine protease inhibitor that preserves post-translational modifications such as SUMOylation during the preparation of cell extracts, led to an increase in the molecular weight of the detected TRIM5α band by ∼15 kDa, which is suggestive of a mono-SUMO modification of TRIM5α in both PBLs and DCs (Figure 5A). Quantification revealed that the proportion of TRIM5α-70 relative to TRIM5α-55 was greater in PBLs than DCs, and higher molecular weight bands were also visible in PBL extracts prepared in the presence of IAA, but less so in DCs (Figure 5A). These findings suggest that TRIM5α may be more SUMO1-modified in PBLs than in DCs. To confirm that higher molecular weight bands correspond to SUMOylated TRIM5α, IAA extracts were prepared from DCs treated or not with GA. GA treatment reduced overall protein SUMOylation, as observed by reduced smearing of SUMO2/3 labeling (Figure S3A) and decreased the detection of the higher molecular TRIM5α weight bands (Figure S3B), confirming that these represent SUMO-modified forms of TRIM5α.
Figure 5.

TRIM5α deSUMOylation in DCs Nucleus Contributes to Its Nuclear Sequestration
(A) Human DCs extracts were prepared in the presence or absence of IAA (10 mM). Arrows point to TRIM5α (TRIM5α-55) and post-translationally modified TRIM5α (TRIM5α-70). The graph shows TRIM5α-70 normalized band intensity as percentage of total (TRIM5α-55 and TRIM5α-70) for three independent experiments ± SD (ImageJ).
(B) TRIM5α and SUMO1 co-localization was assessed by Duolink proximity assay (red dots) in PBLs and DCs ± 10 or 100 μM GA for 24 hr. Scale bars represent 5 μm. Graph shows the average number of spots over the number of Hoechst-stained nuclei in eight random fields per cell type ± SEM (Photoshop CS6). Statistical significance was assessed by non-parametric unpaired t test (∗∗∗p ≤ 0.001; ∗∗p ≤ 0.01; ∗p ≤ 0.05; ns, non-significant).
(C) Lack of TRIM5α (green) and SUMO-1 (red) co-localization in human DC nuclei.
(D) Cytoplasmic and nuclear fractions from human PBLs or DCs were prepared in the presence of IAA and loaded as equal cell equivalents. Tubulin and lamin A/C control for the purity of cytoplasmic and nuclear fractions only and should not be considered as loading control since PBLs and DCs differ substantially in tubulin and lamin A/C levels per cell.
See also Figure S3.
To investigate the compartmentalization of TRIM5α SUMOylation, we performed Duolink in situ proximity ligation assays (PLA), using TRIM5α and SUMO1 primary antibodies, followed by species-specific PLA probes. The results revealed close proximity of TRIM5α with SUMO with an average of 7.1 ± 1.2 (SEM) spots per cell in PBLs, compared with 2.2 ± 0.4 spots per cell in DCs, pointing to reduced TRIM5α-SUMO interactions in DCs (Figure 5B). Treatment with ginkgolic acid (GA), a small molecule inhibitor of SUMO modification that blocks formation of the E1-SUMO intermediate (Fukuda et al., 2009), induced a dose-dependent reduction in TRIM5α-SUMO1 proximity signals, suggesting that endogenous TRIM5α is conjugated by SUMO1 or interacts with SUMOylated proteins in PBLs and DCs. TRIM5α-SUMO1 signals were observed both in the cytoplasm and nucleus, indicating that SUMO-modified TRIM5α can shuttle in and out of the nucleus. However, nuclear signals were rare in DCs (0.8 ± 0.1 spots per nucleus versus 4.8 ± 1.4 in PBLs), suggesting that nuclear-localized TRIM5α no longer associates with SUMO1 in DCs. To test this, we co-labeled TRIM5α and SUMO1 in human DCs (Figure 5C). Both SUMO1 and TRIM5α strongly localized to the nucleus, but the two did not co-localize, indicating that nuclear TRIM5α does not associate with SUMO1 in DCs.
To confirm these observations, we analyzed TRIM5α post-translational modifications in both nuclear and cytoplasmic fractions by western blotting. Cytoplasmic and nuclear fractions from human PBLs or DCs were prepared in the presence of IAA and loaded on gels such that for each subcellular fraction, the lane contained lysate from equal cell equivalents (4 × 106 cells), rather than equal protein amounts (Figure 5D). High molecular weight bands were observed in PBLs and DCs cytoplasmic fractions, suggesting that TRIM5α is SUMOylated in both cell types, although imaging analysis revealed that cytoplasmic TRIM5α is cytosolic in PBLs but Golgi-associated in DCs (Figure 2B). Total nuclear TRIM5α was diminished in PBLs, as in Figure 2. In DCs, nuclear TRIM5α detection was strong, but higher molecular weight bands were consistently reduced, indicating that nuclear-localized TRIM5α undergoes deSUMOylation.
Nuclear Sequestration of TRIM5α Allows Innate Sensing of Retroviral Infection by cGAS
Innate sensing of retroviral infections by DCs is crucial for the establishment of an efficient immune response required for infection control. To gain insight into the relevance of this TRIM5α regulation mechanism, we asked whether inefficient restriction in DCs might correlate with efficient innate immune sensing.
To address this question, we induced cytoplasmic localization of TRIM5α by overexpression of human TRIM5α in human DCs and assessed innate sensing of retroviral infections by measuring cytokine expression and production following infection with viruses that are either restricted by human TRIM5α (N-MLV and EIAV) or not restricted (HIV-1). Human DCs were transfected with the expression plasmid pLPCX-TRIM5α-HA or pLPCX (empty vector [EV]). Transfection was preferred over lentiviral transduction to avoid high experimental background noise due to sensing of the lentiviral vector. After 24 hr, DCs were treated with 100 μM GA to inhibit SUMOylation, 100 μM TDF to block reverse transcription, or left untreated. After a further 24 hr, DCs were transduced with HIV-1, N-MLV, or EIAV eGFP vectors at MOI 5. DCs were harvested at 10 hpi to prepare DNA extracts and to perform qPCR analysis of reverse transcription products and at 24 hpi to prepare RNA extracts for qRT-PCR quantification of TRIM5α, IFNα1, IFNβ, IL6, and housekeeping transcripts. qPCR analysis of TRIM5α transcripts confirmed an ∼40-fold increase in TRIM5α mRNA following overexpression in DCs (Figure 6A). Moreover, quantification of reverse transcription products revealed that all retroviruses underwent reverse transcription and this was specifically blocked by the reverse transcriptase inhibitor TDF or by heat-inactivation of vectors prior to infection (Figure 6B). Overexpression of human TRIM5α led to a 70%–80% reduction in reverse transcription products of N-MLV and EIAV, but had only limited impact on HIV-1 reverse transcription, confirming that overexpression of TRIM5α in DCs restores efficient TRIM5α-mediated restriction of susceptible viruses (Figures 5B and 6B). Similarly, GA treatment, which induces forced re-localization of TRIM5α to the cytoplasm by blocking its SUMOylation-dependent targeting to Cajal bodies (Figure 4C), also reduced reverse transcription of N-MLV and EIAV without affecting HIV-1 (Figure 6B).
Figure 6.

Forced Altered Localization of TRIM5α by Overexpression of TRIM5α or Inhibition of SUMOylation Reduces Type I IFN Induction following Retroviral Infection
Human DCs were DOTAP transfected with pLPCX TRIM5α-HA (T5) or empty pLPCX (EV) at 1 μg DNA/0.1 × 106 DCs, or left untransfected (day 0) ± 100 μM GA or 100 μM TDF (day 1) and infected with indicated retroviruses at MOI 5 (day 2).
(A) TRIM5α mRNA expression normalized for housekeeping transcript RPL13A (60S ribosomal protein L13a). NI EV cells were arbitrarily set to 1.
(B) Infection efficiency was assessed by qPCR of eGFP/eYFP DNA in DCs extracts at 10 hpi. eGFP/eYFP signal was normalized for housekeeping gene GAPDH. NT or EV were arbitrarily set to 100% for each infection. Signal was undetectable for non-infected samples.
(C–E) IFNα1 (C), IFNβ (D), and IL6 (E) mRNA expression levels were assessed by reverse transcription qPCR at 24 hpi, normalized for housekeeping transcript β2M. NI NT cells were arbitrarily set to 1. All panels are representative of at least three independent experiments on different donors.
See also Figure S4.
To assess sensing of retroviral infection by DCs, we measured cytokine expression following infection. IFNα1 and IFNβ were induced 5- to 20-fold by HIV-1, N-MLV, and EIAV infection (Figures 6C and 6D), indicating that these viruses are sensed in human DCs, as previously shown for HIV-1 (Silvin and Manel, 2015, van Montfoort et al., 2014). In contrast, IL6 expression was not significantly induced, suggesting that the host sensor involved likely operates via IRF-3 and not NF-κB (Figure 6E). Infection in the presence of TDF abolished sensing (Figures 6C and 6D), at both 24 hpi and 8 hpi, indicating that the recognized PAMP is reverse transcribed DNA, as previously shown (Gao et al., 2013, Lahaye et al., 2013, Rasaiyaah et al., 2013, Yoh et al., 2015). Overexpression of human TRIM5α in the cytoplasm strongly reduced IFNα and IFNβ expression (5- to 10-fold), but not IL6 levels, following N-MLV and EIAV infection, pointing to inefficient sensing following forced altered localization of restrictive TRIM5α to the cytoplasm (Figures 6C–6E). In contrast, HIV-1 sensing was not diminished by human TRIM5α overexpression, which is concordant with the fact that TRIM5α overexpression did not reduce HIV-1 reverse transcripts (Figure 6B). The defect in type I IFN production was confirmed by measuring levels of type I IFN released in supernatants from infected DCs at 48 hr post-infection using HL116 indicator cells (Figure S3C).
Similarly, the inhibition of SUMOylation by GA treatment of DCs led to a stark reduction in IFNα and IFNβ transcripts (5- to 10-fold), but not IL6, confirming that the restoration of TRIM5α restriction in DCs by blocking the SUMO machinery reduces efficient sensing of retroviral infection (Figures 6C–6E). Combining GA treatment with TRIM5α overexpression generally further accentuated the block in type I IFN expression following N-MLV or EIAV infection. On the other hand, GA treatment did not affect sensing of HIV-1, indicating that non-specific effects of GA treatment other than TRIM5α regulation are unlikely.
TRIM5α has been reported to contribute to innate immune sensing of HIV-1 infection (Pertel et al., 2011). To determine whether endogenous TRIM5α contributes to viral sensing by DCs, we measured IFNα1, IFNβ, and IL6 expression following infection by HIV-1, N-MLV, EIAV in DCs transfected or not with TRIM5α-specific siRNAs (Figure S4). HIV-1, N-MLV, and EIAV all induced type I IFN production as already shown in Figure 6, and knockdown of TRIM5α had no effect on IFNα1, IFNβ, or IL6 expression following infection, indicating that endogenous TRIM5α does not contribute to viral sensing by DCs. This is concordant with our results showing that TRIM5α is sequestered in the nucleus away from incoming retroviruses. Several groups have shown that the pathogen recognition receptor (PRR) for reverse transcribed DNA during retroviral infection is cGAS (Gao et al., 2013, Lahaye et al., 2013, Rasaiyaah et al., 2013, Yoh et al., 2015). To assess whether cGAS is responsible for innate sensing of retroviral infection in human DCs, we transduced DCs with short hairpin RNAs (shRNAs) targeting either cGAS or, as a control, LacZ and monitored CD86 induction by infection. cGAS knockdown prevented induction of CD86 expression by EIAV in DCs, indicating that cGAS is a sensor of EIAV infection (Figure 6F).
Together, these results suggest that DCs cannot both efficiently restrict and efficiently sense an incoming retrovirus. In wild-type DCs, where TRIM5α is not present in the cytoplasm to intercept incoming viruses, retroviruses are not restricted, and the reverse transcribed provirus is efficiently detected by a host sensor that triggers type I IFN production. Conversely, if TRIM5α is forced to the cytoplasm by overexpression or inhibition of the SUMO pathway, retroviruses are restricted but are no longer sensed, since restriction inhibits viral DNA synthesis and all downstream replication events.
Discussion
In this study, we investigated the mechanism responsible for lack of TRIM5α-mediated restriction in human and non-human primate DCs. DCs from rhesus and cynomolgus macaques, African green monkeys, and humans were all found to be permissive to retroviruses that are normally restricted by TRIM5α from these species. Although TRIM5α in lymphocytes is mainly cytoplasmic, it accumulates within NB that partly co-localize with Cajal bodies in rhesus and human DCs. Nuclear sequestration of TRIM5α in DCs was SUMOylation-dependent and correlated with the lack of retroviral restriction. Forced cytoplasmic localization of TRIM5α, either through TRIM5α overexpression or the inhibition of SUMOylation, restored efficient restriction in DCs but abolished sensing of the reverse transcribed viral genome by cGAS. Inefficient restriction in DCs may paradoxically promote effective immune control if it results in effective sensing. Thus, the absence of efficient TRIM5α restriction in DCs might have evolved to ensure appropriate sensing of retroviral infections by host cell sensors.
Our results indicate that TRIM5α can be SUMO modified and undergo nucleo-cytoplasmic shuttling in both PBLs and DCs, but only in DCs does SUMO modification of TRIM5α lead to Cajal body targeting and nuclear sequestration (Figure 7). It is not known how targeting of TRIM5α to Cajal bodies is mediated. Although TRIM5α can be SUMO modified, we cannot exclude that an interaction between TRIM5α SIM domains with a SUMOylated shuttling protein is also involved. Interestingly, the Cajal body marker coilin is both directly SUMOylated and contains putative SIMs and could therefore mediate TRIM5α nuclear targeting. In PBLs, endogenous TRIM5α remains essentially cytoplasmic despite shuttling, and forced accumulation in the nucleus by LMB treatment leads only to nucleoplasmic localization. Further work on differences in Cajal body biology comparing PBLs and DCs may shed light on the differential nuclear targeting of TRIM5α in these cells. Cajal bodies participate in the biogenesis of spliceosomal small nuclear (snRNP) and small nucleolar RNP (snoRNP) (Cioce and Lamond, 2005, Machyna et al., 2013, Morris, 2008), and it is unclear why TRIM5α localizes to Cajal bodies in DCs. One possible indication comes from the observation that TRIM5α was less SUMO-modified and SUMO-associated in the nuclei of DCs, suggesting its targeting to Cajal bodies may lead to its deSUMOylation and consequent nuclear sequestration. Intriguingly, a novel SUMO isopeptidase ubiquitin-specific protease like 1 (USPL1) was recently identified in Cajal bodies (Schulz et al., 2012). It is also noteworthy that only approximately one-third of nuclear TRIM5α co-localized with the Cajal body marker coilin at any one time. Although this may be explained by the dynamic and transient nature of Cajal bodies or by the fact that some Cajal bodies are coilin-negative (Cioce and Lamond, 2005, Machyna et al., 2013, Morris, 2008), it is also possible that following SUMO-dependent targeting of TRIM5α to Cajal bodies, deSUMOylated TRIM5α leaves Cajal bodies and remains in the nucleoplasm. Further work will be required to specifically test the involvement of E3 SUMO ligases and SUMO isopeptidases in TRIM5α SUMO modification. TRIM5α regulation by SUMOylation is likely to be a finely-tuned mechanism since overexpression of TRIM5α led to strong accumulation of TRIM5α within cytoplasmic bodies but negligible nuclear localization, suggesting that the cellular SUMOylation machinery had been overridden.
Figure 7.

Schematic Diagram Showing the Effect of SUMOylation on the Localization of TRIM5α and Its Ability to Restrict Retroviral Infections in Primary Lymphocytes and Dendritic Cells
When SUMOylated, TRIM5α shuttles freely between the cytoplasm and the nucleus, although in lymphocytes its nuclear residency is brief and may only be seen upon LMB treatment. Upon retroviral infection of lymphocytes, cytoplasmic TRIM5α intercepts incoming capsids and induces their degradation. In DCs, SUMOylated TRIM5α is targeted to Cajal bodies where it likely undergoes deSUMOylation by isopeptidases such as USPL1. DeSUMOylated TRIM5α remains trapped in the nucleus and accumulates within Cajal bodies and the nucleoplasm. Retroviral infection of DCs can proceed unhampered in the absence of cytoplasmic TRIM5α, and reverse transcribed retroviral genomes are sensed by the cytosolic sensor cGAS, which signals interferon type I production. Overexpression of TRIM5α, which likely overwhelms the SUMOylation machinery, or GA treatment, which prevents TRIM5α SUMOylation, both prevent import of de novo synthesized TRIM5α to the nucleus, restore TRIM5α-mediated restriction and abrogate sensing by cGAS.
In DCs, nuclear sequestration of TRIM5α was associated with efficient innate sensing of retroviral infection resulting in substantial type I interferon production. Conversely, induced cytoplasmic expression of TRIM5α in DCs restored restriction but impaired viral sensing, suggesting that restriction of incoming retroviral complexes by cytoplasmic TRIM5α prevents the detection of one or more key viral PAMPs. Retroviral infection of DCs may be detected by a number of cell host sensors at different stages of the viral cycle: RIG-I-like receptors (RLR), toll-like receptors (TLR), and DNA sensors such as IFI16 and cGAS (Cerboni et al., 2013, Luban, 2012, Melchjorsen, 2013, van Montfoort et al., 2014). Retroviral capsid recognition by TRIM5α can signal production of pro-inflammatory cytokines (Pertel et al., 2011, Uchil et al., 2013). However, type I IFN production was by far the more potent signaling outcome of retroviral sensing in DCs. The use of the reverse transcriptase inhibitor TDF abolished type I IFN production following infection, indicating that the retroviral PAMP that is recognized in infected DCs is reverse transcribed DNA and is unlikely to be capsid. Several groups have shown that the PRR for reverse transcribed DNA during retroviral infection is cGAS (Gao et al., 2013, Lahaye et al., 2013, Rasaiyaah et al., 2013, Yoh et al., 2015). Our work confirms that cGAS is the PRR that signals sensing in human DCs infected by N-MLV or EIAV when TRIM5α is sequestered in the nucleus (Figure 7).
Since TRIM5α blocks reverse transcription (Stremlau et al., 2004, Stremlau et al., 2006), efficient restriction in DCs would block innate detection of PAMPs generated by incoming retroviruses downstream of reverse transcription, including reverse transcribed viral DNA, RNA transcripts, and de novo synthesized Gag. In addition, efficient restriction would be likely to prevent integration and presentation of endogenous viral peptides on MHC-I molecules for CD8+ T cell priming. Both innate sensing and triggering of adaptive response of retroviral infection by DCs are crucial for an efficient immune response and the control of infection. Given that all tested DCs lacked efficient TRIM5α restriction, we speculate that absence of TRIM5α restriction might have evolved in DCs frequently exposed to retroviral infections in order to ensure both efficient innate detection of incoming retroviruses by host cell sensors and presentation of viral epitopes to cytotoxic T cells. Conversely, preservation of efficient restriction of incoming retroviruses in lymphocytes, which we confirmed in all tested human and non-human primates, makes an important contribution to the defense of this cell population, which is a key target of many retroviruses (Hatziioannou et al., 2009, Soll et al., 2013).
Of note, the phenotypes described in this study for endogenous TRIM5α are entirely abrogated by the expression of exogenous TRIM5α. Overexpression following standard transduction protocols led to a dramatic ∼40-fold increase in TRIM5α mRNA, underlining the non-physiological nature of TRIM5α overexpression. The dichotomy between results obtained with endogenous TRIM5α and overexpressed protein have been described previously (Zhang et al., 2008), confirming that caution must be exercised when interpreting data based on overexpressed TRIM5α protein. In this regard, the seeming discordance between our study and previous work showing potent NF-κB-dependent transcription following the sensing of retroviral capsid by TRIM5α (Pertel et al., 2011) may be explained by the fact that the regulation of TRIM5α function by SUMOylation that we describe here was observed only with endogenous TRIM5α protein and only in primary DCs.
In conclusion, our study reveals that the ability of TRIM5α to restrict incoming retroviruses in human and non-human primates is lost in DCs as a result of its SUMOylation-dependent sequestration in nuclear Cajal bodies. This lack of restriction allows efficient innate sensing of incoming retroviruses by DC host cell sensors and likely contributes to improved viral control in the infected host. This evolutionary trade-off specific to DCs underlines the multi-valence of DC functions at the crossroads between innate and adaptive immunity, and the ability of DCs to fine-tune their antigen-presenting and sensing functions to best confront recurrent viral exposures.
Experimental Procedures
Blood Sampling
Young (3–4 years old) male cynomolgus macaques (Macaca fascicularis) imported from Mauritius, 6- to 10-year-old pigtailed macaques (Macaca nemestrina) originating from the Palmyre Zoo (Les Mathes, France), and 3- to 4-year-old African green monkeys (Chlorocebus sabaeus) were housed at the Institut Pasteur non-human primates facility. Rhesus macaques (Macaca mulatta) were housed at the CEA (Fontenay-aux-Roses). Monkeys were tested negative for SIV, simian herpes B, filovirus, STLV-1, SRV-1, SRV-2, measles virus, hepatitis B-HbsAg, and hepatitis B-HBcAb before inclusion in the study. Blood sampling was conducted in accordance with guidelines established by the French and European regulations for the care and use of laboratory animals (Animal Welfare Assurance Number: A5476-01). Blood from the saphenous vein was collected using heparin tubes or BD Vacutainer CPT cell preparation tube with sodium heparin. Blood volume and bleeding frequency were defined as a function of animal weight according to the GIRCOR recommendations. Human buffy coats and cytopheresis rings were obtained from the Etablissement de Sang Français (EFS Rungis and Hôpital Saint-Louis).
Cells
PBMC were isolated using Ficoll (100% and 95% for human and monkey blood, respectively) (Biochrom). Monocytes were isolated from PBMC using adherence on plastic for 45 min. DCs were derived from monocytes following 4- to 5-day differentiation using 50 ng/ml GM-CSF (Immunotools) and 20 ng/ml recombinant human of RM IL4 (Immunotools). PBLs were activated by treatment with phytohaemagglutinin (PHA, 10 μg/ml) for 2–3 days and concanavalin A (Con A, 5 μg/ml) (Sigma) for human and non-human primate samples, respectively, and induced to proliferate by treatment with human recombinant IL2 (10 ng/ml) (Sigma). The differentiation and activation phenotypes of cells were systematically checked by phenotypic markers using flow cytometry (Figure S1A). The human fibrosarcoma HL116 cell line carries the luciferase gene under control of the IFN-inducible 6-16 promoter (Uzé et al., 1994). HL116 cells were grown in medium supplemented with HAT (hypoxanthine: 20 μg/ml, aminopterin: 0.2 μg/ml, thymidine: 20 μg/ml). P4-CCR5 are HeLa-based cells that express CD4 and CCR5 and have Tat-inducible β-galactosidase activity.
Plasmids, Vectors, Viruses
Lentiviral and retroviral vectors used were HIV-1 (TRIP-CMV-GFP, Pierre Charneau, Institut Pasteur), SIVmac239 (GAE-CAG-GFP, François-Loic Cosset, ENS Lyon), EIAV-eGFP (ONY8.9, Oxford Biomedica), and N-MLV-eYFP expressing vectors (Jonathan Stoye, MRC London). MLV-based retroviral vectors pLPCX-TRIM5αrh-HA and pLPCX-TRIM5αhu-HA were provided by J. Sodroski (Stremlau et al., 2004). RM and human TRIM5α were fused in frame to the mRFP fluorophore or the first 102 aa of coilin in N-ter by strand overlap PCR. Human coilin was amplified from pENTR-COIL plasmid (Addgen #16172). Fusion constructs were cloned within the pTRIP vector within KpnI/XhoI restriction sites to generate the following constructs: pTRIP-CMV-mRFP-TRIM5αrh, pTRIP-CMV-mRFP-TRIM5αhu, pTRIP-CMV-COI-TRIM5αrh, and pTRIP-CMV-COI-TRIM5αhu. Viruses were SIVmac239, HIV-1 LAI, VSV-G pseudotyped LAI, VSV-G pseudotyped NL4-3-IRES-eGFP, and R5-tropic NL4-3-92th014.12-IRES-eGFP (Papkalla et al., 2002). Vectors and viruses were produced in HEK293T cells by calcium phosphate co-transfection of vector/virus plasmid, Gag-Pol expressing plasmid for vectors, and pVSV-G for pseudotyping. Supernatants were collected 48 hr post-transfection, and vector particles were concentrated by ultracentrifugation at 22,000 rpm for 1 hr at 4°C (Beckman SW41). Mus dunni tail fibroblast (MDTF) cells devoid of TRIM5α, APOBEC3G, and tetherin restriction factors, were used to titer vectors (Carthagena et al., 2008). Viral stocks were quantified by p24/p27 ELISA (Perkin Elmer and Zeptometrix).
Transductions, Transfections
Vector transductions of PBLs and DCs were performed at MOI 0.25–5 (TU/cell) in the presence of cytokines. eGFP or eYFP expression was assessed by flow cytometry or qPCR at 3–4 days post-transduction (dpt). As a control, vectors were heat inactivated by incubation at 56°C in a water bath for 2 hr with frequent mixing prior to transduction. Plasmid and siRNA transfections of human DCs were performed using DOTAP (Roche Molecular) at 1 μg DNA or 160 nM siRNA/0.1 × 106 DCs according to manufacturer’s instructions.
Imaging
PBLs were cytospun onto poly-L-lysine (Sigma-Aldrich)-treated coverslips for 5 min at 500 × g to optimize cell attachment and cytoplasmic spread. DCs were seeded onto collagen-treated coverslips to minimize autofluorescence. Imaging was performed using a Zen 2012 LSM700 and LSM780 confocal microscopes (Zeiss) with a Plan-Apochromat 63×/NA 1.4 objective. Duolink proximity ligation assay (PLA) was performed according to manufacturer’s instructions (Olink Biosciences).
Fractionation
Cytoplasmic and nuclear fractions from 4 × 106 PBLs or DCs were prepared using NE-PER isolation kit (Pierce). Fractions were loaded onto 10% SDS-polyacrylamide gel using equal cell equivalents rather than protein amounts. The purity of subcellular fractionations was confirmed using tubulin and lamin A/C for the cytoplasmic and nuclear fractions, respectively. Quantification of protein bands was performed using ImageJ.
Author Contributions
D.M.P., J.F., M.R., A.K.M., A.B., M.M., A.-S.B., S.N., and N.J.A. conducted the experiments. A.-S.B., M.M.T., F.K., S.N., and N.J.A. designed the experiments. All authors contributed to writing the manuscript.
Acknowledgments
This work was supported by grants from the Agence Nationale de Recherches sur le SIDA et les hépatites virales (ANRS), the ATIP-Avenir program (INSERM), the Deutsche Forschungsgemeinschaft (DFG), European FP7 “HIT HIDDEN HIV” (305762), a DFG Leibniz award, and an Advanced ERC Investigator grant (to F.K.). For veterinary support, NHP welfare, and biological specimens sampling, we thank Nathalie Dereuddre-Bosquet and Céline Gommet (CEA Fontenay-aux-Roses) for RM and CM, Thierry Petit (Zoo de la Palmyre, Les Mathes) for PTM, and Anne-Sophie Liovat for AGM. We thank Paul Bieniasz (Aaron Diamond AIDS Research Center) for the rabbit TRIM5α antibody, Nicolas Manel (Institut Curie) for the cGAS shRNA construct, Cyril Pantoli for graphic design, and Ron Hay (University of Dundee) and Allan Hance (Hôpital Saint-Louis) for helpful discussions. Protein diagrams were prepared using Illustrator for Biological Sequences. Microscopy experiments were performed at the Imagopole (Institut Pasteur) and the technology platform of Hôpital Saint-Louis.
Published: December 31, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures and four figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.12.039.
Supplemental Information
References
- Arhel N.J., Nisole S., Carthagena L., Coutant F., Souque P., Brussel A., Estaquier J., Charneau P. Lack of endogenous TRIM5alpha-mediated restriction in rhesus macaque dendritic cells. Blood. 2008;112:3772–3776. doi: 10.1182/blood-2008-04-151761. [DOI] [PubMed] [Google Scholar]
- Arriagada G., Muntean L.N., Goff S.P. SUMO-interacting motifs of human TRIM5α are important for antiviral activity. PLoS Pathog. 2011;7:e1002019. doi: 10.1371/journal.ppat.1002019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger G., Durand S., Goujon C., Nguyen X.N., Cordeil S., Darlix J.L., Cimarelli A. A simple, versatile and efficient method to genetically modify human monocyte-derived dendritic cells with HIV-1-derived lentiviral vectors. Nat. Protoc. 2011;6:806–816. doi: 10.1038/nprot.2011.327. [DOI] [PubMed] [Google Scholar]
- Besnier C., Takeuchi Y., Towers G. Restriction of lentivirus in monkeys. Proc. Natl. Acad. Sci. USA. 2002;99:11920–11925. doi: 10.1073/pnas.172384599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniasz P.D. Intrinsic immunity: a front-line defense against viral attack. Nat. Immunol. 2004;5:1109–1115. doi: 10.1038/ni1125. [DOI] [PubMed] [Google Scholar]
- Brandariz-Nuñez A., Roa A., Valle-Casuso J.C., Biris N., Ivanov D., Diaz-Griffero F. Contribution of SUMO-interacting motifs and SUMOylation to the antiretroviral properties of TRIM5α. Virology. 2013;435:463–471. doi: 10.1016/j.virol.2012.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan G., Kozyrev Y., Hu S.L. TRIMCyp expression in Old World primates Macaca nemestrina and Macaca fascicularis. Proc. Natl. Acad. Sci. USA. 2008;105:3569–3574. doi: 10.1073/pnas.0709511105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carthagena L., Parise M.C., Ringeard M., Chelbi-Alix M.K., Hazan U., Nisole S. Implication of TRIM alpha and TRIMCyp in interferon-induced anti-retroviral restriction activities. Retrovirology. 2008;5:59. doi: 10.1186/1742-4690-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerboni S., Gentili M., Manel N. Diversity of pathogen sensors in dendritic cells. Adv. Immunol. 2013;120:211–237. doi: 10.1016/B978-0-12-417028-5.00008-9. [DOI] [PubMed] [Google Scholar]
- Chelbi-Alix M.K., Pelicano L., Quignon F., Koken M.H., Venturini L., Stadler M., Pavlovic J., Degos L., de Thé H. Induction of the PML protein by interferons in normal and APL cells. Leukemia. 1995;9:2027–2033. [PubMed] [Google Scholar]
- Cioce M., Lamond A.I. Cajal bodies: a long history of discovery. Annu. Rev. Cell Dev. Biol. 2005;21:105–131. doi: 10.1146/annurev.cellbio.20.010403.103738. [DOI] [PubMed] [Google Scholar]
- Cowan S., Hatziioannou T., Cunningham T., Muesing M.A., Gottlinger H.G., Bieniasz P.D. Cellular inhibitors with Fv1-like activity restrict human and simian immunodeficiency virus tropism. Proc. Natl. Acad. Sci. USA. 2002;99:11914–11919. doi: 10.1073/pnas.162299499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Griffero F., Gallo D.E., Hope T.J., Sodroski J. Trafficking of some old world primate TRIM5α proteins through the nucleus. Retrovirology. 2011;8:38. doi: 10.1186/1742-4690-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutrieux J., Portilho D.M., Arhel N.J., Hazan U., Nisole S. TRIM5α is a SUMO substrate. Retrovirology. 2015;12:28. doi: 10.1186/s12977-015-0155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R.D., Boutell C., Hale B.G. Interplay between viruses and host sumoylation pathways. Nat. Rev. Microbiol. 2013;11:400–411. doi: 10.1038/nrmicro3015. [DOI] [PubMed] [Google Scholar]
- Fukuda I., Ito A., Hirai G., Nishimura S., Kawasaki H., Saitoh H., Kimura K., Sodeoka M., Yoshida M. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem. Biol. 2009;16:133–140. doi: 10.1016/j.chembiol.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Gao D., Wu J., Wu Y.T., Du F., Aroh C., Yan N., Sun L., Chen Z.J. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatziioannou T., Ambrose Z., Chung N.P., Piatak M., Jr., Yuan F., Trubey C.M., Coalter V., Kiser R., Schneider D., Smedley J. A macaque model of HIV-1 infection. Proc. Natl. Acad. Sci. USA. 2009;106:4425–4429. doi: 10.1073/pnas.0812587106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecker C.M., Rabiller M., Haglund K., Bayer P., Dikic I. Specification of SUMO1- and SUMO2-interacting motifs. J. Biol. Chem. 2006;281:16117–16127. doi: 10.1074/jbc.M512757200. [DOI] [PubMed] [Google Scholar]
- Laguette N., Sobhian B., Casartelli N., Ringeard M., Chable-Bessia C., Ségéral E., Yatim A., Emiliani S., Schwartz O., Benkirane M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474:654–657. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahaye X., Satoh T., Gentili M., Cerboni S., Conrad C., Hurbain I., El Marjou A., Lacabaratz C., Lelièvre J.D., Manel N. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity. 2013;39:1132–1142. doi: 10.1016/j.immuni.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Lee K., KewalRamani V.N. In defense of the cell: TRIM5alpha interception of mammalian retroviruses. Proc. Natl. Acad. Sci. USA. 2004;101:10496–10497. doi: 10.1073/pnas.0404066101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Yeung D.F., Fiegen A.M., Sodroski J. Determinants of the higher order association of the restriction factor TRIM5alpha and other tripartite motif (TRIM) proteins. J. Biol. Chem. 2011;286:27959–27970. doi: 10.1074/jbc.M111.260406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao C.H., Kuang Y.Q., Liu H.L., Zheng Y.T., Su B. A novel fusion gene, TRIM5-Cyclophilin A in the pig-tailed macaque determines its susceptibility to HIV-1 infection. AIDS. 2007;21(Suppl 8):S19–S26. doi: 10.1097/01.aids.0000304692.09143.1b. [DOI] [PubMed] [Google Scholar]
- Luban J. Innate immune sensing of HIV-1 by dendritic cells. Cell Host Microbe. 2012;12:408–418. doi: 10.1016/j.chom.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukic Z., Goff S.P., Campbell E.M., Arriagada G. Role of SUMO-1 and SUMO interacting motifs in rhesus TRIM5α-mediated restriction. Retrovirology. 2013;10:10. doi: 10.1186/1742-4690-10-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machyna M., Heyn P., Neugebauer K.M. Cajal bodies: where form meets function. Wiley Interdiscip. Rev. RNA. 2013;4:17–34. doi: 10.1002/wrna.1139. [DOI] [PubMed] [Google Scholar]
- Melchjorsen J. Learning from the messengers: innate sensing of viruses and cytokine regulation of immunity - clues for treatments and vaccines. Viruses. 2013;5:470–527. doi: 10.3390/v5020470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minty A., Dumont X., Kaghad M., Caput D. Covalent modification of p73alpha by SUMO-1. Two-hybrid screening with p73 identifies novel SUMO-1-interacting proteins and a SUMO-1 interaction motif. J. Biol. Chem. 2000;275:36316–36323. doi: 10.1074/jbc.M004293200. [DOI] [PubMed] [Google Scholar]
- Morris G.E. The Cajal body. Biochim. Biophys. Acta. 2008;1783:2108–2115. doi: 10.1016/j.bbamcr.2008.07.016. [DOI] [PubMed] [Google Scholar]
- Müller S., Matunis M.J., Dejean A. Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. EMBO J. 1998;17:61–70. doi: 10.1093/emboj/17.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münk C., Brandt S.M., Lucero G., Landau N.R. A dominant block to HIV-1 replication at reverse transcription in simian cells. Proc. Natl. Acad. Sci. USA. 2002;99:13843–13848. doi: 10.1073/pnas.212400099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepveu-Traversy M.E., Berthoux L. The conserved sumoylation consensus site in TRIM5α modulates its immune activation functions. Virus Res. 2014;184:30–38. doi: 10.1016/j.virusres.2014.02.013. [DOI] [PubMed] [Google Scholar]
- Nepveu-Traversy M.E., Bérubé J., Berthoux L. TRIM5alpha and TRIMCyp form apparent hexamers and their multimeric state is not affected by exposure to restriction-sensitive viruses or by treatment with pharmacological inhibitors. Retrovirology. 2009;6:100. doi: 10.1186/1742-4690-6-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman R.M., Hall L., Kirmaier A., Pozzi L.A., Pery E., Farzan M., O’Neil S.P., Johnson W. Evolution of a TRIM5-CypA splice isoform in old world monkeys. PLoS Pathog. 2008;4:e1000003. doi: 10.1371/journal.ppat.1000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papkalla A., Münch J., Otto C., Kirchhoff F. Nef enhances human immunodeficiency virus type 1 infectivity and replication independently of viral coreceptor tropism. J. Virol. 2002;76:8455–8459. doi: 10.1128/JVI.76.16.8455-8459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertel T., Hausmann S., Morger D., Züger S., Guerra J., Lascano J., Reinhard C., Santoni F.A., Uchil P.D., Chatel L. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature. 2011;472:361–365. doi: 10.1038/nature09976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasaiyaah J., Tan C.P., Fletcher A.J., Price A.J., Blondeau C., Hilditch L., Jacques D.A., Selwood D.L., James L.C., Noursadeghi M., Towers G.J. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature. 2013;503:402–405. doi: 10.1038/nature12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma R., Mael A.A., Ikeda Y. Alpha interferon enhances TRIM5alpha-mediated antiviral activities in human and rhesus monkey cells. J. Virol. 2007;81:10201–10206. doi: 10.1128/JVI.00419-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson D.A., Wang M., Matunis M.J. The small ubiquitin-like modifier-1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for SUMO-1 modification. J. Biol. Chem. 2001;276:21664–21669. doi: 10.1074/jbc.M100006200. [DOI] [PubMed] [Google Scholar]
- Sastri J., Campbell E.M. Recent insights into the mechanism and consequences of TRIM5α retroviral restriction. AIDS Res. Hum. Retroviruses. 2011;27:231–238. doi: 10.1089/aid.2010.0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz S., Chachami G., Kozaczkiewicz L., Winter U., Stankovic-Valentin N., Haas P., Hofmann K., Urlaub H., Ovaa H., Wittbrodt J. Ubiquitin-specific protease-like 1 (USPL1) is a SUMO isopeptidase with essential, non-catalytic functions. EMBO Rep. 2012;13:930–938. doi: 10.1038/embor.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvin A., Manel N. Innate immune sensing of HIV infection. Curr. Opin. Immunol. 2015;32:54–60. doi: 10.1016/j.coi.2014.12.003. [DOI] [PubMed] [Google Scholar]
- Soll S.J., Wilson S.J., Kutluay S.B., Hatziioannou T., Bieniasz P.D. Assisted evolution enables HIV-1 to overcome a high TRIM5α-imposed genetic barrier to rhesus macaque tropism. PLoS Pathog. 2013;9:e1003667. doi: 10.1371/journal.ppat.1003667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B., Javanbakht H., Perron M., Park D.H., Stremlau M., Sodroski J. Retrovirus restriction by TRIM5alpha variants from Old World and New World primates. J. Virol. 2005;79:3930–3937. doi: 10.1128/JVI.79.7.3930-3937.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stremlau M., Owens C.M., Perron M.J., Kiessling M., Autissier P., Sodroski J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- Stremlau M., Perron M., Lee M., Li Y., Song B., Javanbakht H., Diaz-Griffero F., Anderson D.J., Sundquist W.I., Sodroski J. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc. Natl. Acad. Sci. USA. 2006;103:5514–5519. doi: 10.1073/pnas.0509996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchil P.D., Hinz A., Siegel S., Coenen-Stass A., Pertel T., Luban J., Mothes W. TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J. Virol. 2013;87:257–272. doi: 10.1128/JVI.01804-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzé G., Di Marco S., Mouchel-Vielh E., Monneron D., Bandu M.T., Horisberger M.A., Dorques A., Lutfalla G., Mogensen K.E. Domains of interaction between alpha interferon and its receptor components. J. Mol. Biol. 1994;243:245–257. doi: 10.1006/jmbi.1994.1651. [DOI] [PubMed] [Google Scholar]
- van Montfoort N., Olagnier D., Hiscott J. Unmasking immune sensing of retroviruses: interplay between innate sensors and host effectors. Cytokine Growth Factor Rev. 2014;25:657–668. doi: 10.1016/j.cytogfr.2014.08.006. [DOI] [PubMed] [Google Scholar]
- Virgen C.A., Kratovac Z., Bieniasz P.D., Hatziioannou T. Independent genesis of chimeric TRIM5-cyclophilin proteins in two primate species. Proc. Natl. Acad. Sci. USA. 2008;105:3563–3568. doi: 10.1073/pnas.0709258105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap M.W., Nisole S., Lynch C., Stoye J.P. Trim5alpha protein restricts both HIV-1 and murine leukemia virus. Proc. Natl. Acad. Sci. USA. 2004;101:10786–10791. doi: 10.1073/pnas.0402876101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoh S.M., Schneider M., Seifried J., Soonthornvacharin S., Akleh R.E., Olivieri K.C., De Jesus P.D., Ruan C., de Castro E., Ruiz P.A. PQBP1 Is a Proximal Sensor of the cGAS-Dependent Innate Response to HIV-1. Cell. 2015;161:1293–1305. doi: 10.1016/j.cell.2015.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yueh A., Leung J., Bhattacharyya S., Perrone L.A., de los Santos K., Pu S.Y., Goff S.P. Interaction of moloney murine leukemia virus capsid with Ubc9 and PIASy mediates SUMO-1 addition required early in infection. J. Virol. 2006;80:342–352. doi: 10.1128/JVI.80.1.342-352.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Perez-Caballero D., Hatziioannou T., Bieniasz P.D. No effect of endogenous TRIM5alpha on HIV-1 production. Nat. Med. 2008;14:235–236. doi: 10.1038/nm0308-235. author reply 236–238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.