

Summary
ATR and CHK1 maintain cancer cell survival under replication stress and inhibitors of both kinases are currently undergoing clinical trials. As ATR activity is increased after CHK1 inhibition, we hypothesized that this may indicate an increased reliance on ATR for survival. Indeed, we observe that replication stress induced by the CHK1 inhibitor AZD7762 results in replication catastrophe and apoptosis, when combined with the ATR inhibitor VE-821 specifically in cancer cells. Combined treatment with ATR and CHK1 inhibitors leads to replication fork arrest, ssDNA accumulation, replication collapse, and synergistic cell death in cancer cells in vitro and in vivo. Inhibition of CDK reversed replication stress and synthetic lethality, demonstrating that regulation of origin firing by ATR and CHK1 explains the synthetic lethality. In conclusion, this study exemplifies cancer-specific synthetic lethality between two proteins in the same pathway and raises the prospect of combining ATR and CHK1 inhibitors as promising cancer therapy.
Keywords: ATR, CHK1, synthetic lethality, cancer, DNA damage, replication stress
Graphical Abstract
Highlights
-
•
Synthetic lethality within the same pathway is via the induction of replication catastrophe
-
•
New origin firing and replication stress induced by CHK1 inhibitor reliance on ATR
-
•
ATR and CHK1 inhibitor combination results in cancer-specific cytotoxicity
-
•
ATR and CHK1 inhibitor combination could be used broadly across cancer indications
Sanjiv et al. report synthetic lethality in cancer cells induced by the inhibition of two kinases, ATR and CHK1, operating in the same signaling pathway. This combination therapy could potentially be used for the treatment of various cancer types due to cancer-specific cytotoxicity.
Introduction
Cancer is a disease of uncontrolled cellular proliferation, driven by oncogenes, leading to unfaithful and uncoordinated DNA replication, genomic instability, and DNA double-strand breaks (DSBs) (Alexandrov et al., 2013, Bartkova et al., 2006, Di Micco et al., 2006). DSBs activate the ATM kinase, which in turn mediates p53-dependent cell-cycle arrest and apoptosis, working as a tumor barrier to cancer development (Bartkova et al., 2005, Gorgoulis et al., 2005, Halazonetis et al., 2008). In contrast, the ATR kinase is activated by single-stranded DNA (ssDNA) present at stalled replication forks (Hekmat-Nejad et al., 2000, Zou and Elledge, 2003). ATR phosphorylates the checkpoint kinase CHK1, which plays a crucial role in preventing origin firing (Feijoo et al., 2001), avoiding premature chromosome condensation and facilitating RAD51-mediated homologus recombination (Cimprich and Cortez, 2008, Sørensen et al., 2005).
Since cancer cells often harbor some degree of replication stress, they upregulate ATR and CHK1 activity to mediate survival (Choi et al., 2011, Toledo et al., 2011). For example, B cell lymphomas are sensitive to CHK1 inhibitors as they have a high degree of MYC-induced replication stress (Höglund et al., 2011, Murga et al., 2011). Cancer cells also commonly lack compensatory DNA damage response proteins that are synthetic lethal with the ATR pathway, including ATM and p53 (Ding et al., 2008, Jiang et al., 2009), which further increases reliance on ATR and CHK1 in damaged tumor cells (Choi et al., 2011, Murga et al., 2009, Reaper et al., 2011). In addition, the cytotoxic mechanism of action of many anti-cancer drugs is to induce replication stress and replication-associated DNA damage. Taken together, ATR or CHK1 inhibition is a promising strategy, and selective inhibitors are undergoing clinical trials in combination with DNA-damaging chemotherapy and ionizing radiation (Brooks et al., 2013, Fokas et al., 2012, Foote et al., 2013, Foote et al., 2015, Jossé et al., 2014, Ma et al., 2011, Mitchell et al., 2010, Tang et al., 2012).
Although the ATR and CHK1 kinases function in the same pathway, they also may exert unique functions. For example, the ATR protein appears to have a more important role than CHK1 in preventing replication collapse after UV damage (Elvers et al., 2012), which is likely related to a unique role of ATR in supplying RPA to protect replication forks (Toledo et al., 2013). We and others previously have found that inhibition or depletion of CHK1 causes replication stress and activation of ATR, which is explained by the role of CHK1 in suppressing replication origin firing (Choi et al., 2011, Gagou et al., 2010, Petermann et al., 2010a, Syljuåsen et al., 2005). Since ATR is critical for replication fork stability under conditions of replication stress (Toledo et al., 2013), which may be independent of CHK1 (Elvers et al., 2012), we hypothesized that ATR may be critical for survival upon CHK1 inhibition in cancer cells. In line with this hypothesis, we demonstrate that sub-toxic concentrations of both the ATR inhibitor VE-821 and the CHK1 inhibitor AZD7762 combine synergistically to induce complete replication collapse and apoptosis specifically in cancer cells. In addition, the combination of the ATR inhibitor VX-970 and AZD7762 markedly improves overall survival in mice bearing lung and breast tumor xenografts at well-tolerated doses. Here we show cancer-specific synthetic lethality using ATR and CHK1 inhibitors in combination. These data demonstrate that synthetic lethality can be obtained by targeting proteins within the same pathway, and they provide compelling evidence that the combination of ATR and CHK1 inhibitors may be used as a promising cancer therapy.
Results
Combined ATR and CHK1 Inhibition Induces Excess ssDNA, JNK-Mediated Pan-nuclear γH2AX, and DNA Damage in Cancer Cells
In our previous study, we demonstrated that the inhibition of CHK1 using the small molecules UCN-01 and CEP-3891 or depletion of CHK1 by small interfering RNA (siRNA) results in increased initiation of DNA synthesis and phosphorylation of ATR substrates (Syljuåsen et al., 2005). Likewise, the CHK1 inhibitor AZD7762 also causes phosphorylation of CHK1 (a marker for ATR activation), which gets suppressed by ATR inhibitor VE-821 (Figures 1A and 1D). We found that these phosphorylation events are dependent on ATR activity, not on other kinases like ATM or DNAPK, as only selective ATR inhibitors VE-821 and VX-970 decrease phosphorylation of CHK1 on Ser345 (Figures 1A and S1A–S1D). Taken together, our current and previously published data on VE-821 and AZD7762 show that the doses used in this study result in nearly complete inhibition of ATR signaling by VE-821 and VX-970 (Figures S1A–S1C; Huntoon et al., 2013, Jossé et al., 2014, Reaper et al., 2011) and CHK1 signaling by AZD7762 (Aris and Pommier, 2012, Zabludoff et al., 2008).
Figure 1.

ATR Target Activation by the CHK1 Inhibitor AZD7762 in U2OS Cancer Cells
(A) Western blot showing activation of ATR targets. U2OS cells were treated with the indicated concentrations for 30 and 60 min, lysed, and probed with anti-phospho (Serine 345) CHK1 and β-actin antibodies.
(B) Induction of pre-apoptotic pan-nuclear γ-H2AX by ATR and CHK1 inhibitor in combination in cancer cells. U2OS cells were treated with the indicated drug concentrations for 24 hr. Cells were probed with anti-phospho (Serine 139) H2AX antibody. Scale bar, 20 μm.
(C) Quantitative data of γH2AX- (nine or more foci per cells) positive cells or pan-nuclear γH2AX signal after indicated treatments are shown (n = 3, mean ± SEM).
(D) Western blot showing increased phosphorylation of H2AX after combination treatment. U2OS cells were treated with the indicated concentrations for 24 hr. At the end of incubation time, western blotting was performed using anti-phospho (Serine 139) H2AX, anti-phospho (Serine 345) CHK1, cleaved PARP, anti-phospho (Serine 10) H3, and β-actin antibodies.
(E) Comet assay showing DNA damage induction by ATR and CHK1 inhibitor in combination. U2OS cells were treated with the indicated drug concentrations for 24 hr. At the end of incubation, cells were harvested and alkaline comet assay was performed.
(F) Quantitative data of the tail moment are shown (n = 3, mean ± SEM, in each experiment ≥100 comets were measured).
(G) Cancer-specific ssDNA formation by VE-821 and AZD7762, either alone or in combination. U2OS cells were treated with the indicated drug concentrations for 24 hr and pre-extracted using CSK buffer before fixation. Cells were stained with anti-RPA32 antibody; images were taken using a confocal microscope and were analyzed using ImageJ software. A mean intensity of ≥70 a.u. per cell was considered as positive. Quantitative data are presented as mean ± SEM from three independent experiments.
(H) ssDNA formation in normal fibroblast VH-10 cells is shown.
(I) Pre-apoptotic pan-nuclear γH2AX induction by combination treatment of ATR and CHK1 inhibitors in U2OS is mediated through the JNK pathway. U2OS cells were treated with the indicated drug concentrations for 24 hr. Cells were probed with anti-phospho (Serine 139) H2AX antibody, and high-throughput microscopy was used to determine the percentage of γH2AX-positive cells (nine or more γH2AX foci per cell) or an average intensity of ≥2,000 a.u. for pan-nuclear γH2AX-positive cells (n = 2 with multiple wells, mean ± SEM). Statistical significance was determined using one-way ANOVA (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001).
Although no significant increase in γH2AX foci or pan-nuclear γH2AX (a marker of widespread replication fork collapse) was observed in cells treated with VE-821 or AZD7762 alone for 24 hr, the combined treatment triggered a dose-dependent induction of pan-nuclear γH2AX in U2OS cells (Figures 1B–1D, S2A, and S2B). Interestingly, all the pan-nuclear γH2AX-positive cells were completely devoid of nuclear 53BP1 foci, indicating irreparable DNA damage. We observed a decrease in S10H3 phosphorylation (marker for mitosis) and a marked increase in γH2AX (DNA damage marker) in U2OS cells treated with the combination of VE-821 and AZD7762, but not in cells treated with either inhibitor alone. This suggests that either ATR or CHK1 inhibitor alone exhibits its effect by abrogating the cell-cycle checkpoint leading to mitotic catastrophe, while combining both results in extensive DNA damage that likely leads to replication catastrophe (Figure 1D).
To directly determine the magnitude of the DNA damage, we used an alkaline comet assay. Only the combination of both drugs significantly induced DNA breaks (Figures 1E, 1F, S2C, and S2D), with a similar proportion of cells affected as were positive for pan-nuclear γH2AX. In contrast to U2OS cells, there was no significant induction of pan-nuclear γH2AX and nuclear 53BP1 in VH-10 normal fibroblasts (Figures S2E–S2G). This suggests that the combination of VE-821 and AZD7762 specifically induces replication fork collapse in cancer cells.
CHK1 inhibition in cells initiates excessive origin firing, replication fork stalling, and ssDNA formation (Syljuåsen et al., 2005, Wilsker et al., 2008). ssDNA is protected by the ssDNA-binding protein RPA, which subsequently activates ATR (Nam and Cortez, 2011, Toledo et al., 2013). We analyzed pre-extracted RPA32 foci formation in U2OS cancer and VH-10 primary cells. There was no statistically significant increase in the RPA-positive cell population when U2OS cells were treated with AZD7762 alone at 30 nM for 24 hr in comparison to DMSO control, while treatment with 60 nM or 10 μM VE-821 significantly increased the RPA-positive cell population (Figure 1G). As expected the combination of AZD7762 (30 or 60 nM) and 10 μM VE-821 for 24 hr caused a large increase in the RPA-positive U2OS cell population (Figure 1G). In contrast to U2OS cells, treatment of VH-10 cells with AZD7762 and VE-821 alone or in combination only marginally increased the RPA-positive cell population (Figure 1H). These results suggest that replication stress was specifically induced by the combination of ATR and CHK1 inhibitors in U2OS cancer cells, but not normal cells. The differential response between cancer cells and normal cells cannot be explained by slower proliferation of the VH-10 cells, as virtually all cells had progressed through the cell cycle within the 24-hr treatment period (Figures S3A and S3B).
UVA-induced pan-nuclear γH2AX in normal fibroblasts is known to be mediated by JNK and ATM (de Feraudy et al., 2010) kinases. We used small molecule JNK (SR-3306) and ATM (KU-55933) inhibitors to investigate if the pan-nuclear γH2AX induced by combined ATR and CHK1 inhibition also is mediated by the same pathway. The JNK inhibitor markedly reduced the pan-nuclear γH2AX-positive cell population after 24-hr treatment with VE-821, AZD7762, or the combination, while the ATM inhibitor had only a minor effect under all conditions (Figure 1I). Conversely, while the JNK inhibitor increased the proportion of cells positive for discrete γH2AX foci (a marker of lower levels of DNA damage), this form of H2AX phosphorylation was fully ablated by ATM inhibition (Figure 1I). These data suggest distinct roles for ATM and JNK in the cellular response to combined inhibition of ATR and CHK1.
ATR and CHK1 Inhibitors Are Synthetic Lethal Selectively in Cancer Cells
The induction of pan-nuclear γH2AX is suggestive of high levels of replication stress (Choi et al., 2011). We therefore asked whether dual inhibition of ATR and CHK1 is sufficient to trigger cancer cell death. Clonogenic survival assays were performed on U2OS, MCF-7 cancer, and VH-10 primary cells treated with a range of concentrations of VE-821 with and without 20 nM AZD7762. AZD7762 clearly potentiated the effect of VE-821 in U2OS and MCF-7 cancer cells (3- to 10-fold shifts in the IC50 of VE-821), but not in the normal VH-10 fibroblast cells (Figure 2A). Notably, we observed similar growth kinetics across the lines without treatment, indicating that proliferation rate alone does not account for this cancer-specific effect (Figures S3A and S3B).
Figure 2.

Combination of the ATR Inhibitor VE-821 and the CHK1 Inhibitor AZD7762 Synergistically Kills Cancer Cells
(A) Clonogenic survival of U2OS, VH-10, and MCF-7 cells. The 500 (U2OS and MCF-7) or 1,000 (VH-10) cells were seeded in 10-cm2 dishes, and, after 5-hr incubation, the inhibitors were added directly to the media. After 72-hr incubation, drug-containing media were replaced with fresh media and cells were kept for another 5–8 days before colonies were stained with methylene blue. Quantitative data: n = 3, mean ± SEM.
(B) Parental and cMYC-transformed cells were treated with the indicated doses for 72 hr. At the end of the incubation period, resazurin was added and cell viability was measured. Quantitative data: n = 3, mean ± SEM.
(C) BJ-hTERT, BJ-hTERT SV40, and BJ SV40 RAS cells were treated with the indicated doses for 72 hr. At the end of the incubation period, resazurin was added and cell viability was measured. Quantitative data: n = 3, mean ± SEM.
(D) CHK1 functionally compromised cells are sensitive to ATR inhibitor. Clonogenic survival of DLD-1, DLD-1 CHK1S317A/−, DLD-1 CHK1+/−, and DLD-1 ATRS/S after ATR inhibitor VE-821 treatment is shown. A similar protocol was used as for U2OS and VH-10 cells. Quantitative data: n = 3, mean ± SEM.
(E) Therapeutic efficacy of combined inhibition of ATR and CHK1 in mouse tumor models. Therapeutic efficacy of VX-970 and AZD7762 in H460 lung cancer xenografted mice is shown. BALB/c nude mice bearing H460 xenograft were divided in four groups (five animals in each group) with a tumor volume of ∼130 mm3 in each group. The first control group of animals was treated with vehicle (orally and intraperitoneally). The second group of animals was treated with 25 mg/kg body weight of CHK1 inhibitor AZD7762 (intraperitoneal route). The third group of animals was treated with 60 mg/kg body weight of ATR inhibitor VE-822 (oral administration), and the fourth group received a combination of both CHK1 and ATR inhibitors. Vehicle and drugs were administered on days 0–3, 10–12, and 18–20 irrespective of no mice survival in each group. Tumor volume was measured with calipers and is shown here as mean ± SEM. Statistical significance was determined using two-way ANOVA with repeated measurement (∗p < 0.05 and ∗∗p < 0.01).
(F) Kaplan-Meier survival curve of H460-xenografted mice. When tumor size reached 1,000 mm3, the animal was sacrificed.
Cancer cells often have elevated levels of oncogene-induced replication stress (Hills and Diffley, 2014), requiring the ATR/CHK1 pathway to prevent cell death (Gabay et al., 2014, Murga et al., 2011, Rohban and Campaner, 2015). To investigate the role of oncogene-induced replication stress in the selective anti-cancer effect of combined ATR and CHK1 inhibition, we compared the effects of this treatment in transformed cMYC-overexpressing cells (HA1EB-GFP-cMYC) versus isogenic non-transformed control cells (HA1EB-GFP). cMYC is a proto-oncogene that frequently is upregulated in cancer and acts as a regulator of multiple biological processes, including cell growth, cell-cycle progression, and apoptosis (Gabay et al., 2014, Rohban and Campaner, 2015). HA1EB-GFP-cMYC cells have a higher replicative rate and higher levels of DNA damage than control cells, as expected from cMYC overexpression (Figures S3C–S3F). HA1EB-GFP and HA1EB-GFP-cMYC cells were treated with VE-821 and AZD7762 either alone or in combination for 72 hr. We found that 30 nM AZD7762 markedly sensitized cMYC-transformed cells, but not the control cells, to VE-821 (Figure 2B).
To extend these observations, we analyzed the effects of dual ATR and CHK1 inhibition in non-transformed BJ cells immortalized with hTERT compared with transformed isogenic lines additionally expressing SV40 large T antigen (that inactivates p53 and RB) and H-RAS V12 (Hahn et al., 1999; Figure S3G). In agreement with previous results, we observed synthetic lethality between the ATR and CHK1 inhibitors in transformed BJ-SV40T and BJ-RASV12 cells, but not in the non-transformed BJ-hTERT cells (Figure 2C). These data indicate that the combined inhibition of ATR and CHK1 is selective against cells with high replicative stress levels, i.e., cancer cells. Importantly, we also carried out cell viability assays on a larger panel of cancer and normal cell lines treated for 72 hr with VE-821 and AZD7762 (Figures S4A–S4J). The drug combination of VE-821 and AZD7762 was synergistic (combination index [CI] < 1) in all cancer cell lines tested (U2OS, MCF-7, HCT116WT, HCT116p53−/−, H460, MX-1, and HL-60), but not in normal cells, including VH-10 (foreskin fibroblast), CCD841 (colon epithelial cells), and HUVEC (endothelial cells).
Potentially, the synergy observed between ATR and CHK1 inhibitors could merely result from insufficient inhibition of the ATR-CHK1 pathway with each compound alone. Alternatively, this synergy could be caused by the inhibition of both CHK1 and CHK2 by AZD7762. To determine whether there is a genetic interaction between ATR and CHK1 that could explain the combined activity (as opposed to the two alternatives above), we exploited DLD-1 cancer cells with one null CHK1 allele and one allele in which a critical ATR substrate residue on CHK1 has been mutated (CHK1S317A/−) (Wilsker et al., 2008, Zhao and Piwnica-Worms, 2001). Interestingly, these genetically CHK1-defective cells were markedly more sensitive to VE-821 than wild-type control cells, CHK1 heterozygous null DLD-1 cells, and ATR-deficient (Seckel) DLD-1 cells (ATRs/s) (Figure 2D). These data clearly demonstrate functionally dead CHK1 cells (CHK1S317A/−) completely rely on ATR activity for survival, and they suggest that the synthetic lethality observed between VE-821 and AZD7762 reflects specific synthetic lethality between ATR and CHK1. In addition, we found that ATR inhibitors synergize with a number of CHK1 inhibitors with varying selectivity over CHK2, including the highly selective CHK1 inhibitor SCH-900776 (Figures S5A–S5G). Importantly, these data demonstrate a genetic interaction between CHK1 and the ATR inhibitor VE-821, rejecting the hypothesis that the observed synthetic lethal effect is merely a hypomorphic effect due to increased inhibition of the same activity in the ATR-CHK1 pathway.
Co-treatment with VX-970 and AZD7762 Extends Survival in Lung Tumor Mouse Xenograft Models
We next further tested our drug combination in a xenograft model. VX-970 (also referred to as VE-822) is an analog of VE-821 with excellent ATR selectivity and absorption, distribution, metabolism, and excretion properties that support in vivo studies (Fokas et al., 2012). Mice bearing H460 lung cancer xenografts were treated with 60 mg/kg VX-970 orally, 25 mg/kg AZD7762 intraperitoneally, or the combination on days 0–3, 10–12, and 18–20. While no effect on tumor growth was observed in mice treated with ATR or CHK1 inhibitor monotherapy, the combined treatment caused a significant reduction in tumor growth (Figure 2E). Furthermore, combination therapy significantly increased overall survival (Figure 2F). Importantly, VX-970 and AZD7762 were well tolerated both alone and in combination, with no body weight loss observed in the xenograft model (Figure S5H). These data suggest that the combination of ATR and CHK1 inhibitors may provide an efficacious and well-tolerated anti-cancer therapy.
Combined ATR and CHK1 Inhibition Decreases Fork Speed Progression in Cancer Cells, Resulting in S Phase Arrest and Apoptosis
It has been shown previously that inhibition of either ATR or CHK1 slows down replication fork progression due to an increase in origin firing (Couch et al., 2013, Wilsker et al., 2008). Consistently, using DNA fiber assay, we found that treatment of U2OS cells with VE-821 or AZD7762 decreases replication fork speed (Figure 3). Additionally, fork speed was further dramatically reduced by combined treatment of U2OS cells with both agents. In contrast, in VH-10 cells, replication fork progression speed was unperturbed by these inhibitors, either alone or in combination. This may indicate that origin firing is unaffected by ATR or CHK1 inhibition in untransformed cells.
Figure 3.

ATR and CHK1 Inhibitors, Alone or in Combination, Decrease Replication Fork Speed Only in Cancer Cells
(A) Treatment regimen is shown. U2OS and VH-10 cells were treated for 60 min with the indicated drug concentrations and sequentially labeled with 5-chlorodeoxyuridine (CldU) and 5-iododeoxyuridine (IdU) for 30/20 min each in the presence of the inhibitors. DNA fibers were stained and replication speed was measured by IdU labeling.
(B and C) Representative images show stained replication fork tracts for each treatment group.
(D) Quantitative data of replication fork speed (kb/min), mean ± SEM, and p values were analyzed with one-way ANOVA for each condition and cell line.
(E and F) Average distribution of replication fork rates. A minimum of 450 forks per condition were analyzed from at least three independent repeats.
Inhibition of ATR or CHK1 abrogates the G2 DNA damage checkpoint (Huntoon et al., 2013). We therefore next investigated the effects of VE-821 and AZD7762 on the cell cycle in U2OS cells. While treatment with either drug alone for 24 hr had little effect on the cell-cycle profiles, the combination treatment resulted in severe S phase arrest (Figures 4A and 4B). These results were supported by a significant decrease in EdU incorporation in U2OS cells treated with VE-821 and AZD7762 in combination, but not alone (Figure 4C). The S phase arrest and decreased DNA replication are in agreement with replication fork collapse and the accumulation of DNA damage in cancer cells co-treated with ATR and CHK1 inhibitors. At later time points (48–72 hr), the S phase population decreased with a corresponding increase in the sub-G1 population, indicating cell death from replication catastrophe. Cleaved PARP, a marker of apoptosis, also was detected under these conditions (Figures S6A–S6C). Taken together, these data demonstrate that combined inhibition of ATR and CHK1 halts replication fork progression in cancer cells, leading to S phase arrest replication collapse and the induction of apoptosis. Consistent with our DNA fiber results, no significant decrease in EdU incorporation was observed in normal cells treated with VE-821 and AZD7762, suggesting that the effects of these inhibitors on replication dynamics are cancer cell specific (Figure 4D).
Figure 4.

Combination Treatment of VE-821 and AZD7762 Results in S Phase Arrest in U2OS Cells
(A) U2OS cells were treated with the indicated drug concentrations for 24 hr and propidium iodide (PI) staining was carried out to measure cell-cycle profile using flow cytometry.
(B) Quantitative data were obtained using Modfit software.
(C) ATR and CHK1 inhibitors in combination decrease EdU incorporation in U2OS cells. U2OS cells were treated for 24 hr with the indicated doses. Images were taken with a confocal microscope and analyzed using ImageJ software. A mean intensity of ≥80 a.u. per cell was considered as EdU-positive cells. Quantitative data: n = 3, mean ± SEM.
(D) No significant decrease in EdU incorporation in normal fibroblast VH-10 cells treated with the ATR and CHK1 inhibitors either alone or in combination. VH-10 cells were treated for 24 hr with the indicated doses. Quantitative data: n = 3, mean ± SEM. Statistical significance was determined using one-way ANOVA (∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001).
CDK-Mediated Excess Origin Firing in Cancer Cells Explains ATR/CHK1 Synthetic Lethality
Phosphorylation of CHK1 by ATR prevents unscheduled origin firing and replication-induced DNA damage (Feijoo et al., 2001). However, CHK1 also can be activated by replication stress through Claspin, which is independent of ATR signaling (Yang et al., 2008). Thus, there are various ways of activating CHK1 to prevent replication origin firing. Previously, we demonstrated that CHK1 inhibition causes overactivation of CDK2-mediated origin firing and, subsequently, increases replication stress (Petermann et al., 2010b). We therefore sought to test if downregulation of replication initiation leads to loss of the synthetic lethality caused by co-treatment with VE-821 and AZD7762 in cancer cells.
U2OS cells were pretreated with CDK inhibitor Roscovitine or CDK/CDC7 inhibitor PHA-767491 (Jones et al., 2013, Petermann et al., 2010b) prior to the addition of VE-821 and AZD7762 to downregulate origin firing. Both CDK inhibitors reduced the pan-nuclear γH2AX-positive cell population mediated by co-treatment with VE-821 and AZD7762 in a dose-dependent manner, but increased the γH2AX foci-positive cell population, converting the irreparable to reparable lesions (Figures 5A, 5B, S7A, S7C, and S7D). Although CDK inhibitors alone have no effect on replication fork speed (Jones et al., 2013, Petermann et al., 2010b), both Roscovitine and PHA-767491 increased replication fork speed in cells co-treated with VE-821 and AZD7762, suggesting alleviation of replication stress (Figures 5C–5E). Furthermore, Roscovitine significantly abrogated the synergistic cytotoxic effect of dual ATR and CHK1 inhibition in U2OS cells (Figures 5F and S7B). No such enhancement of cell survival was observed with PHA-767491, but this was attributed to the single agent cytotoxicity of the compound over prolonged exposures (Figure S7E). Taken together, these data provide a mechanism of action, where inhibition of both ATR and CHK1 induces an increase in CDK-mediated origin firing in tumor cells, ultimately leading to RPA exhaustion, replication catastrophe, and cell death (Figure 5G).
Figure 5.

Synergistic Cytotoxic Effect in U2OS Cancer Cells by Combination Treatment of AZD7762/VE-821 Is Mainly Due to CDK-Mediated Excess Origin Firing
(A) U2OS cells were pretreated with the indicated concentrations of the CDK inhibitor Roscovitine for 1 hr prior to the addition of VE-821 and AZD7762 for 24 hr. Cells were probed with anti-phospho (Serine 139) H2AX antibody and anti-53BP1, and DNA was counterstained with ToPro.
(B) Quantitative data of pan-nuclear γH2AX are shown (mean ± SEM from two independent experiments).
(C) Treatment regimen for DNA fiber assay in U2OS cells is shown.
(D) CDK inhibitors Roscovitine and PHA-767491 enhance the replication fork speed of U2OS cells treated with VE-821 and AZD7762 in combination. U2OS cells were pre-treated with Roscovitine or PHA-767491 for 1 hr prior to the addition of VE-821 and AZD7762. Representative images show stained replication fork tracts for each treatment group.
(E) Average distribution of replication fork rates. A minimum of 450 forks per condition were analyzed from at least three independent repeats.
(F) Roscovitine abolishes the synergistic cytotoxic effect of combination treatment of AZD7762 and VE-821 in U2OS cancer cells. U2OS cells were individually treated with VE-821, AZD7762, or the combination with our without Roscovitine for 24 hr, followed by recovery for another 48 hr. Cell viability was measured by using resazurin at 72 hr.
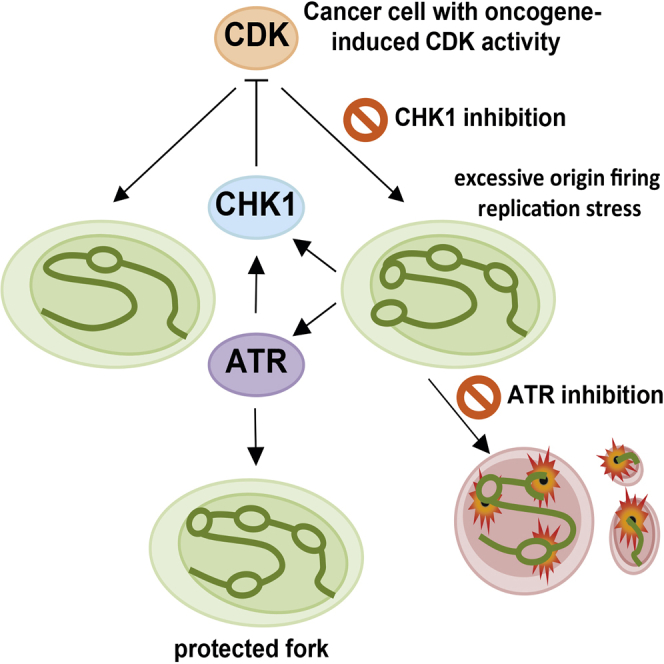
(G) Model for ATR/CHK1 synthetic lethality. CHK1 is activated by replication stress both by ATR-dependent and -independent pathways (Yang et al., 2008) to suppress replication stress in cancer, promoting restart and survival. CHK1 inhibitors increase oncogene-activated CDK activity and origin firing, leading to replication stress and accumulation of stalled replication forks, requiring ATR activity to prevent replication collapse. Red arrows indicate primary route in the presence of both ATR and CHK1 inhibitors.
Combined ATR and CHK1 Inhibition Causes Nuclear Fragmentation in Cancer Cells under Replication Stress
Since DNA-damaging drugs are widely used in cancer therapy, we investigated whether tumor cells responding to DNA-damaging chemotherapy are more susceptible to the combination of VE-821 and AZD7762. Within 24 hr, U2OS cells treated with hydroxyurea (HU), VE-821, and AZD7762 showed a marked increase in nuclear fragmentation and markers of apoptosis compared to cells treated with HU alone. (Figures 6A–6C). In contrast, no increase in nuclear fragmentation was detected in HU-treated normal cells upon the addition of VE-821 and AZD7762 (Figure 6D). These data suggest that concurrent inhibition of ATR and CHK1 may combine productively with DNA-damaging cancer therapy.
Figure 6.

HU-Induced Replication Stress in Combination with VE-821 and AZD7762 Causes Fragmented Nuclei and the Early Onset of Apoptosis Only in U2OS Cells
(A) U2OS cells were treated for 24 hr with the indicated drug concentrations and stained with anti-cleaved caspase 3 and β-actin antibodies. Representative confocal images are shown.
(B) Quantitative data of fragmented nuclei and cleaved caspase-3 positive cells presented as mean ± SEM from three independent experiments.
(C) Western blot showing apoptosis in U2OS cells treated with ATR and CHK1 inhibitors alone or in combination. U2OS cells were treated with the indicated concentrations for 24 hr; lysed; protein extracted; and western blotting was performed with anti-Cleaved PARP, anti-phospho (Serine 10) Histone H3, and anti-β-actin antibodies.
(D) HU-induced replication stress does not cause fragmentation of nuclei or apoptosis in combination with VE-821 and AZD7762 in VH-10 normal fibroblast cells in 24 hr. Etoposide treatment (4 μM) was used to induce apoptosis as a positive control. Scale bar represents 20 μM.
Discussion
Targeting the ATR-CHK1 pathway is a promising strategy for the treatment of cancer. Only a handful of cancers, such as B cell lymphomas, are suggested to have sufficiently high endogenous replication stress to allow the use of ATR or CHK1 inhibitors in monotherapy (Höglund et al., 2011, Murga et al., 2011). Therefore, the overwhelming strategy in the clinic is to use the ATR and CHK1 inhibitors in combination with chemotherapy or radiotherapy (Brooks et al., 2013, Fokas et al., 2012, Foote et al., 2013, Foote et al., 2015, Jossé et al., 2014, Ma et al., 2011, Mitchell et al., 2010, Tang et al., 2012). In our study, we found that sub-toxic doses of the ATR inhibitor VE-821 and the CHK1 inhibitor AZD7762, when combined, show synergistic cytotoxic effects on cancer cell lines that are relatively insensitive to ATR or CHK1 inhibitors alone. We believe that this is not merely due to a hypomorphic effect, as the doses used in this study resulted in nearly complete inhibition of ATR signaling by VE-821 and VX-970 and CHK1 signaling by AZD7762, based on our current and previously published reports. We suggest that this has important implications for widening the potential scope of ATR and CHK1 inhibitors beyond monotherapy in tumors harboring very high levels of replication stress (e.g., B cell lymphomas) or as combinations with DNA-damaging cytotoxic chemotherapy. We demonstrate in vivo efficacy with the drug combination in H460 lung tumor xenograft models (Figures 2E and 2F), without affecting body weight of the animals. The favorable tolerability profile is in line with our important observation of no synthetic lethality between ATR and CHK1 inhibitors in non-transformed cells. This is an important finding as it suggests that a beneficial therapeutic index might be achieved when combining ATR and CHK1 inhibitors in the clinic. More work is now warranted to find candidate biomarkers that might enable prospective identification of cancer patients that will benefit the most from dual ATR and CHK1 inhibition.
Here we show that oncogene-deregulated CDK activity is required to manifest the ATR/CHK1 synthetic lethality. In our model, we propose that CHK1 inhibitors increase overall CDK-mediated replication stress by increased origin firing, which in turn likely depletes the limited dinucleotide triphosphate (dNTP) pool (Bester et al., 2011), resulting in slow or stalled DNA replication (Alexandrov et al., 2013). This generates an increasing amount of ssDNA tracts that are protected by an ATR-mediated supply of RPA to prevent stalled forks from collapsing (Brooks et al., 2013, Gagou et al., 2010, Petermann et al., 2010b, Syljuåsen et al., 2005, Toledo et al., 2013). In the additional presence of ATR inhibitors, the high number of stalled replication forks collapses into toxic, irreparable DSBs, killing the cancer cells. Intuitively, one would not expect inhibitors of CHK1, acting downstream of ATR, to have any synergistic effect with ATR inhibitors. There is, however, much crosstalk in the DNA damage response network, and CHK1 also is activated by Claspin following replication stress, independently of ATR (Yang et al., 2008). Also, there is an ATR-dependent, CHK1-independent, intra-S phase checkpoint that suppresses origin firing (Couch et al., 2013, Luciani et al., 2004). ATR and CHK1 also have distinct functions and may not act linearly in the kinase cascade (Buisson et al., 2015). This underscores the importance of CHK1 in preventing origin firing upon replication stress and a separate role for ATR in preserving replication fork integrity to prevent collapse (Toledo et al., 2013; Figure 5G). The model predicts that CHK1 inhibitor monotherapy may be limited in many cancers due to ATR activation, which will prevent fork collapse and cell death. Similarly, effective ATR-independent suppression of CDK activity by CHK1 could limit the efficacy of ATR monotherapy.
In conclusion, we exemplify a cancer-specific synthetic lethality between the ATR and CHK1 kinase activities, demonstrating that it is possible to obtain synthetic lethality between two proteins within the same pathway. Furthermore, our findings may have important medical implications by raising the prospect that ATR and CHK1 inhibitors may be combined to provide a well-tolerated cancer therapy with the potential to treat a large proportion of cancer patients.
Experimental Procedures
Please refer to the Supplemental Experimental Procedures for additional experimental details.
Cell Lines
U2OS (human bone osteosarcoma cells); VH-10 (human foreskin fibroblast cells); MCF-7 (human breast cancer cells) (ATCC); CCD841 (human colon epithelial cells) (ATCC); HA1EB-GFP (GFP-expressing HA1EB, human immortalized kidney epithelial cells); HA1EB-GFP-cMYC (GFP-cMYC-expressing HA1EB cells); and genetically modified cell lines BJ-hTERT (hTERT-immortalized BJ cells), BJ-SV40T (SV40T-transformed BJ-hTERT cells), and BJ-RASV12 (H-RAS V12-transformed BJ-SV40T cells) (Hahn et al., 1999) were grown in DMEM Glutamax. SW480 (human colorectal adenocarcinoma) was grown in DMEM. HUVEC (human umbilical endothelial cell); HCT116WT (human colon carcinoma) (ATCC); HCT116p53−/− and DLD-1 (human colorectal adenocarcinoma); and genetically modified cell lines DLD-1 CHK1S317A/−, DLD-1 CHK1+/−, and DLD-1 ATRS/S (Wilsker et al., 2008) were grown in McCoy’s media. MX-1 (human breast carcinoma cells) was grown in DMEM-F12 and HL-60 was grown in RPMI 1640. The BJ-hTERT, BJ-SV40T, and BJ-RASV12 cells were generously provided by Dr. Hahn (Dana-Farber Cancer Institute). HCT116p53−/− was also generously provided by Dr. Vogelstein (Johns Hopkins University) (Figure S4H). All media were supplemented with 10% fetal bovine serum (FBS) and 10 U/ml penicillin/streptomycin (PeSt, Invitrogen), and cells were cultured in 37°C with 5% CO2. For further details see the Supplemental Experimental Procedures.
Clonogenic Survival Assay
The 500 (U2OS, MCF-7, DLD-1, DLD-1 CHK1S317A/−, DLD-1 CHK1+/−, and DLD-1 ATRS/S) or 1,000 (VH-10) cells were seeded into 10-cm plates. After 5 hr at 37°C and 5% CO2, cells were treated with either vehicle (maximum 0.05% DMSO) or various concentrations of inhibitors for ATR (VE-821) and/or CHK1 (AZD7762) and incubated for 72 hr. After 72 hr, vehicle- and drug-containing media were replaced with fresh media and further incubated for 5–8 days before fixation and staining with 4% methylene blue in MeOH. Colonies were counted manually.
Viability Assay and Drug Interaction
Drug interaction between inhibitor VE-821 and AZD7762 (CI) was determined using CompuSyn software (version 1.0.1), where CI < 1 indicates synergy, CI = 1 indicates additivity, and CI > 1 indicates antagonism (Chou, 2010). For further details see the Supplemental Experimental Procedures.
DNA Fiber Assay
Fiber assay was done as previously described (Groth et al., 2010); for details see the Supplemental Experimental Procedures.
Microscopy
Confocal images were taken using either an LSM 510 63X or LSM 780 microscope (Zeiss) equipped with a 40× oil objective. For high-throughput microscopy, a PerkinElmer operetta high-content microscope equipped with a 10× objective was used. For details on microscopy and immunofluorescence see the Supplemental Experimental Procedures.
Comet Assay
Comet assay was performed as previously described (Gad et al., 2014); for details see the Supplemental Experimental Procedures.
Cell Cycle-Cell Cycle
Cells (150,000) were seeded in six-well plates and kept for overnight incubation. The next day, cells were treated with DMSO, VE-821, AZD7762, or the combination. At the end of 24-, 48-, or 72-hr incubation, the supernatant as well as the trypsinized single-cell suspension were collected. After washing twice with cold PBS, cells were fixed in 70% chilled methanol and kept at −20°C. On the day of flow cytometry, 400 μl PBS, 50 μl RNaseA (1 mg/ml), and 5 μl propidium iodide (PI, 400 μg/ml) were added to each tube and thereafter incubated at 37°C for 30 min. At the end of the incubation period, the cell suspension was strained through a 40-μm membrane filter. The sample was analyzed by using BD FACScalibur, and Modfit software was used to quantify data.
Western Blotting
For details see the Supplemental Experimental Procedures.
In Vivo Xenograft
Animal experiments were carried out according to the described rules and regulations of the regional animal ethical committee Stockholm and in compliance with EU 2010/63 directive. Before conducting experiments, animals were acclimatized in the animal house for a week with ad libitum food and water, a 12-hr light cycle, and the temperature and humidity set according to laboratory animal guidelines and regulations. Human lung cancer H460 cells (5 million) in PBS were injected subcutaneously in the flank of 6-week-old male nude mice. After 8–9 days of implantation, the animals were divided into four groups based on tumor size, with a mean tumor volume of 130 mm3. The first group received vehicle as follows: 10% D-α-Tocopherol polyethylene glycol 1000 succinate (Sigma) in water administered via oral gavage and 11.3% (2-Hydroxy propyl)-β cyclodextrin in saline administered intraperitoneally. The second group received AZD7762 at 25 mg/kg body weight, dissolved in 11.3% (2-Hydroxy propyl)-β cyclodextrin in saline via intraperitoneal administration. The third group received VX-970 via oral gavage at 60 mg/kg body weight dissolved in 10% D-α-Tocopherol polyethylene glycol 1000 succinate in water. The fourth group received both inhibitors. Body weight and tumor volume (length × width × width × 0.52) were measured twice a week. When tumor volume reached 1,000 mm3, then that particular mouse was sacrificed and considered as endpoint.
Statistical Analysis
Statistical analyses were carried out using GraphPad Prism 5 software. Unless indicated, differences were compared using one-way ANOVA.
Author Contributions
T.H. devised the concept and supervised the project. K.S., A.H., J.M.C.-M., O.M., P.M.R., R.V.K., N.S., U.W.B., M.A., and T.H. designed, performed, and analyzed cell biology experiments. T.K., S.A.J., and M.S. designed, performed, and analyzed medicinal chemistry experiments. P.M.R., P.A.C., and J.R.P. contributed with VX-970 and discussions. K.S., P.M.R., and T.H. wrote the paper. All authors discussed results and approved the manuscript.
Conflicts of Interest
P.M.R., P.A.C., and J.R.P are employees of Vertex Pharmaceuticals (Europe) Ltd., developing ATR inhibitors commercially.
Acknowledgments
We are grateful to Dr. Hahn (Dana-Farber Cancer Institute) and Dr. Vogelstein (Johns Hopkins University) for generously providing cell lines and to Patric Herr for help in making figures. This project was supported by the Swedish Research Council (to M.A. and T.H.), the European Research Council, Swedish Cancer Society, Vinnova, the Göran Gustafsson Foundation, the Swedish Pain Relief Foundation, AFA Insurance, the Torsten and Ragnar Söderberg Foundation (all T.H.), Hållsten Foundation, Åhlen Foundation, StratNeuro (all M.A.), the Helleday Foundation (to K.S. and O.M.), and Vertex Pharmaceuticals (Europe) Ltd. (P.M.R., P.A.C., and J.R.P.).
Published: December 31, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures and seven figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.12.032.
Supplemental Information
References
- Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Børresen-Dale A.L., Australian Pancreatic Cancer Genome Initiative. ICGC Breast Cancer Consortium. ICGC MMML-Seq Consortium. ICGC PedBrain Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aris S.M., Pommier Y. Potentiation of the novel topoisomerase I inhibitor indenoisoquinoline LMP-400 by the cell checkpoint and Chk1-Chk2 inhibitor AZD7762. Cancer Res. 2012;72:979–989. doi: 10.1158/0008-5472.CAN-11-2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J., Horejsí Z., Koed K., Krämer A., Tort F., Zieger K., Guldberg P., Sehested M., Nesland J.M., Lukas C. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Bartkova J., Rezaei N., Liontos M., Karakaidos P., Kletsas D., Issaeva N., Vassiliou L.V., Kolettas E., Niforou K., Zoumpourlis V.C. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- Bester A.C., Roniger M., Oren Y.S., Im M.M., Sarni D., Chaoat M., Bensimon A., Zamir G., Shewach D.S., Kerem B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446. doi: 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks K., Oakes V., Edwards B., Ranall M., Leo P., Pavey S., Pinder A., Beamish H., Mukhopadhyay P., Lambie D., Gabrielli B. A potent Chk1 inhibitor is selectively cytotoxic in melanomas with high levels of replicative stress. Oncogene. 2013;32:788–796. doi: 10.1038/onc.2012.72. [DOI] [PubMed] [Google Scholar]
- Buisson R., Boisvert J.L., Benes C.H., Zou L. Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Mol. Cell. 2015;59:1011–1024. doi: 10.1016/j.molcel.2015.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S., Toledo L.I., Fernandez-Capetillo O., Bakkenist C.J. CGK733 does not inhibit ATM or ATR kinase activity in H460 human lung cancer cells. DNA Repair (Amst.) 2011;10:1000–1001. doi: 10.1016/j.dnarep.2011.07.013. author reply 1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- Cimprich K.A., Cortez D. ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch F.B., Bansbach C.E., Driscoll R., Luzwick J.W., Glick G.G., Bétous R., Carroll C.M., Jung S.Y., Qin J., Cimprich K.A., Cortez D. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev. 2013;27:1610–1623. doi: 10.1101/gad.214080.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Feraudy S., Revet I., Bezrookove V., Feeney L., Cleaver J.E. A minority of foci or pan-nuclear apoptotic staining of gammaH2AX in the S phase after UV damage contain DNA double-strand breaks. Proc. Natl. Acad. Sci. USA. 2010;107:6870–6875. doi: 10.1073/pnas.1002175107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R., Fumagalli M., Cicalese A., Piccinin S., Gasparini P., Luise C., Schurra C., Garre’ M., Nuciforo P.G., Bensimon A. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- Ding L., Getz G., Wheeler D.A., Mardis E.R., McLellan M.D., Cibulskis K., Sougnez C., Greulich H., Muzny D.M., Morgan M.B. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvers I., Hagenkort A., Johansson F., Djureinovic T., Lagerqvist A., Schultz N., Stoimenov I., Erixon K., Helleday T. CHK1 activity is required for continuous replication fork elongation but not stabilization of post-replicative gaps after UV irradiation. Nucleic Acids Res. 2012;40:8440–8448. doi: 10.1093/nar/gks646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feijoo C., Hall-Jackson C., Wu R., Jenkins D., Leitch J., Gilbert D.M., Smythe C. Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J. Cell Biol. 2001;154:913–923. doi: 10.1083/jcb.200104099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fokas E., Prevo R., Pollard J.R., Reaper P.M., Charlton P.A., Cornelissen B., Vallis K.A., Hammond E.M., Olcina M.M., Gillies McKenna W. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012;3:e441. doi: 10.1038/cddis.2012.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foote K.M., Blades K., Cronin A., Fillery S., Guichard S.S., Hassall L., Hickson I., Jacq X., Jewsbury P.J., McGuire T.M. Discovery of 4-4-[(3R)-3-Methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-yl-1H-indole (AZ20): a potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J. Med. Chem. 2013;56:2125–2138. doi: 10.1021/jm301859s. [DOI] [PubMed] [Google Scholar]
- Foote K.M., Lau A., Nissink J.W. Drugging ATR: progress in the development of specific inhibitors for the treatment of cancer. Future Med. Chem. 2015;7:873–891. doi: 10.4155/fmc.15.33. [DOI] [PubMed] [Google Scholar]
- Gabay M., Li Y., Felsher D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014;4:a014241. doi: 10.1101/cshperspect.a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gad H., Koolmeister T., Jemth A.S., Eshtad S., Jacques S.A., Ström C.E., Svensson L.M., Schultz N., Lundbäck T., Einarsdottir B.O. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature. 2014;508:215–221. doi: 10.1038/nature13181. [DOI] [PubMed] [Google Scholar]
- Gagou M.E., Zuazua-Villar P., Meuth M. Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Mol. Biol. Cell. 2010;21:739–752. doi: 10.1091/mbc.E09-07-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis V.G., Vassiliou L.V., Karakaidos P., Zacharatos P., Kotsinas A., Liloglou T., Venere M., Ditullio R.A., Jr., Kastrinakis N.G., Levy B. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- Groth P., Ausländer S., Majumder M.M., Schultz N., Johansson F., Petermann E., Helleday T. Methylated DNA causes a physical block to replication forks independently of damage signalling, O(6)-methylguanine or DNA single-strand breaks and results in DNA damage. J. Mol. Biol. 2010;402:70–82. doi: 10.1016/j.jmb.2010.07.010. [DOI] [PubMed] [Google Scholar]
- Hahn W.C., Counter C.M., Lundberg A.S., Beijersbergen R.L., Brooks M.W., Weinberg R.A. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- Halazonetis T.D., Gorgoulis V.G., Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- Hekmat-Nejad M., You Z., Yee M.C., Newport J.W., Cimprich K.A. Xenopus ATR is a replication-dependent chromatin-binding protein required for the DNA replication checkpoint. Curr. Biol. 2000;10:1565–1573. doi: 10.1016/s0960-9822(00)00855-1. [DOI] [PubMed] [Google Scholar]
- Hills S.A., Diffley J.F. DNA replication and oncogene-induced replicative stress. Curr. Biol. 2014;24:R435–R444. doi: 10.1016/j.cub.2014.04.012. [DOI] [PubMed] [Google Scholar]
- Höglund A., Nilsson L.M., Muralidharan S.V., Hasvold L.A., Merta P., Rudelius M., Nikolova V., Keller U., Nilsson J.A. Therapeutic implications for the induced levels of Chk1 in Myc-expressing cancer cells. Clin. Cancer Res. 2011;17:7067–7079. doi: 10.1158/1078-0432.CCR-11-1198. [DOI] [PubMed] [Google Scholar]
- Huntoon C.J., Flatten K.S., Wahner Hendrickson A.E., Huehls A.M., Sutor S.L., Kaufmann S.H., Karnitz L.M. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013;73:3683–3691. doi: 10.1158/0008-5472.CAN-13-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Reinhardt H.C., Bartkova J., Tommiska J., Blomqvist C., Nevanlinna H., Bartek J., Yaffe M.B., Hemann M.T. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009;23:1895–1909. doi: 10.1101/gad.1815309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones R.M., Mortusewicz O., Afzal I., Lorvellec M., García P., Helleday T., Petermann E. Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene. 2013;32:3744–3753. doi: 10.1038/onc.2012.387. [DOI] [PubMed] [Google Scholar]
- Jossé R., Martin S.E., Guha R., Ormanoglu P., Pfister T.D., Reaper P.M., Barnes C.S., Jones J., Charlton P., Pollard J.R. ATR inhibitors VE-821 and VX-970 sensitize cancer cells to topoisomerase i inhibitors by disabling DNA replication initiation and fork elongation responses. Cancer Res. 2014;74:6968–6979. doi: 10.1158/0008-5472.CAN-13-3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciani M.G., Oehlmann M., Blow J.J. Characterization of a novel ATR-dependent, Chk1-independent, intra-S-phase checkpoint that suppresses initiation of replication in Xenopus. J. Cell Sci. 2004;117:6019–6030. doi: 10.1242/jcs.01400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.X., Janetka J.W., Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol. Med. 2011;17:88–96. doi: 10.1016/j.molmed.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell J.B., Choudhuri R., Fabre K., Sowers A.L., Citrin D., Zabludoff S.D., Cook J.A. In vitro and in vivo radiation sensitization of human tumor cells by a novel checkpoint kinase inhibitor, AZD7762. Clin. Cancer Res. 2010;16:2076–2084. doi: 10.1158/1078-0432.CCR-09-3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga M., Bunting S., Montaña M.F., Soria R., Mulero F., Cañamero M., Lee Y., McKinnon P.J., Nussenzweig A., Fernandez-Capetillo O. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat. Genet. 2009;41:891–898. doi: 10.1038/ng.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga M., Campaner S., Lopez-Contreras A.J., Toledo L.I., Soria R., Montaña M.F., D’Artista L., Schleker T., Guerra C., Garcia E. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 2011;18:1331–1335. doi: 10.1038/nsmb.2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam E.A., Cortez D. ATR signalling: more than meeting at the fork. Biochem. J. 2011;436:527–536. doi: 10.1042/BJ20102162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E., Orta M.L., Issaeva N., Schultz N., Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E., Woodcock M., Helleday T. Chk1 promotes replication fork progression by controlling replication initiation. Proc. Natl. Acad. Sci. USA. 2010;107:16090–16095. doi: 10.1073/pnas.1005031107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaper P.M., Griffiths M.R., Long J.M., Charrier J.D., Maccormick S., Charlton P.A., Golec J.M., Pollard J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011;7:428–430. doi: 10.1038/nchembio.573. [DOI] [PubMed] [Google Scholar]
- Rohban S., Campaner S. Myc induced replicative stress response: How to cope with it and exploit it. Biochim. Biophys. Acta. 2015;1849:517–524. doi: 10.1016/j.bbagrm.2014.04.008. [DOI] [PubMed] [Google Scholar]
- Sørensen C.S., Hansen L.T., Dziegielewski J., Syljuåsen R.G., Lundin C., Bartek J., Helleday T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- Syljuåsen R.G., Sørensen C.S., Hansen L.T., Fugger K., Lundin C., Johansson F., Helleday T., Sehested M., Lukas J., Bartek J. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol. Cell. Biol. 2005;25:3553–3562. doi: 10.1128/MCB.25.9.3553-3562.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y., Hamed H.A., Poklepovic A., Dai Y., Grant S., Dent P. Poly(ADP-ribose) polymerase 1 modulates the lethality of CHK1 inhibitors in mammary tumors. Mol. Pharmacol. 2012;82:322–332. doi: 10.1124/mol.112.078907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo L.I., Murga M., Fernandez-Capetillo O. Targeting ATR and Chk1 kinases for cancer treatment: a new model for new (and old) drugs. Mol. Oncol. 2011;5:368–373. doi: 10.1016/j.molonc.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo L.I., Altmeyer M., Rask M.B., Lukas C., Larsen D.H., Povlsen L.K., Bekker-Jensen S., Mailand N., Bartek J., Lukas J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 2013;155:1088–1103. doi: 10.1016/j.cell.2013.10.043. [DOI] [PubMed] [Google Scholar]
- Wilsker D., Petermann E., Helleday T., Bunz F. Essential function of Chk1 can be uncoupled from DNA damage checkpoint and replication control. Proc. Natl. Acad. Sci. USA. 2008;105:20752–20757. doi: 10.1073/pnas.0806917106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.H., Shiotani B., Classon M., Zou L. Chk1 and Claspin potentiate PCNA ubiquitination. Genes Dev. 2008;22:1147–1152. doi: 10.1101/gad.1632808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabludoff S.D., Deng C., Grondine M.R., Sheehy A.M., Ashwell S., Caleb B.L., Green S., Haye H.R., Horn C.L., Janetka J.W. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol. Cancer Ther. 2008;7:2955–2966. doi: 10.1158/1535-7163.MCT-08-0492. [DOI] [PubMed] [Google Scholar]
- Zhao H., Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L., Elledge S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.