

Abstract
CARMILs regulate capping protein (CP), a critical determinant of actin assembly and actin-based cell motility. Vertebrates have three conserved CARMIL genes with distinct functions. In migrating cells, CARMIL2 is important for cell polarity, lamellipodial assembly, ruffling, and macropinocytosis. In cells, CARMIL2 localizes with a distinctive dual pattern to vimentin intermediate filaments and to membranes at leading edges and macropinosomes. The mechanism by which CARMIL2 localizes to membranes has not been defined. Here, we report that CARMIL2 has a conserved membrane-binding domain composed of basic and hydrophobic residues, which is necessary and sufficient for membrane localization, based on expression studies in cells and on direct binding of purified protein to lipids. Most important, we find that the membrane-binding domain is necessary for CARMIL2 to function in cells, based on rescue expression with a set of biochemically defined mutants. CARMIL1 and CARMIL3 contain similar membrane-binding domains, based on sequence analysis and on experiments, but other CPI motif proteins, such as CD2AP, do not. Based on these results, we propose a model in which the membrane-binding domain of CARMIL2 tethers this multidomain protein to the membrane, where it links dynamic vimentin filaments with regulation of actin assembly via CP.
Keywords: actin, cell migration, cell polarity, invadopodia, plasma membrane, vimentin, RLTPR, capping protein, lamellipodia, lipid binding
Introduction
Malignant tumors invade surrounding tissues and metastasize to distant locations, constituting the major causes of morbidity and mortality in patients with cancer (1). Invasive carcinomas undergo an epithelial to mesenchymal transition, a hallmark of which is the expression of vimentin intermediate filaments (2, 3). Indeed, vimentin expression correlates with the potential for migration and invasion of cells (4–7). Vimentin filaments have been functionally linked to the formation of focal adhesions and actin-rich protrusions, including invadopodia, which drive cell migration and invasion (8–11). Invadopodia also promote cell migration and invasion by degrading the surrounding extracellular matrix (12). The force driving membrane protrusion comes from the polymerization of actin filaments at their barbed ends, which face the plasma membrane. Regulation of polymerization of actin at the barbed end is thus critical for assembly and architecture of these actin filament networks (13–15).
Actin filaments in leading-edge protrusions are organized into branched networks of short filaments along with bundles of unbranched long filaments, which depend on Arp2/3 complex and formins, respectively, for assembly (13, 16). In both cases, capping protein (CP)3 regulates the growth of barbed ends. For Arp2/3-nucleated networks, capping of barbed ends by CP restricts filament length, thus increasing the branching density and strength of the filament network (13–16). For bundles of unbranched filaments, formins prevent the binding of CP, which keeps barbed ends free and thus promotes their continued growth (17). Cells lacking CP exhibit loss of Arp2/3-based lamella and lamellipodia, and they have increased numbers of filopodia (18).
CARMIL (CP, Arp2/3, myosin-I linker) family proteins are large (∼1400-aa) multidomain proteins found in all metazoa. Their domain architecture (Fig. 1A) includes an N-terminal non-canonical pleckstrin homology (PH) domain, followed by a leucine-rich repeat domain, a helical homodimerization domain, and an intrinsically disordered region containing a CP-binding region and a proline-rich domain (19, 20). The CP-binding region is composed of two conserved motifs: the CP-interaction (CPI) motif and the CARMIL-specific interaction (CSI) motif.
FIGURE 1.
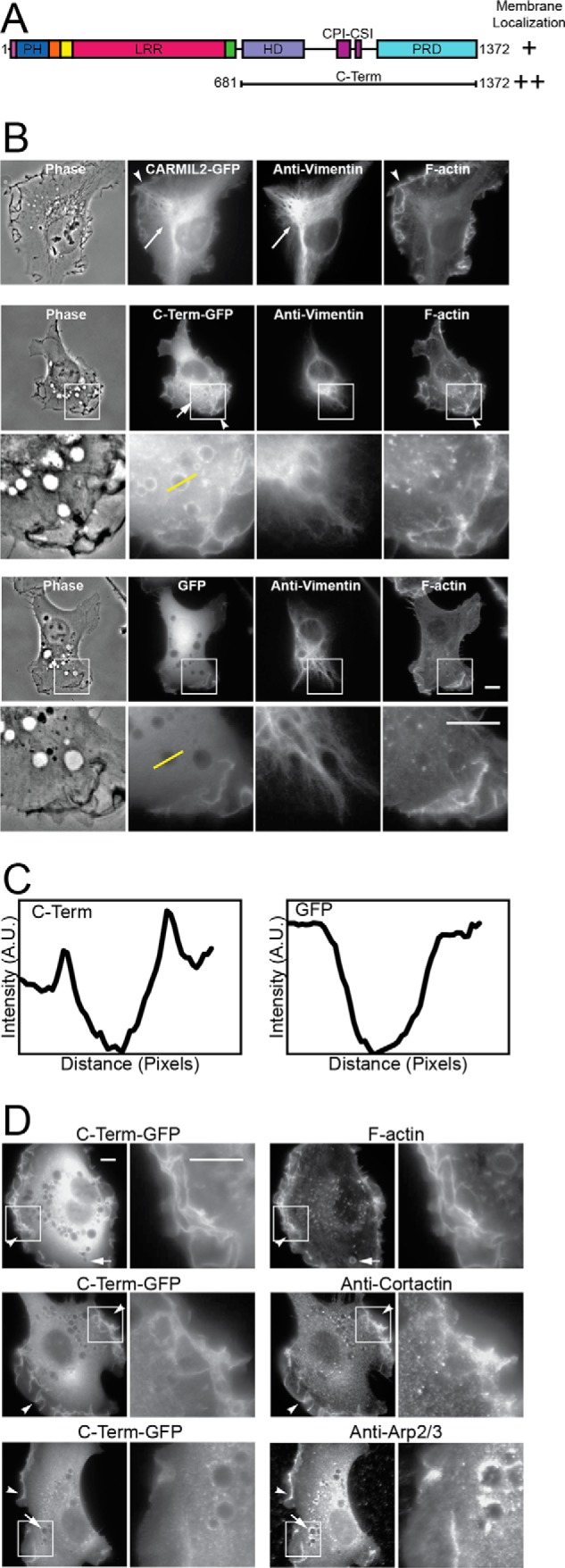
C-terminal half of CARMIL2 localizes to membranes. A, domain architecture of CARMIL2, indicating the CARMIL2 C-terminal portion (C-Term) used here. B, localization of full-length CARMIL2 (upper panels) and C-term (middle panels), as GFP fusions, compared with vimentin and F-actin, in HT1080 cells. Plain GFP (lower panels) is a negative control. Boxed regions are shown at higher magnification. Long arrows, co-localization of CARMIL2 with vimentin. Short arrows, puncta of CARMIL2 C-Term-GFP on macropinosomes. Arrowheads, ruffling membranes at the leading edge. Yellow lines indicate location of the line scans for panel C. Scale bars, 10 μm. C, line scans through macropinosomes in images from panel B. CARMIL2 C-Term-GFP fluorescence intensity shows peaks at the edges of macropinosomes. GFP, a negative control, does not. D, dual localization of CARMIL2 C-Term-GFP with F-actin and actin regulators. F-actin and Arp2/3 complex co-localize with CARMIL2 C-Term at leading-edge membranes and macropinosomes. Cortactin co-localizes at leading-edge membranes, but not macropinosomes. Boxed regions are shown at higher magnification. Scale bars, 10 μm.
Vertebrates have three conserved isoforms of CARMIL, whereas lower organisms have only one (19). Evidence to date, which is not complete, indicates that the three vertebrate isoforms, called CARMIL1, CARMIL2, and CARMIL3, have distinct functions (19, 21).
We have investigated the roles of vimentin and actin in cell migration and invadopodia formation, addressing the hypothesis that CARMIL2 links the two systems structurally and functionally at the molecular level (22). Loss of CARMIL2 in cultured fibrosarcoma cells (HT1080) leads to loss of macropinocytosis, defective lamellipodial assembly, impaired collective-cell migration, and a distinctive multi-polar phenotype (21, 22). Most intriguing, CARMIL2 localizes to vimentin filaments in cells (21). We recently demonstrated that both vimentin localization and CP binding are necessary for the function of CARMIL2 (22).
Here, we report that CARMIL2 also localizes to the plasma membrane and macropinosomes derived from the plasma membrane, via a domain composed of basic and hydrophobic residues. We investigated the physiological significance of this membrane-binding domain for the function of CARMIL2. We determined that the membrane-binding domain is necessary and sufficient for membrane localization and necessary for all cellular functions of CARMIL2, including cell migration in wound healing and invadopodia-mediated matrix degradation.
Experimental Procedures
Antibodies and Reagents
Chemical and biological reagents and supplies were from Sigma or Fisher Scientific (Pittsburgh, PA), unless stated otherwise. Plasmids used in this study are listed in Table 1. To detect human CARMIL2 by immunoblot, we produced a rabbit polyclonal (pAb) against a peptide consisting of amino acid residues 665–678 (HPTRARPRPRRQHH). To detect vimentin by immunostaining, we used mouse monoclonal antibody (mAb) 3B4 (Millipore, Billerica, MA). Other antibodies were as follows: rabbit pAb anti-ARPC2/p34 (Millipore); mouse mAb anti-cortactin 4F11; mouse mAb anti-CP 2A3 (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA); rabbit anti-caveolin (BD Biosciences, San Jose, CA); rabbit pAb anti-GFP 6C5 and mouse mAb anti-GAPDH (Abcam, Cambridge, MA); horseradish peroxidase-conjugated secondary antibodies (Sigma); Alexa Fluor-conjugated secondary antibodies (Life Technologies). To stain for F-actin, we used Alexa Fluor-conjugated phalloidin (Sigma).
TABLE 1.
Plasmids used in this study
| Plasmid | pBJ Number |
|---|---|
| pAcGFP1-C1 | 1987 |
| pAcGFP1-C1-CARMIL2 | 2017 |
| pAcGFP1-C1-CARMIL2 C-Term | 2019 |
| pAcGFP1-C1-CARMIL2-27-aa | 2286 |
| pAcGFP1-C1-CARMIL2-11-aa | 2285 |
| pAcGFP1-C1-CARMIL11059-1085 | 2439 |
| pAcGFP1-C1-CARMIL31043-1069 | 2440 |
| pAcGFP1-C1-CD2AP96-122 | 2441 |
| pAcGFP1-C1-CKIP124-50 | 2442 |
| pAcGFP1-C1-CKIP177-103 | 2443 |
| pAcGFP1-C1-Cin8585-111 | 2444 |
| pAcGFP1-C1-CapZIP153-179 | 2445 |
| pAcGFP1-C1-Fam211002-1028 | 2446 |
| pGEX-KG-CARMIL2-27-aa | 2447 |
| pAcGFP1-C1-CARMIL2 Δ11-aa | 2308 |
| pAcGFP1-C1-CARMIL2 GGG | 2312 |
| pAcGFP1-C1-CARMIL2 EEE | 2316 |
| pAcGFP1-C1-CARMIL2 C-Term Δ11-aa | 2271 |
| pAcGFP1-C1-CARMIL2 C-Term GGG | 2272 |
| pAcGFP1-C1-CARMIL2 C-Term EEE | 2273 |
| pFLRu-FH-GFP-Scramble | 1967 |
| pFLRu-FH-GFP-CARMIL2 knockdown | 1971 |
| pBOB-GFP CARMIL2 shRNA-resistant | 2299 |
| pBOB-GFP CARMIL2 Δ11-aa shRNA resistant | 2387 |
| pBOB-GFP CARMIL2 GGG shRNA resistant | 2388 |
| pBOB-GFP CARMIL2 EEE shRNA resistant | 2389 |
| tdTomato-N1 | 2090 |
Immunoblots
Immunoblots were probed with primary and secondary antibodies listed above, developed with SuperSignal West Pico Chemiluminescent substrate (Thermo-Scientific, Rockford, IL), and exposed to autoradiography film.
Membrane-binding Domain Search
The CARMIL2 membrane-binding domain was initially identified with a computer program using an algorithm that detects basic and hydrophobic residues (helixweb.nih.gov/bhsearch) (23). NCBI accession numbers for sequences of human CPI motif proteins used in searches were as follows: CARMIL2 (ACI49710); CARMIL1a (ACI49709); CARMIL3 (NP_612369); CD2AP (NP_036252.1); CKIP-1 (NP_057358.2); Cin85 (NP_114098.1); CapZIP (NP_443094.3); WASHCAP/FAM21 (NP_05677.2). The algorithm identified the following regions: CARMIL2-(1060–1070); CARMIL1-(1067–1071); CARMIL3-(1050–1062); CD2AP-(105–114); CKIP1-(89–92); Cin85-(93–103); CapZIP-(159–173); and Fam21-(1009–1021).
PM Index Calculation
We quantitated targeting of GFP fusions to the plasma membrane from fluorescence images by calculating a plasma-membrane index as described (24). To account for volume effects, td-Tomato was co-expressed from a second plasmid (pBJ 2090, Table 1). The equation for the PM index was as follows:
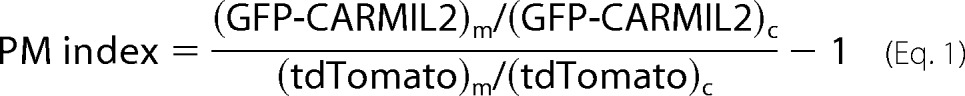 |
(GFP-CARMIL2)m, (GFP-CARMIL2)c, tdTomatom, and tdTomatoc are average fluorescence intensities of GFP constructs and tdTomato at the membrane (m) at the leading edge of the migrating and in the cytoplasm (c) behind the leading edge, measured using ImageJ.
Sequence Alignments
Sequence alignments for CARMIL family membrane-binding domains and PH domains were made with ClustalW and MegAlign (DNAStar, Madison, WI).
For CARMIL2, NCBI database sequences were as follows: human ACI49710; chimpanzee XP_523395; mouse Q3V3V9; dog XP_536814; cow XP_003587270; cat XP_003998218; goat XP_005692517; guinea pig XP_003472064; white rhinoceros XP_004431792; long-tailed chinchilla XP_005403791; star-nosed mole 004690278; nine-banded armadillo XP_004459744; horse XP_005608389; Western lowland gorilla XP_00405788; thirteen-lined ground squirrel XP_005318381; lesser Egyptian jerboa XP_004661621; golden hamster XP_005076355; prairie vole XP_005345516; gray short-tailed opossum XP_003339802; ferret XP_004802006; American pika XP_004584220; Degu XP_004625917; walrus XP_004393431; orca XP_004280894; bonobo XP_003812265; olive baboon XP_003917099; black-capped squirrel monkey XP_003936397; Tasmanian devil XP_003758471; common shrew XP_004600966; West Indian manatee XP_004371706; bottlenose dolphin XP_004326832; chicken XP_414033; collared flycatcher XP_005052671; budgerigar XP_005152479; rock dove XP_005512719; ground tit XP_005526471; zebra finch XP_002196131; white-throated sparrow XP_005490793; and zebrafish XP_009291885.
For CARMIL1, NCBI database sequences were as follows: human ACI49709; chimpanzee JAA04583; mouse NP_081101; dog XP_003434241; cow NP_001179832; cat XP_006931462; goat XP_005696872; giant panda XP_002915852; water buffalo XP_006051290; cape golden mole XP_006872885; star-nosed mole XP_004693386; wild bactrian camel XP_006182263; Chinese hamster XP_003502236; common marmoset JAB45009; long-tailed chinchilla XP_005403404; guinea pig XP_003468819; white rhinoceros XP_004432118; nine-banded armadillo XP_004462123; horse XP_003363795; cape elephant shrew XP_006888705; Western lowland gorilla XP_004043459; naked mole rat XP_004847643; lesser Egyptian jerboa XP_004670003; African bush elephant XP_003416520; Weddell seal XP_006741155; golden hamster XP_005066503; gray short-tailed opossum XP_001366647; crab-eating macaque XP_005553941; Rhesus macaque AFJ70756; prairie vole XP_005355024; ferret XP_004787996; Northern white-cheeked gibbon XP_003263275; platypus XP_001517071; sheep XP_004019144; Degu XP_004640197; Northern greater galago XP_003788390; orca XP_004273560; American pika XP_004593141; walrus XP_004406550; black flying fox XP_006919853; olive baboon XP_003897186; sperm whale XP_007116292; Tibetan antelope XP_005982838; bonobo XP_003823256; Siberian tiger XP_007074546; rat NP_001178621; common shrew XP_004614029; black-capped squirrel monkey XP_003927340; Tasmanian devil XP_003760172; wild boar XP_005665699; Chinese tree shrew XP_006162756; West Indian manatee XP_004384033; alpaca XP_006198657; thirteen-lined ground squirrel XP_005337086; mallard XP_005028957; rock dove XP_005505337; collared flycatcher XP_005041860; saker falcon XP_005432081; peregrine falcon XP_005240376; medium ground finch XP_005419441; chicken NP_001152842; turkey XP_003204958; budgerigar XP_005153647; ground tit XP_005517383; zebra finch XP_002191117; white-throated sparrow XP_005483038; American alligator XP_006260613; Chinese alligator XP_006031165; green sea turtle XP_007060452; painted turtle XP_005281477; Chinese soft-shelled turtle XP_006134063; and West Indian ocean coelacanth XP_006009837.
For CARMIL3, NCBI database sequences were as follows: human NP_612369; chimpanzee XP_509860; mouse NP_001019816; dog XP_005623303; cow NP_001179393; cat XP_003987543; yak XP_005904956; water buffalo XP_006077774; common marmoset JAB47510; wild Bactrian camel XP_006172904; guinea pig XP_003474512; white rhinoceros XP_004421263; long-tailed chinchilla XP_005377115; cape golden mole XP_006835477; star-nosed mole XP_004694678; Chinese hamster XP_003502140; nine-banded armadillo XP_004463410; small Madagascar hedgehog XP_004698968; cape elephant shrew XP_006883224; horse XP_001918362; naked mole rat XP_004864201; thirteen-lined ground squirrel XP_005338803; lesser Egyptian jerboa XP_004661869; Weddell seal XP_006738700; crab-eating macaque XP_005560962; rhesus macaque XP_001103904; golden hamster XP_005085644; prairie vole XP_005370511; ferret XP_004801153; Brandt's bat XP_005859679; mouse-eared bat XP_006760947; little brown bat XP_006102334; Northern white-cheeked gibbon XP_003260996; American pika XP_004584764; Degu XP_004647501; walrus XP_004402123; Orca XP_004283259; Northern greater galago XP_003801988; sheep XP_004010949; bonobo XP_003809113; olive baboon XP_003901663; prairie deer mouse XP_006988583; sperm whale XP_007130155; Sumatran orangutan XP_002824679; black flying fox XP_006913630; rat Q5XHY1; black-capped squirrel monkey XP_003924686; Tasmanian devil XP_003755927; common shrew XP_004609733; wild boar XP_003128578; West Indian manatee XP_004390554; bottlenose dolphin XP_004314696; alpaca XP_006217377; American alligator XP_006259804; Chinese alligator XP_006036594; and painted turtle XP_005290215.
Lipid Co-sedimentation Assays
To prepare GST-27-aa to use in co-sedimentation assays, a pair of complementary DNA oligonucleotides encoding CARMIL2 residues Thr1052-Arg1078 (IDT, Coralville, IA) were cloned into pGEX-KG (ATCC). DNA sequencing of the insert and junctions verified the identity and integrity of the plasmid. Protein was expressed in BL21(DE3) Escherichia coli induced with isopropyl β-d-thiogalactoside (0.3 mm final concentration) at 25 °C for 3 h. GST-27-aa was purified with glutathione Fast-Flow Sepharose resin (GE Healthcare, Piscataway, NJ); elution buffer was 20.0 mm Tris-HCl, pH 7.4 (22.0 °C), 1.0 mm EDTA, 100 mm NaCl, 1.0 mm NaN3, 5.0 mm dithiothreitol, 1.0 mm glutathione. The eluate was concentrated, dialyzed into storage buffer (20.0 mm Tris-HCl, pH 7.5, 1 mm Tris-(2-carboxyethyl)phosphine, 50 mm KCl, 1.0 mm NaN3), snap frozen with liquid N2, and stored at −70 °C. Protein concentration was determined from A280 using calculated extinction coefficients, confirmed by SDS-polyacrylamide gels stained with Coomassie Blue.
Assays for co-sedimentation of CARMIL2 with lipids were performed as described (25). Folch fraction I lipids (Sigma) suspended at 2 mg/ml in liposome buffer (20 mm HEPES, pH 7.4, 150 mm NaCl, 1 mm DTT) were sonicated and passed through a 200-nm filter nine times. Resulting liposomes (100 μl) were mixed with GST-27-aa at 5 μm, incubated at room temperature for 15 min, and pelleted at 150,000 × g for 30 min. The pellets were analyzed by SDS-PAGE with Coomassie Blue staining.
Cell Culture, Transfection, Knockdown, and Rescue of CARMIL2
Human CARMIL2b cDNA (NCBI: FJ026014) was the starting material for all subcloning and mutagenesis. GFP fusions were created by subcloning CARMIL2 and fragments into pAcGFP1-C1 (Clontech) at BglII and HindIII sites. For fragments, truncation sites were chosen based on secondary structure predictors (DomPred) and alignments with the mouse CARMIL1 structure of Zwolak and colleagues (20). Drs. Adam Zwolak and Roberto Dominguez of the University of Pennsylvania provided critical advice on selecting sites to yield stable protein.
Human HT1080 cells (ATCC, Manassas, VA) were grown in DMEM (Gibco BRL, Grand Island, NY) supplemented with 10% fetal bovine serum (Sigma) at 37 °C with 5% CO2. Cells were transfected using Transit-LT1 (Mirus, Madison, WI). HT1080 cells were used for all assays because they are highly motile and express almost exclusively the CARMIL2b isoform used in the rescue experiments. These cells also contain CARMIL1, but lack CARMIL3 (21).
To deplete endogenous CARMIL2 from cells, we used shRNA in a lentiviral vector, pFLRu-FH-GFP, as described (21). The target sequence was GCAAAGATGGCGAGATCAAG, with the sequence CAGTCGCGTTTGCGACTGG serving as a scrambled non-targeting control. For expression rescue of CARMIL2-depleted cells, an shRNA-resistant CARMIL2 cDNA was cloned into pBOB-GFP and introduced into cells with lentivirus as described (26). Expression rescue plasmids were made resistant to shRNA with three codon-silent nucleotide changes (lowercase): GCAAgGAcGGgGAGATCAAG, using QuikChange mutagenesis. CARMIL2 membrane-binding domain mutants for expression rescue were also created using QuikChange site-directed mutagenesis (Stratagene, La Jolla, CA). Levels of CARMIL2 depletion and rescue expression from immunoblots were quantified using ImageJ from three independent replicates.
Membrane Fractionation Assays
HT1080 cells grown on 10-cm plates were lysed 24 h post-transfection with 200 μl of fractionation buffer (250 mm sucrose, 20 mm HEPES, pH 7.4, 10 mm KCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 1 mm DTT) by cell scraping. Unlysed cells were removed by centrifugation at 100 × g for 10 min at 4 °C. The membrane fraction was then pelleted at 10,000 × g for 1 h at 4 °C. The membrane and cytoplasmic fractions were then analyzed by SDS-PAGE and immunoblotting.
Immunofluorescence and Live-cell Imaging
For immunofluorescence, HT1080 cells were grown on glass coverslips coated with 30 μg/ml of fibronectin (Sigma) and infected with lentivirus or transfected with plasmid. Lentivirus-infected cells were fixed 72 h post-infection, and cells transfected with plasmid were fixed 24 h post-transfection. For staining with phalloidin or most antibodies, cells were fixed with 2.5% paraformaldehyde, 0.25% glutaraldehyde in PIPES buffer (125 mm NaCl, 5 mm KCl, 1.25 mm NaH2PO4, 0.4 mm KH2PO4, 1 mm MgCl2, 5.5 mm glucose, 0.5 mm EGTA, 20 mm PIPES, pH 7.1) at 37 °C for 10 min and then quenched with 1 mg/ml of NaBH4 in Dulbecco's phosphate-buffered saline, pH 7.1. Cells were permeabilized with 0.1% Triton X-100 in PIPES buffer at room temperature for 10 min. For vimentin staining, cells were fixed in ice-cold MeOH at −20 °C for 10 min. For CP staining, cells were fixed in GlyoFixx (Thermo-Scientific) at 37 °C for 10 min followed by permeabilization with 0.1% Triton X-100 in PIPES buffer at room temperature for 10 min. Cells were imaged using ×60/1.4 NA and ×100/1.4 NA objectives on an Olympus IX70 inverted microscope (Olympus, Melville, NY) equipped with a Coolsnap HQ camera (Photometrics, Woburn, MA). Images were collected and initially processed with Micromanager (27) and ImageJ. Pseudocoloring was performed with the Fire look-up Table in ImageJ.
For live-cell fluorescence movies, cells were grown on glass-bottom culture dishes (MatTek, Ashland, MA) coated with 30 μg/ml of fibronectin (Sigma) in DMEM (Gibco) supplemented with 10% fetal bovine serum (Sigma), and imaged 24 h post-transfection. Temperature and CO2 were maintained at 37 °C and 5.0%, respectively, during movie collection. Images were captured every 6 s for 5 min using a ×100/1.4NA objective. Fluorescence was pseudocolored with the Fire look-up Table in ImageJ.
For phase-contrast movies, cells were grown and handled in a similar manner. Movies of 30 migrating single cells at 72 h post-infection were acquired, with frames collected every 60 s for 1 h using a ×60/1.4 NA objective on an Olympus IX70 inverted microscope. To avoid observer bias in selecting cells for movie analysis, we imaged the first 30 isolated cells encountered when scanning the dish in a systematic grid pattern.
Circularity and Macropinocytosis
Circularity was defined as (Areacell × 4π)/(Perimetercell2) (28), using the area and perimeter of cells from the initial frame of phase-contrast time-lapse movies (n = 30). Area and perimeter were measured with ImageJ. Macropinosomes were counted as phase-bright vesicles in the initial frame of phase-contrast time-lapse movies (n = 30) of migrating single cells.
Cell Migration in Wound Healing
HT1080 cells were infected with lentivirus for depletion of endogenous CARMIL2 and expression rescue by CARMIL2 mutants. Cells grew to a confluent monolayer on glass-bottom culture dishes (MatTek, Ashland, MA) coated with 30 μg/ml of fibronectin (Sigma). At 72 h post-infection, a wound was scratched with a pipette tip. Measurements were made every hour for 4 h using an ocular micrometer.
Invadopodia Matrix-degradation Assay
CARMIL2-depleted and rescue expression cells prepared as for wound-healing experiments were plated sparsely onto coverslips coated with fluorescent gelatin (Life Technologies) as described (29). After 3 h, the preparations were fixed and stained with fluorescent phalloidin. To quantitate matrix degradation, we created a binary image from fluorescent-gelatin images by setting the threshold at 3 S.D. below mean pixel intensity. ImageJ was used to measure cell area from phalloidin-stained images and area of matrix degradation from thresholded fluorescent-gelatin images. To prevent counting stray pixels as areas of degradation, we constrained the particle analysis to areas greater than 10 pixels in size.
Statistical Analysis
We performed Student's t test on population means to determine whether parameters from depletion and rescue cells were significantly different from those for uninfected HT1080 cells. Data analysis and graphical representations were done using Prism 6 (GraphPad, La Jolla, CA).
Results
Membrane-binding Activity of CARMIL2
CARMIL2 displays two localization patterns in cells. One coincides with vimentin intermediate filaments, and the other coincides with the membrane at the leading edge of migrating cells. The membrane localization pattern includes lamellipodia and ruffles, along with macropinosomes, which are derived from ruffles (30). The molecular mechanism by which CARMIL2 localizes to these membranes is not known. To address this question, we localized a GFP fusion of the C-terminal half of CARMIL2 (C-Term, residues 681–1372) in HT1080 fibrosarcoma cells (Fig. 1A). The pattern of membrane localization was enhanced, and the pattern of vimentin localization was lost (Fig. 1, B and C). In similar experiments reported elsewhere (22), we found that the N-terminal half of CARMIL2 (residues 1–688) localized only to vimentin and not to membranes.
Lamellipodia, ruffles, and macropinosomes are associated with dynamic networks of actin filaments that are nucleated by the Arp2/3 complex, stabilized by cortactin, and regulated by CP (15, 17). CARMIL2 C-Term contains two motifs that bind to and regulate CP (22), so we asked whether CARMIL2 C-Term localized with components of dynamic actin networks (Fig. 1D). Indeed, fluorescence imaging revealed co-localization of CARMIL2 C-Term with F-actin, Arp2/3 complex, and cortactin at lamellipodia and ruffles. For macropinosomes, which are derived from ruffles, a lower level of co-localization was observed; many CARMIL2 C-Term puncta did not co-stain for F-actin or Arp2/3, and no puncta co-stained for cortactin. This difference suggests that CARMIL2 may be more intimately associated with the plasma membrane than are other actin cytoskeleton components, which are lost in the transition from ruffle to macropinosome.
Identification of a Domain Sufficient for Membrane Binding
We next sought to identify CARMIL2 domains necessary and sufficient for membrane targeting. CARMIL1 contains a non-canonical PH domain at its N terminus that mediates membrane localization (20); however, sequence alignments among vertebrate CARMILs reveals that the residues necessary for membrane binding in CARMIL1 are not conserved in CARMIL2 (Fig. 2A). In addition, localization experiments reported elsewhere demonstrate that the CARMIL2 PH domain does not localize to the membrane (22). The amino acid sequence of CARMIL2 does not reveal any other well known membrane-binding domains.
FIGURE 2.
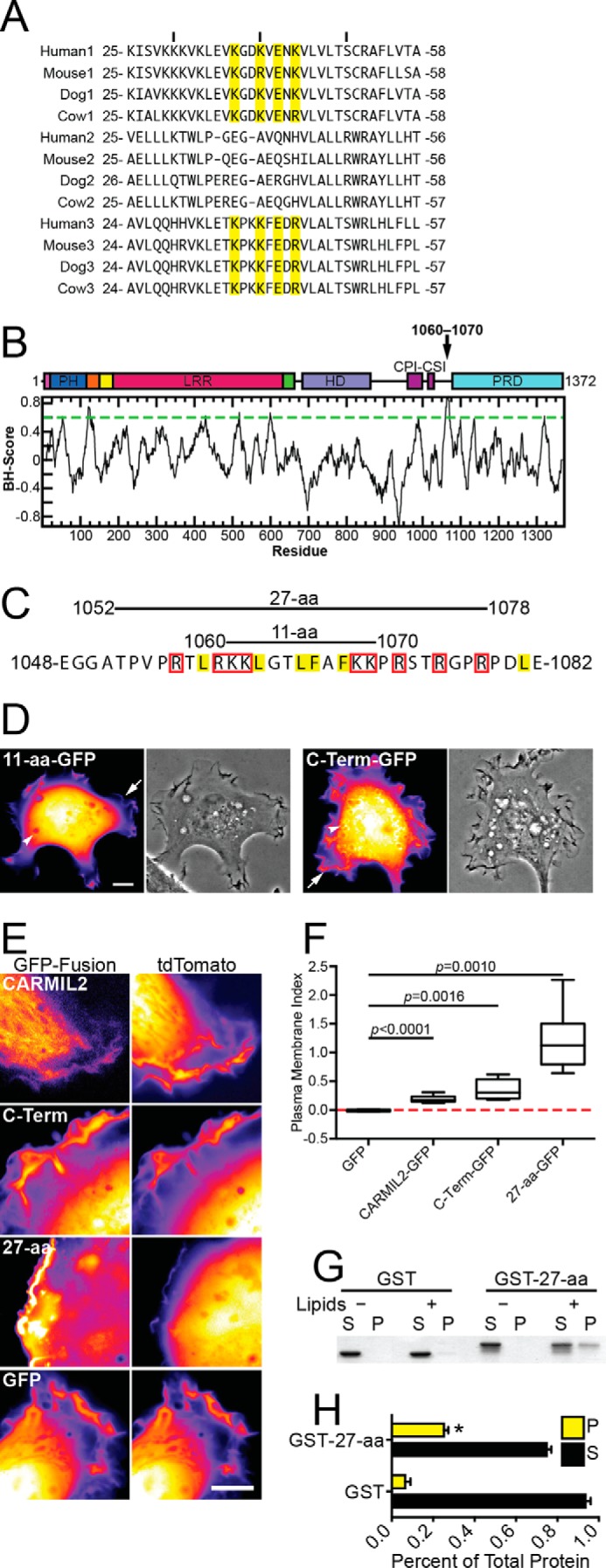
Domain of CARMIL2 sufficient for membrane localization. A, alignment of PH domains of vertebrate CARMILs. Residues necessary for membrane binding in CARMIL1 are highlighted in yellow (20). B, predicted membrane-binding residues (1060–1070), identified as described under “Results,” on a domain map and a plot of basic/hydrophobic algorithm scores for the CARMIL2 aa sequence. C, amino acid residue sequence of the region from panel A. Residues likely to contribute to lipid binding through hydrophobic or electrostatic interactions are highlighted in yellow or boxed in red, respectively (23). Fragments of CARMIL2 used for localization experiments are indicated. D, localization of the CARMIL2 11-aa fragment from panel C, compared with the C-terminal half of CARMIL2. Both are expressed as GFP fusion proteins. Representative fluorescence and phase-contrast of single HT1080 cells are shown. Arrows points to ruffling membranes at the cell edge, and arrowheads point to macropinosomes. Scale bar, 10 μm. E, CARMIL2 GFP fusion proteins in HT1080 cells, with tdTomato as a control for cytoplasmic volume. Initial frames from supplemental Movies S1–S4. Scale bar, 10 μm. F, quantitation of PM localization from fluorescence images similar to those in panel E. The PM index normalizes the membrane-associated GFP signal to the tdTomato signal (see “Experimental Procedures”). Box and whisker plots indicate median, interquartile range, and extremes. p values as indicated. Number of data points as follows: GFP, 5; CARMIL2-GFP, 7; C-Term-GFP, 6; 27-aa-GFP, 6. G, purified GST-27-aa co-sediments with liposomes composed of bovine brain lipids. S, supernatant; P, pellet. H, quantification of co-sedimentation assays from three independent experiments. Mean values are plotted. Error bars are S.E. The pellet fraction is significantly larger for GST-27-aa than for GST alone (asterisk, p = 0.0053).
To investigate the molecular mechanism for membrane targeting, we started with a computer-based algorithm that identifies clusters of basic and hydrophobic residues, as a tool to find potential intrinsically disordered membrane-binding domains (23). An 11-aa region (residues 1060–1070) was identified (Fig. 2B), which contains a high density of basic and hydrophobic residues, along with its surrounding residues (Fig. 2C). We expressed and localized a GFP fusion of the 11-aa region in cells; the fluorescence pattern was diffuse and cytoplasmic, similar to that of GFP alone, indicating that the 11-aa region is not sufficient for membrane localization (Fig. 2D).
Next, we localized a larger 27-aa region (Fig. 2C), which included the 11 residues in question, as a GFP fusion. We observed strong localization to membranes, using co-expression of tdTomato as a volume marker. In dual-fluorescence movies, comparing GFP with tdTomato, the localization of the 27-aa fragment to the membrane was noticeably stronger than that of CARMIL2 full-length and C-terminal half-constructs (Fig. 2E, supplemental Movies S1–S4). To quantify membrane localization, we used the digital images to calculate a parameter called the plasma-membrane (PM) index (24), which controls for expression level differences and volume effects. The value of the PM index values is zero for a cytoplasmic protein, and a 100% increase in fluorescence intensity at the membrane increases the value of the PM index by 1 unit. For the 27-aa fragment, the PM index was 1.21 ± 0.23, compared with 0.35 ± 0.07 for the C-terminal half and 0.19 ± 0.02 for full-length CARMIL2 (Fig. 2F), confirming the conclusions from qualitative assessment of the images. All three of these PM index values were statistically significantly higher than the value for plain GFP, as a negative control. In addition, each of the three index values differed from the other values by statistically significant amounts, with probability values as follows: CARMIL2 versus C-Term, p = 0.046; CARMIL2 versus 27-aa, p = 0.0006; C-Term versus 27-aa, p = 0.0053.
To investigate the mechanism of membrane binding and extend the analysis of sufficiency, we asked whether the 27-aa domain could bind directly to membrane lipids in vitro, using purified components. We assayed for co-sedimentation of purified GST-27-aa with liposomes composed of brain lipids (Fig. 2G), using GST as a negative control. GST-27-aa sedimented with liposomes, but GST did not. In the absence of lipids, GST-27-aa did not sediment. Results of co-sedimentation assays were quantified from three independent experiments (Fig. 2H). We conclude that the CARMIL2 27-aa region is sufficient to bind to membranes in cells and directly to membrane lipids in vitro.
Necessity of the Membrane-binding Domain for Membrane Localization
To test if the domain of CARMIL2 identified as sufficient for membrane binding is necessary for CARMIL2 localization to membranes in cells, we generated a set of mutants (Fig. 3A). In the context of full-length CARMIL2 and the C-terminal half of CARMIL2, we deleted the 11-aa core of the membrane-binding domain, residues 1060–1070. In addition, we changed those 11 residues to glycine, to remove the positive charge, and to glutamic acid, to change the positive charge to negative. This domain is intrinsically disordered, based on secondary structure predictions and on biochemical studies (31, 32), so we did not expect overall protein stability to be altered dramatically by these mutations.
FIGURE 3.
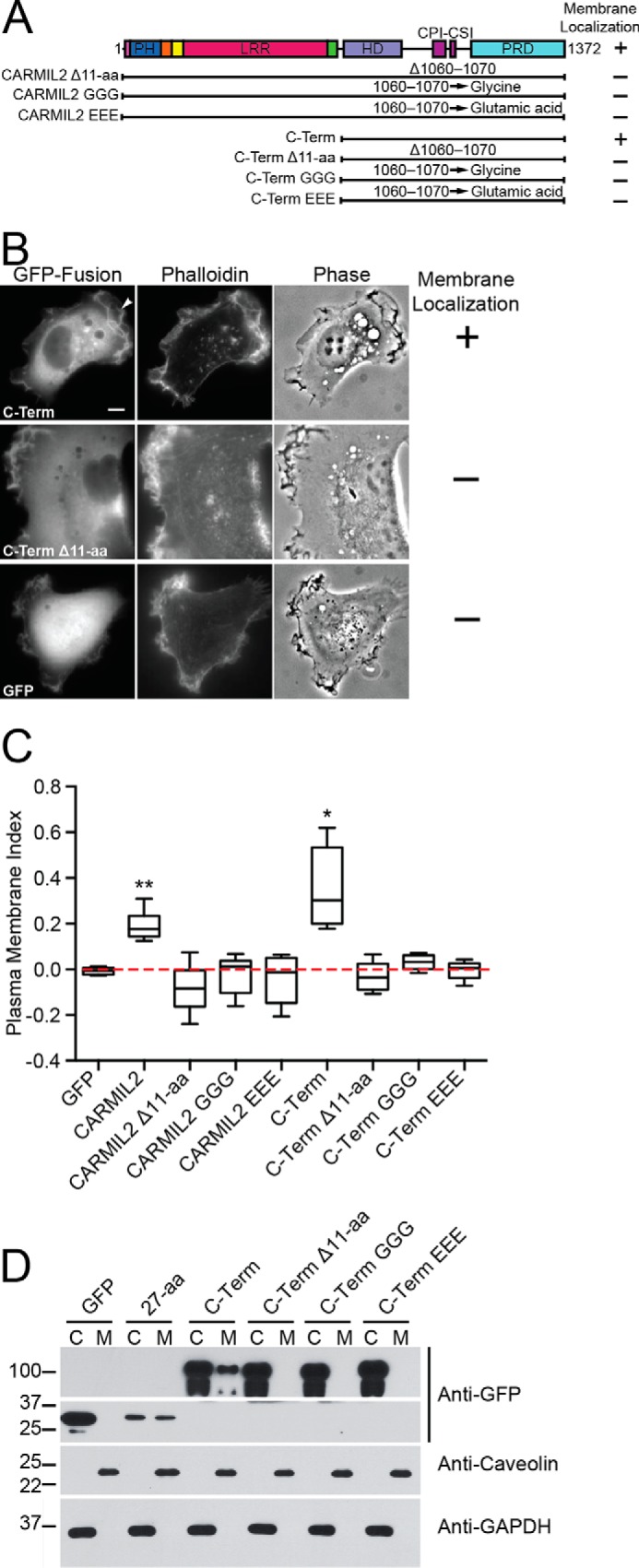
Membrane-binding domain of CARMIL2 is necessary for membrane localization. A, domain map and summary of results for CARMIL2 constructs with membrane-binding domain mutations. The 11-amino acid residues, 1060–1070 (Fig. 2), were mutated as follows: Δ11-aa, indicates deletion; GGG, indicates substitution by glycine; and EEE, indicates substitution by glutamic acid. B, representative images of CARMIL2-GFP fusions in HT1080 cells, co-stained with fluorescent phalloidin. CARMIL2 C-Term localizes to the membrane. Deletion of the membrane-binding domain led to loss of leading-edge localization. Similar images were collected for the other mutants listed in panels A and C. Scale bar, 10 μm. C, quantitation of PM localization from fluorescence images of cells expressing GFP fusion proteins from panel A, with tdTomato as a control for cytoplasmic volume. The PM index normalizes the membrane-associated GFP signal to the tdTomato signal (see “Experimental Procedures”). Box and whisker plots indicate median, interquartile range, and extremes. Calculations were made on single frames from live-cell movies (supplemental Movies S5–S10). Statistical significance as follows: double asterisk, p < 0.0001; and single asterisk, p = 0.0016. Number of data points as follows: GFP, 5; CARMIL2, 7; CARMIL2 Δ11-aa, 5; CARMIL2 GGG, 6; CARMIL2 EEE, 5; C-Term, 6; C-Term Δ11-aa, 5; C-Term GGG, 5; C-Term EEE, 5. D, membrane fractionation experiment of HT1080 cells expressing CARMIL2-GFP C-Term constructs. C, cytoplasm; M, membrane. CARMIL2-GFP C-Term is observed in the membrane fraction, whereas the mutants are not.
First, we tested the ability of the mutants to localize to the membrane, using GFP fusions expressed in HT1080 cells co-stained with fluorescent phalloidin. Deletion of the 11-aa core led to loss of accumulation at the membrane with an increase in the diffuse cytoplasmic distribution, based on qualitative assessment of the images (Fig. 3B). Similar results were obtained for the mutants in which the 11 residues were changed to glycine or glutamic acid. For all the mutants, the level of GFP fluorescence was comparable with that of wild-type CARMIL2, confirming the stability of the expressed mutant proteins.
In addition, we quantified the loss of membrane accumulation of the mutants by calculating the PM index, as discussed above, with co-expression of tdTomato as a volume marker (Fig. 3C, supplemental Movies S5–S10). In the context of full-length CARMIL2 and the C-terminal half of CARMIL2, the mutants had PM indexes similar to that of GFP alone, reflecting their complete failure to localize to the membrane. The mutants made in full-length CARMIL2 localized to vimentin filaments, and the mutants in CARMIL2 C-term were diffuse in the cytoplasm.
To extend the analysis, we performed membrane fractionation experiments using the membrane-binding mutants in the context of the C-terminal half of CARMIL2 (Fig. 3D). Although both the 27-aa domain and the CARMIL2 C-term were observed in the membrane fraction, the mutants were only present in the cytoplasm. Thus, we conclude that the 11-aa core of the membrane-binding domain is necessary for CARMIL2 to localize to the membrane in cells.
Physiological Functions of the CARMIL2 Membrane-binding Domain
To determine the physiological significance of the CARMIL2 membrane-binding domain, we tested the effect of these mutations on the ability of full-length CARMIL2 to rescue phenotypes caused by depletion of endogenous CARMIL2. Endogenous CARMIL2 was depleted with shRNA-expressing plasmids introduced into cells with lentivirus. The CARMIL2 mutants were expressed from a separate lentiviral plasmid, which was designed as resistant to the CARMIL2 shRNA by codon-silent mutations. Endogenous CARMIL2 was depleted by 82 ± 5%, as determined by immunoblot from three independent replicates. Levels of rescue expression did not significantly differ from each other (Fig. 4A).
FIGURE 4.

Membrane-binding domain mutants fail to rescue CARMIL2-depletion phenotypes. A, immunoblot showing depletion of endogenous CARMIL2 and expression of CARMIL2-GFP mutants. B, representative images reveal shapes of single migrating cells. Initial frames from 1-h movies (supplemental Movies S11–S17). Scale bar, 10 μm. C, quantitation of polarity of cells from images similar to those in panel B. Circularity of cell border calculated as described under “Experimental Procedures.” Box and whisker plots indicate the median, interquartile range, and 10th and 90th percentiles. Statistical analysis: n = 30 cells per sample, double asterisk, indicates p < 0.0001, and single asterisk, indicates p < 0.001. Values are not significantly different between CARMIL2-shRNA and mutant-rescue cells. D, macropinosomes in CARMIL2-depleted and rescue expression cells, counted in initial frames of movies (n = 30 cells). Plotted values are mean ± S.E. Asterisks indicate p < 0.0001. Values are not significantly different between CARMIL2-shRNA and mutant rescue cells. E, leading edges of CARMIL2-depleted and rescue expression cells stained with anti-vimentin, fluorescent phalloidin, or anti-CP. Arrowheads indicate F-actin and arrows indicate CP in lamellipodia. Scale bar, 10 μm.
First, we examined phenotypes related to cell polarity, lamellipodial dynamics, ruffling, and macropinocytosis (21). To assess cell polarity, phase-contrast movies of sparsely plated cells were collected (Fig. 4B, supplemental Movies S11–S13). CARMIL2-depleted cells displayed multiple leading edges, with cells attempting to move in several directions at once, instead of having a unipolar shape characteristic of persistent motion in one direction. In addition, the leading edges were smooth and flat, lacking lamellipodia and ruffles. Consistent with the lack of ruffles, the number of macropinosomes that formed was greatly decreased. In contrast, untreated cells and cells infected with negative-control scrambled-sequence shRNA exhibited a unipolar shape, with a single leading edge, and the leading edge showed prominent lamellipodia with ruffles and macropinosomes.
We quantified the cell polarity and macropinocytosis phenotypes (Fig. 4, C and D). For cell polarity, we calculated circularity, defined as the ratio of cell area to the area of a circle with the same perimeter as the cell (see “Experimental Procedures”). For macropinosomes, we simply counted their number, defining them as phase-bright vesicles that arose from ruffles. Both phenotypes were fully rescued by expression of wild-type CARMIL2, based on the images and the quantitative measures (Fig. 4, B–D, supplemental Movies S14–S17), demonstrating specificity of the CARMIL2 shRNA. In striking contrast, none of the three CARMIL2 membrane-binding mutants provided any level of rescue in either quantitative assay (Fig. 4, C and D). We conclude that the membrane-binding activity of CARMIL2 is absolutely and completely necessary for all functions of CARMIL2 in cells.
Hierarchy of Molecular Interactions and Cellular Functions
To investigate the hierarchy of the interactions of CARMIL2 with vimentin, actin, CP, and membranes, we examined the vimentin filament and lamellipodial F-actin networks in CARMIL2-depleted and mutant rescue expression cells by fluorescence imaging. Loss of function of CARMIL2 had no effect on the vimentin filament network, in any case (Fig. 4E). In contrast, F-actin networks were disrupted in all cells with loss of CARMIL2 function, including CARMIL2 membrane-binding mutants. Control cells showed strong F-actin staining in lamellipodia, ruffles, and actin bundles at the leading edge. Control cells also showed strong anti-CP staining at the leading edge and in puncta around macropinosomes. In CARMIL2-depleted cells, F-actin and anti-CP staining were severely decreased at these locations. Expression of wild-type CARMIL2 rescued the staining patterns; however, expression of CARMIL2 membrane-binding mutants had no discernable effect. Thus, the vimentin network is functionally “upstream” of CARMIL2, and actin assembly dynamics are functionally “downstream” of CARMIL2.
Cell Migration during Wound Healing
Cell migration defects in wound-healing assays have been described for cells with decreased levels of CARMIL2 (21). To investigate if the CARMIL2 membrane-binding domain is important for the role of CARMIL2 in cell migration, we performed wound-healing assays with the CARMIL2 membrane-binding mutants using the approaches described above (Fig. 5, A and B). CARMIL2-depleted cells migrated to fill a wound more slowly than controls, as expected. Expression of shRNA-resistant wild-type CARMIL2 completely rescued the migration defect; however, expression of CARMIL2 mutants provided essentially no level of rescue. Thus, CARMIL2 membrane-binding activity is necessary for CARMIL2 function in cell migration during wound healing.
FIGURE 5.

Membrane-binding domain mutants do not function for cell migration in wound healing or for matrix degradation by invadopodia. A, migration of cells to fill a scratch wound. Distance traveled by cells from the edge of the wound versus time. B, average speed of cells. Panels A and B show results from three independent experiments performed on different days. Each experiment includes nine repetitions. Mean values are plotted. Error bars are S.E. Asterisks indicate p < 0.0001. Values are not significantly different between CARMIL2-shRNA and mutant rescue cells. C, images of cells plated on fluorescent gelatin. Invadopodia (arrowheads) are identified with fluorescent phalloidin. Dark areas in fluorescent-gelatin image correspond to sites of matrix degradation. For quantification, the fluorescent-gelatin image was processed with a threshold set at 3 S.D. below mean pixel intensity. Scale bar, 10 μm. D, quantification of matrix degradation. Mean of percent of cell area with matrix degradation. Error bars are S.E. n = 20 cells. Asterisks indicate p < 0.0001. Values are not significantly different between CARMIL2-shRNA and mutant rescue cells.
Invadopodia Formation and Matrix Degradation
Cancer cells invade the surrounding tissue and degrade the extracellular matrix using specialized protrusions called invadopodia, which elongate and extend into the tissue. Invadopodia formation and elongation depend on both the actin and vimentin networks (10–12, 33–35). We found that CARMIL2 provides a molecular connection between vimentin filaments and actin assembly and that CARMIL2 is necessary for invadopodia formation and matrix degradation (22). Therefore, we investigated the importance of the membrane-binding activity of CARMIL2 for invadopodia formation.
To assess invadopodia formation, we performed a matrix degradation assay. We plated cells on fluorescent gelatin, and then imaged the formation of invadopodia and the loss of fluorescent matrix under the cells. Control cells formed F-actin-rich puncta at locations where matrix was degraded (Fig. 5C), which are the features that define invadopodia (36). Cells depleted of CARMIL2 showed neither F-actin-rich puncta nor matrix degradation. We quantified matrix degradation using a method that converted the fluorescent-gelatin image to a binary format and measured areas of matrix degradation and total cell area (Fig. 5D, see “Experimental Procedures”). Matrix degradation was significantly decreased in the CARMIL2-depleted cells. Expression of shRNA-resistant wild-type CARMIL2 rescued the matrix-degradation phenotype, whereas expression of the membrane-binding mutants did not. Thus, CARMIL2 membrane-binding activity is necessary for CARMIL2 function in invadopodia formation and matrix degradation.
Membrane-binding Domains in CARMILs and Other CPI Motif Proteins
For CARMIL1, two different domains have been implicated in membrane targeting. One is the PH domain (20); the other is a basic and hydrophobic domain in a position analogous to the one described here for CARMIL2. The basic and hydrophobic domain was predicted from sequence analysis, and a relatively large fragment containing that region was found to bind liposomes (23).
We examined the ability of the putative CARMIL1 membrane-binding domain to localize to membranes in cells, which was not examined in the previous study (23), and we asked if CARMIL3 has a membrane-binding domain in an analogous position. The computer algorithm identified the analogous regions of CARMIL1 and CARMIL3 (Fig. 6A). We expressed 27-aa fragments containing the predicted domains of CARMIL1 (residues 1059–1085) and CARMIL3 (residues 1043–1069) as GFP fusion proteins in cells. Both CARMIL1 and CARMIL3 fragments localized to the membrane (Fig. 6B), with PM index values significantly larger than that of the GFP negative control (Fig. 6C). The PM index of the CARMIL3 membrane-binding domain was similar to that of CARMIL2, and both were significantly higher than that of CARMIL1 (CARMIL1 versus CARMIL2, p = 0.0042; CARMIL1 versus CARMIL3, p = 0.0085).
FIGURE 6.
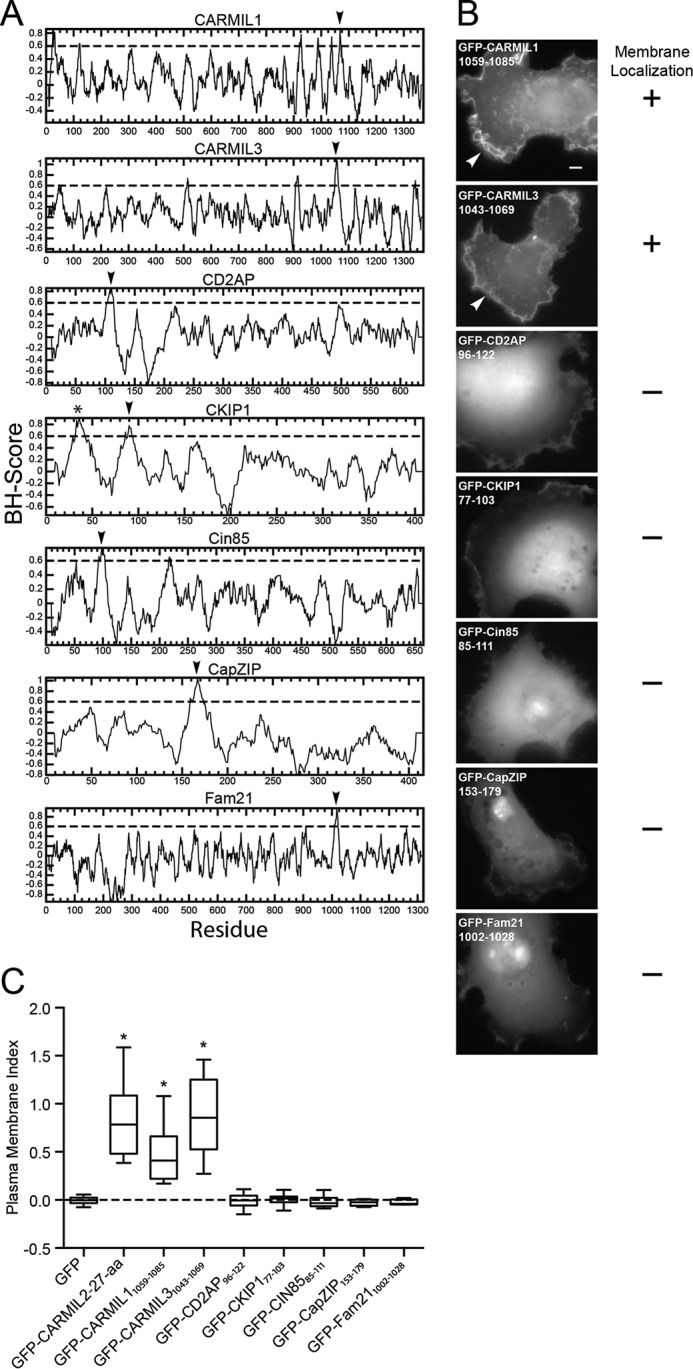
Assays for membrane localization by selected regions of CPI motif proteins. A, basic/hydrophobic plots of CPI motif proteins suggest possible membrane-binding domains (arrowheads). For CARMIL1 and CARMIL3, membrane-binding domains are predicted in locations analogous to that for CARMIL2. Asterisk denotes the known PH domain of CKIP-1. B, tests of the predicted membrane-binding domains from panel A for membrane localization. GFP fusions were expressed in HT1080 cells. Only the predicted membrane-binding domains of CARMIL1 and CARMIL3 localize to leading edge ruffles, similar to CARMIL2. Scale bar, 10 μm. C, plasma membrane index values for GFP fusions, with TdTomato as a cytoplasmic volume marker. Box and whisker plots show median, interquartile range, and the extremes. Number of data points as follows: GFP, 19; CARMIL2-(1052–1078), 19; CARMIL1-(1059–1085), 12; CARMIL3-(1043–1069), 14; CD2AP-(96–122), 15; CKIP1-(77–103), 16; Cin85-(85–111), 10; CapZIP-(153–179), 9; Fam21-(1002–1028), 5. Asterisks indicate p < 0.0001 relative to GFP.
For all three CARMILs, the membrane-binding domains lie in close proximity to the CP-binding region, which is composed of two CP-binding motifs in tandem, the CPI motif and the CSI motif. A number of proteins other than CARMILs contain CPI motifs, but not CSI motifs, and many of these proteins are associated with membranes (19). Therefore, we asked if non-CARMIL CPI motif proteins possess similar membrane-binding domains. We used the same computer algorithm to analyze the sequences of CD2AP, CKIP-1, Cin85, CapZIP, and Fam21 (Fig. 6A). Regions with high scores falling within previously defined membrane-binding domains, including the N-terminal PH domain of the CPI motif protein CKIP-1, were ignored. Regions with high scores were observed in all the other CPI motif proteins, including CD2AP, Cin85, CKIP-1, CapZIP, and Fam21. We expressed a 27-aa fragment of each protein containing the high-score region as a GFP fusion in cells, but we found that none of them localized to membranes (Fig. 6B). PM index calculations quantitated membrane localization and confirmed the complete lack of membrane localization (Fig. 6C). Thus, we conclude that unstructured basic/hydrophobic membrane-binding domains are unique to CARMILs among CPI motif proteins and that the computer algorithm is sensitive but not specific for their identification.
Evolutionary Conservation of the Membrane-binding Domain
The fact that all three CARMIL family members have membrane-binding domains in the same location suggests evolutionary conservation of function. To determine whether the amino acid residues in these domains are conserved, we aligned the sequences and identified basic and hydrophobic residues in the membrane-binding domains of each CARMIL isoform. Within each isoform, the locations of basic and hydrophobic residues were conserved (CARMIL2, Fig. 7; CARMIL1, Fig. 8; CARMIL3, Fig. 9). The membrane-binding domains of birds, fish, and reptiles differed slightly from those of mammals; however, the amino acid substitutions of basic and hydrophobic residues were largely conservative. However, comparing the different isoforms (1, 2, and 3) with each other, we found no conservation of location of basic or hydrophobic residues, indicating that the membrane-binding domains have diverged during evolution and suggesting that they may have distinct functions.
FIGURE 7.

Sequences of CARMIL2 membrane-binding domains in vertebrates. Sequences were aligned using ClustalW. All residues, except the ones highlighted in blue, are identical to the consensus. Consensus residues likely to contribute to lipid binding through hydrophobic or electrostatic interactions are highlighted in yellow or boxed in red, respectively (23). Colored bars indicate strength of consensus.
FIGURE 8.

Sequences of CARMIL1 membrane-binding domains in vertebrates. Similar to Fig. 7, for CARMIL1 sequences.
FIGURE 9.

Sequences of CARMIL3 membrane-binding domains in vertebrates. Similar to Fig. 7, for CARMIL3 sequences.
Discussion
In this study, we discovered that CARMIL2 binds to the plasma membrane via a conserved membrane-binding domain that interacts directly with lipids. This domain is necessary and sufficient for localization of CARMIL2 to leading-edge lamellipodia, ruffles, and macropinosomes. Most important, we discovered that the membrane-binding activity of CARMIL2 is completely necessary for the function of CARMIL2 in cell migration during wound healing and invadopodia formation.
A Novel Membrane-binding Domain Necessary and Sufficient for Localization
We have determined the molecular mechanism for targeting of CARMIL2 to the membrane, which was observed in our previous studies (22), along with localization to vimentin (21). By analogy to CARMIL1 (20), one might have expected the non-canonical PH domain to play this role; however, the residues necessary for lipid binding in the CARMIL1 PH domain are not conserved in CARMIL2 (20), and expression of the CARMIL2 PH domain as a GFP fusion protein does not localize to the membrane (22). Instead, we found that the C-terminal half of CARMIL2, which does not contain the PH domain, localized exclusively and strongly to the membrane, more robustly than did markers of the lamellipodial actin network.
Within the C-terminal half of CARMIL2, we identified a 27-aa conserved membrane-binding domain composed of hydrophobic and basic residues. Similar membrane-binding domains have been described in other proteins (23, 24, 37–40). These domains are thought to be intrinsically disordered and to interact with membrane lipids through electrostatic and hydrophobic interactions. The interaction depends on the density of basic and hydrophobic residues but is independent of the sequence. We demonstrated that the CARMIL2 membrane-binding domain is necessary and sufficient for membrane localization in cells and that purified protein interacts with membrane lipids in vitro.
The activity of the membrane-binding domain may be regulated in cells. The 27-aa membrane-binding fragment is more enriched at the membrane than either full-length CARMIL2 or the C-terminal half of CARMIL2 (Fig. 2, E and F). Much of full-length CARMIL2 is targeted to vimentin filaments by the leucine-rich repeat domain (22). Disruption of targeting to vimentin, either by truncation or mutation, increases membrane targeting, but not to the degree seen with expression of the 27-aa membrane-binding domain alone. One might imagine that the 27-aa membrane-binding domain is buried within a fold of the full-length protein, however, previous studies have shown that the membrane-binding domain is found in an intrinsically disordered region of CARMIL2 (31, 32). Thus, this option seems unlikely. Alternatively, the membrane-binding domain may be subject to post-translational modification in the context of larger CARMIL2 fragments, but not when expressed alone. Intrinsically disordered regions are known targets of phosphorylation (41). The resulting electrostatic disruptions would ablate binding to membranes, and provide a potential level of regulation of localization. In support of this hypothesis, proteomic analysis of post-translation modifications has identified Thr1052 and Thr1057 as phosphorylation sites in Jurkat T-cells (42). Indeed, in preliminary experiments we found that phosphomimic mutations (T1052D, T1057D, T1064D, S1073D, and T1074D) did abolish membrane targeting, in the context of the 27-aa membrane-binding domain expressed alone as a GFP fusion (data not shown). Further study is needed to understand the physiological relevance of this observation.
Physiological Relevance of Membrane-binding Localization
We investigated the physiological relevance of the membrane-binding domain for CARMIL2 function by generating a set of CARMIL2 mutants that are not able to localize to the membrane. We tested the ability of these mutants to rescue the phenotypes that result from the loss of CARMIL2. Expression of the mutants did not rescue defects in lamellipodial formation, ruffles, macropinocytosis, or cell polarity. The lamellipodial regions showed loss of F-actin and CP, consistent with defective lamellipodial actin assembly. The CARMIL2 mutants also had no ability to rescue cell migration and invadopodia formation defects in wound-healing and matrix degradation assays. In contrast to the wound-healing assays, previous studies from our lab have shown that loss of CARMIL2 does not affect random migration of individual cells, although the mechanism behind this difference is not understood (22). From these results, we conclude that the membrane-binding domain is completely necessary for all aspects of CARMIL2 function in collective cell migration and invadopodia-mediated matrix degradation.
The membrane-binding domain is in close proximity to the CP-binding region, which targets and regulates CP, suggesting that CARMIL2 targets actin assembly to sites on membranes where lamellipodia form (14, 15). Lamellipodial actin assembly supports the creation of ruffles and the process of macropinocytosis (30). Indeed we found that all these biological processes require the CARMIL2 membrane-binding domain. In addition, the CP-binding ability of CARMIL2 is essential for these functions (22). We conclude that CARMIL2 provides a novel link between membrane phospholipids and regulators of the actin network.
Roles for Membrane Binding and Lamellipodia in Cell Migration
Lamellipodia are not important for cell migration in wound healing and several other settings (43–46). Examples include CP-binding mutants of CARMIL1 or CARMIL2, which show little or no defect in collective cell migration despite the loss of lamellipodia (22, 43). Here, the membrane-binding mutants of CARMIL2 did not support cell migration during wound healing, indicating that the membrane-binding domain has a separate function distinct from lamellipodial assembly and CP regulation. CARMIL2 mutants that cannot interact with vimentin also fail to function for cell migration (22). Therefore, one possibility is that the membrane-binding domain of CARMIL2 brings the vimentin-interacting domain of CARMIL2 to the membrane and some vimentin-related function may be necessary for collective cell migration.
Evolution of the Membrane-binding Domain
The sequences of the membrane-binding domains of the three CARMIL isoforms are similar but distinct, suggesting that their functions may have evolved in different ways. The membrane-binding domains of the three isoforms are in similar positions, near their CP-binding CPI and CSI motifs. Although all three have basic and hydrophobic residues, their sequences are not similar across isoforms, but they are similar among CARMILs of one isoform. The membrane-binding domains are only present in the vertebrate CARMIL isoforms; sequence analysis does not reveal them in CARMILs of lower eukaryotes, including amoebazoans and invertebrates. In addition, the membrane-binding domains found in CARMIL family proteins are not found in other proteins with CPI motifs. Thus, these membrane-binding domains, along with the CSI motif, may play a role in separating the function of CARMILs from those of other CPI motif proteins.
For CARMIL1, a predicted membrane-binding domain was previously identified (23) in the same location as the one we identified here in CARMIL2. Here, we showed that this domain is sufficient to bind to membranes in cells. The presence of this second membrane-binding domain in CARMIL1, in addition to the membrane-binding PH domain, explains the observation that CARMIL1 fragments lacking the PH domain maintain some ability to localize to the membrane (20). Although the physiological relevance of this membrane-binding domain for the function of CARMIL1 function has not been examined directly, we know that the membrane-binding domain of CARMIL1 is able to replace the membrane-binding domain of CARMIL2, based on expression rescue experiments using chimeras (22).
Model for CARMIL2 Function
Based on the results here, combined with those from other studies (22), we propose a model for CARMIL2 function in which dynamic vimentin filaments target CARMIL2 to critical membrane locations, where CARMIL2 then regulates CP and actin assembly (Fig. 10). The induced actin assembly causes the formation of cell protrusions involved in lamellipodia, ruffles, and macropinosome formation, as well as invadopodia. Elucidating the properties of CARMIL2 as a novel connection between vimentin, actin, and the leading edge lends new insight into our understanding of how cancer cells migrate and invade.
FIGURE 10.
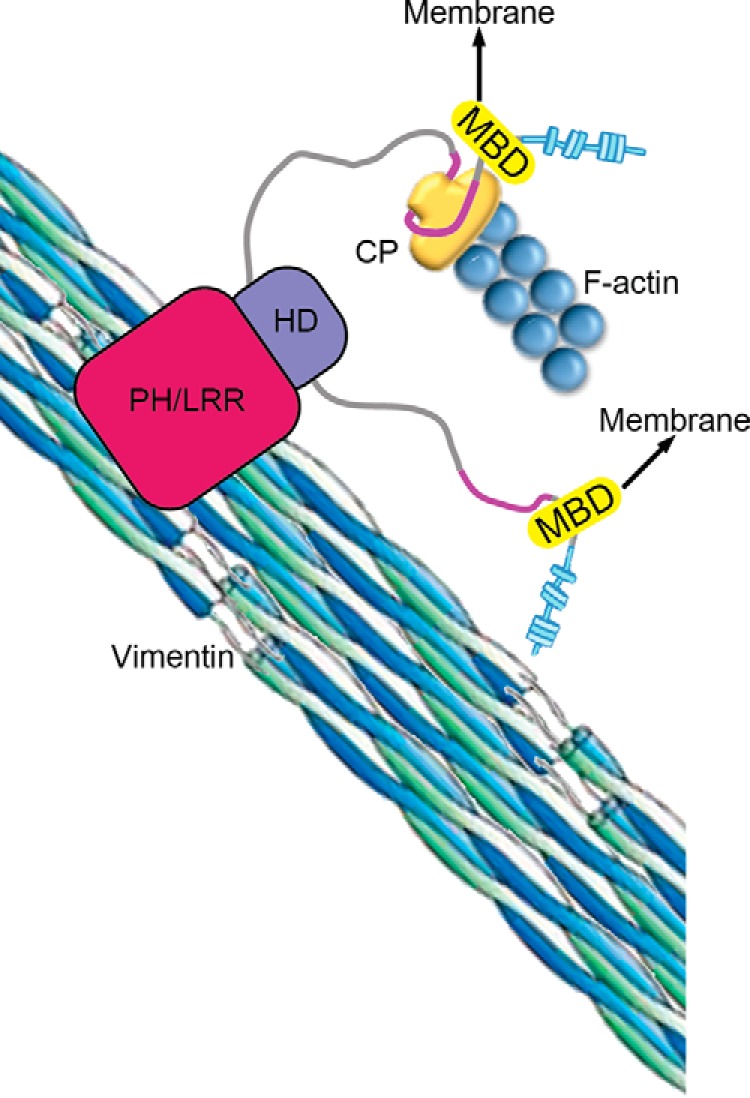
Model for CARMIL2 function in cells. The membrane-binding domain (MBD) tethers CARMIL2 to the membrane, which targets the essential functions of CP regulation and vimentin interaction to specific locations involved in lamellipodia and ruffle formation, cell migration, and invasion.
Author Contributions
M. H. L. designed and performed experiments, analyzed data and wrote the manuscript. P. M. assisted in setting up and performing lipid co-sedimentation experiments (Fig. 2). J. A. C. assisted with design of experiments and analysis of data and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Drs. Roberto Dominguez and Adam Zwolak, University of Pennsylvania, for their advice with this project. We also thank our lab colleagues for their comments and assistance, specifically Dr. Yun Liang for providing plasmids and Jinmei Li for making lentivirus.
This work was supported, in whole or in part, by National Institutes of Health Grant GM95509 (to J. A. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Movies S1–S17.
- CP
- capping protein
- PH
- pleckstrin homology
- CPI
- CP interaction
- CSI
- CARMIL-specific interaction
- PM
- plasma membrane
- aa
- amino acid
- C-Term
- C-terminal.
References
- 1.Weigelt B., Peterse J. L., and van't Veer L. J. (2005) Breast cancer metastasis: markers and models. Nat. Rev. Cancer 5, 591–602 [DOI] [PubMed] [Google Scholar]
- 2.Satelli A., and Li S. (2011) Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol. Life Sci. 68, 3033–3046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Savagner P. (2010) The epithelial-mesenchymal transition (EMT) phenomenon. Ann. Oncol. 21, vii89-vii92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eckes B., Colucci-Guyon E., Smola H., Nodder S., Babinet C., Krieg T., and Martin P. (2000) Impaired wound healing in embryonic and adult mice lacking vimentin. J. Cell Sci. 113, 2455–2462 [DOI] [PubMed] [Google Scholar]
- 5.Gilles C., Polette M., Zahm J. M., Tournier J. M., Volders L., Foidart J. M., and Birembaut P. (1999) Vimentin contributes to human mammary epithelial cell migration. J. Cell Sci. 112, 4615–4625 [DOI] [PubMed] [Google Scholar]
- 6.Menko A. S., Bleaken B. M., Libowitz A. A., Zhang L., Stepp M. A., and Walker J. L. (2014) A central role for vimentin in regulating repair function during healing of the lens epithelium. Mol. Biol. Cell 25, 776–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rogel M. R., Soni P. N., Troken J. R., Sitikov A., Trejo H. E., and Ridge K. M. (2011) Vimentin is sufficient and required for wound repair and remodeling in alveolar epithelial cells. FASEB J. 25, 3873–3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Havel L. S., Kline E. R., Salgueiro A. M., and Marcus A. I. (2015) Vimentin regulates lung cancer cell adhesion through a VAV2-Rac1 pathway to control focal adhesion kinase activity. Oncogene 34, 1979–1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Helfand B. T., Mendez M. G., Murthy S. N., Shumaker D. K., Grin B., Mahammad S., Aebi U., Wedig T., Wu Y. I., Hahn K. M., Inagaki M., Herrmann H., and Goldman R. D. (2011) Vimentin organization modulates the formation of lamellipodia. Mol. Biol. Cell 22, 1274–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schoumacher M., Goldman R. D., Louvard D., and Vignjevic D. M. (2010) Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J. Cell Biol. 189, 541–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sutoh Yoneyama M., Hatakeyama S., Habuchi T., Inoue T., Nakamura T., Funyu T., Wiche G., Ohyama C., and Tsuboi S. (2014) Vimentin intermediate filament and plectin provide a scaffold for invadopodia, facilitating cancer cell invasion and extravasation for metastasis. Eur. J. Cell Biol. 93, 157–169 [DOI] [PubMed] [Google Scholar]
- 12.Weaver A. M. (2006) Invadopodia: specialized cell structures for cancer invasion. Clin. Exp. Metastasis 23, 97–105 [DOI] [PubMed] [Google Scholar]
- 13.Nürnberg A., Kitzing T., and Grosse R. (2011) Nucleating actin for invasion. Nat. Rev. Cancer 11, 177–187 [DOI] [PubMed] [Google Scholar]
- 14.Pollard T. D., and Borisy G. G. (2003) Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465 [DOI] [PubMed] [Google Scholar]
- 15.Pollard T. D., and Cooper J. A. (2009) Actin, a central player in cell shape and movement. Science 326, 1208–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooper J. A., and Sept D. (2008) New insights into mechanism and regulation of actin capping protein. Int. Rev. Cell Mol. Biol. 267, 183–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pollard T. D. (2007) Regulation of actin filament assembly by Arp2/3 complex and formins. Annu. Rev. Biophys. Biomol. Struct. 36, 451–477 [DOI] [PubMed] [Google Scholar]
- 18.Mejillano M. R., Kojima S., Applewhite D. A., Gertler F. B., Svitkina T. M., and Borisy G. G. (2004) Lamellipodial versus filopodial mode of the actin nanomachinery: pivotal role of the filament barbed end. Cell 118, 363–373 [DOI] [PubMed] [Google Scholar]
- 19.Edwards M., Zwolak A., Schafer D. A., Sept D., Dominguez R., and Cooper J. A. (2014) Capping protein regulators fine-tune actin assembly dynamics. Nat. Rev. Mol. Cell Biol. 15, 677–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zwolak A., Yang C., Feeser E. A., Ostap E. M., Svitkina T., and Dominguez R. (2013) CARMIL leading edge localization depends on a non-canonical PH domain and dimerization. Nat. Commun. 4, 2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang Y., Niederstrasser H., Edwards M., Jackson C. E., and Cooper J. A. (2009) Distinct roles for CARMIL isoforms in cell migration. Mol. Biol. Cell 20, 5290–5305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lanier M. H., Kim T., and Cooper J. A. (2015) CARMIL2 is a novel molecular connection between vimentin and actin essential for cell migration and invadopodia formation. Mol. Biol. Cell., 10.1091/mbc.E15-08-0552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brzeska H., Guag J., Remmert K., Chacko S., and Korn E. D. (2010) An experimentally based computer search identifies unstructured membrane-binding sites in proteins: application to class I myosins, PAKS, and CARMIL. J. Biol. Chem. 285, 5738–5747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gorelik R., Yang C., Kameswaran V., Dominguez R., and Svitkina T. (2011) Mechanisms of plasma membrane targeting of formin mDia2 through its amino terminal domains. Mol. Biol. Cell 22, 189–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peter B. J., Kent H. M., Mills I. G., Vallis Y., Butler P. J., Evans P. R., and McMahon H. T. (2004) BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science 303, 495–499 [DOI] [PubMed] [Google Scholar]
- 26.Mooren O. L., Li J., Nawas J., and Cooper J. A. (2014) Endothelial cells use dynamic actin to facilitate lymphocyte transendothelial migration and maintain the monolayer barrier. Mol. Biol. Cell 25, 4115–4129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edelstein A., Amodaj N., Hoover K., Vale R., and Stuurman N. (2010) Computer control of microscopes using μManager. Curr. Protoc. Mol. Biol. Chapter 14, Unit 14.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thurston G., Jaggi B., and Palcic B. (1988) Measurement of cell motility and morphology with an automated microscope system. Cytometry 9, 411–417 [DOI] [PubMed] [Google Scholar]
- 29.Artym V. V., Yamada K. M., and Mueller S. C. (2009) ECM degradation assays for analyzing local cell invasion. Methods Mol. Biol. 522, 211–219 [DOI] [PubMed] [Google Scholar]
- 30.Kerr M. C., and Teasdale R. D. (2009) Defining macropinocytosis. Traffic 10, 364–371 [DOI] [PubMed] [Google Scholar]
- 31.Yang C., Pring M., Wear M. A., Huang M., Cooper J. A., Svitkina T. M., and Zigmond S. H. (2005) Mammalian CARMIL inhibits actin filament capping by capping protein. Dev. Cell 9, 209–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zwolak A., Uruno T., Piszczek G., Hammer J. A. 3rd, and Tjandra N. (2010) Molecular basis for barbed end uncapping by CARMIL homology domain 3 of mouse CARMIL-1. J. Biol. Chem. 285, 29014–29026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buccione R., Orth J. D., and McNiven M. A. (2004) Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat. Rev. Mol. Cell. Biol. 5, 647–657 [DOI] [PubMed] [Google Scholar]
- 34.Lorenz M., Yamaguchi H., Wang Y., Singer R. H., and Condeelis J. (2004) Imaging sites of N-wasp activity in lamellipodia and invadopodia of carcinoma cells. Curr. Biol. 14, 697–703 [DOI] [PubMed] [Google Scholar]
- 35.Yamaguchi H., Lorenz M., Kempiak S., Sarmiento C., Coniglio S., Symons M., Segall J., Eddy R., Miki H., Takenawa T., and Condeelis J. (2005) Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J. Cell Biol. 168, 441–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen W. T. (1989) Proteolytic activity of specialized surface protrusions formed at rosette contact sites of transformed cells. J. Exp. Zool. 251, 167–185 [DOI] [PubMed] [Google Scholar]
- 37.Heo W. D., Inoue T., Park W. S., Kim M. L., Park B. O., Wandless T. J., and Meyer T. (2006) PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science 314, 1458–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson J. L., Erickson J. W., and Cerione R. A. (2012) C-terminal di-arginine motif of Cdc42 protein is essential for binding to phosphatidylinositol 4,5-bisphosphate-containing membranes and inducing cellular transformation. J. Biol. Chem. 287, 5764–5774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Magalhaes M. A., and Glogauer M. (2010) Pivotal advance: phospholipids determine net membrane surface charge resulting in differential localization of active Rac1 and Rac2. J. Leukoc. Biol. 87, 545–555 [DOI] [PubMed] [Google Scholar]
- 40.Papayannopoulos V., Co C., Prehoda K. E., Snapper S., Taunton J., and Lim W. A. (2005) A polybasic motif allows N-WASP to act as a sensor of PIP2 density. Mol. Cell 17, 181–191 [DOI] [PubMed] [Google Scholar]
- 41.Iakoucheva L. M., Radivojac P., Brown C. J., O'Connor T. R., Sikes J. G., Obradovic Z., and Dunker A. K. (2004) The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 32, 1037–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mertins P., Qiao J. W., Patel J., Udeshi N. D., Clauser K. R., Mani D. R., Burgess M. W., Gillette M. A., Jaffe J. D., and Carr S. A. (2013) Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 10, 634–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edwards M., Liang Y., Kim T., and Cooper J. A. (2013) Physiological role of the interaction between CARMIL1 and capping protein. Mol. Biol. Cell 24, 3047–3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gupton S. L., Anderson K. L., Kole T. P., Fischer R. S., Ponti A., Hitchcock-DeGregori S. E., Danuser G., Fowler V. M., Wirtz D., Hanein D., and Waterman-Storer C. M. (2005) Cell migration without a lamellipodium: translation of actin dynamics into cell movement mediated by tropomyosin. J. Cell Biol. 168, 619–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suraneni P., Rubinstein B., Unruh J. R., Durnin M., Hanein D., and Li R. (2012) The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J. Cell Biol. 197, 239–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu C., Asokan S. B., Berginski M. E., Haynes E. M., Sharpless N. E., Griffith J. D., Gomez S. M., and Bear J. E. (2012) Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell 148, 973–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.