

Abstract
The recent classification of glycoside hydrolase family 5 (GH5) members into subfamilies enhances the prediction of substrate specificity by phylogenetic analysis. However, the small number of well characterized members is a current limitation to understanding the molecular basis of the diverse specificity observed across individual GH5 subfamilies. GH5 subfamily 4 (GH5_4) is one of the largest, with known activities comprising (carboxymethyl)cellulases, mixed-linkage endo-glucanases, and endo-xyloglucanases. Through detailed structure-function analysis, we have revisited the characterization of a classic GH5_4 carboxymethylcellulase, PbGH5A (also known as Orf4, carboxymethylcellulase, and Cel5A), from the symbiotic rumen Bacteroidetes Prevotella bryantii B14. We demonstrate that carboxymethylcellulose and phosphoric acid-swollen cellulose are in fact relatively poor substrates for PbGH5A, which instead exhibits clear primary specificity for the plant storage and cell wall polysaccharide, mixed-linkage β-glucan. Significant activity toward the plant cell wall polysaccharide xyloglucan was also observed. Determination of PbGH5A crystal structures in the apo-form and in complex with (xylo)glucan oligosaccharides and an active-site affinity label, together with detailed kinetic analysis using a variety of well defined oligosaccharide substrates, revealed the structural determinants of polysaccharide substrate specificity. In particular, this analysis highlighted the PbGH5A active-site motifs that engender predominant mixed-linkage endo-glucanase activity vis à vis predominant endo-xyloglucanases in GH5_4. However the detailed phylogenetic analysis of GH5_4 members did not delineate particular clades of enzymes sharing these sequence motifs; the phylogeny was instead dominated by bacterial taxonomy. Nonetheless, our results provide key enzyme functional and structural reference data for future bioinformatics analyses of (meta)genomes to elucidate the biology of complex gut ecosystems.
Keywords: carbohydrate chemistry, enzyme inhibitor, enzyme kinetics, microbiome, plant cell wall, polysaccharide, structural biology, structure-function, Bacteroidetes, affinity labeling
Introduction
The chemical and structural complexity of plant cell walls poses a challenge to organisms, from bacteria to humans, in extracting energy from biomass via polysaccharide saccharification and further metabolism. A diversity of amorphous polysaccharides (“hemicelluloses” and “pectins”), structural (glyco)proteins, and polyphenolics (“lignin”) associate with paracrystalline cellulose microfibrils within the plant cell wall to form a composite framework that is both strong and dynamic (1). Among the many matrix glycans in land plants, the diverse family of xyloglucans and the mixed-linkage glucans predominate in varying ratios, depending on the plant lineage and tissue type (2–5). Mixed-linkage glucans have a general structure composed of short stretches of β(1,4)-linked glucosyl residues (typically 3–5 residues) that are linked together by β(1,3)-linkages (Fig. 1B) (6). In contrast, xyloglucans are composed of a linear backbone of β(1,4)-linked glucosyl residues decorated with a regular pattern of α(1,6)-linked xylosyl residues, which are further extended with galactosyl, fucosyl, and/or arabinosyl residues (Fig. 1A) (7). The β(1,3) “kinks” of mixed-linkage glucan and the complex branches of xyloglucan appear to serve a similar function of inducing structural disorder, thereby endowing these polysaccharides with significant water solubility and hydrogellation properties, while at the same time maintaining affinity to cellulose.
FIGURE 1.
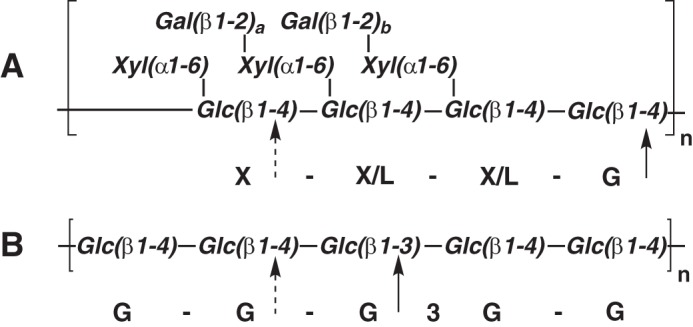
Polysaccharide structures. A, structure of tamarind xyloglucan depicting the repeating Glc4 oligosaccharide moiety and variable galactosylation (a, b = 0 or 1). The primary site of PbGH5A attack (and canonical cleavage site of XyG) is marked with a solid arrow, and the secondary site is marked with a dashed arrow. B, structure of barley β(1,3)/β(1,4)-mixed linkage glucan (depicting GGG3GG). The primary and secondary sites of PbGH5A attack are indicated as for tXyG.
The vast diversity of glycoside hydrolases (GHs)6 directed toward plant cell walls is a testament to the importance, and the challenge, of biomass degradation in the biosphere. Indeed, hundreds of thousands of GHs have been annotated in over 130 structurally related families in the Carbohydrate-Active Enzymes (CAZy) classification, the majority of which are directed to plant polysaccharides (8–10). Moreover, considerable divergent evolution has occurred within individual GH families giving rise to substrate specificity differences among members. Mapping functional diversity in such polyspecific families has been enabled by further division into phylogenetic subfamilies in some cases (11–14).
Glycoside hydrolase family 5 (GH5) is a key example of family diversity, with members demonstrating over 20 known specificities. GH5 members are united by a canonical double-displacement, anomeric configuration-retaining mechanism of hydrolysis, which involves two key catalytic carboxylic acid side chains presented on a conserved (β/α)8 protein fold (15). The recent division of GH5 into subfamilies has shown that many of these activities cluster into phylogenetic clades (11). Among these, GH5 subfamily 4 (GH5_4) constitutes one of the largest, which generally encompasses endo-β(1,4)-glucanases, including cellulases (EC 3.2.1.4), mixed-linkage endo-β(1,3)/β(1,4)-glucanases (EC 3.2.1.73), and highly specific endo-xyloglucanases (EC 3.2.1.151) evolved for the saccharification of plant biomass. GH5_4 endo-xyloglucanases (16, 17) are particularly distinguished by their ability to accommodate and harness the numerous extended α(1,6)-xylosyl branches in diverse xyloglucans (18). Unfortunately, the current level of functional characterization of GH5_4, which includes the observation that most of the characterized members have not been tested consistently on the same panel of substrates (e.g. including xyloglucan) (19, 20), means that clear delineation of polysaccharide specificity in this subfamily is not straightforward. This presents a significant difficulty for in silico analysis of (meta)genomes for functional prediction, as well as for the selection and application of specific enzymes for industrial biomass utilization.
To address this issue, we present here the characterization of a novel GH5_4 member, PbGH5A, from the symbiotic gut bacterium Prevotella bryantii B14 involved in dietary polysaccharide breakdown (21–23). Locus PBR_0368 of the P. bryantii B14 genome encodes a bi-modular gene product composed of a predicted N-terminal, Signal Peptidase I-cleavable signal peptide, followed by a GH26 module and a C-terminal GH5 module (PbGH5A) (23). Early efforts to clone PBR_0368 and characterize its product (equivalent to GenBankTM AAC97596, also known as ORF4, CMCase, or Cel5A) revealed general endo-glucanase activity via assay on carboxymethylcellulose (24, 25). However, detailed specificity data are currently lacking, especially in light of the identified bimodularity of this protein and diverse specificities found within GH26 and GH5 (26). Notably, PBR_0368 is located in a predicted Polysaccharide Utilization Locus encoding hallmark SusD- and SusC-like proteins and at least two other GHs whose collective function is currently unknown (27). In this study, kinetic analyses on a range of natural and artificial substrates, together with tertiary structures of enzyme variants in complex with oligosaccharides and an active-site affinity label, yielded molecular level insight into interactions along the entire active-site cleft responsible for the specificity of recombinant PbGH5A for mixed-linkage glucan over xyloglucan.
Experimental Procedures
Analytical Methods
HPAEC-PAD Carbohydrate Analysis
HPAEC-PAD was performed on a Dionex ICS-5000 system equipped with an AS-AP auto-sampler with a temperature-controlled sample tray run in a sequential injection configuration using Chromeleon 7 control software. The injection volume was 10 μl unless otherwise specified. A 3 × 250-mm Dionex CarboPac PA200 column with a 3 × 50-mm guard column was used for all HPAEC separations. Solvent A was ultrapure H2O; solvent B was 1.0 m NaOH prepared from a carbonate-free 50–52% stock, and solvent C was 1.000 m NaOAc prepared from anhydrous BioUltra-grade solid (Sigma). The gradients used were as follows: gradient A: 0–5 min; 10% B, 2% C; 5–12 min 10% B, 2–30% C linear gradient; 12–12.1 min 50% B; 50% C; 12.1–13 min return to initial conditions (exponential profile 9); 13–17 min, initial conditions; and gradient B: gradient A with 0% C initially; gradient C: gradient A with 6% C and a 12-min linear gradient; gradient D: gradient A with 3.5% C initially.
Mass Spectrometry
Intact protein masses were determined on a Waters Xevo Q-TOF with a nanoACQUITY UPLC system, according to the method described by Sundqvist et al. (28). MALDI-TOF analysis was performed on a Bruker Autoflex MALDI-TOF equipped with a Bruker Smartbeam-II 355-nm laser system. Samples dissolved in ultrapure water (0.1–10 mg/ml) were mixed 1:1 with 10 mg/ml 2,5-dihydroxybenzoic acid dissolved in 1:1 H2O/MeOH. The samples were left to dry under ambient conditions. MALDI spectra were acquired in positive reflectron mode, averaging 500 laser shots per spectrum. External calibration was performed using the standard mix of XXXG, XXLG/XLXG, and XLLG obtained from canonical enzymatic hydrolysis of tamarind tXyG (18).
Substrates and Inhibitors
Oligosaccharides and their derivatives are abbreviated using the general shorthand for xyloglucan oligosaccharides, in which G represents Glcp, X represents [Xylp(α1,6)]Glcp, and L represents [Galp(β1,2)Xylp(α1,6)]Glcp, with β(1,4) linkages between backbone glucosyl units as the default (7). In mixed-linkage gluco-oligosaccharides, β(1,3)-linked glucosyl residues are denoted as G3 (e.g. G3G is laminaribiose and GG is cellobiose).
Commercial Substrates
High purity (>94%) mixed-linkage glucan (β-glucan (barley; high viscosity)) (bMLG), carboxymethylcellulose (CMC), konjac glucomannan (kGM), carob galactomannan, tamarind xyloglucan (tXyG), wheat arabinoxylan, beechwood xylan, cello-oligosaccharides (Glc3-Glc6), mixed-linkage-glucan oligosaccharides (G3GGG, GG3GG, and GGG3G), mannohexaose (MMMMMM), and 2-chloro-4-nitrophenyl β-cellotrioside (GGG-CNP) were purchased from Megazyme International (Ireland) and used for all activity measurements and HPAEC-PAD experiments. Hydroxyethylcellulose (HEC) was purchased from Fluka (Sigma). 4-nitrophenyl β-glucoside and 4-nitrophenyl β-cellobioside (GG-PNP) were purchased from Sigma. 2-chloro-4-nitrophenyl β-cellobioside (GG-CNP) was purchased from Carbosynth (UK). Phosphoric acid-swollen cellulose (PASC) (29) was prepared according to Ref. 30.
Synthetic Substrates and Inhibitors
4-Nitrophenyl β-cellotrioside (GGG-PNP) was a kind gift from Prof. S. Withers (University of British Columbia). The 2-chloro-4-nitrophenyl glycoside of XXXG (XXXG-CNP) (31) and xyloglucan-derived inhibitors (32) were synthesized as described previously.
Xyloglucan Oligosaccharides (tXyGOs)
The tetradecasaccharide XXXGXXXG was prepared by partial digestion of xyloglucan from de-oiled tamarind kernel powder (dTKP, Premcem Gums) with His6-PpXG5 (16) followed by degalactosylation with Aspergillus niger β-galactosidase (Megazyme International, Bray, Ireland). XXXG was prepared similarly, with CjBgl35A replacing the A. niger β-galactosidase (33). Briefly, 100 g of dTKP were slowly added to 1 liter of 10 mm NH4OAc (pH 5.5) containing 500 units (∼2.5 mg) of PpXG5 (where 1 unit is defined as the amount of enzyme that releases 1 μmol of glucose-eq reducing ends per min). The reaction was stirred at 50 °C until a smooth, tan opaque suspension formed (∼30 min). The reaction was sampled regularly. The samples were filtered, run over Dowex 1X2 Cl, and analyzed using HPAEC-PAD (gradient C) until the population of Glc8-tXyGOs was maximal (∼4 h). The pH was raised to 8 (using 1 m NH4OH) to stop the reaction, and the solution was centrifuged for 15 min at 4000 × g. The translucent yellow supernatant was then decolorized by passage through Dowex 1X2 Cl and passed through a HisTrap FF crude column (GE Healthcare) to fully remove His6-PpXG5. The pH was then returned to 5.5 with 1 m AcOH, and 1400 units of β-galactosidase was added and stirred at 30 °C overnight. The degalactosylated tXyGOs were lyophilized for storage. 500 mg of this was then dissolved in 5 ml of ultrapure water, 0.45 μm filtered, and purified using a 90-cm P6 BioGel (Bio-Rad) column (XK 26/100, GE Healthcare), and run at 6 cm/h at room temperature. Fractions were monitored by HPAEC-PAD (gradient C) and homogeneous fractions of XXXG, XXXGXXXG, and XXXGXXXGXXXG were pooled and lyophilized to give a white foam (final yield: 200 mg of XXXG, 55 mg of XXXGXXXG, and 30 mg of XXXGXXXGXXXG).
Mixed-linkage Glucan Oligosaccharides (bMLGOs)
Glcβ(1,3)-Glcβ(1,4)-Glcβ(1,4)-Glcβ(1,3)-Glcβ(1,4)-Glcβ(1,4)-Glc(G3GGG3GGG) was prepared by the digestion of oat β-glucan (B-CAN, Garuda International) with Vitis vinifera family 16 endo-glucanase (VvEG16) (expressed and purified in-house)7 to give a mixture of oligosaccharides with the formula G3GGG(3GGG)n. 10 g of B-CAN was initially swelled in 500 ml of deionized H2O at 25 °C for 15 min. The B-CAN was then collected by centrifugation at 1000 × g for 2 min, and the supernatant was discarded. The material was washed in this manner three times to extract glucose, unidentified oligosaccharides, fines, and colored material. The swelled particles were then resuspended in 500 ml of 10 mm NH4OAc (pH 5.5) and heated to 80 °C. The solution was stirred until dissolved (∼15 min) and allowed to cool to 37 °C. 50 units (∼10 mg) of VvEG16 was then added, and the reaction was stirred at 37 °C overnight. 30 min into the digestion, the now significantly less viscous solution was centrifuged at 4000 × g for 5 min to remove a small amount of insoluble matter. The reaction completion was confirmed based on the oligosaccharide distribution observed by HPAEC-PAD (gradient A) and the opaque tan solution was centrifuged at 4000 × g for 5 min at room temperature to separate insoluble bMLG from soluble bMLG. The now clear and faintly yellow solution was then adjusted to pH 8 using 1 m NH4OH and decolorized by running through a 5-g plug of Dowex 1X2 Cl. The product was then precipitated from the clear colorless solution by the addition of 1 liter of acetone. After cooling to −20 °C in the freezer, a well flocculated white precipitate was collected by centrifugation (in a high density polyethylene bottle) at 1000 × g for 2 min. The product was dried under vacuum for several hours to give 1.52 g of a white powder. 500 mg of this was then dissolved in 5 ml of ultrapure H2O and purified using a 90-cm P2 BioGel (Bio-Rad) column (XK 26/100, GE Healthcare) run at 6 cm/h at room temperature. Fractions were monitored by HPAEC-PAD (gradient A), and homogeneousfractions of G3GGG and G3GGG3GGG were pooled and lyophilized to give white foam (final yield: 41 mg of G3GGG and 62 mg of G3GGG3GGG).
Enzyme Cloning and Expression
A DNA fragment of P. bryantii B14 locus PBR_0368 encoding amino acid residues 425–776, corresponding to the GH5 catalytic domain (PbGH5A), was received from the Joint Genome Institute (jgi.doe.gov) in a pET101 plasmid and subcloned into a cloning vector p15Tv-LIC (34) providing an N-terminal His6-tagged fusion with a tobacco etch virus protease cleavage site between the tag and the enzyme. PbGH5A was expressed in E. coli BL21(DE3) grown in auto-induction media (35) for 3 h at 37 °C and continued overnight growth at 18 °C. Cells were harvested via centrifugation at 5000 × g. The resulting pellet was resuspended in a binding buffer (50 mm HEPES (pH 7.5), 500 mm NaCl, 5 mm imidazole, and 14% glycerol (v/v)) and lysed via sonication, and cell debris was removed via centrifugation at 30,000 × g for 30 min. Cleared lysate was loaded onto a 5-ml nickel-nitrilotriacetic acid column (Qiagen) pre-equilibrated with the binding buffer, and the column was washed with the binding buffer containing 30 mm imidazole. Bound proteins were eluted using the binding buffer with 250 mm imidazole. The His6 tag was removed by cleavage with tobacco etch virus protease (expressed and purified in-house per Ref. 36) overnight at 4 °C during dialysis against 0.5 m NaCl, 10 mm HEPES (pH 7.5), and 0.5 mm tris[2-carboxyethyl]phosphine. The sample was passed over nickel-nitrilotriacetic acid resin and the flow-through was collected. Fractions containing the protein of interest were identified by SDS-PAGE.
Enzyme Kinetics and Product Analysis
Polysaccharide Hydrolysis
Polysaccharide hydrolysis was quantified using either the BCA (37) or DNSA (38) assays. For BCA assays, reactions were prepared to a final volume of 100 μl and heated to the incubation temperature for 0, 15, and 30 min before being quenched by the addition of fresh BCA reagent (100 μl). A glucose series (10–500 μm) was run with each assay. Color was developed by heating to 80 °C for 10 min before reading the absorbance at 563 nm. For DNSA assays, 100 μl of the reaction was quenched by adding 100 μl of DNSA reagent. The reaction was then heated to 95 °C for 10 min to develop color, cooled to room temperature, and centrifuged for 1 min. The absorbance was read at 540 nm.
The pH optimum of the enzyme was initially determined using the BCA assay to quantify reducing ends over 15 min of incubation of 0.05 nm native enzyme with 1 mg/ml bMLG at 37 °C using 50 mm MES (pH 5.3–7.9), MOPS (pH 7.4–8.5), Tris (pH 7.2–8.8), acetate (pH 3.75–5.5), citrate (pH 3.5–6.5), phosphate (pH 6.1–7.9), and glycine (pH 8.4–9.4) buffers. However, using the polysaccharide substrate, different kinetic pKa values were observed for different buffers (Fig. 2B). The pH optimum of the enzyme was further determined using citrate (pH 3–6), acetate (pH 3.75–5.5), MES (pH 5.3–7.9), MOPS (pH 7.4–8.5), and Tris (pH 7.2–8.4) buffers (50 mm) (Fig. 2A) with a chromogenic oligosaccharide substrate, giving two consistent kinetic pKa values. The native enzyme (0.33 nm) was incubated for 30 min with GGG-CNP (0.5 mm) at 37 °C, and then free CNP was determined by diluting 5:1 into 100 mm Na2CO3 and measurement of A405. Rates in Tris buffer were barely detectable at all pH values indicating that Tris is strongly inhibitory.
FIGURE 2.
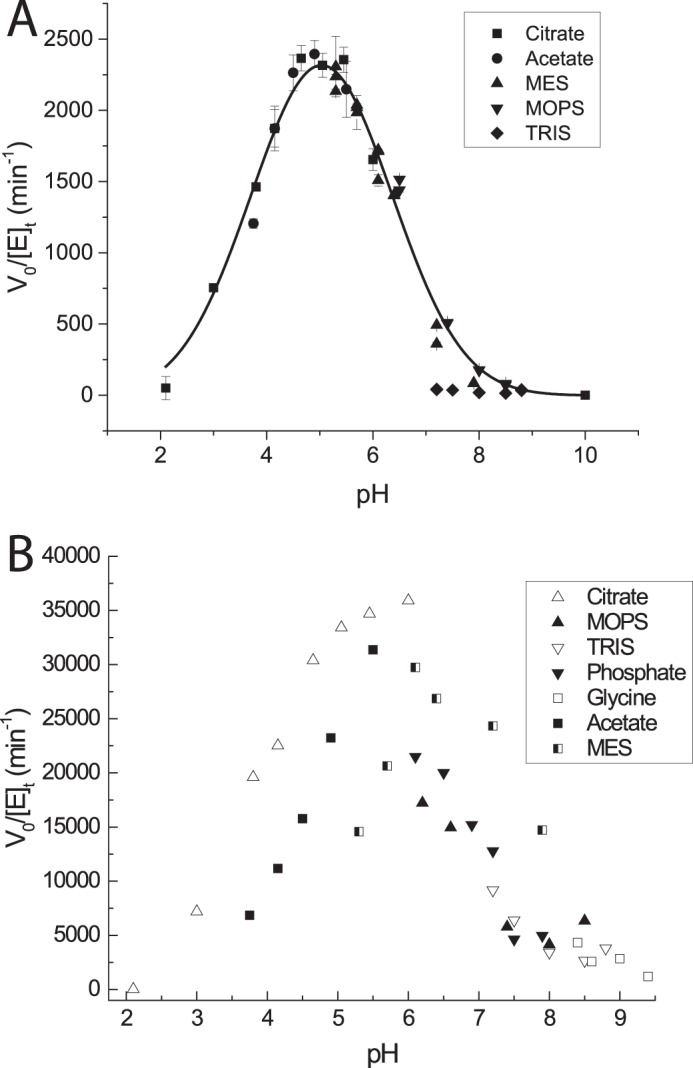
pH-rate profiles. A, activity of PbGH5A on 0.5 mm GGG-CNP in various buffers across a range of pH at 37 °C. Error bars are the standard deviation of three single-point replicates. The values (excluding the Tris series) were fit to determine the pH optimum (pH 5) and apparent kinetic pKa values (3.5 and 6.5). B, activity of PbGH5A on 1 mg/ml bMLG in various buffers across a range of pH at 37 °C. Each point is the average of two replicates.
The temperature optimum was determined in 50 mm (pH 5.5) sodium citrate buffer using 1 mg/ml bMLG as substrate (Fig. 3A) with 0.02 nm enzyme. The reaction was mixed at 4 °C and incubated at a temperature ranging from 30 to 55 °C for 30 min before reducing ends were quantified using the BCA assay. The specific activity of PbGH5A was standardized with 1 mg/ml bMLG substrate at 37 °C in 10 mm (pH 5.5) sodium citrate buffer. The thermal stability of PbGH5A was determined by incubating the enzyme (1 μg/ml in 20 mm (pH 5.5) citrate) at temperatures ranging from 30 to 74 °C. At regular time intervals, samples were taken, diluted into room temperature citrate buffer (pH 5.5), and assayed using 200 μm XXXG-CNP.
FIGURE 3.
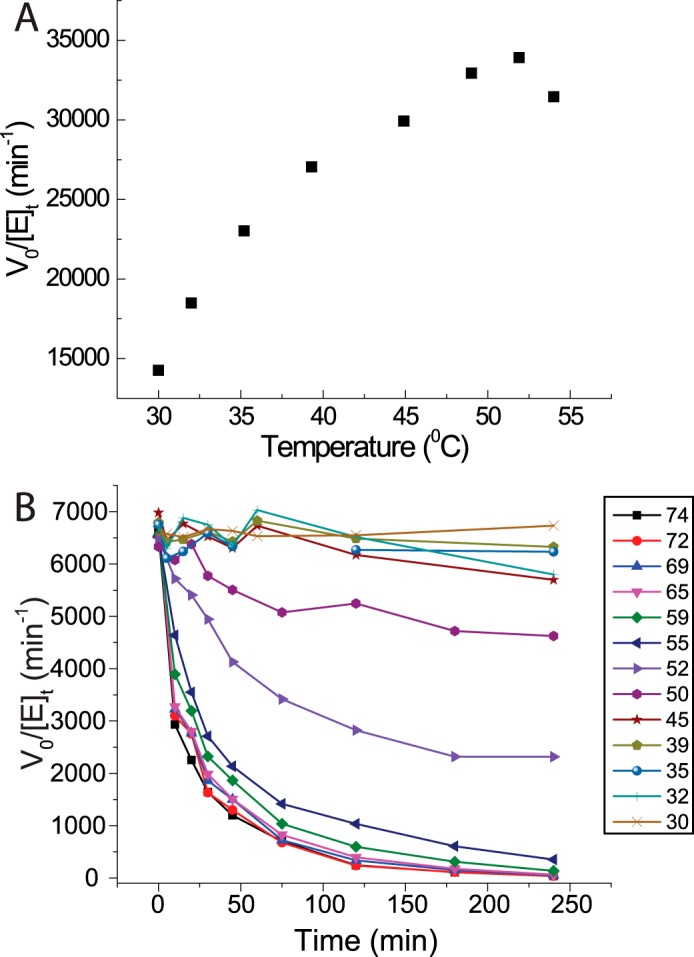
Thermal stability. A, activity of PbGH5A on 1 mg/ml bMLG in pH 5.5 NH4OAc as a function of temperature. The reaction was incubated for 15 min to minimize instability effects. Each point is the average of two single-point replicates after subtraction of a boiled-enzyme control. B, long term thermal stability of PbGH5A in pH 5 citrate buffer across a range of temperatures over 4 h. After incubation at temperature, the enzyme was diluted and assayed using 0.2 mm XXXG-CNP.
To determine limit-digestion products, PbGH5A (10 μg) was added to 1 ml of 0.1 mg/ml substrate in 50 mm NaOAc (pH 5.5) and incubated for 4 h at 37 °C. 10 μl of the reaction was then analyzed by HPAEC-PAD directly using gradient A.
Chromogenic Oligosaccharide Hydrolysis
4-Nitrophenyl glycoside hydrolysis kinetics were determined by mixing enzyme (20–1000 nm), buffer (50 mm (pH 5.5) citrate), and substrate (0.1–25 mm) to a final volume of 200 μl. At 5-min intervals, 60 μl of the reaction was diluted into 540 μl of 50 mm Na2CO3, and A405 was measured on a Cary 60 UV-visible spectrometer with a 1-cm path length quartz cuvette. An extinction coefficient of 18.2 mm−1 cm−1 was used to quantify 4-nitrophenol release. 1 unit was defined as the amount of enzyme that releases 1 μmol of 4-nitrophenol/min.
CNP substrate kinetics were determined by preheating 180 μl of 1.11× substrate stock (to give 0.02–10 mm final concentration) and adding 20 μl of 10× enzyme stock to give 0.01–100 nm final concentration in 20 mm NaOAc (pH 5.5). The change in absorbance at 405 nm was followed continuously over 10 min at 37 °C in 200-μl quartz cuvettes using a Cary 300 UV-visible spectrometer with an 8-cell sample changer and thermostat. The extinction coefficient for CNP was determined to be 10.7 mm−1 cm−1 in the buffer used. For the XXXG-CNP substrate, the assay was optimized to obtain conditions compatible for residual activity measurement. The hydrolysis was monitored in 50 mm citrate buffer at pH 5.5; absorbance was measured at 405 nm, and the extinction coefficient for CNP was determined to be 11.2 mm−1 cm−1 in the buffer used. Specific activity measurements for wild-type enzyme and the three mutants, E280A, S119A, H112A, were determined using GGG-CNP at 500 μm and XXXG-CNP at 200 μm in 50 mm citrate buffer at pH 5.5.
Native Oligosaccharide Hydrolysis
HPLC-based enzyme kinetics were determined by mixing a 10× enzyme in buffer stock (to give 0.02–10 nm and 20 mm (pH 5.5) citrate final enzyme and buffer concentration) with a 1.11× substrate stock (to give 0.005–1 mm final substrate concentration) preheated to 37 °C. For example, 10 μl of 0.2 nm PbGH5A in 200 mm sodium citrate buffer (pH 5.5) was added to 90 μl of 1.11 mm GGGGG in ultrapure H2O preheated to 37 °C. The reaction was then injected four times (10 μl each) at regular time intervals, and the change in peak area over time was quantified. Gradient A was used for monitoring cello-oligosaccharide and mixed-linkage glucan oligosaccharide degradation; gradient D was used for monitoring XXXGXXXG degradation. An 8-point linear calibration series from 0.4 to 100 μm was run for each product quantified. Rates were fit to the Michaelis-Menten model (39, 40) using OriginPro graphing software (Origin Lab).
To determine the regiospecificity of cellopentaose hydrolysis, 18O incorporation from [18O]water was determined by mass spectrometry (41). 1 μl of PbGH5A (0.10 μg/ml in 20 mm NH4OAc (pH 5.5)) and 1 μl of 0.5 m NH4OAc (pH 5.5) containing 5 mm NaOAc (to control adduct formation) were added to 22 μl of 97% [18O]water (Cambridge Isotope Laboratories) and mixed thoroughly by reciprocal pipetting. To this was then added 1 μl of 10 mm cellopentaose. The reaction was then mixed thoroughly again to give an estimated final 18O concentration of 85%. The reaction was drawn into a 50-μl gas-tight Hamilton syringe (Hamilton, model 1705) and infused into a Waters Xevo QTof at 2 μl/min using a syringe pump (Harvard Apparatus 11 Plus). The degree of isotopic labeling was quantified as the area ratio of [M + Na]+(16O-1) to [M + Na]+(18O-1).
Inhibition Kinetics
Inhibition kinetic parameters were determined at 37 °C using 0.038 μm PbGH5A in 25 mm citrate buffer at pH 5.5, containing 1% bovine serum albumin (BSA), and incubation with various putative inhibitor concentrations (0.1–3.5 mm, ca. 1/5 Ki to 5 Ki). Ten μl of enzyme/inhibitor solution was added to 190 μl of 0.13 mm XXXG-CNP in 5 mm sodium citrate buffer (pH 5.5), and the reaction was monitored at 405 nm over 1–2 min in a 1-cm quartz cuvette maintained at 37 °C. The inhibition data were fit according to the Kitz-Wilson model (42), and apparent inactivation rate constants (kapp) were determined by fitting the exponential decay function as shown in Equation 1,
to the residual activity data. Ki and ki values were determined by fitting plots of inactivation rate constants versus putative inhibitor concentrations as shown in Equation 2,
by nonlinear regression using OriginPro graphing software.
Enzyme Crystallization
PbGH5A wild-type enzyme was crystallized at room temperature using the hanging drop method, with 1.8 μl of protein solution at 28 mg/ml mixed with 1.8 μl of reservoir solution (0.1 m sodium cacodylate (pH 6.3 to 7.1), 0.2 m calcium acetate, 25% PEG8K). The PbGH5A(E280A) mutant was crystallized in the same crystallization solution and using serial dilution seeding with wild-type crystals. The PbGH5A·XXXG and PbGH5A(E280A)·GGGG complexes were obtained by soaking apoenzyme crystals in reservoir solution supplemented with 20 mm XXXG or 10 mm GGGG for 3.5 and 2 h, respectively. Prior to data collection, crystals were cryoprotected with Paratone-N oil and flash-frozen in liquid nitrogen.
The PbGH5A·XXXG-NHCOCH2Br and PbGH5A(E280A)·XXXGXXXG complexes were obtained through co-crystallization with the tag-less protein at a final concentration of 28 mg/ml. For the PbGH5A·XXXG-NHCOCH2Br complex, 6.6 mm EDTA was added to the protein, and the mixture was incubated at 4 °C overnight. The inhibitor was then added to a final concentration of 8 mm, and the mixture was further incubated at 37 °C for 3 h. For the PbGH5A(E280A)·XXXGXXXG complex, the protein solution was incubated with 2.4 mm ligand at 37 °C for 3 h. The crystals were grown at room temperature using the sitting drop method, with 0.5 μl of complex solution mixed with 0.5 μl of reservoir solution: 0.2 m calcium chloride, 20% (w/v) PEG3350 for the XXXG·NHCOCH2Br complex, and 0.2 m magnesium acetate, 20% (w/v) PEG3350 for the XXXGXXXG complex. All complex crystals were cryoprotected by Paratone-N oil and flash-frozen in liquid nitrogen.
X-ray Crystal Structure Determination
Diffraction data at 100 K were collected using a Rigaku Micromax-007 HF rotating copper anode source with a Rigaku R-AXIS IV image plate detector (for the apoenzyme and PbGH5A·XXXG complexes) or using a Rigaku Micromax-007 HF rotating copper anode source with a Rigaku Saturn A200 CCD (at the Structure Genomics Consortium, for the PbGH5A(E280A)·GGGG, PbGH5A·XXXG-NHAcBr, and PbGH5A(E280A)·XXXGXXXG complexes). All x-ray data were reduced with HKL-3000 (43). First, the apoenzyme structure was determined by molecular replacement using a model generated by the Phyre2 server (44) of the PbGH5A sequence with the Paenibacillus pabuli GH5 structure as a template (PDB code 2JEQ (16)) and Phenix.phaser (45) and followed by automated model building using Phenix.autobuild. The PbGH5A ligand complex structures were determined by molecular replacement using the apoenzyme structure as the search model and Phenix.phaser to obtain phasing information. All refinement was performed with Phenix.refine with manual editing in Coot (46). During refinement B-factors were defined as anisotropic for all non-hydrogen atoms, and TLS parameterization was utilized. The final atomic model of the structures included chain A residues 7–352 and chain B residues 6–352. Average B-factor and bond angle/length root mean square deviation (r.m.s.d.) values were calculated using Phenix.b_factor_statistics. All geometry was verified using the Phenix Molprobity and Coot validation tools plus the wwPDB Deposition server. The data collection and refinement statistics are listed in Table 2.
TABLE 2.
X-ray diffraction data statistics
| PbGH5A apoenzyme | PbGH5A-E280A·XXXGXXXG complex | PbGH5A·XXXG complex | PbGH5A-E280A·cellotetraose complex | PbGH5A·XXXG-NHCOCH2 Br complex | |
|---|---|---|---|---|---|
| PDB code | 3VDH | 5D9M | 5D9N | 5D9O | 5D9P |
| Data collection | |||||
| Space group | P21 | P212121 | P212121 | P21 | P1 |
| Cell dimensions | |||||
| a, b, c, Å | 57.91, 82.25, 74.56 | 77.43, 81.68, 108.49 | 75.56, 84.11, 138.63 | 49.17, 85.06, 74.96 | 48.19, 49.02, 85.97 |
| β, ° | 109.15 | 101.41 | 76.39, 89.98, 66.80 | ||
| Resolution, Å | 50.0- 1.60 | 36.5–1.90 | 25.0–1.86 | 15.0–1.63 | 31.9–1.80 |
| Rsyma | 0.039 (0.443)b | 0.048 (0.50) | 0.051 (0.437) | 0.060 (0.526) | 0.046 (0.309) |
| I/σ(I) | 38.95 (4.67) | 23.26 (3.88) | 41.71 (5.98) | 21.86 (2.09) | 11.47 (2.13) |
| Completeness, % | 84.0 (61.6) | 99.2 (98.3) | 99.9 (99.7) | 97.8 (95.0) | 92.8 (86.5) |
| Redundancy | 3.4 (4.0) | 7.5 (7.2) | 6.2 (6.0) | 4.0 (4.0) | 2.0 (1.7) |
| Refinement | |||||
| No. of reflections | |||||
| Working, test | 79,400, 1951 | 54,601, 1994 | 74,720, 2000 | 85,156, 4269 | 60,096, 1850 |
| R-factor/free R-factorc | 15.2/19.9 (29.4/31.6) | 17.3/20.4 (22.9/30.0) | 13.9/16.8 (19.4/22.2) | 15.3/18.0 (22.7/24.5) | 16.3/20.4 (25.9/35.0) |
| No. of refined atoms, molecules | |||||
| Protein | 5493 | 5499 | 5542 | 5510 | 5503 |
| Ligand | NAd | 215 | 144 | 90 | 300 |
| Solvent | 939 | 728 | 962 | 987 | 750 |
| B-factors | |||||
| Protein | 24.4 | 26.1 | 25.2 | 11.8 | 25.5 |
| Ligand | NA | 26.7 | 49.1 | 28.0 | 30.6 |
| Solvent | 28.5 | 34.5 | 37.6 | 24.7 | 34.6 |
| r.m.s.d. | |||||
| Bond lengths, Å | 0.010 | 0.005 | 0.015 | 0.007 | 0.008 |
| Bond angles, ° | 1.222 | 0.915 | 1.370 | 1.118 | 1.189 |
| Ramachandran plot | |||||
| Allowed, % | 89.4 | 96.3 | 95.9 | 96.5 | 96.6 |
| Additionally allowed, % | 10.1 | 3.4 | 3.7 | 3.1 | 4.0 |
| Disallowed, % | 0.4e | 0.3 | 0.4 | 0.4 | 0.4 |
a Rsym = ΣhΣi|Ii(h) − 〈I(h)〉/ΣhΣiIi(h), where Ii(h) and 〈I(h)〉 are the ith and mean measurement of the intensity of reflection h.
b Figures in parentheses throughout the table indicate the values for the outer shells of the data.
c R = Σ|Fpobs − Fpcalc|/ΣFpobs, where Fpobs and Fpcalc are the observed and calculated structure factor amplitudes, respectively.
d NA means not applicable.
e Residues in the disallowed region are Ala-116 (which comprises the active site cleft) and/or Thr-272.
Phylogenetic Analysis
Sequences from glycoside hydrolase family 5 (11) from bacteria were fetched from the CAZy Database (10) and aligned with MAFFT (47). The distances between sequences were calculated by FastTree (48) with this multiple sequence alignment. The resulting tree is displayed with Dendroscope (49).
Results
Polysaccharide Kinetics
In light of the diverse specificities observed in GH5, we tested recombinant PbGH5A for activity on a library of linear β-glycan polysaccharides, including HEC, CMC, PASC, tXyG, kGM, and bMLG. As anticipated from its membership in GH5 subfamily 4, significant activity toward tamarind tXyG (kcat = 6800 min−1, Km = 1.1 mm; Table 1) was observed at the pH optimum of 4.5–5.5 (Fig. 2A) and at 37 °C. However, our kinetic analysis revealed that PbGH5A is significantly more selective for barley mixed-linkage glucan, with a kcat of almost 3.5 × 104 min−1 and Km of 0.12 mg/ml (Fig. 4 and Table 1). PbGH5A was poorly active on the synthetic soluble cellulose mimics CMC and HEC, perhaps reflecting detrimental interactions of the pendant groups in the active-site cleft. Interestingly, PASC was a worse substrate for PbGH5A than CMC (Fig. 4). Low activity was also measured for the hydrolysis of kGM, which suggested some tolerance of β(1,4)-linked mannosyl residues in the polysaccharide backbone. Although very poor activity was observed for PbGH5A acting on xylans, no activity was observed with either galactomannan polysaccharide or mannohexaose, confirming the glucan specificity of the enzyme.
TABLE 1.
Kinetic parameters for the hydrolysis of various substrates by PbGH5A
ND, not determinable.
| Substrate | kcat | Km | kcat/Km | Assay |
|---|---|---|---|---|
| min−1 | mm | m−1s−1 | ||
| bMLG | 3.49 ± 0.05 × 104 | 0.122 ± 0.007a | 4.8 × 103a | BCA |
| tXyG | 6.8 ± 0.4 × 103 | 1.1 ± 0.2a | 103a | BCA |
| kGM | 3.2 ± 0.2 × 103 | 0.90 ± 0.17a | 59a | BCA |
| cGM | ND | ND | ND | BCA |
| CMC | 1.83 ± 0.14 × 103 | 2.3 ± 0.4a | 13a | BCA |
| HEC | 2.0 ± 0.2 × 103 | 0.62 ± 0.13a | 54a | BCA |
| PASC | 2.0 ± 0.2 × 103 | 10.8 ± 1.3a | 3.1a | BCA |
| wAX | ND | ND | 0.23 ± 0.04 | BCA |
| bX | ND | ND | 0.23 ± 0.03 | BCA |
| G-PNP | ND | ND | ND | PNP |
| GG-PNP | ND | ND | 4.4 ± 0.2 | PNP |
| GG-CNP | 460 ± 30 | 11.4 ± 1.3 | 670 | CNP |
| GGG-PNP | 360 ± 25 | 9.2 ± 1.2 | 650 | PNP |
| GGG-CNP | 9.1 ± 0.3 × 103 | 1.5 + 0.1 | 1.01 × 105 | CNP |
| GGGG-CNP | 8.1 ± 0.4 × 103 | 0.7 ± 0.1 | 1.94 × 105 | CNP |
| XXXG-CNP | 9.7 ± 0.8 × 103 | 0.064 ± 0.013 | 2.53 × 106 | CNP |
| G3GGG3GGG→G3GGG+GGG | 1.97 ± 0.13 × 104 | 0.570 ± 0.040 | 5.8 × 105 | HPAEC-PAD |
| G3GGG | ND | ND | ND | HPAEC-PAD |
| GG3GG→GG+GG | ND | ND | 1.46 ± 0.2 × 103 | HPAEC-PAD |
| GGG3G→GGG+G | ND | ND | 3.13 ± 0.4 × 103 | HPAEC-PAD |
| GGG3G→GG+G3G | ND | ND | 600 ± 200 | HPAEC-PAD |
| GGGG→GG+GG | 106 ± 4 | 0.77 ± 0.05 | 2.3 × 103 | HPAEC-PAD |
| GGGG→GGG+G | ND | ND | 300 ± 50 | HPAEC-PAD |
| GGGGG→GGG+GG | 1.21 ± 0.02 × 104 | 0.753 ± 0.026 | 2.68 × 105 | HPAEC-PAD |
| GGGGGG→GGGG+GG | 9.3 ± 1.5 × 103 | 0.79 ± 0.22 | 1.96 × 105 | HPAEC-PAD |
| GGGGGG→GGG+GGG | 6.3 ± 1.1 × 103 | 0.84 ± 0.26 | 1.25 × 105 | HPAEC-PAD |
| XXXGXXXG→2XXXXG | 422 ± 10 | 0.037 ± 0.009 | 1.9 × 105 | HPAEC-PAD |
| MMMMMM | ND | ND | ND | HPAEC-PAD |
a Concentrations are expressed as g/liter. Apparent Km values are reported.
FIGURE 4.
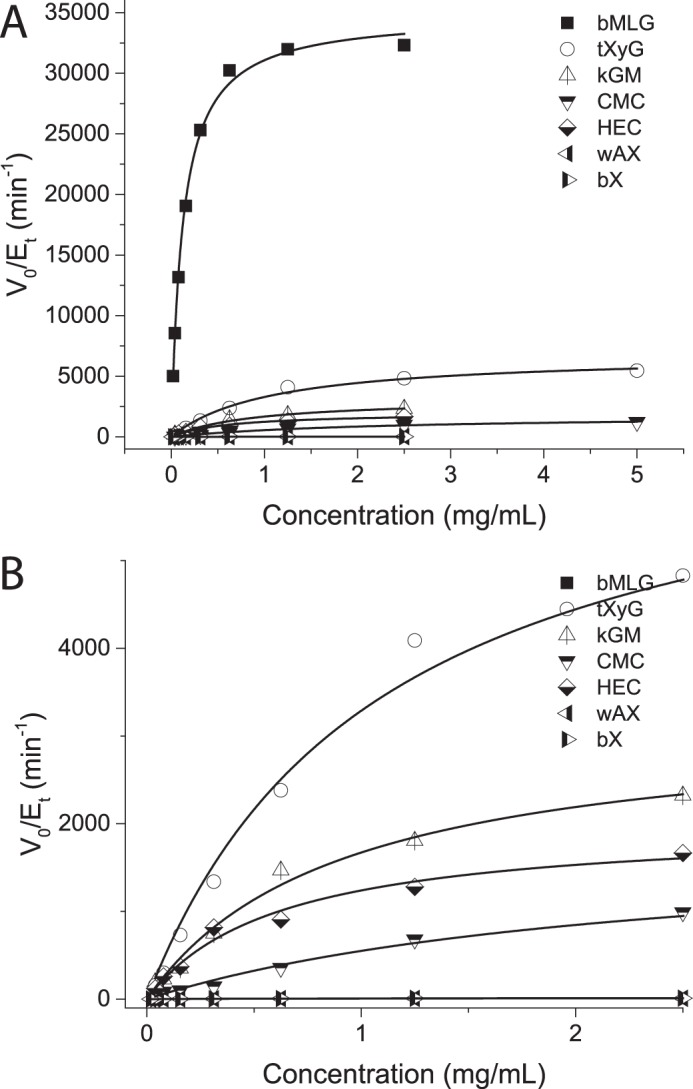
Polysaccharide specificity. A, Michaelis-Menten plots of PbGH5A acting on various β-glucan substrates. Activities were measured as rates of reducing-end production using the BCA assay. B, graphical expansion of data for tXyG, kGM, CMC, HEC, PASC, bX, and wheat arabinoxylan (wAX) from A.
The pH-rate profile of PbGH5A was affected by the substrate used for its determination (Fig. 2). The pH-rate profile of PbGH5A with XXXG-CNP gave a pH optimum of 5 with kinetic pKa values of 3.5 and 6.5; however, the pH-rate profile with MLG demonstrated different kinetic pKa values depending on the buffer used. The activity-temperature profile of PbGH5A on bMLG substrate indicates that the enzyme has limited activity enhancement above 37 °C (Fig. 3A); hence, kinetic measurements were routinely performed at 37 °C in pH 5.5 buffer. The enzyme is stable below 45 °C (Fig. 3B) but exhibits rapid (t½ = 15 min) and permanent inactivation at elevated temperatures.
Polysaccharide Hydrolysis Product Distributions
Analysis of the limit-digestion products was subsequently performed to determine the cleavage specificity of PbGH5A. HPAEC-PAD analysis of the initial digest of bMLG (Fig. 5A) contained three peaks with short retention times, corresponding to primarily cellotriose and cellotetraose with a small amount of cellobiose. When allowed to run significantly longer, the limit digestion of bMLG gave glucose, cellobiose, and cellotriose (data not shown). Interestingly, a small number of peaks with longer retention times was also generated but not further degraded in longer incubations. The major late-eluting peak was determined to be G3GGG based on retention time and standard addition; the other peaks were not identified (see “Substrates and Inhibitors” under the “Experimental Procedures” for oligosaccharide nomenclature).
FIGURE 5.
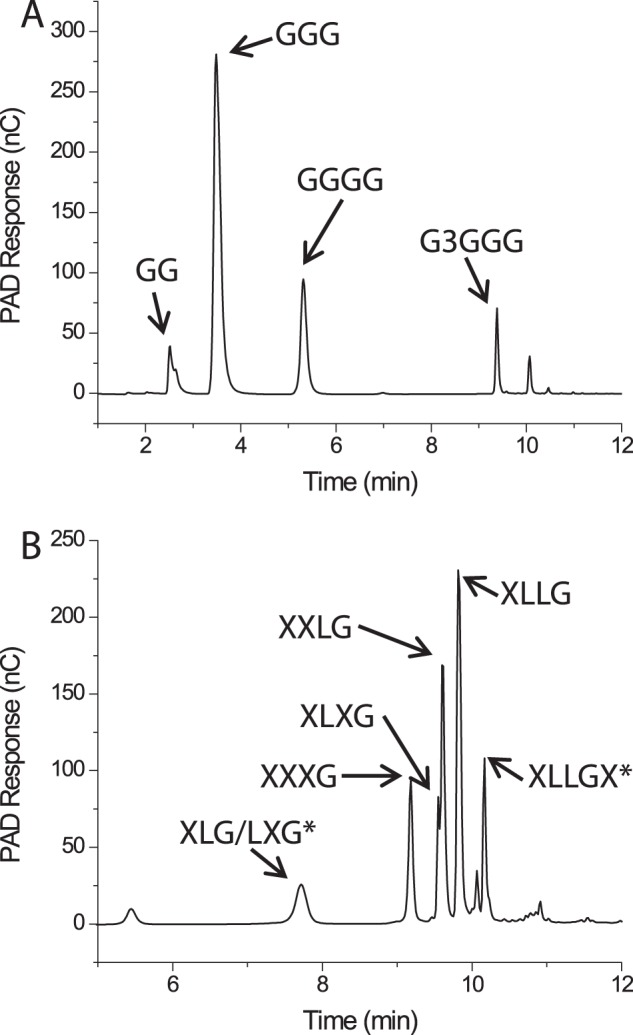
HPAEC-PAD chromatograms of the limit digests of bMLG (A) and tXyG (B) hydrolyzed by PbGH5A. Identifiable hydrolysis products are labeled. XLLGX and XLG/LXG have been identified by mass spectrometry but have not been unambiguously assigned as the chromatographic peaks specified.
The presence of more than the canonical four peaks corresponding to XXXG, XLXG, XXLG, and XLLG (ratio ∼13:9:28:50 (18, 50)) in the limit digest of tamarind tXyG (Fig. 5B) indicates that the enzyme is able to cut at sites other than the unbranched glucosyl residues. Indeed, MALDI-MS analysis of the digest revealed the presence of fragments with masses corresponding to XLLGX and XLG/LXG, which confirmed an alternate cleavage mode in which xylosyl-branched glucosyl units bind in the −1 and +1 subsites (active-site nomenclature according to Ref. 51).
Chromogenic Substrate Kinetics
To map the negative enzyme subsites and determine their specific contributions to catalysis, we employed a series of initial-rate kinetic experiments measuring the release of the aglycone from the CNP and PNP β-glycosides of glucose (G), cellobiose (GG), cellotriose (GGG), cellotetraose (GGGG), and the xyloglucan heptasaccharide XXXG. Hydrolysis of G-CNP was undetectable, and only weak activity (kcat/Km = 670 m−1 s−1) was observed with GG-CNP (Fig. 6 and Table 1). GGG-CNP was a significantly better substrate (kcat/Km = 1.01 × 105 m−1 s−1), thus indicating a significant contribution to catalysis due to binding of the additional Glc residue in a −3 subsite; a similar trend was observed for the PNP congeners (Table 1). The specificity constant for GGGG-CNP hydrolysis (kcat/Km = 1.94 × 105 m−1 s−1) was only 2-fold higher than that of GGG-CNP, suggesting little to no contribution from a −4 subsite. In keeping with the poorer leaving-group ability of the aglycone (31, 52), GG-PNP and GGG-PNP were hydrolyzed significantly more slowly than the CNP congeners. Comparison of the kinetic constants for XXXG-CNP (Xyl3Glc4-CNP, composed of a GGGG backbone) with GGGG-CNP revealed a similar kcat value but a significantly (10-fold) lower Km value, yielding a corresponding increase in specificity constant (kcat/Km = 2.53 × 106 m−1 s−1, Table 1) for the branched substrate. However, the observation of significant substrate inhibition and deviation from classical Michaelis-Menten kinetics with XXXG-CNP (Fig. 6) suggests that caution is warranted in interpreting the apparent positive effects of xylosyl branches in the negative subsites.
FIGURE 6.

Michaelis-Menten plots for the hydrolysis of various chromogenic substrates by PbGH5A. Activities are measured as CNP or PNP release rates measured by monitoring A405 over time. Error bars represent the fitting error of each measured rate. A–F, Michaelis-Menten plots for the hydrolysis of cellobiose-CNP, cellotriose-CNP, cellotetraose-CNP, XXXG-CNP, cellobiose-PNP, and cellotriose-PNP by PbGH5A, respectively.
Native Oligosaccharide Kinetics
To gain insight into the contribution of the positive subsites to substrate binding and catalysis, we determined the initial-rate kinetics of PbGH5A on cello-oligosaccharides, mixed-linkage β(1,3)/β(1,4)-glucan oligosaccharides, and xyloglucan oligosaccharides, using an HPLC-based assay. No activity was observed with laminaribiose (G3G), cellobiose (GG), or cellotriose (GGG), suggesting that PbGH5A requires the occupancy of at least four subsites for initiation of the glycosidic bond cleavage. Indeed, cellotetraose (GGGG) was readily hydrolyzed through two modes, one yielding two molecules of cellobiose (2×GG), and one yielding glucose (G) plus cellotriose (GGG); Michaelis-Menten analysis revealed that the symmetric cleavage mode was favored by a 7-fold greater kcat/Km value (Fig. 7 and Table 1). Notably, cellohexaose (GGGGGG) was degraded to GG, GGG, and GGGG with similar kinetic constants to GGGGG, which was exclusively converted to cellotriose (GGG) and cellobiose (GG), with a kcat/Km value 130-fold higher than that for symmetric cleavage of cellotetraose (Figs. 7 and 8 and Table 1). Exclusive isotopic labeling of the product cellotriose (GGG) in H218O revealed that recognition across the −3→+2 subsites was responsible for this cleavage mode (Fig. 9). Specifically, the M + 2 peak of cellobiose did not increase in relative intensity above the natural abundance, whereas the intensity of the M + 2 peak of cellotriose indicated 74% 18O labeling (theoretical, 85%).
FIGURE 7.

Michaelis-Menten plots for the hydrolysis of various model oligosaccharides by PbGH5A. Activities are measured as the rate of increase in product peak integration using HPAEC-PAD. Error bars represent the fitting error of each measured rate. A–C, Michaelis-Menten plots for the hydrolysis of cello-oligosaccharides (GGGG, GGGGG, and GGGGGG) by PbGH5A. D–F, Michaelis-Menten plots for the hydrolysis of mixed-linkage glucan oligosaccharides (GG3GG, GGG3G, and G3GGG3GGG) by PbGH5A. G, Michaelis-Menten plot for the hydrolysis of XXXGXXXG by PbGH5A.
FIGURE 8.
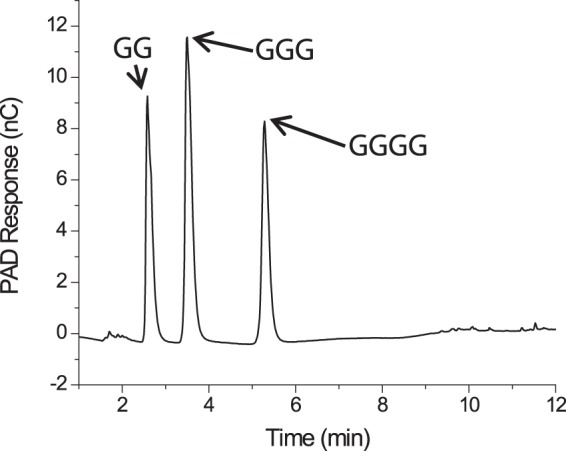
HPAEC-PAD product analysis of the digestion of cellohexaose by PbGH5A.
FIGURE 9.

Mass spectrum of the products of cellopentaose degradation by PbGH5A in H218O. A, isotopic distribution of produced cellobiose. No increase in the M + 2 peak indicates no 18O incorporation. B, isotopic distribution of produced cellotriose showing significant increase in the M + 2 peak intensity.
Turning our attention to mixed-linkage β(1,3)/β(1,4)-glucan oligosaccharides, we observed that G3GGG was not hydrolyzed, which suggested that β(1,3) bonds are not tolerated between the first three negative subsites. In contrast, GG3GG was a competent substrate, yielding cellobiose as the only product (Fig. 7 and Table 1). This recapitulated the rejection of β(1,3) bonds between the negative subsites, and furthermore, it highlighted the importance of −2 subsite binding. The specificity constant of the GG3GG degradation is only ∼1.5-fold lower than that of cellotetraose (Table 1), which indicated a lack of selectivity for β(1,3) or β(1,4) bonds in the cleavage site. Interestingly, GGG3G was hydrolyzed via two modes, in which the production of cellotriose (GGG) plus glucose, via binding in the −3→+1 subsites and cleavage of the β(1,3)-linkage, was favored by a factor of 5 in kcat/Km values over the production of cellobiose (GG) plus laminaribiose (G3G), via binding in the −2→+2 subsites and cleavage of the β(1,4)-linkage and −2→+2-binding subsites (Fig. 7 and Table 1). The extended mixed-linkage heptasaccharide G3GGG3GGG was hydrolyzed most rapidly of all the substrates tested, with a kcat/Km value exceeding that of cellopentaose or cellohexaose by 2-fold, to give only cellotriose (GGG) and G3GGG as products (Fig. 7 and Table 1).
Inhibition and Covalent Labeling with an Active-site-directed Inhibitor
We have previously introduced N-bromoacetylglycosylamines and bromoketone C-glycoside derivatives of xyloglucan oligosaccharides as specific active-site affinity labels for endo-xyloglucanases (Fig. 10) (32). Incubation of PbGH5A with the N-bromoacetylglycosylamine derivative of XXXG (XXXG-NHCOCH2Br, compound 1) led to a rapid time- and concentration-dependent inactivation (Fig. 10), with a dissociation constant, Ki, of 0.63 ± 0.03 mm, and an irreversible inactivation constant, ki, of 0.0364 ± 0.0006 min−1 (ki/Ki = 0.06 mm−1 min−1). Intact protein MS after a 3-h incubation of PbGH5A with compound 1 at 1.4 mm and 37 °C revealed exclusive single labeling of the enzyme (Fig. 11). The bromoketone C-glycoside isostere (XXXG-CH2COCH2Br, compound 2) was a less potent, but nonetheless effective, inhibitor of PbGH5A, with a 3-fold lower ki value and 2.5-fold lower Ki (Ki = 0.27 ± 0.07 mm, ki = 0.0113 ± 0.0008 min−1, and ki/Ki = 0.04 mm−1 min−1). Intact protein MS of PbGH5A under conditions similar to those giving essentially complete inactivation (7.3 μm PbGH5A, 1.4 mm inhibitor compound 2, 3-h incubation at 37 °C) indicated near-complete labeling of the enzyme, also at 1:1 stoichiometry (Fig. 11).
FIGURE 10.

Inhibition of PbGH5A with active-site affinity labels. A and C, inhibition of PbGH5A with compound 1. A, plot of reaction velocity versus time at different inhibitor concentrations. C, plot of pseudo first-order rate constants versus inhibitor concentration. B and D, inhibition of PbGH5A with compound 2. B, plot of reaction velocity versus time at different inhibitor concentrations. D, plot of pseudo first-order rate constants versus inhibitor concentration. Error bars represent the fitting error of the exponential decay function. E, chemical structure of inhibitor compound 1. F, chemical structure of inhibitor 2.
FIGURE 11.

A, intact MS of wild-type PbGH5A at 7.7 μm, expected mass, 41,649.0; found, 41,650.6. B, MS of PbGH5A at 7.7 μm and inhibitor 1 at 1.4 mm, 3-h incubation at37 °C. The peak at 42,753.3 corresponds to the mono-labeled enzyme adduct (expected mass, 42,751.7; found, 42,753.3). C, MS of PbGH5A at 7.4 μm and inhibitor 2 at 1.4 mm, 3-h incubation at 37 °C. The peak at 42,751.2 corresponds to the mono-labeled enzyme (expected mass, 42,750.7; found, 42,751.2). D, MS of the PbGH5A mutant E280A at 6.4 μm, 3-h incubation at 37 °C with 1.4 mm inhibitor 1 XXXG-NHCOCH2Br. The peak at 42,693.3 corresponds to mono-labeled protein. Expected mass, 42,693.7; found, 42,693.3.
Structural Characterization of PbGH5A Variants in the Apoform and in Complexes with Oligosaccharides
To provide molecular level insight into substrate recognition by PbGH5A, we determined the crystal structure of this protein to 1.65 Å resolution (PDB code 3VDH). We also obtained high resolution (1.6–1.9 Å) structures of enzyme variants with four different ligands (Table 2). The complex structures of the catalytically inactive PbGH5A(E280A) site-directed mutant with the tetradecasaccharide substrate XXXGXXXG (PDB code 5D9M) and that of the wild-type enzyme with the covalent inhibitor XXXG-NHCOCH2Br (compound 1, PDB code 5D9P) contained clear electron density corresponding to ligand molecules that spanned the length of the active-site cleft for both complexes (Fig. 12). Together, these complexes provide the most complete view of enzyme-substrate interactions across the entire active site of a GH5 member to date. In the complex structures between the wild-type enzyme and heptasaccharide XXXG (PDB code 5D9N) and between the E280A variant and the linear glucan cellotetraose GGGG (PDB code 5D9O), the respective ligands occupied the positive subsites of the PbGH5A active site, thereby providing a unique opportunity to directly compare binding for branched and unbranched ligands in the GH5_4 subfamily.
FIGURE 12.
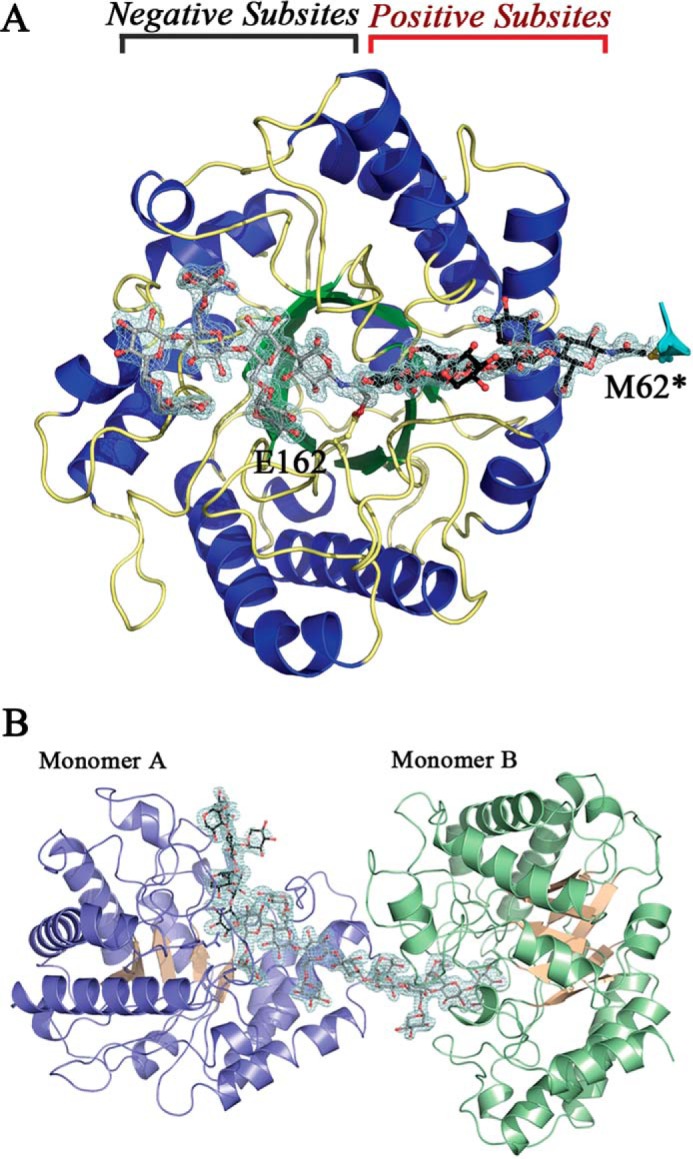
Overall structure of PbGH5A. A, PbGH5A in complex with inhibitor 1. Secondary structure of PbGH5A is shown in schematic representation and color-coded with strands in green, helices in blue, and loops in yellow. Two ligand molecules, XXXG-NHCOCH2Br, are shown in ball-and-stick, in gray and black. The active site positive and negative subsites are indicated. B, asymmetric unit for the PbGH5A(E280A)·XXXGXXXG complex. Secondary structure of PbGH5A is shown in schematic representation and color-coded blue and pink for monomer A and green and pink for monomer B. Two ligand molecules are shown in ball-and-stick, in gray and black.
Overall Structure of PbGH5A
The PbGH5A apoenzyme structure was determined by molecular replacement using the structure of P. pabuli GH5 (PDB code 2JEQ) as a search model. The asymmetric unit contained two polypeptide chains corresponding to PbGH5A residues 7–352. According to analytical gel filtration analysis (data not shown) the PbGH5A protein predominantly exists as a monomer in solution, suggesting that intermolecular contacts observed in the crystal structure are most probably a result of crystal packing. The overall structure of PbGH5A is a (β/α)8-barrel fold typical of the GH5 family (Fig. 12A). Structural comparison using the Dali server (53) identified other characterized GH5 subfamily 4 enzymes as the closest structural homologues of PbGH5A. The best match was the structure of endoglucanase A from Piromyces rhizinflata (PDB 3AYS) (54), which superimposed with PbGH5A with an r.m.s.d. value of 1.7 Å over 323 of 357 Cα positions (Table 3).
TABLE 3.
Dali (53) search results
| PDB code | Organism | Protein name | Z score | r.m.s.d. | % ID | Active-site pocket area | Active-site pocket volume | Activity spectrum | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Å | Å2 | Å3 | |||||||
| 3VDH | P. bryantii | PbGH5A | – | – | – | 600 | 1020 | bMLG, tXyG, kGM, CMC, HEC | This work |
| 3AYS | P. rhizinflata | PrGH5 | 43 | 1.7 | 34 | 600 | 990 | Avicel, MLG, CMC, lichenin | 72 |
| 2JEQ | P. pabuli | PpXG5 | 43 | 1.8 | 33 | 880 | 1620 | XyG | 16 |
| 4W87 | Uncultured bacterium | XEG5A | 42 | 1.8 | 34 | 500 | 740 | XyG, lichenin, CMC | 64 |
| 3ZMR | B. ovatus | BoGH5 | 42 | 2.0 | 30 | 830 | 1630 | XyG | 17 |
| 4V2X | B. halodurans | BhGH5 | 42 | 1.9 | 28 | 866 | 537 | XyG, MLG | 63 |
| 1EDG | Clostridium cellulolyticum | Cel5A | 41 | 1.8 | 34 | 617 | 943 | Cellulose | 73 |
| 4W8B | Uncultured bacterium | XEG5B | 40 | 2.1 | 33 | 680 | 1140 | XyG | 64 |
The active site in the homologous enzymes is located in a large solvent-accessible cavity formed by loop regions at the top of the barrel. As inferred from comparison of primary and tertiary structures of GH5 members, Glu-280 and Glu-162 are the catalytic active-site nucleophile and general acid/base, respectively, in PbGH5A (55–57). Accordingly, the mutation of Glu-280 to alanine resulted in a >18,000-fold reduction in activity (below the limit of detection) compared with the wild type on both chromogenic substrates XXXG-CNP and GGG-CNP. Hence, the catalytically inactive PbGH5A(E280A) variant was used for co-crystallization with tetradecasaccharide and heptasaccharide substrates.
Non-covalent Complexes of PbGH5A Variants with Branched and Unbranched Ligands
The PbGH5A(E280A) variant in complex with tetradecasaccharide substrate exhibited unambiguous, well ordered electron density corresponding to a single XXXGXXXG molecule spanning the active sites of both monomers found in the asymmetric unit (Fig. 12B); one-half of the substrate occupied the positive subsites of one monomer (monomer A), and the other half localized to the negative subsites of the second monomer (monomer B). In addition, the negative subsites of monomer A featured density corresponding to the XXXG moiety of the second substrate molecule, the rest of which was apparently disordered in the solvent channel. The positive subsites of monomer B did not contain any additional electron density.
The conformation of the XXXGXXXG substrate molecule in the negative subsites of both PbGH5A(E280A) monomers was well defined and virtually identical. Detailed analysis of interactions between PbGH5A(E280A) monomers and the substrate molecule showed that subsite −1 of the enzyme forms by far the most direct interactions with the glucosyl moieties of the substrate (Fig. 13A). More specifically, this subsite was occupied by the α-anomer of the reducing-end glucose unit, with the C1-hydroxyl forming a hydrogen bond with the side chain of Tyr-240 (Fig. 14). This glucose unit is also positioned by a stacking interaction with Trp-324, and hydrogen bonds between C2-, C3-, and C6-hydroxyls and the side chains of Asn-161, His-112, and Asp-288, respectively. It is interesting to note that the C1-carbon atom itself is located 3.1 Å away from the C4-hydroxyl of the xylosyl-branched glucosyl unit bound in the +1 subsite, implying that little movement would be necessary to bring the two ligands within the distance required for formation of an intact β-1,4 bond in the reverse reaction. These observations suggested that PbGH5A(E280A)·XXXGXXXG complex structure represents a good proxy for wild-type enzyme-product interactions, despite having been generated from a catalytically inactive enzyme variant.
FIGURE 13.

PbGH5A active site. A, stereo view of the negative subsites of XXXGXXXG complex. The ligand in the active site is in gray, in ball-and-stick, whereas PbGH5 side chains, involved in binding, are in yellow; water molecules are shown as red spheres. Fo − Fc electron density (3σ level) is contoured in green around the ligand. The ligand in adjacent positive subsites is shown in line representation in black. B, stereo view of the positive subsites of XXXGXXXG complex. The ligand is in black ball-and-stick. The ligand shown in gray lines is bound in the negative subsites of the same monomer. The rest as in A. C, stereo view of the positive subsites of XXXG complex. The same representation as for B. D, stereo view of the positive subsites of cellotetraose complex. The same representation as for B.
FIGURE 14.

Schematic representation of the PbGH5A active site bound to two XXXG oligosaccharide units, based on the structure of the PbGH5A in complex with the XXXGXXXG tetradecasaccharide. Key hydrogen bonds between the enzyme and the ligands are represented by dashed lines, and key stacking interactions are represented by curved lines, and the residues involved are in gray. Interactions with the solvent were omitted for visual clarity.
The −2 subsite is formed by the side chains of Asn-28 and Asp-288, which hydrogen bonds to the C2- and C3-hydroxyls of the corresponding glucosyl residue, respectively (Fig. 14). Binding of the −2′ xylosyl unit is water-mediated, with the exception of a hydrogen bond between the C4-hydroxyl and the backbone of Ala-117. In subsite −3, the main interaction is stacking of the glucosyl residue against Trp-48, whereas in subsite −4, the stacking is against Phe-47. These structural observations (see also the inhibitor complex below) are in line with the kinetic analysis presented above, which likewise suggest the existence of a total of four negative subsites in PbGH5A.
As mentioned above, the PbGH5A(E280A)·XXXGXXXG complex features one substrate molecule extending from the positive subsites of one monomer into the negative subsites of another monomer in the asymmetric unit. In the positive subsites, the substrate binding maps from the +1 subsite, in which the glucosyl moiety is closely stacked against Trp-170 (Fig. 13B). The glucosyl moiety forms a hydrogen bond with the side chain of Glu-162 via the C4-hydroxyl, although the other hydroxyls are solvent-coordinated. The xylosyl unit in the +1 subsite occupies the space between the active site cleft and the glucan backbone and forms an intricate network of hydrogen bonds with the protein, making use of all the available hydroxyls of the sugar moiety. The glucosyl unit in the +2 subsite stacks against Trp-243, and its C2-hydroxyl forms hydrogen bonds with the side chain of Asp-171. The xylosyl unit in the +2 position is bound away from the active site cleft and is completely solvent-exposed, as are the ligand units in the +3 and +4 positions.
In the wild-type PbGH5A·XXXG heptasaccharide complex, clear electron density corresponding to all seven monosaccharide residues bound in the positive subsites was observed (Fig. 13C). Although the general orientation of the ligand molecule in this complex structure is similar to that observed in the PbGH5A(E280A)·XXXGXXXG complex, it nevertheless has a distinct conformation in which the glucan backbone of the substrate is rotated ∼15° away from the enzyme catalytic center (Fig. 15B). This alternative conformation of the ligand results in a more parallel binding along the bottom of the active site cleft, and it is observed for both PbGH5A monomers found in the asymmetric unit. Because of this shift, a unique set of interactions is formed between the enzyme and the ligand as compared with the tetradecasaccharide complex structure (Fig. 15B). The interactions conserved between the two structures include Trp-170 and Trp-243 stacking with the ligand in the +1 and +2 subsites, respectively, and the hydrogen bond between Lys-214 and the C4-hydroxyl of the +1′ xylosyl residue. The latter moiety forms four unique hydrogen bond interactions not observed in the complex with XXXGXXXG, demonstrating that although different, the recognition of XXXG is as elaborate as for the tetradecasaccharide. The xylosyl units for the rest of the ligand are completely solvent-exposed. The presence of available hydrogen bonding partners in the PbGH5A active site, and lack of conserved interactions with the side chain xylose moieties in the two complexes, implies that ligand binding is dominated by overall accommodation of the xyloglucan polymer rather than specific recognition of the pendant groups.
FIGURE 15.

Comparison of PbGH5A complexed structures. A, overall shape of the active-site pocket and conformation of ligand binding within. Surface representation of PbGH5A complexes with ligands from the four complexes are shown superimposed. Enzyme regions in direct contact with the ligand are colored dark blue for the XXXGXXXG complex, and additional interactions are in gray for the XXXG complex and in pink for cellotetraose complex. Ligands are in ball-and-stick; in black (XXXGXXXG), in pink (XXXG-NHCOCH2Br), in orange (XXXG), and in light blue (cellotetraose). B, pairwise comparison, binding of branched ligands. Stereo view of XXXGXXXG and XXXG ligand superposition is shown in ball-and-stick, ligands are color-coded as in A. PbGH5A residues shown for orientation are in yellow, and key differences in binding are indicated by gray lines. C, pairwise comparison, binding of branched versus unbranched ligand. Stereo view of XXXG and GGGG ligand superposition is shown in ball-and-stick, and ligands color-coded as in A. PbGH5A residues shown for orientation are in yellow, and key differences in binding indicated by gray lines.
As observed for the complexes with branched ligands, the PbGH5A(E280A)·cellotetraose (GGGG) complex contained the ligand bound in the positive subsites, although clear electron density was present only for glucosyl residues +1 to +3 (Fig. 13D). In contrast to the xylogluco-oligosaccharide-bound structures, the binding conformation of the cellotetraose ligand is dramatically different. In particular, the glucosyl residue in the +1 subsite in the cellotetraose complex is flipped ∼180° about the C1-C4 axis vis à vis the XXXG complex (Fig. 15C). In this orientation, the glucan backbone is pushed deeper into the core of the protein and results in all four glucosyl moieties of cellotetraose forming direct interactions with the protein. In the +2 subsite, the C6-hydroxyl of the glucosyl moiety occupies the space equivalent to the +1′-xylosyl residue of the branched ligands and forms hydrogen bonds with the side chain of Asp-241 and the main chain amide of Tyr-240. In the +3 subsite, the glucosyl moiety forms direct interactions with the protein not seen in the other complexes via hydrogen bonding interactions of the C3-hydroxyl with the side chain of Asp-241 and the main chain amide of Trp-243. The limited electron density of the glucosyl unit in the +4 position also suggests hydrogen bonding with the protein. The more extensive hydrogen bonding network of the unbranched glucan vis à vis the xylose-branched congeners parallels the observed catalytic preference of PbGH5A for bMLG, whereas the flip in the binding conformation of this ligand provides structural rationalization for the lack of discrimination in hydrolysis of β(1,3) versus β(1,4) bonds (as discussed in greater detail below).
Active-site Affinity-labeled Complex of PbGH5A
In keeping with the anticipated reactivity of the electrophilic affinity label XXXG-NHCOCH2Br, the XXXG-NHCOCH2- moiety was observed in the negative subsites, covalently bound to the side chain oxygen of the catalytic acid base, Glu-162, via displacement of the bromide nucleofuge (Fig. 16A). The specific labeling of Glu-162 was consistent with the observation by MS of a single protein·inhibitor covalent complex of both the wild-type and the E280A catalytic nucleophile mutant in solution (Fig. 11). Likewise, labeling of the general-acid base residue in a cellulase by a homologous N-bromoacetylcellobiosylamine has previously been observed (58). As with the PbGH5A(E280A)·XXXGXXXG complex structure, clear electron density for the entire ligand indicates well ordered binding. The position of the oligosaccharide moiety of the label in the negative subsites superimposes remarkably well with that of XXXGXXXG in the corresponding complex structure (Fig. 15A). This observation confirmed that these inhibitors retain their full ability to interact with the negative subsites of PbGH5A and are thus accurate substrate mimics. The key difference between these structures results from accommodation of the inhibitor's “handle” in the −1 subsite. Here, the strictly conserved His-328 orients the amide moiety through a hydrogen bond, although the active site nucleophile, Glu-280, forms a hydrogen bond with the C2-hydroxyl of the glucose moiety (Fig. 16A).
FIGURE 16.
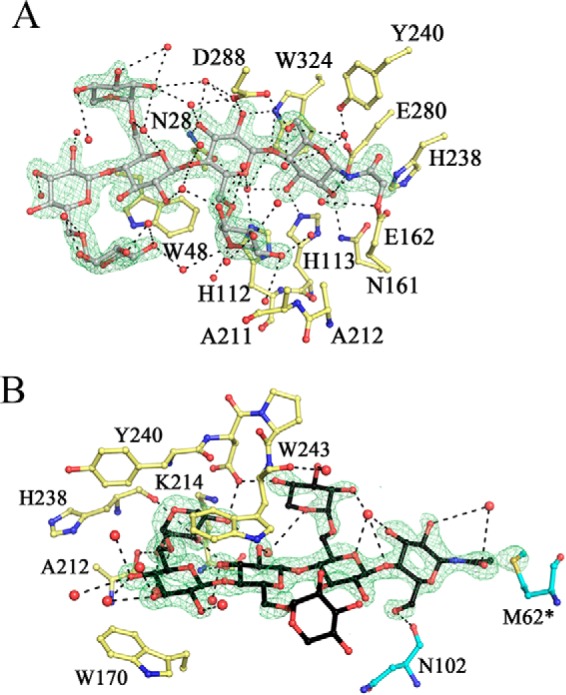
PbGH5A active site in complex with XXXG-NHCOCH2Br. A, negative subsites. The ligand in the active site is in gray, in ball-and-stick, and PbGH5A side chains, involved in binding are in yellow; water molecules are shown as red spheres. Fo − Fc electron density (3.5σ level) is contoured in green around the ligand. B, positive subsites. The ligand is in black, and the residues from adjacent symmetry-related monomer are in cyan. Other coloring is as in A.
An unexpected second inhibitor molecule occupied the positive subsites of the enzyme in crystallo. This inhibitor molecule was covalently linked to Met-62 of a neighboring enzyme molecule within the crystal packing, suggesting that in this orientation the terminal group of the inhibitor was solvent-exposed and suitably poised to react with the nucleophilic thioether. Multiple labeling of PbGH5A in the presence of this inhibitor in solution was not observed by MS, indicating that this was a fortuitous event, prompted by the particular crystal packing. Notably, this result follows a previous observation made by Black et al. (59), who suggested that nonspecific labeling could occur at solvent-exposed methionine residues.
This covalent pinning via the N-acetyl moiety resulted in well ordered binding for all four glucosyl units of the second inhibitor moiety (Fig. 16B). The oligosaccharide portion is oriented similarly to this in the PbGH5A·XXXG and PbGH5A(E280A)·XXXGXXXG complexes and is likewise distinct from the PbGH5A(E280A)·cellotetraose complex. Subtle differences in the conformation of XXXG-NHCOCH2- in the positive subsites vis à vis XXXG and XXXGXXXG again suggests that the recognition of the XXXG motif is plastic. Here, the hydrogen-bonding network represents a mixture of that observed for these other branched complexes. The xylosyl residue in the +1′ subsite retains the hydrogen bond with Lys-214 and forms an additional hydrogen bond with the side chain of Asp-241 (Fig. 16B). Similar to the XXXG complex, the +1′ xylosyl residue of the inhibitor forms a hydrogen bond with the backbone of Ala-212, yet similar to the XXXGXXXG complex, the same moiety forms a hydrogen bond to a hydroxyl of the +2 glucosyl residue. The xylosyl unit in the +3′ position is hydrogen-bonded to the main chain of Trp-243 and the side chain of Asp-241, reminiscent of glucose binding in the +3 subsite of the cellotetraose complex. The remainder of the interactions are entirely water-mediated.
In summary, the comparative analysis of PbGH5A·ligand complex structures revealed a striking variation in ligand-protein interactions within the positive but not the negative subsites. This, however, does not translate into conformational changes in the protein structure itself. The enzyme backbones in all four complex structures superimposed with an average r.m.s.d. of 0.3 Å for the protein Cα atoms. The binding of the various oligosaccharides in approximately the same location but with a drastically different orientation therefore points to the great versatility of the PbGH5A active site with respect to accommodation of different ligands within the positive subsites (Fig. 15A).
Discussion
The combination of detailed kinetic analysis together with new insight brought by novel crystallographic complexes of PbGH5A provides a unique opportunity to explore key enzyme-substrate interactions that define substrate specificity within GH5_4 and to further elucidate the roles of this subfamily in glucan catabolism.
PbGH5A Is a Predominant Mixed-linkage Endo-glucanase but Also a Competent Endo-xyloglucanase
Polysaccharide kinetics reveal that although PbGH5A is a competent endo-xyloglucanase (EC 3.2.1.151), with an activity (kcat = 6800 min−1, Km = 1.1 mm) similar to that of the highly specific bacterial GH5_4 endo-xyloglucanases from P. pabuli (PpXG5, vo/[E]t = 8700 min−1 at 0.5 mg/ml substrate) (16) and Bacteroides ovatus (BoGH5, kcat = 2.61 × 104 min−1, Km = 0.82 mm) (17), it is a superior bMLG hydrolase (EC 3.2.1.73), with kcat = 3.5 × 104 min−1 and Km = 0.12 mg/ml. PbGH5A thus has significant catalytic flexibility, having an ability to tolerate branching xylosylation on β-glucan chains. This activity profile clearly distinguishes PbGH5A from the strict endo-xyloglucanases of GH5_4 (16, 17) and provides further direct evidence of the polyspecificity of subfamily GH5_4, which also includes a number of characterized carboxymethylcellulases and mixed-linkage endo-glucanases (60–62).
Hence, we compared PbGH5A to other well characterized GH5_4 enzymes as follows: P. pabuli XG5 (PpXG5, PDB code 2JEQ) (16); B. ovatus (BoGH5, PDB code 3ZMR) (17); Bacillus halodurans GH5 (BhGH5, PDB code 4V2X) (63); and Xeg5A (PDB code 4W88) and Xeg5B (PDB code 4W8B) (64). These were chosen as they have been subjected to detailed structure-function characterization and have been specifically tested for both mixed-linkage endo-glucanase and endo-xyloglucanase activities. Of these, BhGH5 and Xeg5A, like PbGH5A, accept both bMLG and xyloglucan substrates, whereas PpXG5, BoGH5, and Xeg5B are highly specific for xyloglucan.
Examination of the overall shape of the active site of these enzymes reveals a substantially shallower and narrower cleft of the predominant mixed-linkage endo-β-glucanases versus the predominant endo-xyloglucanases (Fig. 17A). Quantitation of this difference using the CASTp server indicated that both the active site surface area and volume are greater by approximately one-third for the former enzymes (Table 3 (65)). The contrast is particularly dramatic at the catalytic center (the region surrounding subsites −1 and +1), where PbGH5A, BhGH5, and Xeg5A possess a shallow cleft with a constriction in the middle, whereas BoGH5, PpXG5, and Xeg5B are more open with extra space visible both at the top and the bottom of the catalytic site.
FIGURE 17.

Comparison of GH5_4 structural homologs. A, overall shape of the active site pocket. The active sites are shown as a semi-transparent blue surface representation for six structures as follows: three MLG active enzymes PbGH5A, Xeg5A (PDB code, 4W88), and BhGH5 (PDB code, 4V2X); and three XyG-specific enzymes, Xeg5B (PDB code, 4W8B), PpXG5 (PDB code, 2JEQ), and BoGH5 (PDB code, 3ZMR). Ligands, if present, are shown in cyan ball-and-stick representation. Highlighted in red are the two catalytic glutamate residues present in all of the compared structures. Highlighted in green are two regions that contribute the most to the differences in the active-site shape between the compared structures: the narrowing at the top of the −1 subsite in MLG-active enzymes (absent in XyG-specific enzymes), and the presence of a bulky aromatic residue making up the binding platform for the −2′-xylose in XyG (absent in MLG-active enzymes). For emphasis, a white circle contours the binding surface available at the −1 and −2 subsites of the XyG-specific enzymes and points to the lack thereof for the MLG-active enzymes. B, superposition of the MLG-active enzymes from A. For clarity, only the secondary structure of PbGH5A is shown. The loops making up the active site are shown in cyan (top four loops) and blue (bottom three loops). Shown in ball-and-stick are the residues responsible for the unique shape of the active site: top acidic residues narrowing the −1 subsite and the bottom His residue forming the −2′ subsite. PbGH5A residues are in green, Xeg5A in gray, and BhGH5 in wheat. For general orientation, the XXXGXXXG ligand in PbGH5A structure is shown in line representation. C, comparison between PbGH5A and XyG-specific enzymes. The representation is the same as in B. Distinct residues in the −1 and −2′-subsites are in the following color code: PbGH5A, green; BoGH5, orange; Xeg5B, violet; PpGH5, pink.
Seven loop regions combine to form this distinct shape of the PbGH5A active site as follows: four at the top (residues 27–48, 238–262, 280–295, and 324–339), and three at the bottom (113–121, 152–165, and 210–214) (Fig. 17). There is a great variability in the overall conformation of these loops compared with the other discussed GH5_4 enzymes; however, key features are conserved. The conserved regions encompass functionally equivalent residues participating in key protein-ligand interactions, which are generally found at the base of the loops and include catalytic residues Glu-162 and Glu-280, stacking residues Trp-48, Tyr-240, Trp-243, and Trp-324, and hydrogen bonding partners His-112 and Asn-161.
The constriction at the top of the catalytic center in PbGH5A is mainly formed by the loop residues 280–295, of which Asp-288 forms a direct interaction with the glucosyl moiety bound in the −1 subsite. This feature, which is conserved in the bMLG-specific enzymes (Fig. 17B), is absent in the xyloglucanases (Fig. 17C). At the bottom of the active center, the shallow pocket of PbGH5A is formed in part by a well conserved histidine residue, His-113. Here, the additional depth of the xyloglucan-specific enzymes is due to the presence of a bulky aromatic residue found in the −2 subsite and responsible for stacking of the −2′-xylosyl (Fig. 17, A and C). The distinct conservation in this region has been observed previously and reported as a potential signature motif (16). The active-site pocket widens beyond the −1 and +1 subsites, and although the branching residues of xyloglucan saccharides can be accommodated by the protein here, there are no obvious pockets that appear to be specifically tailored for this purpose. In the negative subsites, it is the glucan backbone that is intricately bound via stacking and hydrogen-bonding interactions, whereas the majority of the xylosyl moieties are solvent-exposed. This is distinct from the specific endo-xyloglucanases of GH5_4, in which aromatic residues have been identified to provide binding platforms for −2′ and −3′ xylosyl residues (Fig. 17, B and C) (16). Likewise in the positive subsites, PbGH5A appears to accommodate, rather than specifically harness, branched oligosaccharide in an open cleft. It is particularly striking that the glucan backbones of cellotetraose (GGGG) and its triple-xylosylated congener XXXG are bound with different trajectories through the positive subsite region (Fig. 15), which again implies significant flexibility in substrate binding.
In this context, it is notable that PbGH5A hydrolyzes xyloglucans at non-canonical backbone cleavage sites. Of the GH5 endo-xyloglucanases characterized to date, all cleave the dicot xyloglucan polysaccharide (exemplified by Tamarindus indica xyloglucan) at the unbranched backbone glucosyl unit (Fig. 1) to generate oligosaccharides based on a Glc4 backbone (16, 17, 64). This cleavage pattern is also typical for GH7, GH9, GH12, and GH16 members, with known exceptions of certain GH44 and GH74 members (16, 18, 66–71). Although the heptasaccharide XXXG was not hydrolyzed in the presence of high enzyme concentrations (0.1 mg/ml of PbGH5A), the limit-digestion products of tamarind xyloglucan hydrolysis contained oligosaccharides consistent with cleavage via binding “X” ([Xylα(1,6)]Glc-) units at subsite −1 (Fig. 15). Initial-rate kinetic analysis of the hydrolysis of the tetradecasaccharide XXXGXXXG revealed that cleavage of this substrate at the internal unbranched glucosyl residue predominated, although it was slow (kcat = 422 min−1, Km = 32 μm, Table 1). However, analysis of the limit-digest (data not shown) showed alternative cleavage modes resulting in the formation of XXG and XXXGX. Taken together, the data indicate that glucan chain branching is generally not well tolerated at the cleavage site due to constriction at subsites −1/+1 (Fig. 17A), although an overall lack of specificity for xyloglucan motifs allows variable substrate positioning in the active-site cleft.
Active Site of PbGH5A Comprises Seven Subsites in Total
Mapping the PbGH5A active site using chromogenic and native substrates, together with crystallographic analysis of enzyme·oligosaccharide complexes, suggests the presence of seven well defined subsites, four negative subsites and three positive subsites, in an open active-site cleft. Indeed, the highest activity was observed for a mixed-linkage heptasaccharide, G3GGG3GGG (closely followed by cellopentaose and cellohexaose), whereas unbranched tetrasaccharides represent the smallest competent naturally occurring substrates for PbGH5A (Table 1). The mode of hydrolysis of the minimal substrate cellotetraose defined the smallest subset of subsites utilized for activity on linear glucans, with the −2→+2 binding/hydrolysis mode significantly favored over −3→+1. When two positive subsites are occupied, the importance of the −3 subsite contribution is emphasized by the 130-fold increase in the kcat/Km value of −3→+2, the binding/hydrolysis mode for cellopentaose versus the −2→+2 mode (Table 1). An essentially identical increase in kcat/Km values for release of the aglycones from GGG-CNP versus GG-CNP and GGG-PNP versus GG-PNP was observed. Collectively, these data indicate that binding in the −3 subsite of PbGH5A contributes a ΔΔG of −12 kJ/mol to catalysis.
Comparison of the −3→+1 binding/hydrolysis mode for cellotetraose with the −3→+2 binding/hydrolysis mode for cellopentaose reveals that binding in the +2 subsite contributes −17 kJ/mol to catalysis. As such, interactions in the +2 subsite (stacking with Trp-243 and hydrogen bonding with Asp-171) make a significantly greater contribution to catalysis than the interactions in the −3 subsite (stacking with Trp-48).
Moving beyond the five core subsites spanning −3→+2, crystallographic complexes provide compelling evidence for an additional negative subsite that may explain the slight kinetic enhancement observed for the catalysis of GGGGGG→GGGG+GG over GGGGGG→GGG+GGG (Fig. 15 and Table 1). Specifically, the non-covalent PbGH5A(E280A)·XXXGXXXGstructure (Fig. 13A) and the affinity-labeled PbGH5A·XXXG-NHCOCH2- structure (Fig. 12A) reveal a glucose-phenylalanine stacking interaction constituting subsite −4 (Fig. 13A). In contrast, differential binding of XXXG and GGGG ligands in the positive subsites makes clear definition of an additional positive subsite, +3, difficult. The apparent length of the active-site cleft (Fig. 15) and well ordered electron density for the +3 glucose residue in all ligands (Fig. 13, B–D) imply that a binding surface may exist, although the breadth of the cleft at this point is not sufficient to restrict the backbones of all ligands to lie on the same trajectory. The absence of a +4 subsite is less ambiguous (Fig. 15). Unfortunately, a lack of a sufficient diversity of higher oligosaccharide substrates precludes detailed kinetic dissection of these more distal subsites; yet the observation that the heptasaccharide G3GGG3GGG is cleaved exclusively through a −4→+3 binding/hydrolysis mode (Table 1) strongly supports the definition of seven subsites (Fig. 14).
PbGH5A Exhibits Subtle Discrimination of β-Glucan Linkage Regiochemistry in the Active Site
Kinetic analysis of the hydrolysis of mixed-linkage oligoglucosides revealed that PbGH5A was essentially equally competent at hydrolyzing β(1,3)- and β(1,4)-linkages at the catalytic center but demonstrated differential preference for these linkages in the positive and negative subsites. The tetrasaccharides GGGG (cellotetraose), GG3GG, and GGG3G are all hydrolyzed with similar kcat/Km values for the −2→+2 binding/hydrolysis mode, yet G3GGG was not cleaved by PbGH5A (Table 1).
The kcat/Km value of GGGG is only 1.5-fold greater than that of GG3GG (−2→+2 binding/hydrolysis mode). This effective lack of linkage specificity can be rationalized in light of the oligosaccharide orientation in the positive subsites of the GGGG and XXXG heptasaccharide ligand complexes. The dramatic difference in the binding mode of these two ligands results in the close superposition of the C3-hydroxyl of cellotetraose and the C4-hydroxyl of XXXG due to a 180° rotation of the glucan backbone (Fig. 15C). Assuming that these structures reflect both the EP (enzyme/product) and corresponding ES (Michaelis) complexes, both C3- and C4-hydroxyl moieties can be suitably positioned as nucleofuges at the catalytic center. Furthermore, the apparent breadth of the active site toward the positive subsites readily accommodates the different binding orientations distal to the catalytic center required for longer β-glucan substrates (Fig. 15). Here, a substantial number of ordered water molecules are present (data not shown), which can potentially be displaced variably during the binding of alternative substrates.
Further reflecting an ambivalence to linkage regiochemistry at the catalytic center, both GGGG and GGG3G were cleaved via the −3→+1 binding/hydrolysis mode to produce cellotriose (GGG) and glucose (G). However, the loss of +2 subsite binding and gain of −3 subsite binding had a large negative effect on the kcat/Km value of GGGG (7.7-fold lower than that of the −2→+2 mode). In contrast, the kcat/Km value for GGG3G hydrolyzed via the −3→+1 mode is increased 5.2-fold versus the −2→+2 mode. The results highlight the delicate balance between the contributions of subsite binding and glycosidic bond specificity to catalysis. Although it is difficult to fully disentangle these competing effects given the available kinetic and structural data, it is clear that +2 subsite binding is particularly important for catalysis of all-β(1,4)-linked substrates; based on kcat/Km values (Table 1), cellopentaose is hydrolyzed nearly 900-fold better in the −3→+2 mode (the exclusive hydrolysis mode) than cellotetraose is hydrolyzed in the −3→+1 mode (which, again, is 7.7-fold poorer than in the −2→+2 mode).
The observation that GGG3G is efficiently hydrolyzed to GG and laminaribiose (G3G) indicates that β(1,3) glucosidic bonds are tolerated between the +1 and +2 subsites. Comparison with the kcat/Km value for the −2→+2 mode of hydrolysis of cellotetraose (GGGG), indicates that β(1,3) bonds are slightly disfavored in this position by a factor of 4 (Table 1), although this equates to less than 2 kJ/mol of lost transition-state stabilization. The recognition of β(1,3)-linkages between subsites +1 and +2 is likely to be responsible for the generation of G3GGG in the limit digest of barley bMLG (Fig. 5). The complexes of PbGH5A with GGGG and XXXG in the positive subsites suggests that the presence of a β(1,3)-linkage between the +1 and +2 subsites would necessarily cause the saccharide chain to adopt a different conformation, possibly disrupting the +2 hydrogen bonding interaction with Asp-171, but stacking with Trp-243 in subsite +2 would be anticipated to remain, due to the plasticity of this interaction (Fig. 15).
Turning to the negative subsites, binding in subsite −2 is essential for catalysis; no substrates, including G-PNP, were hydrolyzed to release glucose via −1→+n modes (Table 1). Notably, kinetic analyses revealed that β(1,3)-linkages are not tolerated between three of four negative subsites. In particular, G3GGG is not hydrolyzed through possible −1→+3, −2→+2, or −3→+1 modes (Table 1). The lack of −2→+2 and −3→+1 activity vis à vis the three other mixed-linkage tetrasaccharides provides clear evidence that β(1,3)-linkages are not accepted between subsites −2 and −1, as well as −3 and −2. Furthermore, GG3GG is not hydrolyzed via the −3→+1 mode, unlike GGGG and GGG3G, which also indicates intolerance of β(1,3)-linkages between subsites −2 and −1. Similarly, the heptasaccharide G3GGG3GGG is only cleaved at the internal β(1,3)-glycosidic bond. The two β(1,3)-linkages prevent productive binding and cleavage at the four possible β(1,3)-glycosidic bonds, whereas the non-reducing-end β(1,3)-linkage is tolerated in subsite −4. The inability of PbGH5A to accept β(1,3) bonds in the negative subsites is partially substantiated by the structures of complexes with xyloglucan oligosaccharides bound in these subsites. As discussed above, the xylosyl residues of these XXXG-based ligands are mostly solvent-exposed, such that the observed binding of the backbone (Figs. 3A and 13A) might be anticipated to closely approximate that of the unbranched cellotetraosyl unit (GGGG). In the −1 subsite, the enzyme forms intimate contacts with each ligand, with the C1-hydroxyl hydrogen bonding to the catalytic acid/base Glu-162 and the C3-hydroxyl directly interacting with conserved residues His-112 and His-113. As such, accommodating a β(1,3) link to the −2 subsite would break this interaction and require a major change in substrate orientation, likely altering the position of the scissile bond relative to the catalytic center. Beyond the −2 subsite, the active-site cleft widens significantly, such that there are no obvious steric factors that would hinder substrate binding in this region. Binding of β(1,3)-linked glucose across subsites −3 and −2 may be disfavored because the resulting kink in the glucan backbone could disrupt key stacking interactions with Trp-48 and Phe-47, which are the main contributors to the well ordered ligand binding seen in the −3 and −4 subsites, respectively. Regardless, the presence of a β(1,3)-linkage between subsites −4 and −3 would appear to be structurally accommodated, as underscored by the superior kinetics of G3GGG3GGG (Table 1).
Implications for Specificity Prediction in GH5 Subfamily 4
Subfamily 4 is one of the largest GH5 subfamilies, which resulted from the merger of the previous cellulase subfamilies A3 and A4 (11). To explore the possibility of delineating the known “cellulase,” mixed-linkage endo-glucanase, and endo-xyloglucanase activities within specific clades, we performed a new phylogenetic analysis of GH5_4 using all sequences in the public CAZy Database (supplemental Fig. S1). Bootstrap analysis revealed several well defined clades; however, endo-glucanase and endo-xyloglucanase activities were not absolutely segregated.
A lack of systematic enzymological data further hampers efforts to delineate specificities by phylogeny. Although a generally low coverage of biochemical characterization is a ubiquitous problem for all GH families, a further significant issue arises from the use of CMC as a proxy to measure cellulase activity. As the present reanalysis of PbGH5A activity shows (Table 1), the original use of CMC as a substrate to characterize this enzyme was misleading (24); in fact, the amorphous, phosphoric acid-swollen cellulose is an even poorer substrate for PbGH5A. Analogously, it is therefore unclear how many of the 56 GH5_4 members currently assigned as cellulases or endo-β(1,4)-glucanases, often solely on the basis of activity toward this unnatural, anionic, polysaccharide derivative, have been incorrectly annotated. When assaying new GH5_4 members, a wider panel of soluble polysaccharide substrates must be tested, and more detailed re-evaluation of currently characterized members is certainly warranted. More broadly, it could be argued that CMC should be abandoned as a substrate altogether.
Regardless, a growing body of data suggests that GH5_4 members are more likely to be active on the amorphous cross-linking glycans of the composite plant cell wall, rather than on the para-crystalline cellulose component. Testing this hypothesis will require further characterization of this large and historically significant subfamily via structure-function analyses that are at the same time systematic and deep. As our work here shows, such endeavors are likely to be fruitful in uncovering unanticipated specificities, thereby increasing the library of biocatalysts for potential applications.
Author Contributions
N. M. prepared the XXXG, XXXGXXXG, G3GGG, and G3GGG3GGG substrates and measured and analyzed kinetic data for the breakdown of polymers, chromogenic substrates, and model oligosaccharides. M. M. determined the crystal structures of the PbGH5A complexes with ligands. T. H. F. synthesized the inhibitor molecules and performed and analyzed all inhibition kinetics and labeling experiments. T. H. F. also performed some preliminary hydrolysis kinetics analysis. P. S. determined the crystal structure of the apo-PbGH5A. N. L. performed protein sequence phylogenetic analysis. V. Y. collected and analyzed all protein intact mass spectra and worked with N. M. on H218O isotopic labeling experiments. X. X. crystallized PbGH5A variants with ligand molecules. E. E. crystallized apo-PbGH5A. H. C. expressed and purified PbGH5A for crystallization trials. B. H., A. S., and H. B. provided project guidance and analyzed data. N. M., M. M., A. S., and H. B. co-wrote the manuscript, with input from the other co-authors.
Supplementary Material
Acknowledgments
The Waters Corp. is gratefully acknowledged for the provision and skilled maintenance of the LC-MS system used in this study. We thank Aiping Dong for data collection of the PbGH5A-cellotetraose complex. We thank Prof. Stephen Withers and the Withers laboratory at University of British Columbia for the generous gift of 4-nitrophenyl β-cellotrioside for activity measurements.
This work was supported in part by the Natural Sciences and Engineering Research Council of Canada (Discovery Grant), the Canada Foundation for Innovation, and the British Columbia Knowledge Development Fund (to H. B.) and by National Institutes of Health Grants U54-GM074942 and U54-GM094585 (to A. S. through the Midwest Center for Structural Genomics). H. B. and A. S. also acknowledge joint funding to their laboratories from the Natural Sciences and Engineering Research Council via the Industrial Biocatalysis Network. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Fig. S1.
The atomic coordinates and structure factors (codes 3VDH, 5D9M, 5D9N, 5D9O, and 5D9P) have been deposited in the Protein Data Bank (http://wwpdb.org/).
N. McGregor, V. Yin, C.-C. Tung, F. Van Petegem, and H. Brumer, manuscript in preparation.
- GH
- glycoside hydrolase
- PASC
- phosphoric acid-swollen cellulose
- PDB
- Protein Data Bank
- CMC
- carboxymethylcellulose
- bMLG
- barley mixed-linkage glucan
- kGM
- konjac glucomannan
- tXyG
- tamarind xyloglucan
- tXyGO
- tamarind xyloglucan oligosaccharide
- bX
- beechwood xylan
- HEC
- hydroxyethylcellulose
- HPAEC-PAD
- high performance anion-exchange chromatography with pulsed amperometric detection
- PNP
- p-nitrophenyl
- GG-PNP
- 4-nitrophenyl β-cellobioside
- GGG-PNP
- 4-nitrophenyl β-cellotrioside
- GG-CNP
- 2-chloro-4-nitrophenyl β-cellobioside
- GGG-CNP
- 2-chloro-4-nitrophenyl β-cellotrioside
- GGGG
- cellotetraose
- GGGGG
- cellopentaose
- GGGGGG
- cellohexaose
- r.m.s.d.
- root mean square deviation
- G
- Glcp
- X
- (Xylp(α1–6))Glcp
- L
- (Galp(β1–2)Xylp(α1–6))Glcp
- CNP
- 2-chloro-4-nitrophenyl.
References
- 1.Albersheim P., Darvill A., Roberts K., Sederoff R., and Staehelin A. (2010) Plant Cell Walls, pp. 1–363, Garland Science, New York [Google Scholar]
- 2.Fry S. C. (1989) The structure and functions of xyloglucan. J. Exp. Bot. 40, 1–11 [Google Scholar]
- 3.Hayashi T. (1989) Xyloglucans in the primary cell wall. Annu. Rev. Plant Physiol. Plant Mol. Biol. 40, 139–168 [Google Scholar]
- 4.Fry S. C., Nesselrode B. H., Miller J. G., and Mewburn B. R. (2008) Mixed-linkage (1→3,1→4)-β-d-glucan is a major hemicellulose of Equisetum (horsetail) cell walls. New Phytologist 179, 104–115 [DOI] [PubMed] [Google Scholar]
- 5.Xue X., and Fry S. C. (2012) Evolution of mixed-linkage (1 → 3, 1 → 4)-β-d-glucan (MLG) and xyloglucan in Equisetum (horsetails) and other monilophytes. Ann. Bot. 109, 873–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burton R. A., and Fincher G. B. (2009) (1,3;1,4)-β-d-Glucans in cell walls of the Poaceae, lower plants, and fungi: a tale of two linkages. Mol. Plant 2, 873–882 [DOI] [PubMed] [Google Scholar]
- 7.Tuomivaara S. T., Yaoi K., O'Neill M. A., and York W. S. (2015) Generation and structural validation of a library of diverse xyloglucan-derived oligosaccharides, including an update on xyloglucan nomenclature. Carbohydr. Res. 402, 56–66 [DOI] [PubMed] [Google Scholar]
- 8.Davies G. J., and Sinnott M. L. (2008) Sorting the diverse: the sequence-based classifications of carbohydrate-active enzymes. Biochem. J. 10.1042/BJ20080382 [DOI] [Google Scholar]
- 9.Davies G. J., Gloster T. M., and Henrissat B. (2005) Recent structural insights into the expanding world of carbohydrate-active enzymes. Curr. Opin. Struct. Biol. 15, 637–645 [DOI] [PubMed] [Google Scholar]
- 10.Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., and Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aspeborg H., Coutinho P. M., Wang Y., Brumer H. 3rd, and Henrissat B. (2012) Evolution, substrate specificity and subfamily classification of glycoside hydrolase family 5 (GH5). BMC Evol. Biol. 12, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garron M. L., and Cygler M. (2010) Structural and mechanistic classification of uronic acid-containing polysaccharide lyases. Glycobiology 20, 1547–1573 [DOI] [PubMed] [Google Scholar]
- 13.Lombard V., Bernard T., Rancurel C., Brumer H., Coutinho P. M., and Henrissat B. (2010) A hierarchical classification of polysaccharide lyases for glycogenomics. Biochem. J. 432, 437–444 [DOI] [PubMed] [Google Scholar]
- 14.Stam M. R., Danchin E. G., Rancurel C., Coutinho P. M., and Henrissat B. (2006) Dividing the large glycoside hydrolase family 13 into subfamilies: towards improved functional annotations of α-amylase-related proteins. Protein Eng. Des. Sel. 19, 555–562 [DOI] [PubMed] [Google Scholar]
- 15.Barras F., Bortoli-German I., Bauzan M., Rouvier J., Gey C., Heyraud A., and Henrissat B. (1992) Stereochemistry of the hydrolysis reaction catalyzed by endoglucanase Z from Erwinia chrysanthemi. FEBS Lett. 300, 145–148 [DOI] [PubMed] [Google Scholar]
- 16.Gloster T. M., Ibatullin F. M., Macauley K., Eklöf J. M., Roberts S., Turkenburg J. P., Bjørnvad M. E., Jørgensen P. L., Danielsen S., Johansen K. S., Borchert T. V., Wilson K. S., Brumer H., and Davies G. J. (2007) Characterization and three-dimensional structures of two distinct bacterial xyloglucanases from families GH5 and GH12. J. Biol. Chem. 282, 19177–19189 [DOI] [PubMed] [Google Scholar]
- 17.Larsbrink J., Rogers T. E., Hemsworth G. R., McKee L. S., Tauzin A. S., Spadiut O., Klinter S., Pudlo N. A., Urs K., Koropatkin N. M., Creagh A. L., Haynes C. A., Kelly A. G., Cederholm S. N., Davies G. J., et al. (2014) A discrete genetic locus confers xyloglucan metabolism in select human gut Bacteroidetes. Nature 506, 498–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eklöf J. M., Ruda M. C., and Brumer H. (2012) Distinguishing xyloglucanase activity in endo-β(1→4)glucanases. Methods Enzymol. 510, 97–120 [DOI] [PubMed] [Google Scholar]
- 19.Faure E., Belaich A., Bagnara C., Gaudin C., and Belaich J.-P. (1989) Sequence analysis of the Clostridium cellulolyticum endoglucanase-A-encoding gene, celCCA. Gene 84, 39–46 [DOI] [PubMed] [Google Scholar]
- 20.Hamamoto T., Foong F., Shoseyov O., and Doi R. H. (1992) Analysis of functional domains of endoglucanases from Clostridium cellulovorans by gene cloning, nucleotide sequencing and chimeric protein construction. Mol. Gen. Genet. 231, 472–479 [DOI] [PubMed] [Google Scholar]
- 21.Avgustin G., Wallace R. J., and Flint H. J. (1997) Phenotypic diversity among ruminal isolates of Prevotella ruminicola: proposal of Prevotella brevis sp. nov., Prevotella bryantii sp. nov., and Prevotella albensis sp. nov., and redefinition of Prevotella ruminicola. Int. J. Syst. Bacteriol. 47, 284–288 [DOI] [PubMed] [Google Scholar]
- 22.Flint H. J., Bayer E. A., Rincon M. T., Lamed R., and White B. A. (2008) Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat. Rev. Microbiol. 6, 121–131 [DOI] [PubMed] [Google Scholar]
- 23.Purushe J., Fouts D. E., Morrison M., White B. A., Mackie R. I., North American Consortium for Rumen Bacteria, Coutinho P. M., Henrissat B., and Nelson K. E. (2010) Comparative genome analysis of Prevotella ruminicola and Prevotella bryantii: insights into their environmental niche. Microb. Ecol. 60, 721–729 [DOI] [PubMed] [Google Scholar]
- 24.Gardner R. G., Wells J. E., Fields M. W., Wilson D. B., and Russell J. B. (1997) A Prevotella ruminicola B14 operon encoding extracellular polysaccharide hydrolases. Curr. Microbiol 35, 274–277 [DOI] [PubMed] [Google Scholar]
- 25.Matsushita O., Russell J. B., and Wilson D. B. (1990) Cloning and sequencing of a Bacteroides ruminicola B(1)4 endoglucanase gene. J. Bacteriol 172, 3620–3630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cantarel B. L., Coutinho P. M., Rancurel C., Bernard T., Lombard V., and Henrissat B. (2009) The carbohydrate-active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martens E. C., Kelly A. G., Tauzin A. S., and Brumer H. (2014) The devil lies in the details: how variations in polysaccharide fine-structure impact the physiology and evolution of gut microbes. J. Mol. Biol. 426, 3851–3865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sundqvist G., Stenvall M., Berglund H., Ottosson J., and Brumer H. (2007) A general, robust method for the quality control of intact proteins using LC-ESI-MS. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 852, 188–194 [DOI] [PubMed] [Google Scholar]
- 29.Walseth C. S. (1952) The influence of the fine structure of cellulose on the action of cellulases. Tappi 35, 233–238 [Google Scholar]
- 30.Zhang Y. H., Cui J., Lynd L. R., and Kuang L. R. (2006) A transition from cellulose swelling to cellulose dissolution by o-phosphoric acid: evidence from enzymatic hydrolysis and supramolecular structure. Biomacromolecules 7, 644–648 [DOI] [PubMed] [Google Scholar]
- 31.Ibatullin F. M., Baumann M. J., Greffe L., and Brumer H. (2008) Kinetic analyses of retaining endo-(xylo)glucanases from plant and microbial sources using new chromogenic xylogluco-oligosaccharide aryl glycosides. Biochemistry 47, 7762–7769 [DOI] [PubMed] [Google Scholar]
- 32.Fenger T. H., and Brumer H. (2015) Synthesis and analysis of specific covalent inhibitors of endo-xyloglucanases. ChemBioChem 16, 575–583 [DOI] [PubMed] [Google Scholar]
- 33.Larsbrink J., Thompson A. J., Lundqvist M., Gardner J. G., Davies G. J., and Brumer H. (2014) A complex gene locus enables xyloglucan utilization in the model saprophyte Cellvibrio japonicus. Mol. Microbiol. 94, 418–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eschenfeldt W. H., Lucy S., Millard C. S., Joachimiak A., and Mark I. D. (2009) A family of LIC vectors for high-throughput cloning and purification of proteins. Methods Mol. Biol. 498, 105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 36.Waugh D. S. (2011) An overview of enzymatic reagents for the removal of affinity tags. Protein Expr. Purif. 80, 283–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doner L. W., and Irwin P. L. (1992) Assay of reducing end-groups in oligosaccharide homologues with 2,2′-bicinchoninate. Anal. Biochem. 202, 50–53 [DOI] [PubMed] [Google Scholar]
- 38.Miller G. L. (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Chem. 31, 426–428 [Google Scholar]
- 39.Michaelis L., Menten M. L., Johnson K. A., and Goody R. S. (2011) The original Michaelis constant: translation of the 1913 Michaelis-Menten paper. Biochemistry 50, 8264–8269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michaelis L., and Menten M. L. (1913) Die kinetik der invertinwirkung. Biochem. Z. 49, 352 [Google Scholar]
- 41.Schagerlöf H., Nilsson C., Gorton L., Tjerneld F., Stålbrand H., and Cohen A. (2009) Use of 18O water and ESI-MS detection in subsite characterisation and investigation of the hydrolytic action of an endoglucanase. Anal. Bioanal. Chem. 394, 1977–1984 [DOI] [PubMed] [Google Scholar]
- 42.Kitz R., and Wilson I. B. (1962) Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J. Biol. Chem. 237, 3245–3249 [PubMed] [Google Scholar]
- 43.Minor W., Cymborowski M., Otwinowski Z., and Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 44.Kelley L. A., and Sternberg M. J. (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protocols 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 45.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 47.Katoh K., and Standley D. M. (2014) MAFFT: iterative refinement and additional methods. Methods Mol. Biol. 1079, 131–146 [DOI] [PubMed] [Google Scholar]
- 48.Price M. N., Dehal P. S., and Arkin A. P. (2010) FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huson D. H., and Scornavacca C. (2012) Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 61, 1061–1067 [DOI] [PubMed] [Google Scholar]
- 50.York W. S., van Halbeek H., Darvill A. G., and Albersheim P. (1990) Structural analysis of xyloglucan oligosaccharides by 1H-NMR spectroscopy and fast-atom-bombardment mass spectrometry. Carbohydr. Res. 200, 9–31 [DOI] [PubMed] [Google Scholar]
- 51.Davies G. J., Wilson K. S., and Henrissat B. (1997) Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 321, 557–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kempton J. B., and Withers S. G. (1992) Mechanism of Agrobacterium β-glucosidase: kinetic studies. Biochemistry 31, 9961–9969 [DOI] [PubMed] [Google Scholar]
- 53.Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tseng C.-W., Ko T.-P., Guo R.-T., Huang J.-W., Wang H.-C., Huang C.-H., Cheng Y.-S., Wang A. H. J., and Liu J.-R. (2011) Substrate binding of a GH5 endoglucanase from the ruminal fungus Piromyces rhizinflata. Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun. 67, 1189–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miao S., McCarter J. D., Grace M. E., Grabowski G. A., Aebersold R., and Withers S. G. (1994) Identification of Glu340 as the active-site nucleophile in human glucocerebrosidase by use of electrospray tandem mass spectrometry. J. Biol. Chem. 269, 10975–10978 [PubMed] [Google Scholar]
- 56.Withers S. G., Rupitz K., Trimbur D., and Warren R. A. (1992) Mechanistic consequences of mutation of the active site nucleophile Glu 358 in Agrobacterium β-glucosidase. Biochemistry 31, 9979–9985 [DOI] [PubMed] [Google Scholar]
- 57.Yuan J., Martinez-Bilbao M., and Huber R. E. (1994) Substitutions for Glu-537 of β-galactosidase from Escherichia coli cause large decreases in catalytic activity. Biochem. J. 299, 527–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tull D., Burgoyne D. L., Chow D. T., Withers S. G., and Aebersold R. (1996) A mass spectrometry-based approach for probing enzyme active sites: identification of Glu 127 in Cellulomonas fimi exoglycanase as the residue modified by N-bromoacetyl cellobiosylamine. Anal. Biochem. 234, 119–125 [DOI] [PubMed] [Google Scholar]
- 59.Black T. S., Kiss L., Tull D., and Withers S. G. (1993) N-Bromoacetyl-glycopyranosylamines as affinity labels for a β-glucosidase and a cellulase. Carbohydr. Res. 250, 195–202 [DOI] [PubMed] [Google Scholar]
- 60.Berger E., Jones W. A., Jones D. T., and Woods D. R. (1989) Cloning and sequencing of an endoglucanase (end1) gene from Butyrivibrio fibrisolvens H17c. Mol. Gen. Genet. 219, 193–198 [DOI] [PubMed] [Google Scholar]
- 61.Foong F., Hamamoto T., Shoseyov O., and Doi R. H. (1991) Nucleotide sequence and characteristics of endoglucanase gene engB from Clostridium cellulovorans. J. Gen. Microbiol. 137, 1729–1736 [DOI] [PubMed] [Google Scholar]
- 62.Palackal N., Lyon C. S., Zaidi S., Luginbühl P., Dupree P., Goubet F., Macomber J. L., Short J. M., Hazlewood G. P., Robertson D. E., and Steer B. A. (2007) A multifunctional hybrid glycosyl hydrolase discovered in an uncultured microbial consortium from ruminant gut. Appl. Microbiol. Biotechnol. 74, 113–124 [DOI] [PubMed] [Google Scholar]
- 63.Venditto I., Najmudin S., Luís A. S., Ferreira L. M., Sakka K., Knox J. P., Gilbert H. J., and Fontes C. M. (2015) Family 46 carbohydrate-binding modules contribute to the enzymatic hydrolysis of xyloglucan and β-1,3–1,4-glucans through distinct mechanisms. J. Biol. Chem. 290, 10572–10586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dos Santos C. R., Cordeiro R. L., Wong D. W., and Murakami M. T. (2015) Structural basis for xyloglucan specificity and α-d-Xylp(1 → 6)-d-Glcp recognition at the −1 subsite within the GH5 family. Biochemistry 54, 1930–1942 [DOI] [PubMed] [Google Scholar]
- 65.Dundas J., Ouyang Z., Tseng J., Binkowski A., Turpaz Y., and Liang J. (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 34, W116–W118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eklöf J. M., Shojania S., Okon M., McIntosh L. P., and Brumer H. (2013) Structure-function analysis of a broad specificity Populus trichocarpa endo-β-glucanase reveals an evolutionary link between bacterial licheninases and plant XTH gene products. J. Biol. Chem. 288, 15786–15799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ariza A., Eklöf J. M., Spadiut O., Offen W. A., Roberts S. M., Besenmatter W., Friis E. P., Skjøt M., Wilson K. S., Brumer H., and Davies G. (2011) Structure and activity of Paenibacillus polymyxa xyloglucanase from glycoside hydrolase family 44. J. Biol. Chem. 286, 33890–33900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gilbert H. J., Stålbrand H., and Brumer H. (2008) How the walls come crumbling down: recent structural biochemistry of plant polysaccharide degradation. Curr. Opin. Plant Biol. 11, 338–348 [DOI] [PubMed] [Google Scholar]
- 69.Ravachol J., Borne R., Tardif C., de Philip P., and Fierobe H.-P. (2014) Characterization of all family-9 glycoside hydrolases synthesized by the cellulosome-producing bacterium Clostridium cellulolyticum. J. Biol. Chem. 289, 7335–7348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou Q., Rutland M. W., Teeri T. T., and Brumer H. (2007) Xyloglucan in cellulose modification. Cellulose 14, 625–641 [Google Scholar]
- 71.Desmet T., Cantaert T., Gualfetti P., Nerinckx W., Gross L., Mitchinson C., and Piens K. (2007) An investigation of the substrate specificity of the xyloglucanase Cel74A from Hypocrea jecorina. FEBS J. 274, 356–363 [DOI] [PubMed] [Google Scholar]
- 72.Sato S., Kato T., Kakegawa K., Ishii T., Liu Y.-G., Awano T., Takabe K., Nishiyama Y., Kuga S., Sato S., Nakamura Y., Tabata S., and Shibata D. (2001) Role of the putative membrane-bound endo-1,4-β-glucanase KORRIGAN in cell elongation and cellulose synthesis in Arabidopsis thaliana. Plant Cell Physiol. 42, 251–263 [DOI] [PubMed] [Google Scholar]
- 73.Ducros V., Czjzek M., Belaich A., Gaudin C., Fierobe H. P., Belaich J. P., Davies G. J., and Haser R. (1995) Crystal structure of the catalytic domain of a bacterial cellulase belonging to family 5. Structure 3, 939–949 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.